
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly crystalline and efficient red-emissive π-conjugated polymer film: tuning of macrostructure for light-emitting properties†
Shotaro
Hayashi

Research Center for Molecular Design, School of Environmental Science and Engineering, Kochi University of Technology, Kami, Kochi 782-8502, Japan. E-mail: hayashi.shotaro@kochi-tech.ac.jp
First published on 26th June 2020
Abstract
Solid-state red-emissive π-conjugated molecules and polymers have attracted significant attention. In this work, the optical properties of alternating π-conjugated copolymers based on phenylene–bithiophene were investigated. It was found that the alternating 3,3′-dihexylbithiophene and tetrafluorophenylene structure was the primary one in the highly crystalline polymer film, while the other bithiophene–phenylene alternating copolymers did produce crystalline films. The number of fluorine atoms on the phenylene unit critically affected the film crystallinity; this could be attributed to the rigidity of the π-structure based on intramolecular H–F and S–F interactions. The crystalline polymer films demonstrated efficient red-emission. A red-shift of the emission band (+∼150 nm) and a twofold increase of the quantum yield for films compared with the 1,2-dichloroethane solution state were observed. This is likely due to crystallization-induced enhanced emission by the π-conjugated polymers. Additionally, significant tuning of the light-emission (from yellow, orange to red-colored emission) of the films was realized by controlling the crystallinity. The casting solvents and annealing treatment for film formation strongly affected the macrostructure (crystalline domain). X-ray diffraction patterns and the optical properties of the π-conjugated polymer films were used to investigate the dependence of the macrostructure on the fabrication parameters.
Introduction
Supramolecular self-assembly (macrostructural formation) of π-conjugated polymers influences the optoelectronic properties of polymer films and powders.1 The performance of optoelectronic polymer thin-film devices depends on the crystalline structure of the active polymer layer.2 Therefore, the development of crystalline π-conjugated polymers is needed for continued progress in the field of flexible organic optoelectronics.3 The macrostructure of such polymers can be tuned via self-assembly, leading to a resultant tuning of the polymer's light absorption, light emission, and carrier mobility.3 For example, films of regioregular poly(3-hexylthiophene)s, rr-P3HTs, (Scheme 1a) are highly crystalline and their light-absorption and carrier-mobility can be tuned.4 Interestingly, rr-P3HT can form single-crystals (monoclinic form II).4c Poly(9,9-dioctylfluorene), PF8, (Scheme 1b) is also an interesting polymer structure; annealing and vapor treatments of these polymer films can produce the crystalline α- and β-phase from the amorphous state.5 Changes in the planarity of the π-conjugated structure between fluorene units in PF8 can produce deep blue- and sky blue-emitting films. The tuning of such properties is important for the improvement of device performance.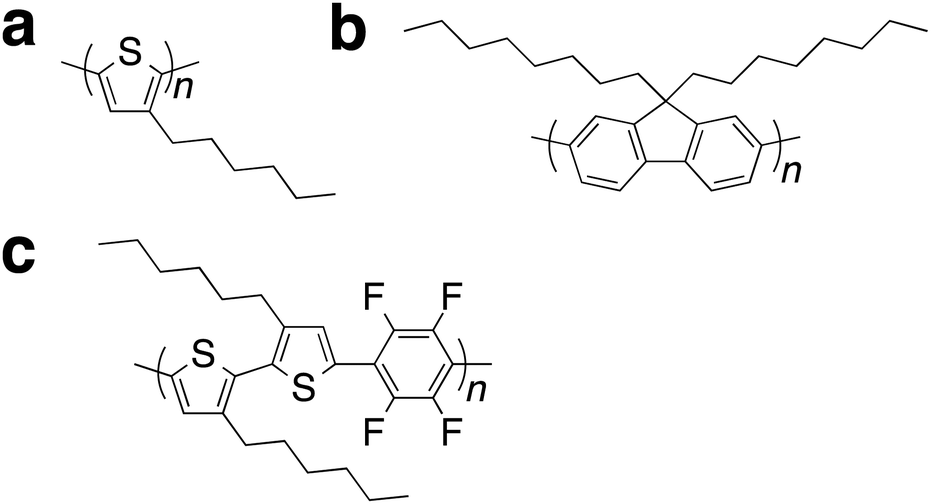 | ||
| Scheme 1 Typical examples of π-conjugated polymers for crystalline films. (a) P3HT. (b) PF8. (c) PF4. | ||
Efficient solid-emitters with low-energy emission have attracted significant attention because of their potential applications in organic devices.6 Red-emission requires a narrow HOMO–LUMO band gap based on an extended planar π-system and/or a donor–acceptor system. However, the aggregation-caused quenching because of intermolecular stacking and/or strong dipole–dipole interactions is an issue for solid emitters. To achieve efficient red-emitting solid materials, the design of π-conjugated molecules with bulky groups that have large steric effects has generally been adopted to suppress undesired intermolecular interactions.6 Similarly, solid red-emitting materials based on π-conjugated polymers are generally designed by introducing bulky groups.6 These approaches have disadvantages, such as a tedious synthetic process and complex structures. In addition, because intermolecular interactions in π-conjugated polymers induce crystallinity in the film state, the addition of large groups to the main polymer chain inhibit the formation of crystalline films. The author recently reported a π-conjugated polymer that shows high crystallinity and efficient red-emitting properties in the solid state.7 Supramolecular gel formation of a simple alternating copolymer composed of tetrafluorophenylene and 3,3′-dihexylbithiophene, PF4, (Scheme 1c) resulted in a lamellar crystalline macrostructure based on π–π stacking and interdigitated packing of the alkyl side chains. The properties of the gels could be controlled using organic solvents. To understand the chemical structure relationship between the crystallinity and efficient red-emission, the author report here the studies on films of PF4, its model molecule (i.e., repeating units of the alternating copolymers), and series of similar π-conjugated phenylene–bithiophene alternating copolymers. Moreover, typical crystalline polymers (for example see Scheme 1a and b) show film processing dependent properties,4,5 but these do not show film dependent fluorescence properties in the low energy visible range (>500 nm). Thus, the author also reports on the film processing dependent light-emitting properties through macrostructural tuning of PF4 film.
