
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVapor phase infiltration of zinc oxide into thin films of cis-polyisoprene rubber†
Julian
Pilz
 ab,
Anna Maria
Coclite
a and
Mark D.
Losego
*b
ab,
Anna Maria
Coclite
a and
Mark D.
Losego
*b
aInstitute of Solid State Physics, NAWI Graz, Graz University of Technology, 8010 Graz, Austria
bSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: losego@gatech.edu
First published on 21st July 2020
Abstract
Elastomers are an important class of polymers for many applications. Often, additives are added to the polymer matrix of elastomers to promote vulcanization or enhance physical or chemical properties. In this study, vapor phase infiltration (VPI) is investigated for transforming unvulcanized cis-polyisoprene (from natural rubber) into an organic/inorganic hybrid material. Specifically, we examine single-cycle infiltration with diethylzinc (DEZ) and water to form infiltrated zinc oxide species. Interestingly, low-temperature pre-heating of the cis-polyisoprene acutely affects the processes of infiltration, including diffusivity, maximum solubility, and chemical reactivity. We attribute these effects to a combination of film relaxation and oxidation. Independent of thermal pre-treatments, all infiltration processes exhibited consistent zinc oxide loading irrespective of purge time between the DEZ and water doses, indicating the presence of a strongly bound intermediate state between the DEZ precursor and the cis-polyisoprene polymer. Increasing infiltration process temperature accelerates diffusion and lowers the maximum solubility, in accordance with Fick's law and gas phase sorption equilibrium. Resulting organic–inorganic hybrid films show enhanced resistance to dissolution in toluene, a good solvent for the pure polymer.
I. Introduction
Cis-Polyisoprene is the main component of natural rubber.1 In commercial applications, cis-polyisoprene is often vulcanized (i.e. cross-linked; in the case of natural rubbers often driven by sulfur compounds) and different components such as activators, accelerators, or fillers2 are added to the polymer to achieve desired properties. Activators and accelerators lower the process temperatures and accelerate the vulcanization process. ZnO is widely used as an activator in rubber manufacturing. An even distribution in the polymer matrix as well as the morphology of ZnO particles is key to its effectiveness in modulating vulcanization.3,4 Fillers are added to reinforce physical properties (mechanical, electrical, optical,…) and prominent examples are carbon black5 and silica.6 Similar to activators and accelerators, the size and distribution of fillers impacts the resulting properties.7As such, vulcanizates (i.e. cross-linked elastomer polymers with additives) are a prominent example of composite and often hybrid organic–inorganic materials. Polymers that make up these vulcanizates are a new opportunity for vapor phase infiltration (VPI), a solvent-free method for infusing inorganic materials into polymers. Unlike many other methods currently used, VPI incorporates the inorganic species homogeneously at the atomic level, and the species may be chemically bound or unbound to the polymer host.8
VPI is capable of distributing the inorganic material uniformly inside polymers and the amount of loading can be tuned by infiltration hold times, pressures, and cycle numbers.9,10 While VPI originated from the atomic layer deposition (ALD) community and shares similar precursor chemistries, the process mechanisms are substantially different. In ALD, precursors follow self-limiting surface reactions on the exterior of a material. In VPI, the gas-phase precursors are expected to sorb into the material (polymer), diffuse throughout that material, and eventually become entrapped within the material through either chemical reactions or other means. The mechanism for entrapment is important as it determines whether VPI processing can be run as an equilibrium or non-equilibrium process and whether the inorganic species form primary chemical bonds to the polymer host. These final hybrid materials are intermixed organic–inorganic hybrid structures with properties distinctly different from the original polymer.11,12 For example, VPI has been used to increase the toughness of spider silk,13 increase the electrical conductivity of semiconducting polymers,14 and increase the solvent stability and separation performance of membranes.15
A recent investigation examined the use of vapor phase infiltration to incorporate inorganic materials into block co-polymers containing a rubber (elastomeric) block. In this recent study, VPI with diethyl zinc and water was used to infiltrate ZnO into epoxidized and unepoxidized polystyrene-block-poly(1,4-isoprene) copolymers (PS-b-PI).16 The authors found that even PS-b-PI (lacking strong functional groups) were effective templates for selective infiltration of the polyisoprene block, attributing it to factors such as increased miscibility of DEZ in the polyisoprene block. However, a concrete mechanism of infiltration of polyisoprene has not been reported in literature, despite the technological importance of this polymer.
Depending on a polymer's chemical structure, different mechanisms of infiltration may happen.17 In the case of non-reactive polymers, precursor molecules can diffuse into the polymer matrix and become weakly entrapped by physisorption or steric hindrance. However, by applying long purges, these weakly bound precursors should diffuse out of the matrix, leaving no loading within the polymer. For polymers with reactive functional groups, precursor molecules chemically bind to the available reactive groups and are not removed by long purges. This mechanism may further lead to cross-linking of the polymer chains. It is unclear to what extent the alkene functional groups in polyisoprene may play in entrapping metalorganic precursors in a VPI process.
This study aims to elucidate the fundamental kinetic and chemical mechanisms of zinc oxide VPI into cis-polyisoprene using DEZ and water precursors. The results expand the currently limited knowledge about VPI in rubber and provide a new possible technical application for VPI in introducing inorganic constituents to rubber polymers.