Results and discussion
π-Conjugated polymers were synthesized by direct (C–H) arylation polycondensation8 of 3,3′-dihexyl-2,2′-bithiophene, BT6, with various 1,4-dibromophenylenes, PXs, using PITS-Cl (palladium-immobilized on thiol-modified silica gel) catalyst,8 to form the series of poly(bithiophene-alt-phenylene)s, PXs (Scheme 2 and Fig. 1a). PM, PF0, and PF2 had good solubility in common organic solvents (such as dichloromethane, chloroform, tetrahydrofuran, and toluene), however, PF4 had poor solubility in these solvents. Characterization of PF4 was performed under heating at ∼40–50 °C. 1H NMR analyses of the polymers provided information on the intramolecular interactions between the thiophene and phenylene units (Fig. S1, ESI†). The thiophene signal of PM was at a higher magnetic field than PF0.9 The 1H NMR signal of the thiophene unit was shifted to a lower magnetic field on the introduction of fluorine atoms: PF4 > PF2 > PF0. This is because of intramolecular interaction between hydrogen atom of thiophene unit and neighbor electron-withdrawing fluorine atom of phenylene unit.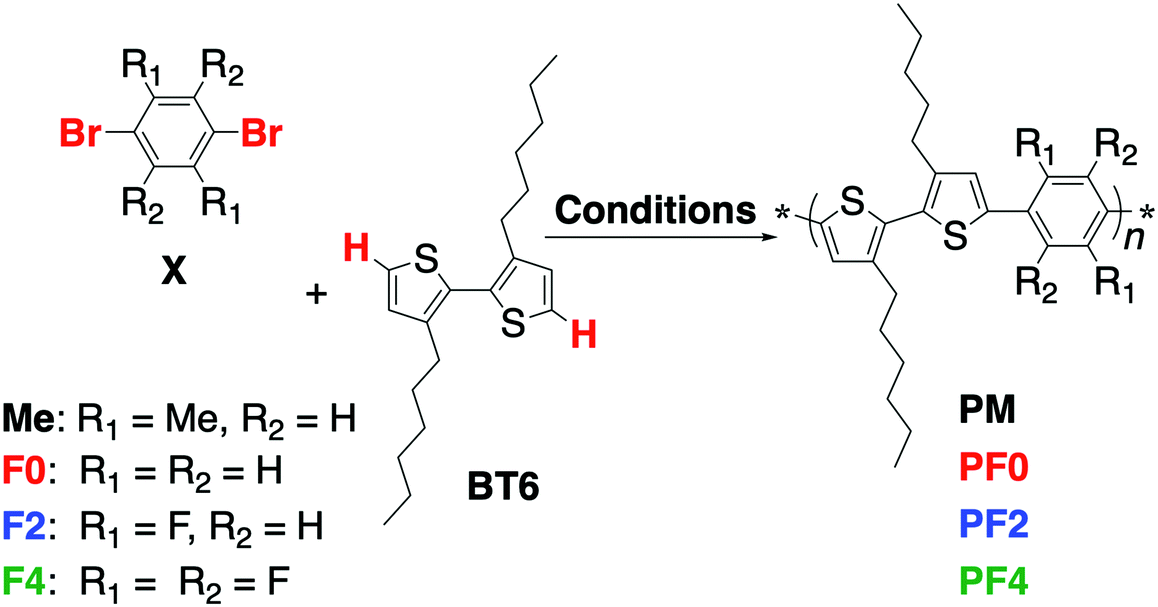 | ||
| Scheme 2 Direct arylation polymerization synthesis of bithiophene–phenylene alternating copolymers. Conditions: monomers (0.30 mmol), 1-adamantane carboxylic acid (30 mol%), K2CO3 (300 mol%), PITS-Cl (1.0 mol%) in DMAc (0.30 M) for 120 min at 120 °C under argon. | ||
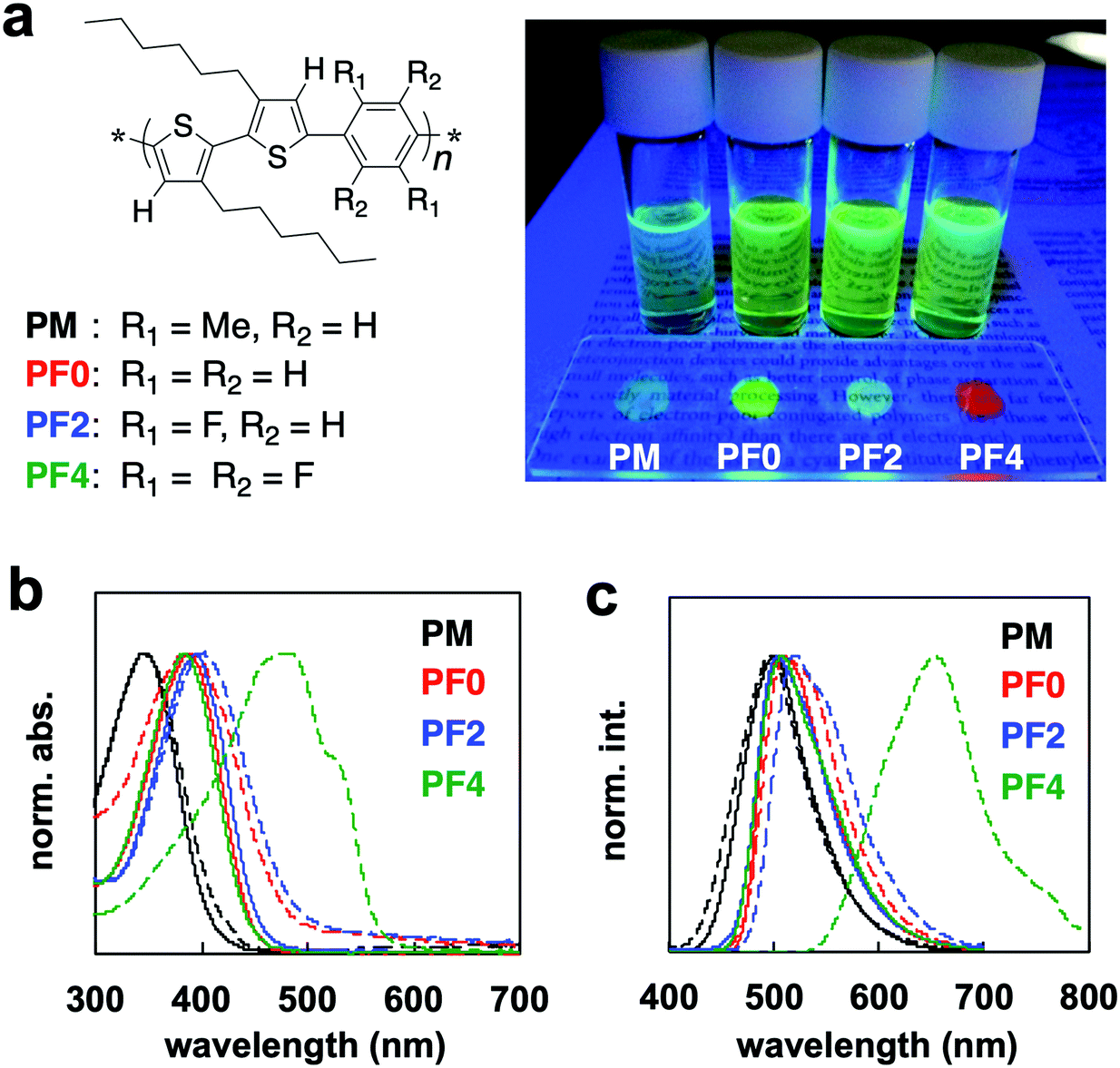 | ||
| Fig. 1 (a) Structure of PXs. Photographic fluorescence images of PBT6PFXs in DCE and film under UV irradiation. (b) UV-vis absorption spectra of PXs in DCE (solid line) and film (dotted line). (c) Fluorescence spectra of PXs in DCE (solid line) and film (dotted line). | ||
The UV-Vis absorption spectra of PBT6PFXs in 1,2-dichloroethane (DCE) and films are shown in Fig. 1b. The film was prepared by drop-casting on a glass substrate from a DCE solution and then dried at room temperature for 2 days. PM in DCE showed a higher energy band because steric hindrance would cause a twist along the polymer backbone by the methyl group at the phenylene unit (λmax = 340 nm). PF0, PF2, and PF4 showed absorption bands centered at ∼380, 391, and 382 nm, respectively. The spectra of PM, PF0, and PF2 films showed similar feature compared with their corresponding DCE solutions. Only PF4 showed a red-shift on moving from DCE solution to film. In addition to the red-shift of λmax (+100 nm), a characteristic shoulder band was observed between 500–600 nm. The red-shift is due to the planarization of the polymer backbone, suggesting that it forms crystalline domains. rr-P3HT, which forms crystalline films, also showed a significant red-shift (∼70–100 nm) with a characteristic shoulder band at lower energy.
The absorption peak (around 560 nm) and shoulder band (610 nm) of rr-P3HT film have been reported to be indicative of strong intermolecular π–π stacking and the planarization of the polymer backbone, respectively.4g The specific absorption peak (around 480 nm) and shoulder (500–610 nm) of PF4 film is probably because of planarization and intermolecular π–π stacking. X-ray diffraction (XRD) measurements were carried out on a PF4 film prepared by drop-casting on a silicon wafer from DCE solution, followed by thermal annealing (200 °C → 30 °C). The XRD results showed peaks derived from both interdigitated packing of the hexyl side chain (0.69 and 1.38 nm) and π–π stacking of the main chain (0.44 nm) (Fig. S2, ESI†). These peaks were not observed for the other polymer films. These results are supported by differential scanning calorimetry (DSC) measurements. The melting peaks for polymers PBT6PFMe, PF0, and PF2 were not observed, while a peak of the melting point could be observed for PF4 (Fig. S3, ESI†). Therefore, PBT6PFMe, PF0, and PF2 were determined to form amorphous films.