II. Experimental
Cis-Polyisoprene was purchased from Sigma-Aldrich (431257, average Mw ∼ 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by GPC, made from natural rubber). The purity of the polyisoprene was investigated with ATR-FTIR, which did not reveal any obvious impurities such as phosphates (see Fig. S1, ESI†). 2 wt% solutions were prepared in toluene (Sigma-Aldrich) and stirred overnight. Thin films of polyisoprene (thickness 78 ± 2 nm (n = 100) after coating) were deposited on single-side polished Si wafers (p-type, WRS Materials) and glass slides by spin coating (10 s, 2000 rpm). No cleaning procedures were applied to the substrates prior to coating except for nitrogen flow. Pre-heating of the films was performed at 80 °C on a hot plate in air (thickness after 5 h pre-heating 68 ± 6 nm (n = 180), thickness after 24 h pre-heating 47 ± 6 nm (n = 100)). Note that the glass transition temperature for this polymer is around −64 °C.18
000 by GPC, made from natural rubber). The purity of the polyisoprene was investigated with ATR-FTIR, which did not reveal any obvious impurities such as phosphates (see Fig. S1, ESI†). 2 wt% solutions were prepared in toluene (Sigma-Aldrich) and stirred overnight. Thin films of polyisoprene (thickness 78 ± 2 nm (n = 100) after coating) were deposited on single-side polished Si wafers (p-type, WRS Materials) and glass slides by spin coating (10 s, 2000 rpm). No cleaning procedures were applied to the substrates prior to coating except for nitrogen flow. Pre-heating of the films was performed at 80 °C on a hot plate in air (thickness after 5 h pre-heating 68 ± 6 nm (n = 180), thickness after 24 h pre-heating 47 ± 6 nm (n = 100)). Note that the glass transition temperature for this polymer is around −64 °C.18
Vapor phase infiltration (VPI) of the polymer films was performed in a custom-built system described earlier,15 and was controlled via a tree-based control software.19 The chamber was operated at a temperature of 60, 80, or 100 °C. Diethylzinc (STREM Chemicals, 95% purity, DANGER: pyrophoric) was used as the metalorganic precursor, deionized water as the oxidizing co-reactant, and nitrogen (99.995% purified from air) as the purging gas (flow rate 250 sccm). One cycle of VPI was applied to the polymer films which consisted of purging for 3600 s (to release residual water and other vapors trapped within the polymer), pump down to base pressure (∼60 mTorr), chamber isolation, DEZ dose for 1 s (through a valve with Cv = 0.27, providing a chamber pressure of ∼0.2 Torr DEZ), hold in this static DEZ atmosphere for 20 s to 7200 s, purge for 300 s, pump down, chamber isolation, water dose for 5 s (reaching ∼2 Torr H2O), hold in this static H2O atmosphere for 1800 s, and purge for 300 s. For investigating the influence of purge time, the DEZ hold was fixed at 800 s and the purge time after DEZ was varied from 100 s to 232200 s (∼2.5 days).
Attenuated total reflectance infrared spectroscopy (ATR-FTIR) was performed to monitor the chemical changes of spin-coated films upon pre-heating and infiltration. For these studies, films were prepared on glass substrates to minimize background intensities. Spectra were collected on a Thermo Scientific™ Nicolet™ iS5 FTIR spectrometer with an iD7 ATR accessory with a diamond crystal. The resolution was 4 cm−1 and the spectra were averaged over 128 scans.
Thicknesses and optical constants of pristine, infiltrated, and calcinated films were obtained by spectroscopic ellipsometry (SE; J. A. Woolam alpha-SE). Spectra were obtained in a wavelength range 380–900 nm at 70° (additionally 75° for calcinated films). The model to fit the data consisted of a (semi-infinite) Si substrate, 1.7 nm fixed native oxide, and a Cauchy layer with thickness, A, B, and k as fit parameters. For better comparison of layer thickness and due to correlation of fit parameters, the generic oscillator library model for ZnO (Complete Ease 5.08 software) was used for calcinated films and only the thickness was fitted.
Inorganic loading within the polymer was determined with both X-ray fluorescence spectroscopy (XRF; Thermo Scientific Niton FXL FM-XRF) and calcination of the infiltrated films. XRF was used to measure the relative amount of Zn in the films after infiltration. For this purpose, the amplitude of the Zn Kα peak at 8.637 keV was obtained by fitting a Gaussian to the spectrum. Spectra were recorded in general metals mode with a measurement time of 30 s. Infiltrated films were also calcined in a furnace (Barnstead Thermolyne 1400; ramp up to 650 °C for 20 min, hold for 30 min) to remove the polymeric template and leave only the ZnO fraction. This residual quantity of ZnO was then evaluated with spectroscopic ellipsometry.
X-ray photoelectron spectroscopy was conducted with a Thermo Scientifc K-Alpha system using a monochromatic Al-Kα X-ray source (1486.6 eV) with a 60° incident angle and a 90° emission collection geometry (pass energy 200 eV) for survey elemental analysis.
Zinc concentration depth profiles of the infiltrated polyisoprene films were measured with time-of-flight secondary ion mass spectroscopy (ToF-SIMS, IONTOF, 5 Series). SIMS spectra were collected using oxygen ion sputtering (150 μm × 150 μm, 2 kV accelerating voltage) and bismuth analyzer (50 μm × 50 μm) beams. Positive polarity was used to detect the Zn+, Si+, and C+ signals from the infiltrated polymer films.
Chemical stability was tested by immersing pre-heated and infiltrated films (on Si) in 10 ml of toluene. The thickness was monitored intermittently by ex situ spectroscopic ellipsometry.