Fluorescence of PBT6PFXs solutions and films under UV irradiation are shown in Fig. 1a. The red-colored fluorescence was only observed for PF4 films. It should be noted that tetrafluorophenylene is not acting as an electron-accepting unit in PF4. The similar spectra of PF0, PF2, and PF4 in a DCE solution support that charge transfer interactions between the phenylene and bithiophene did not occur. The optical properties of PF4 can be attributed to the planarization and macrostructure formation for extension of the effective-conjugated system. Compared with similar bithiophene–phenylene alternating copolymers, the chemical structure of tetrafluoroarene with thiophene is important for macrostructure-induced fluorescence.
Crystalline polymers such as rr-P3HT generally show fluorescence in solution, but they display an aggregation-caused quenching (ACQ) behavior in film. Thus, crystalline polymers are not expected to display fluorescence in the aggregated state. However, the quantum yield of PF4 increased on moving from a DCE solution to film. The absolute quantum yields (Φ) of the solution and the crystals were measured using an integrating sphere equipped with a multichannel spectrometer. The Φ values increased from 13.1 (DCE solution) to 23.3% (film). The PF4 film exhibited a low-energy fluorescence band centered at 653 nm, while other PXs (PM, PF0, and PF2) films showed similar energy bands at ∼499, 505, and 511 nm, respectively (Fig. 1c). The electrochemical bandgap of the PF4 film via cyclic voltammetry analysis was not narrower than the other PXs (Fig. S5, ESI†). Efficient interpolymer energy transfer could be realized by packing of the thiophene–tetrafluorophenylene–thiophene units. These results indicate the formation of a unique macrostructure for PF4. These optical behaviors were also observed in 1,4-bis[2-(4-methylthienyl)]-2,3,5,6-tetrafluorobenzene, BT1PF4.10
BT1PF4 is a model compound for the PF4 repeating unit (Fig. 2a). The compound the author developed formed a large single crystal, which showed flexibility and fluorescence based on crystallization-induced enhanced emission (CIEE)11 and mechanochromic behavior.12 The macrostructure and properties of the molecule are related to that of PF4. To understand the specific macrostructure of PF4 in the film state, the properties of BT1PF4 in solution, as a crystal, and as ground powder were compared.
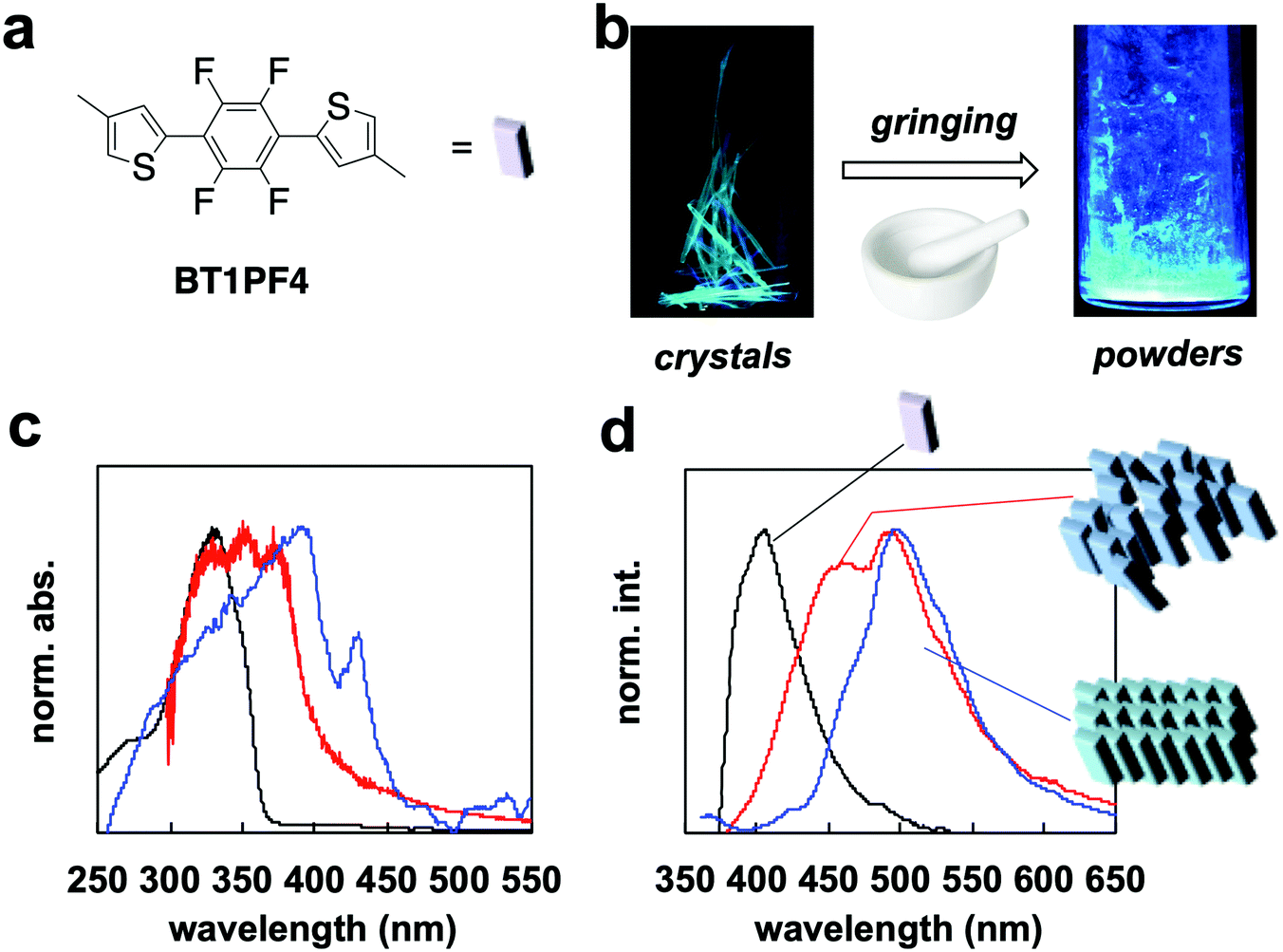 | ||
| Fig. 2 (a) Chemical structure of BT1PF4. (b) Photographic images of crystals and powders under UV irradiation. (c) UV-vis absorption spectra in dichloromethane (black line), powder (red line), and crystal (blue line). (d) Fluorescence spectra in dichloromethane (black line), powder (red line), and crystal (blue). Schematic illustration of molecular assembles. | ||
The fluorescence changed from green to greenish-blue when the crystal was ground (Fig. 2b). The absorption spectrum of the crystal showed a red-shifted band (λab = 395 nm) with a characteristic peak at 440 nm compared with that in dichloromethane (λab = 325 nm). The peak at 440 nm is probably due to delocalization of π-orbitals on face-to-face molecular slip-stacking (Fig. S6, ESI†). Overlap between the π-orbitals results through conjugation-like behavior in the solid state. The spectrum of the ground powders lost the characteristic band at 440 nm and shifted to a higher energy compared with the crystal (Fig. 2c). Powder XRD analysis demonstrated disordering on moving from the crystal to powder (Fig. S7, ESI†); the broad and unclear XRD pattern indicated that the structure was disordered. There was no effective face-to-face slip-stacking between the BT1PF4 molecules, which likely causes the disappearance of the peak at 440 nm. The appearance of this specific absorption peak is similar to that of rr-P3HT4 and PF4 (Fig. 1b) on going from solution to film. The red-shift of the absorption band of PF4 indicates intermolecular packing similar to that in BT1PF4 may be occuring in the film.
The fluorescence spectrum of BT1PF4 in crystal also showed a red-shifted band (λfl = 500 nm) compared with that in dichloromethane (λfl = 405 nm). The spectrum of BT1PF4 in powder showed a lower energy fluorescence band at 448 nm, which was different in crystal and in solution (Fig. 2d). The quantum yield (Φ) of the crystal (25.2%) was higher than in dichloromethane (14.3%) but decreased on grinding (11.2%). The compound showed typical crystallization-induced enhanced emission (CIEE)-like behavior. These results indicated that the molecular packing (crystal structure) of BT1PF4 critically affected the solid-state fluorescence properties.
The optical properties, red-shift, and CIEE of BT1PF4 were caused by its crystal structure (Fig. S6, ESI†).10 Intramolecular H–F and S–F interactions occur between the thiophene and tetrafluorophenylene units. The rigidity induced by the intramolecular interactions between neighboring units may cause the highly crystallinity of PF4. Following supramolecular gel formation of PF4,7 the author hypothesized that macrostructural crystal structure formation would occur in the film state and could be controlled by the casting solvent and thermal treatment. Depending on the film preparation and processing methods shown below, the crystalline and red-fluorescence π-conjugated polymer (PF4) also showed a similar color change behavior for the model compound (BT1PF4).
PF4 showed tunable optical properties via film processing. The results of the absorption and emission spectra are displayed in Fig. 3a and b. The processing pathway is illustrated in Fig. 3c. The polymer in solution, such as in 1,2-dichloroethane (DCE) and mesitylene (MES) showed a green-colored fluorescence (λab = 384 nm, λfl = 507 nm, Φ = 13.1% in DCE, 12.9% in MES). Drop-casting from a DCE solution gave a film with red-fluorescence (λab = 481 nm, λfl = 652 nm, Φ = 23.2%). The drop-cast film from an MES solution resulted in a film with orange fluorescence (λab = 480 nm, λfl = 642 nm, Φ = 9.6%). The absorption bands of FilmDCE and FilmMES had similar peaks and a characteristic shoulder peak at 515 nm (Fig. 1a). However, the broader fluorescence band for FilmMES than FilmDCE could be attributed to the inhibition of the self-assembly by π–π stacking (Fig. 1b). Both films were tuned via thermal processing (Fig. 1c). DSC analysis of the polymer provided information for thermal annealing. A broad melting peak for PF4 was observed (Fig. S3, ESI†). The onset, peak, and end points of melting were at 160, 182, and 211 °C, respectively. From the information obtained by TGA analysis, the decomposition of the polymer began at 350 °C (Fig. S4, ESI†). Thus, melting of the crystalline state occurred at 220 °C. The film that was annealed at 220 °C for 1 min and then slowly cooled at 30 °C for 5 min, had a bright red emission (λab = 478 nm, λfl = 654 nm, Φ = 25.1%). The rapid cooling of the annealed film at 0 °C for 1 min gave a film with yellow fluorescence (λab = 398 nm, λfl = 569 nm, Φ = 19.7%). The tuning of the emission can likely be attributed to the change of the macrostructure in the film.