III. Results and discussion
A. Demonstration of DEZ + H2O VPI in cis-polyisoprene
Fig. 1 illustrates that DEZ + H2O VPI can successfully infuse ZnO into cis-polyisoprene films. Fig. 1(a) shows an X-ray fluorescence spectrum (XRF) of a hybrid film and a blank Si wafer. The hybrid film shows a clear signal from the Zn Kα transition (enlarged spectrum shown in the inset), whereas all the other peaks stem either from the Si wafer or the sample holder (the fluorescence signal from organic elements [Z < 12] are undetectable by this XRF). The strong XRF signal is indicative of the Zn being infiltrated throughout the entire film and not just a single atomic layer on the surface. Investigations on the morphology of polyisoprene and hybrid films by atomic force microscopy (Fig. S2, ESI†) support this argument as no major changes in the morphology are observed upon infiltration. Furthermore, no signs of phase separation between the organic and inorganic compounds are observed. Fig. 1(b) shows X-ray photoelectron spectra (XPS) of a polyisoprene and a hybrid film. The polyisoprene spectrum does not display significant amounts of impurities (except for oxygen due to oxidation; see next paragraph) and the spectrum of the hybrid film shows clear Zn signals. The shown spectra serve as a preview on the infiltration of polyisoprene; in the following paragraphs the infiltration mechanisms as well as the influence of the pretreatment of the polyisoprene films are examined in detail.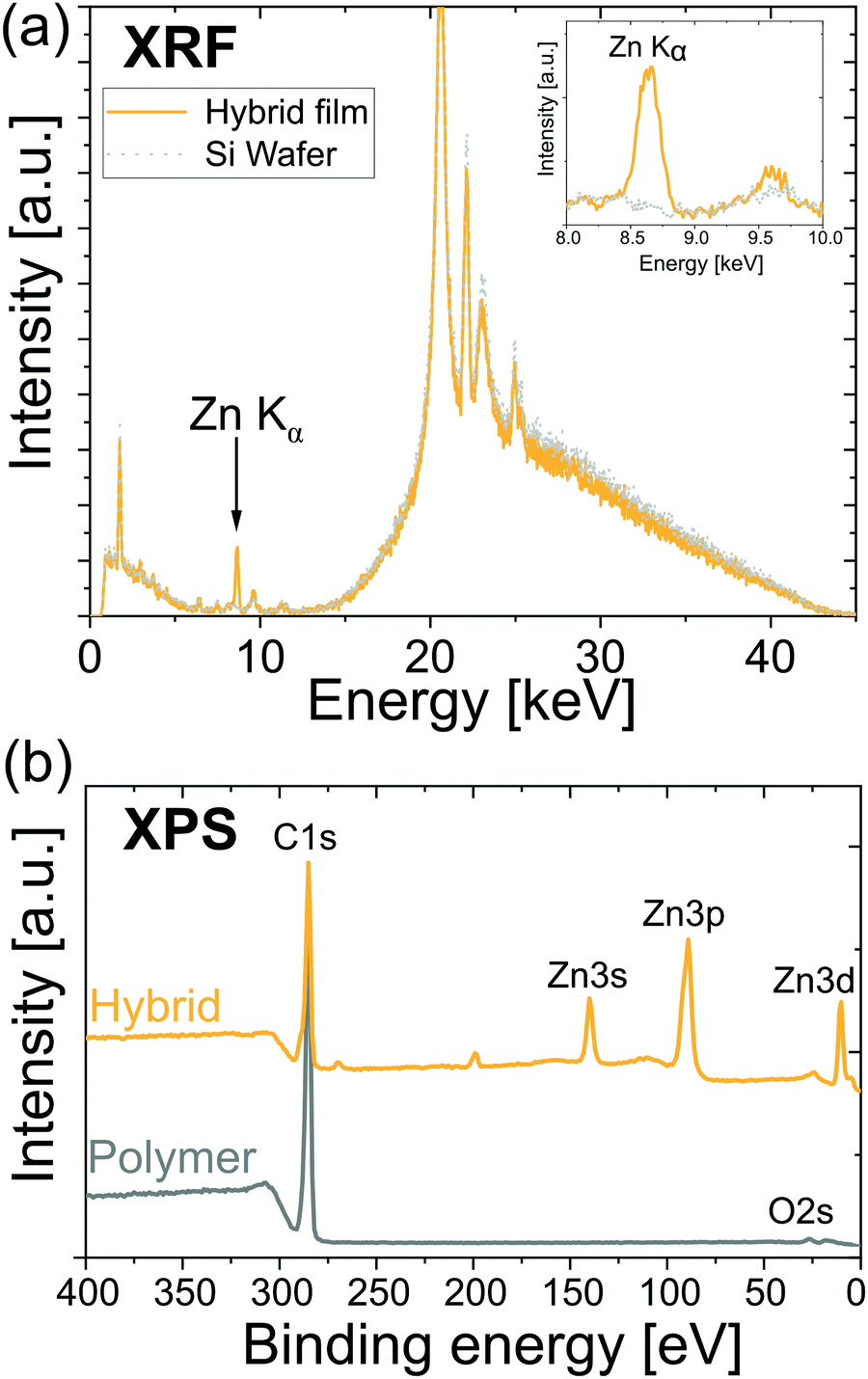 | ||
| Fig. 1 Exemplary X-ray spectra of hybrid films demonstrating that ZnO was successfully loaded into the polyisoprene films. (a) X-ray fluorescence spectrum of a hybrid film and a blank Si wafer. The hybrid film displays a significant Zn Kα signal whereas all the other peaks stem either from the Si wafer or the sample holder. (b) X-ray photoelectron survey spectra (for low binding energy) from the surface of a polymer and hybrid film. The polymer film does not display impurities such as phosphorus and the hybrid film displays clear Zn signals. The spectra are stacked for clarity. | ||
B. Pre-heating of polyisoprene films
Early in our investigations of the VPI process on polyisoprene, we noted some unusual variations in the polymer relaxation when being pre-heated on the hotplate. A pre-heating step is typically used after spin coating a polymer film to remove solvent and relax the polymer structure. For polyisoprene, we found that this relaxation process appears to occur over a fairly long period of time, potentially many hours. Fig. 2(a) plots the polyisoprene film thickness as a function of drying time (80 °C) for four independent trials (For trials 1–3, the thickness of the same individual film was monitored. For trial 4, each datapoint corresponds to the mean thickness of two individual samples, which were removed from the hotplate after the respective pre-heating times). While the thickness immediately after spin coating is around 80 nm, all samples show a rapid thickness decrease for the first 5 to 10 h. After about 20 h, the thickness decrease slows and approaches a value between 40 and 50 nm (50 to 65% of the initial thickness). Initially, we attribute this reduction in thickness to a reduction in free volume of the polymer. Rapidly drying the polymer from solution via spin casting at room temperature often results in a metastable glass with high free volume. Even though the polymer is above Tg at room temperature, heating it should accelerate chain motion, relaxation, and reduce free volume, resulting in a reduction of the film thickness. However, as shown subsequently, it is likely that the changes in film thickness may be a combination of changes to both the free volume and polymer chemistry (e.g., an increase in cross-linking).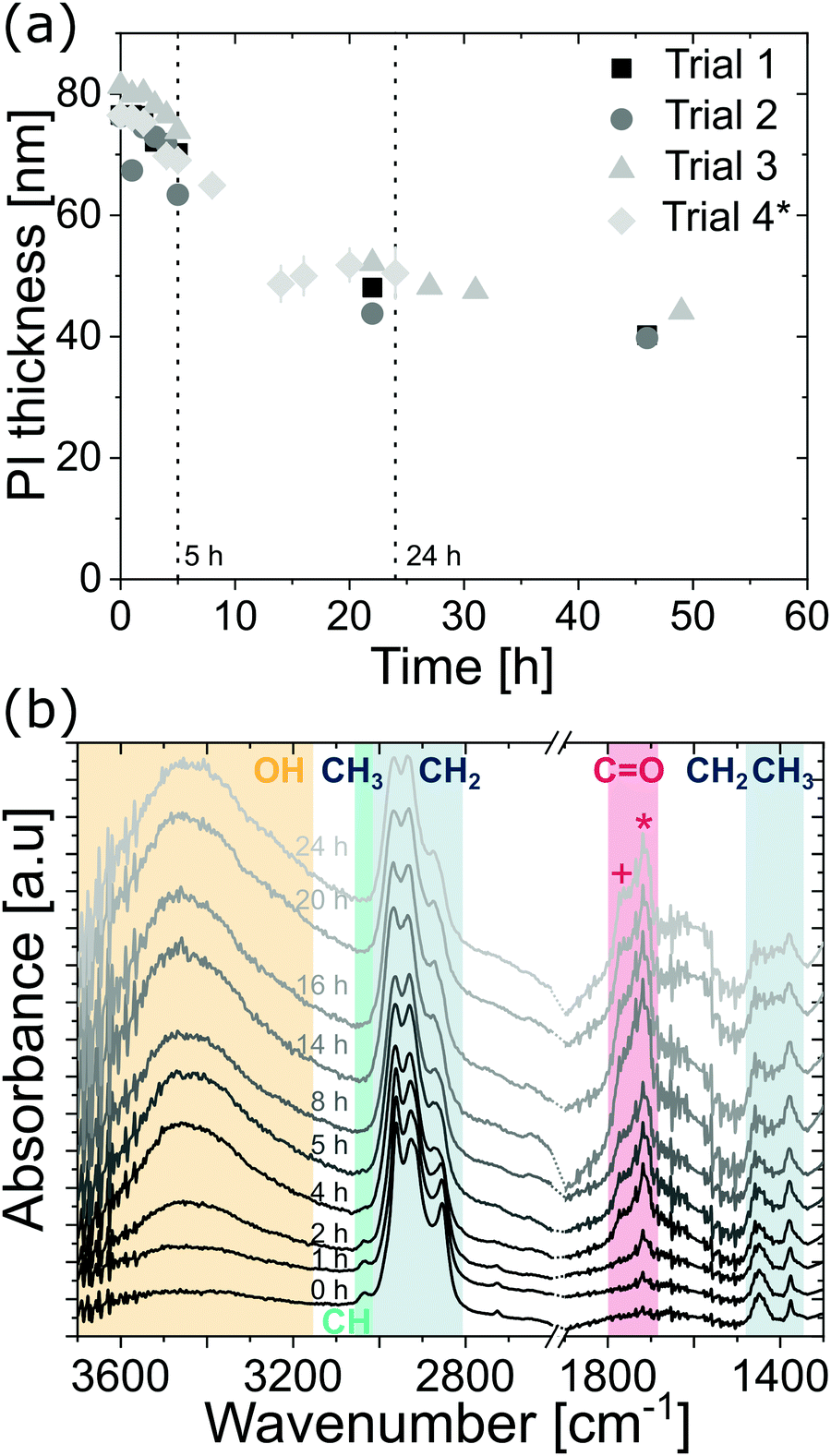 | ||