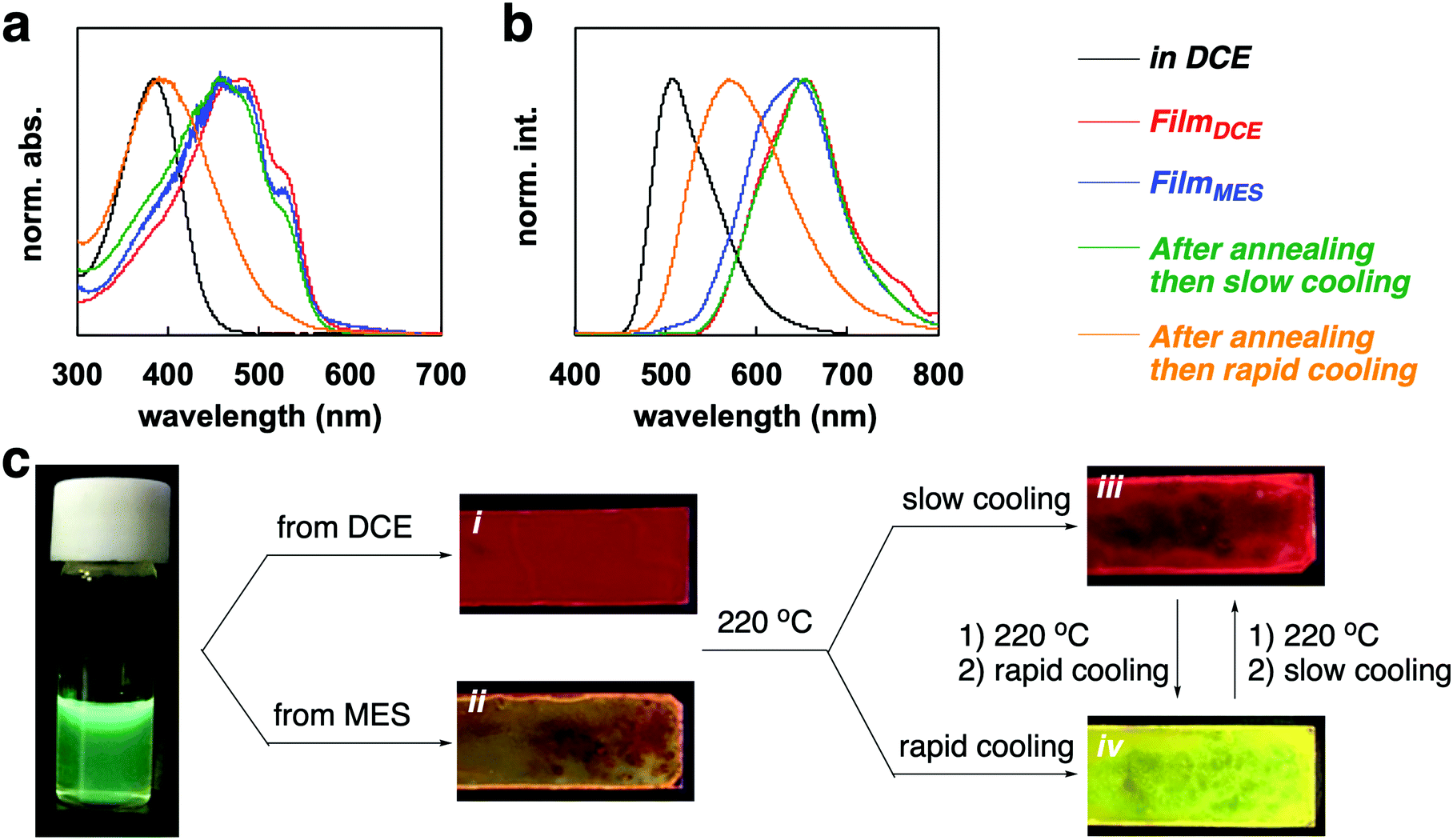 | ||
| Fig. 3 (a) UV-vis absorption spectra of PF4 in various states. (b) Emission spectra of PF4 in various states. (c) Film fabrication pathway from different cast solvents and annealing treatments. Photographic images of the solution and film under UV (365 nm) irradiation. | ||
Powder XRD analysis of the polymer films provided information on the macrostructural change in the film (Fig. 4). XRD patterns of the FilmDCE showed peaks derived from interdigitated packing of the hexyl side chain (0.69 and 1.38 nm), which could be attributed to d100 and d200 (Fig. 4a). π–π stacking of the main chain (ca. 0.4 nm) was not clearly observed. Because π–π stacking of the main chain causes characteristic absorption and emission behaviors (Fig. 3), it can be considered that ordered assembly of the main chain occurs but it was not highly ordered. In addition, the macrostructure was probably affected by the π-conjugated main chain and the hexyl side chain and the formation of many amorphous domains (Fig. S8i, ESI†). The XRD patterns for the FilmMES showed peaks derived from interdigitated packing of the hexyl side chain (0.34, 0.69, and 1.38 nm, which are attributed to d100, d200, and d300, respectively); however, the peak for π–π stacking of the main segments were not clearly detected (Fig. 4b). From the spectra (Fig. 3), there is a domain forming structure as shown in Fig. S8i (ESI†). However, π–π stacking of the main chains were inhibited by the solvent, resulting in an interdigitated packing rich (π–π stacking) film (Fig. S8ii, ESI†). XRD analysis of the film after annealing and slow cooling gave peaks associated with both the interdigitated packing of the hexyl side chain (0.69 and 1.38 nm) and π–π stacking of the main chain (∼0.4 nm), which are attributed to d100, d200, and d010 (Fig. 4c). An increased peak intensity and a narrowed full width half maximum compared with FilmDCE were clearly observed. Thus, a highly crystalline film was obtained (Fig. S8iii, ESI†). The film after annealing and rapid cooling did not show peaks associated with assembly (Fig. 4d). This film had an amorphous state (Fig. S8iv, ESI†). Therefore, dynamic tuning of the light-emission properties and macrostructure of the PF4 film were successfully demonstrated by drop-casting and facile treatment of the polymer film.
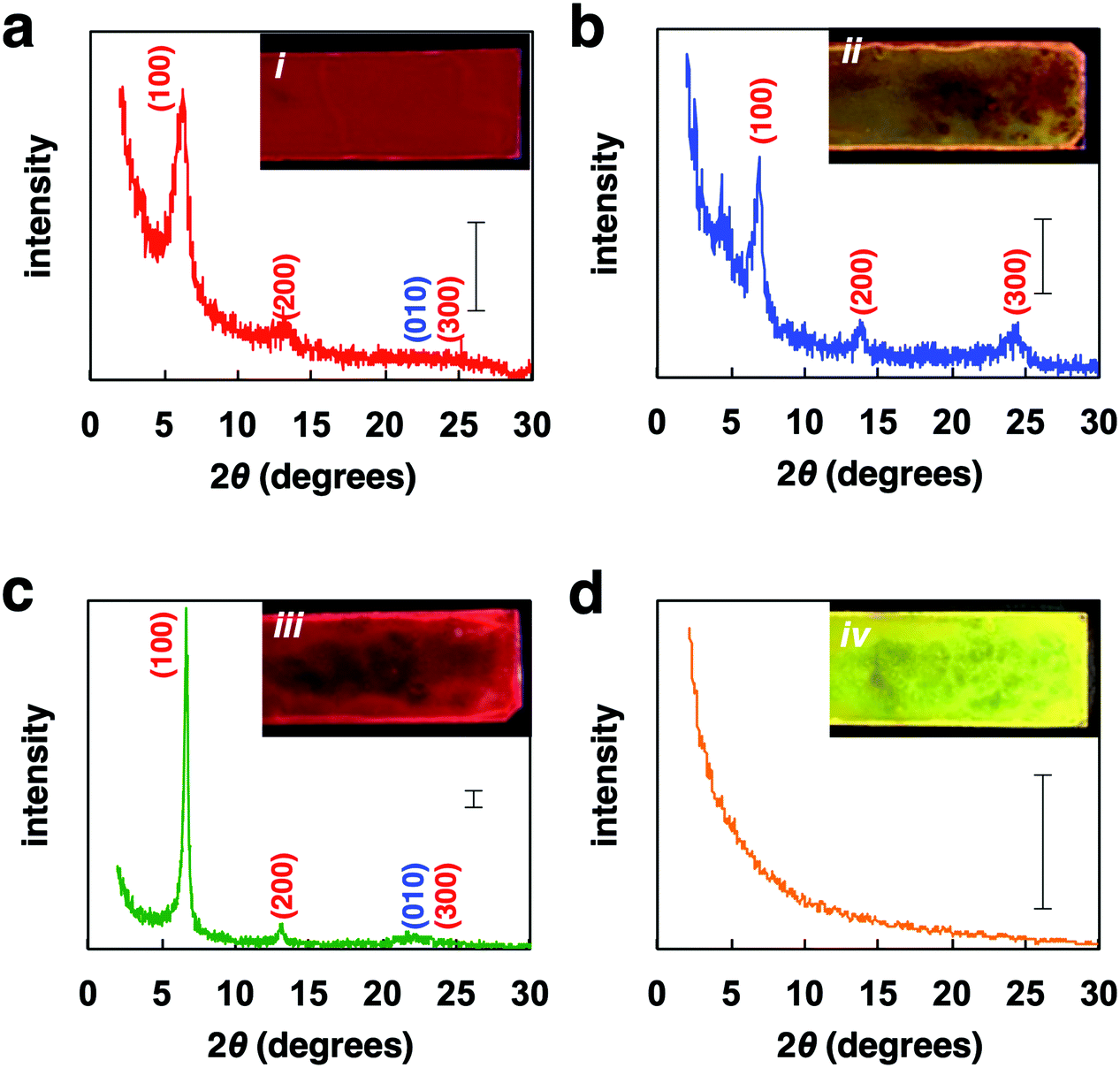 | ||
| Fig. 4 XRD patterns of (a) FilmDCE, (b) FilmMES, (c) film after annealing then slow cooling, (d) film after annealing then rapid cooling. Scale bar: 100 counts. | ||
Conclusion
In conclusion, the author has shown the key structure for highly crystalline and red-fluorescent polymer film by comparing with 3,3′-dihexylbithiophene-phenylene alternating copolymers, poly(bithiophene-alt-tetrafluorophenylene). Fluorine atoms at phenylene unit induced intramolecular interactions. Tetrafluorophenylene structure in the alternating polymer showed macrostructural structure formation and aggregation (crystallization)-induced enhanced low energy emission. Moreover, dramatic structure and properties tuning of poly(bithiophene-alt-tetrafluorophenylene) film was successfully demonstrated by film processing of both cast-coating and thermal annealing. Fluorescent properties could be changed by changing the crystalline structure in film. Further study to light-emission device and transistor was now under consideration.Experimental
Chemicals
Solvents and reagents were used as received. 1,4-Bis[2-(4-methylthienyl)]-2,3,5,6-tetrafluorobenzene, BT1PF4, (CCDC deposition no. 1847366) was used as same product in previously report.10a Space group: P21/c (14), cell: a = 10.306(3) Å, b = 4.3965(11) Å, c = 15.490(4) Å, α = 90°, β = 92.080(3)°, γ = 90°. Polymers were synthesized by following procedure.Synthetic procedure
A procedure for the direct arylation polycondensation of BT6 with dibromoarene is as follows. A mixture of BT6 (100 mg, 0.30 mmol), dibromoarene (0.30 mmol), 1-adamantanecarboxylic acid (16 mg, 30 mol%), K2CO3 (125 mg, 0.90 mmol) and PITS-Cl (1.0 mol%) was stirred in dry N,N-dimethylacetoamide (1.0 mL) for 120 min at 120 °C under argon. The reaction mixture was diluted by toluene, rapidly cooled to room temperature, and then filtered to remove insoluble salts. The filtrate was pored into a large amount of methanol. The resulting polymer was collected by filtration, and then washed with a large amount of methanol. The polymer was dried under vacuum to give as an orange powder. The measurements of the polymer were performed without further treatment. The yield was estimated by the weight of the polymer, which was insoluble in methanol. Molecular weight (Mn) and polydispersity (Mw/Mn) of the polymer were estimated by size exclusion chromatography (SEC, polystyrene standard).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300; Mw/Mn = 3.78.