Fig. 2 (a) Thickness decrease as a function of time during pre-heating of polyisoprene thin films at 80 °C. Trial 1 to 3 constitute thickness data for the same films, respectively, whereas for trial 4 each datapoint constitutes the mean value of 2 individual samples. (b) ATR-FTIR absorbance spectra of spin coated polyisoprene films on glass. Films were pre-heated at 80 °C for different durations (indicated by time in hours). Colored regions refer to O–H stretching (∼3400 cm−1), ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching (∼3036 cm−1), CH3 and CH2 stretching (2800–2900 cm−1), CO stretching (+ and * referring to carboxylic acids (∼1766 cm−1) and ketones (∼1714 cm−1), respectively), and CH2 and CH3 deformation (1370–1450 cm−1). Spectra are scaled to their respective maximum value, offset, and cut to the regions where the main absorptions occur (strong absorption of glass substrate below 1300 cm−1). Dotted lines at the axis break serve as a guide for the eye. C–H stretching (∼3036 cm−1), CH3 and CH2 stretching (2800–2900 cm−1), CO stretching (+ and * referring to carboxylic acids (∼1766 cm−1) and ketones (∼1714 cm−1), respectively), and CH2 and CH3 deformation (1370–1450 cm−1). Spectra are scaled to their respective maximum value, offset, and cut to the regions where the main absorptions occur (strong absorption of glass substrate below 1300 cm−1). Dotted lines at the axis break serve as a guide for the eye. | ||
ATR-FTIR spectra were collected as a function of pre-heating time to interrogate potential chemical changes to the cis-polyisoprene films. The (scaled) IR spectra are shown in Fig. 2(b). For unheated films (0 h), the FTIR spectrum shows the characteristic C–H stretching, and CH3 and CH2 stretching and deformation modes of polyisoprene.20,21 The weak CC stretching mode (∼1665 cm−1) is not visible, which we attribute to the rather small thickness of the films. After 1 h of heating, an absorption peak attributed to CO stretching (ketones, ∼1714 cm−1) is apparent, indicative of partial oxidation. Longer heating durations (i.e. further oxidation) leads to more ketone groups as well as an increase in O–H stretching absorbance. At 4 h heating, an additional peak appears, which can be attributed to carboxylic acids (∼1766 cm−1). Longer heating times lead to an increased absorbance in the O–H and CO regions until a heating duration of 14 h, after which the oxidation appears to reach saturation (i.e. no further increase in O–H and CO absorbance). These studies suggest continued oxidation of the polymer from ketones to alcohols to carboxylic acids with longer heat treatments. Thermal treatment of unvulcanized and vulcanized elastomers has been a field of significant research.22–26 In a study by Colin et al.,22trans-polyisoprene is shown to oxidize in atmosphere similar to the behavior observed here via mechanisms such as chain-scission and cross-linking. Using molecular dynamics simulations, Li et al.27 show that (sulfur) cross-linking leads to shrinkage of cis-polyisoprene chains. While the exact mechanisms of oxidation and film shrinkage are difficult to ascertain, oxidation and subsequent cross-linking could be one factor in addition to polymer relaxation for the film shrinkage observed in Fig. 2(a).