700; Mw/Mn = 4.29.
300; Mw/Mn = 3.78.
700; Mw/Mn = 4.29.
Measurements
1H NMR spectra were recorded on a JEOL EX-300 spectrometer. SEC (GPC) analyses were performed by a Toso GPC system (HLC-8220), using tetrahydrofuran as the eluent after calibration with polystyrene standards. UV-vis absorption spectra were obtained on an Ocean Optics USB4000-XR1 fiber spectrometer with DH2000-BAL tungsten halogen light source. Solid data was recorded using thin crystal and powder coated on glass plate and silicon wafer substrates. The substrate was used after ultrasonic treatment in acetone and heat treatment at 300 °C using hot plate. Fluorescence spectra were obtained on an Ocean Optics USB4000 fiber spectrometer with light-emitting diode light source (365 nm). Absolute quantum yield was obtained by Hamamatsu C9920-02. Cyclic voltammetry measurements were performed by ALS 611. A three-electrode system equipped with the glassy carbon or indium-tin-oxide-coated polyethylene terephthalate electrode, platinum counter electrode and Ag/AgCl as a reference electrode was used in an electrolytic solution of acetonitrile containing 0.1 M tetraethylammonium tetrafluoroborate (Et4NBF4). Powder X-ray diffraction analysis was performed by JEOL JDX-3530 X-ray diffractometer system. The single crystal X-ray diffraction data for single crystal BT1PF4 were collected at room temperature using SMART APEX II (Bruker AXS) with a CCD detector. The crystal structures were solved by the direct method and refined by full matrix least-squares using SHELXTL. DSC analysis was performed by a Shimadzu DS-60, which measured during heating from room temperature to 400 °C (for PF0 and PF4) and 350 °C (for PM and PF2) at heating rate of 10 °C min−1 in nitrogen. TGA analysis was performed by a Shimadzu TA-60, which measured during heating from room temperature to 600 °C at heating rate of 20 °C min−1 in nitrogen.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. H. acknowledged a KAKENHI (Grant-in-Aid for Scientific Research B: no. 18H02052, Grant-in-Aid for Scientific Research on Innovative Areas ‘π-figuration’: no. 17H05171, ‘coordination asymmetry’: no. 19H04604, and ‘soft crystal’: no. 20H04684) of the Japan Society for the Promotion of Science (JSPS). This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”.Notes and references
-
(a) G. M. Newbloom, K. M. Weigandt and D. C. Pozzo, Macromolecules, 2012, 45, 3452–3462 CrossRef CAS
; (b) J.-H. Chen, C.-S. Chang, Y.-X. Chang, C.-Y. Chen, H.-L. Chen and S.-A. Chen, Macromolecules, 2009, 42, 1306–1314 CrossRef CAS
-
(a) A. T. Kleinschmidt, S. E. Root and D. J. Lipomi, J. Mater. Chem. A, 2017, 5, 11396–11400 RSC
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed
-
(a) M. Brinkmann, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1218–1233 CrossRef CAS
-
(a) L. Xu, J. Zhang, J. Peng and F. Qiu, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 633–639 CrossRef CAS
-
(a) M.-J. Lin, J. JimØnez, C. Burschka and F. Würthner, Chem. Commun., 2012, 48, 12050–12052 RSC
-
(a) S. Hayashi, A. Takigami and T. Koizumi, Chem. – Asian J., 2018, 13, 2014–2018 CrossRef CAS PubMed
- J. Pei, W.-L. Yu and W. Huang, Macromolecules, 2000, 33, 2462–2471 CrossRef CAS
-
(a) S. Hayashi, Y. Kojima and T. Koizumi, Polym. Chem., 2015, 6, 881–885 RSC
-
(a) S. Hayashi and T. Koizumi, Angew. Chem., Int. Ed., 2016, 55, 2701–2704 CrossRef CAS PubMed
- P. Xue, B. Yao, J. Sun, Z. Zhang, K. Li, B. Li and R. Lu, Dyes Pigm., 2015, 112, 255–261 CrossRef CAS
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00218f |
| This journal is © The Royal Society of Chemistry 2020 |