C. Quantifying inorganic loading
Because pre-heating steps significantly affect the polyisoprene chemistry and structure, they are also expected to affect the infiltration process. To better understand this influence, we study infiltration of unheated, 5 h pre-heated (partially relaxed and oxidized), and 24 h pre-heated (close to fully relaxed and oxidized) films. Fig. 3 summarizes the variations in zinc oxide loading depending upon pre-heating conditions for VPI at 80 °C and DEZ hold times of 7200 s (5 individual samples per pre-heating condition). Most of the polyisoprene films pre-heated for 0 h and 5 h show a decrease in thickness (−1.6% and −4.3% on average, respectively) upon infiltration while most films pre-heated for 24 h show an increase in thickness (+5.6% on average). Presuming that volume change is indicative of inorganic loading, these differences in film thickness upon infiltration suggest that the films pre-heated for 0 h or 5 h have less inorganic loading than the films heated for 24 h. However, after calcining these films to leave just the inorganic ZnO component, we find the opposite trend. Fig. 3(b) plots this result, showing the ZnO layer thickness after calcination for each of the trials in Fig. 3(a). On average the polyisoprene films pre-heated for only 5 h have about twice the ZnO thickness (∼8 nm) as those pre-heated for 0 h or 24 h (∼4 nm). While the swelling of polymer films is often used as an indicator for inorganic loading in VPI literature,11,17,28 it is not a direct measure of the mass loading, and as shown here, film swelling is not always a good indicator of inorganic loading. The noncorrelation of the swelling and the ZnO thickness after calcination is attributed to a competition between the relaxations/shrinking occurring within the film due to a reduction of the polymer's free volume (thickness decrease) with the swelling due to inorganic mass uptake (thickness increase).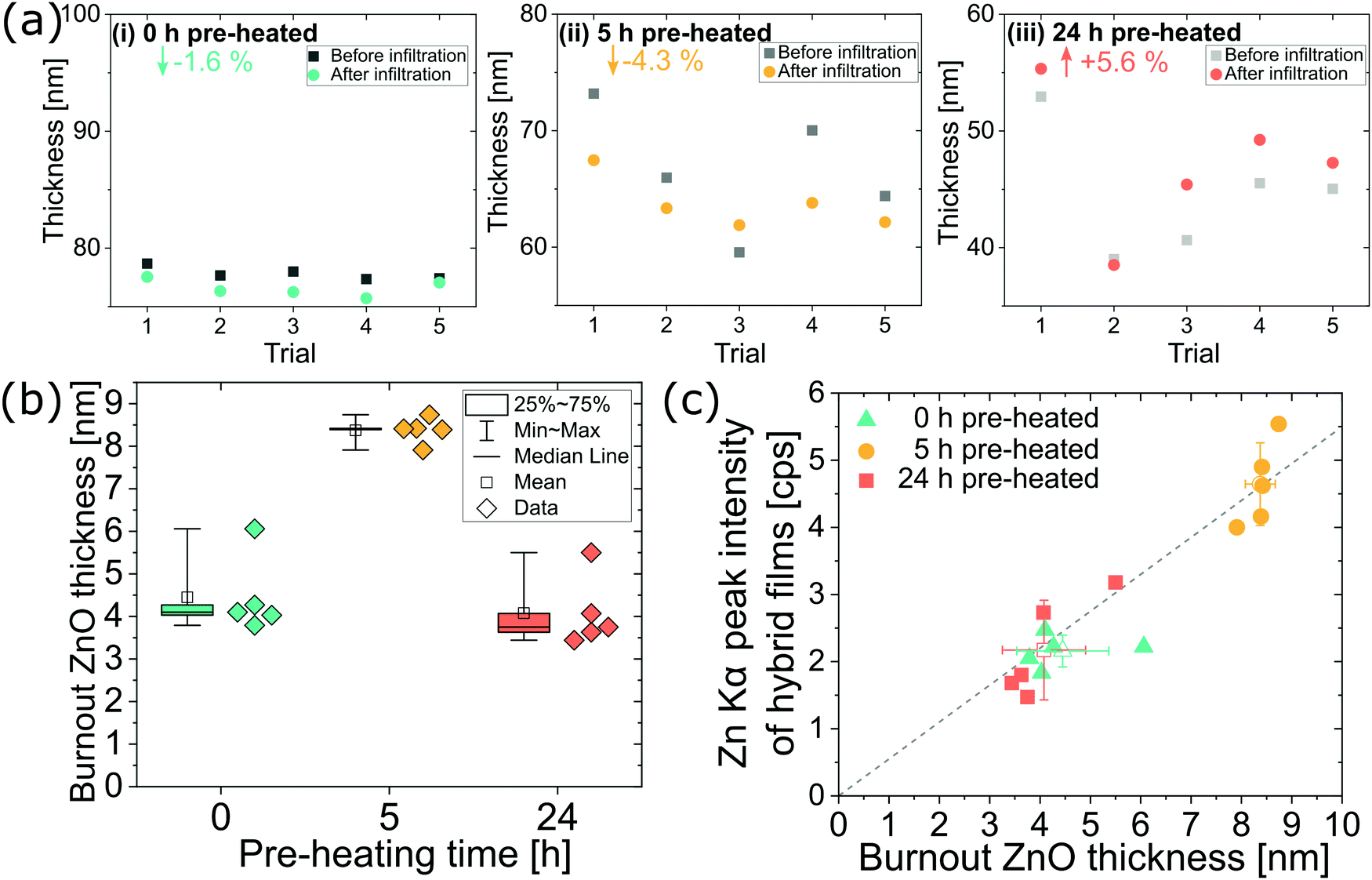 | ||
| Fig. 3 (a) Thicknesses of cis-polyisoprene films before and after zinc oxide infiltration for 5 separate but equivalent trials. Films were pre-heated at 80 °C for (i) 0 h, (ii) 5 h, and (iii) 24 h in air prior to infiltration. (Note the changing abscissa in (i) to (iii) for film thickness but same unit spacing). (b) ZnO film thickness after calcination of the infiltrated films at 650 °C for 30 min in air, and (c) correlation of the XRF Zn Kα peak height of the hybrid films and the ZnO film thickness after calcination; mean values with their standard deviation of the respective data are indicated with open symbols and the linear slope serves as a guide for the eye. All films were infiltrated at 80 °C using a 7200 s, 0.2 Torr DEZ exposure step. | ||
These results suggest that spectroscopic ellipsometry is not always a reliable non-destructive characterization tool for assessing inorganic loading in VPI synthesized hybrid materials. Thus, we also explored the use of XRF as an alternative method for inorganic loading quantification. XRF has not previously had wide use in the VPI community.11 XRF is a non-destructive and fast (measurement times ∼1 min) characterization technique, that can readily be used to provide relative quantities of inorganics within a hybrid material. (Note: absolute amounts can be measured by calibrating the XRF detector to standards; within the scope of this paper, relative amounts are sufficient, although good standards for infiltrated materials should be developed.) Fig. 3(c) plots the correlation of the measured XRF Zn Kα peak height (of the hybrid film) against the calcined ZnO film thickness determined by spectroscopic ellipsometry in Fig. 3(b). Good correlation is found between these two methods, validating XRF's use for non-destructive quantification of Zn loading in VPI hybrid thin films.
D. Kinetics of zinc oxide infiltration into cis-polyisoprene
Next, we examine the kinetics of inorganic loading as a function of VPI process temperature (60 °C, 80 °C, and 100 °C). Fig. 4 plots the amount of zinc uptake (measured by XRF) into cis-polyisoprene during a 1-cycle VPI process for varying 0.2 Torr DEZ exposure times (20 s to 4 h). These Zn uptake plots are shown for both unheated (Fig. 4(a)), 5 h pre-heated (Fig. 4(b)) and 24 h pre-heated (Fig. 4(c)) cis-polyisoprene films. (Fig. S3 (ESI†) plots the calcined ZnO film thickness as a consistent comparison).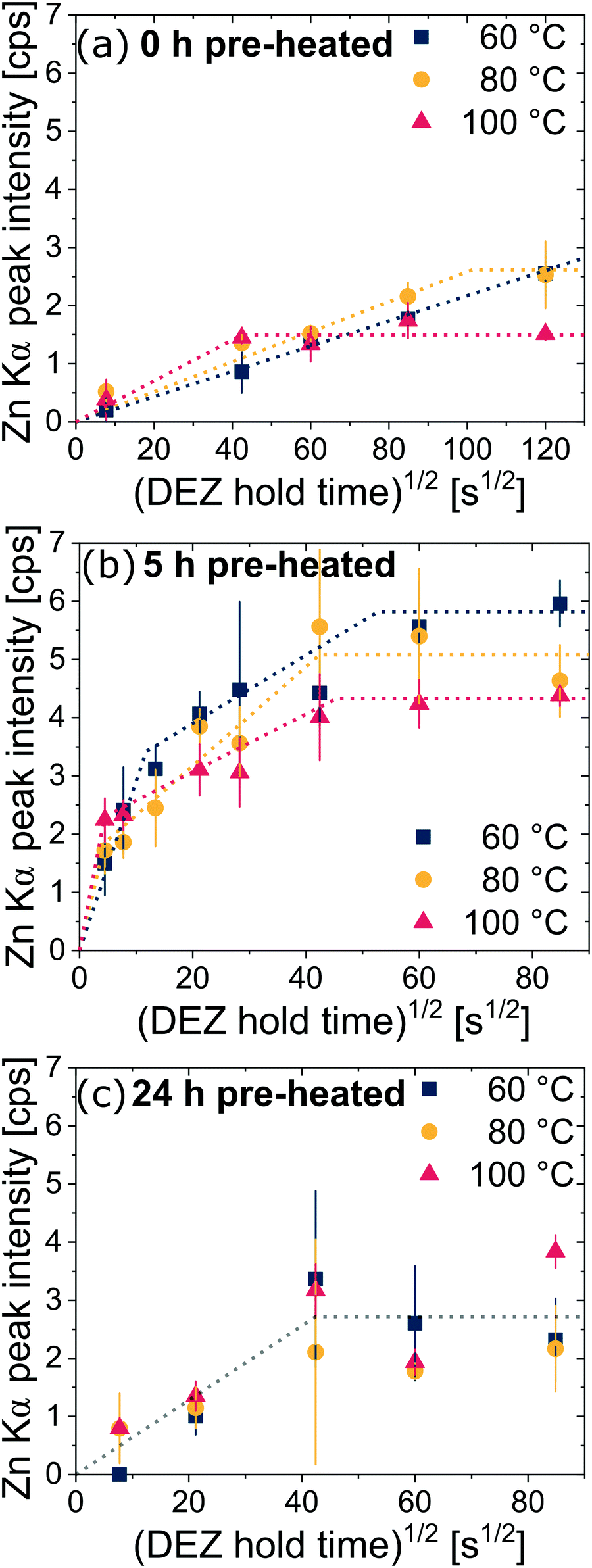 | ||
| Fig. 4 XRF measurements of zinc uptake after VPI as a function of DEZ exposure time for various process temperatures using cis-polyisoprene thin films pre-heated at 80 °C in air for (a) 0 h, (b) 5 h, and (c) 24 h. Note the wider ordinate scale for (a). Dotted lines serve as a guide for the eye. Most data points are an average of at least 5 independently processed hybrid films. Error bars represent the standard deviation from measurements on multiple films. | ||
When the cis-polyisoprene is not pre-heated prior to infiltration (Fig. 4(a)), the sorption behavior is consistent with standard models. The inorganic loading increases with increasing hold time until it saturates (for 80 °C and 100 °C). Higher temperatures seem to promote faster saturation (i.e., faster diffusion); in fact, at 60 °C, saturation is not realized within the time scales studied (4 h). The saturation loading (i.e. maximum solubility) decreases with increasing process temperature. These observations are consistent with the kinetic model for VPI presented by Leng and Losego.28 From a derivation of Fick's 2nd law, the normalized mass uptake (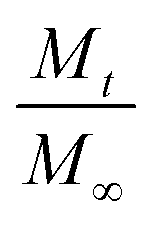 , where M∞ is the saturation value) shows a square-root dependence on time (t), with a slope that depends upon the diffusivity (D) and thickness (L) of the polymer matrix
, where M∞ is the saturation value) shows a square-root dependence on time (t), with a slope that depends upon the diffusivity (D) and thickness (L) of the polymer matrix
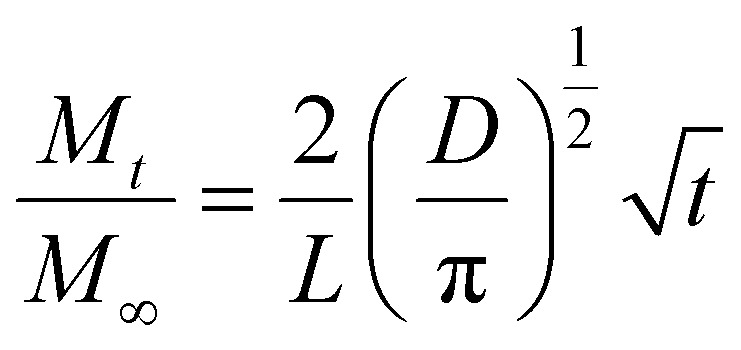
The diffusivity furthermore follows an Arrhenius relationship
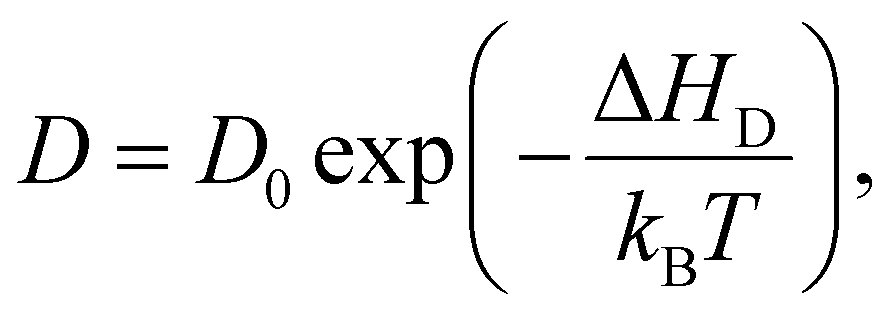
Using these equations and the data presented in Fig. 4(a), we estimate the diffusivity to be about 0.5 × 10−14 cm2 s−1 and 3 × 10−14 cm2 s−1 at 80 °C and 100 °C, respectively. These values are within the same order of magnitude as effective diffusivity values reported for infiltration of trimethylaluminum into poly(methyl methacrylate)28 and can give valuable information about infiltration depth when upscaling the process to thicker films or macroscopic objects.8,28 Diffusivity values in the order of 10−6 cm2 s−1 have been reported for the diffusion of gas molecules such as O2, N2, or CO2 in (vulcanized) natural rubber, i.e. 8 orders of magnitude higher than the current findings; however, it was shown that the diffusivity rapidly decreases with increasing gas molecule size for natural rubber and other polymers.29,30 Diffusivity of the much larger DEZ molecule is thus expected to be much lower than for the above mentioned diatomic and triatomic gases.
A close examination of the mass uptake curves for the 5 h pre-heated cis-polyisoprene films presented in Fig. 4(b) suggests that two separate diffusion regimes may exist (indicated by two linear slopes). For short hold times, precursor can diffuse faster whereas the diffusion slows down for longer hold times (estimates of the diffusivity are given in the ESI†). A possible reason for this behavior might be ongoing relaxation of polyisoprene films during infiltration, which comes into effect for longer hold times. Furthermore, from FTIR analysis shown in Fig. 2(b), films pre-heated for 5 h appear to be in a state in which the oxidation is not yet complete. This might lead to the more complex diffusion kinetic curves as the precursor molecules diffuse through both oxidized and unoxidized regions.
For the films pre-heated for 24 h (shown in Fig. 4(c)), the infiltration kinetics are less differentiable with respect to temperature. While an increase of loading can be observed by increasing the hold time, the relative changes (between measurements for different temperatures) are rather small and difficult to differentiate within measurement error. A reason might be that on average these films are thinner than the films pre-heated for 5 h and thus reach full infiltration faster, limiting the temporal resolution of the diffusion process.28 However, it appears that for these films only one diffusion regime exists (indicated by an averaged slope), which agrees with the fact that the films were much longer pre-heated prior to infiltration. Also, the maximum loading is on average smaller than for the films pre-heated for 5 h, which can be attributed to the decreased thickness.
E. Entrapment of DEZ precursor in cis-polyisoprene
Next we explore the entrapment of DEZ precursors within cis-polyisoprene films. Films undergoing 0 h, 5 h, and 24 h pre-heating at 80 °C were all investigated. Infiltration was conducted at 80 °C for 800 s at ∼2 Torr DEZ. Purge times between the DEZ exposure and water exposure were varied between 100 s and 232200 s (∼2.5 days). If the DEZ precursor does not strongly bind to the polyisoprene, Zn loading should decrease with increasing purge time.31 If DEZ forms a permanent or semi-permanent bond to the cis-polyisoprene, then Zn loading should remain nearly invariant with this purge time. As shown in Fig. 5, the latter situation is true, Zn concentration is relatively invariant with purge time irrespective of pre-heating conditions, even up to a purge time of ∼2.5 days. This suggests that the precursor DEZ molecules are strongly bound (entrapped) within the polymer and do not easily diffuse out upon removal of the DEZ overpressure.
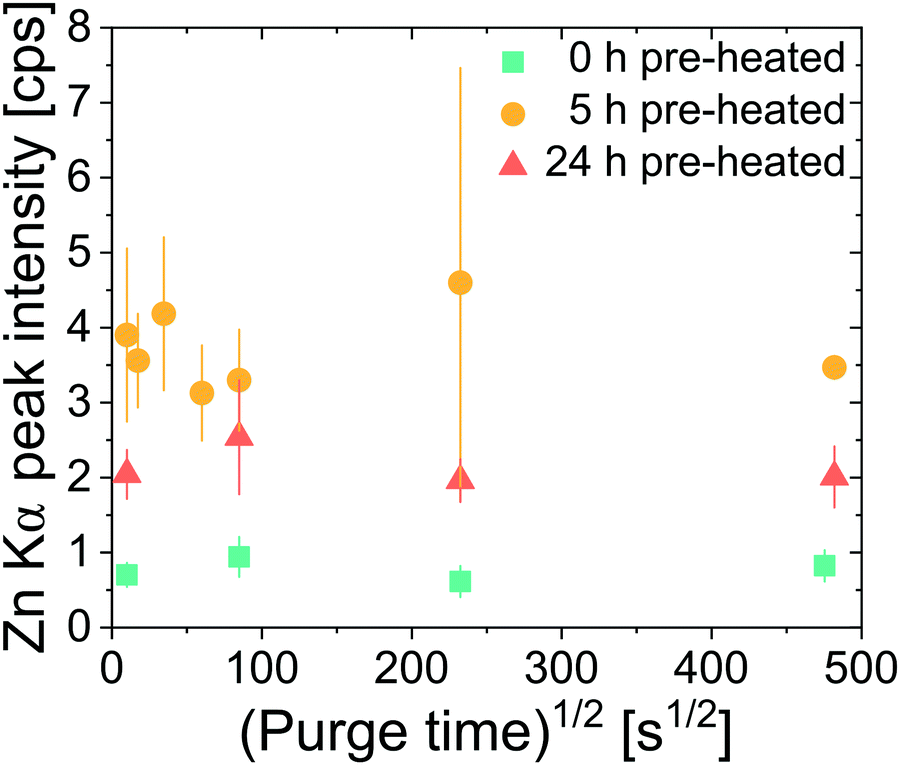 | ||
| Fig. 5 XRF measurements of zinc concentration in hybrid films as a function of purge time between DEZ exposure and H2O exposure. Polyisoprene films are pre-heated at 80 °C for 0 h, 5 h, or 24 h, DEZ hold time is 800 s, and infiltration temperature is 80 °C. Most data points are an average of at least 5 independently processed hybrid films. Error bars represent the standard deviation from measurements on multiple films. | ||
To further understand the entrapment mechanisms, ATR-FTIR spectroscopy is used to understand the chemical structure of the ZnO-polyisoprene hybrid after infiltration. These ATR-FTIR spectra are shown in Fig. 6, together with the spectra of the polyisoprene films before infiltration (from Fig. 2(b)). In comparison to the spectra of pure polymer films, several changes can be observed. The contribution of O–H is increased for all films which can be attributed to the water co-reaction in the infiltration process and the likely formation of metal oxyhydroxide inorganic clusters. The reduction in CO peak intensity with DEZ exposure time indicates a consumption of these groups, likely due to chemical reaction with DEZ. For all the hybrid films, a broad absorption emerges at ∼1550–1650 cm−1. In VPI literature, this mode has been attributed to metalorganic precursors binding to carbonyl groups, both for DEZ32 and trimethylaluminum.8,33,34 This suggests that DEZ entrapment is at least partially explained by binding and reaction with these carbonyl groups, which are not inherent to the pure polyisoprene structure but are rather easily created via pre-heating oxidation. This proposed mechanism is furthermore supported by the analysis of high-resolution XPS spectra of oxygen and carbon (Fig. S5 (ESI†) and Discussion). However, based on Fig. 5, the unheated polyisoprene also retains constant Zn loading with purging despite having a negligible concentration of carbonyl or hydroxyl groups. This suggest that DEZ molecules can also become permanently or semi-permanently entrapped within the pure cis-polyisoprene polymer. While the Lewis-acidic precursor DEZ35 may potentially form an adduct to the alkene functional group, the weak absorption of alkene in IR spectroscopy makes this difficult to ascertain. Thus, further investigations, including a computational investigation and/or in situ spectroscopy are likely necessary.
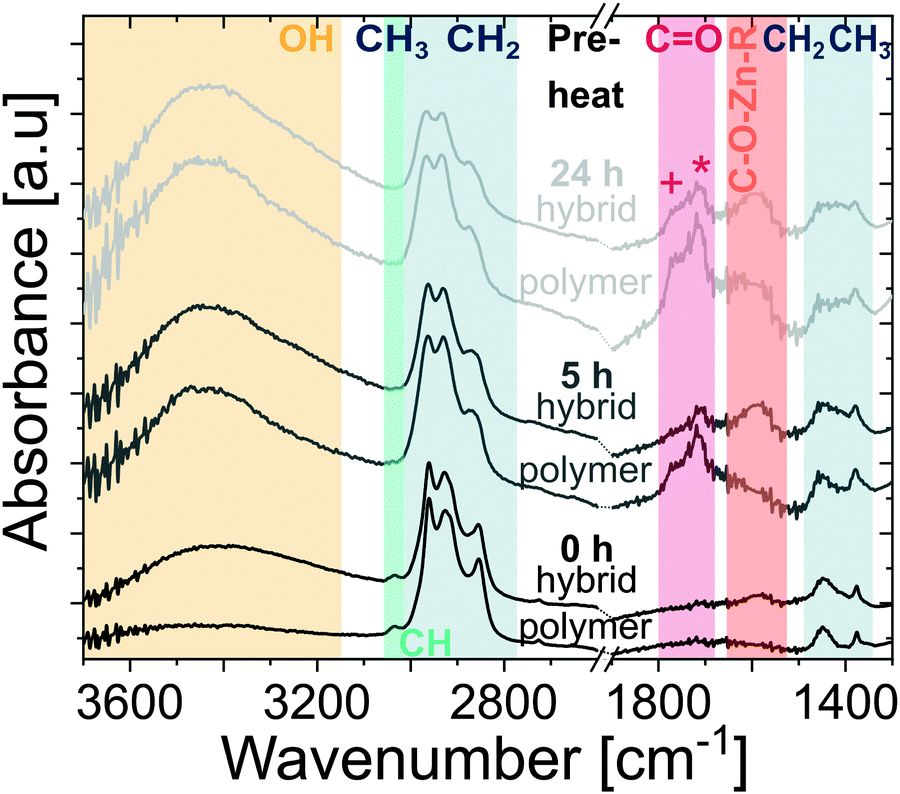 | ||
| Fig. 6 ATR-FTIR spectra of polyisoprene films before (same data as in Fig. 2(b)) and after infiltration, for polyisoprene films pre-heated at 80 °C in air for different durations (indicated above the curves) prior to infiltration. Colored regions refer to O–H stretching (∼3400 cm−1), C–H stretching (∼3036 cm−1), CH3 and CH2 stretching (2800–2900 cm−1), CO stretching (+ and * referring to carboxylic acids (∼1766 cm−1) and ketones (∼1714 cm−1), respectively), C–O–Zn–R (∼1550–1650 cm−1), and CH2 and CH3 deformation (1370–1450 cm−1). Spectra are scaled to their respective maximum value, offset, and cut to the regions where the main absorptions occur (strong absorption of glass substrate below 1300 cm−1). Dotted lines at the axis break serve as a guide for the eye. | ||
Upon further inspection of this entire IR data set (see Fig. S6, ESI†), it appears that the relative intensity of remaining CO groups after infiltration is higher for films pre-heated for a longer duration (see Fig. S6, ESI†). One explanation is that these groups are inaccessible for reaction with DEZ. A possible reason is that these more oxidized polymers react with more DEZ molecules in the near surface region, creating a dense hybrid near-surface layer that hinders further DEZ diffusion. If true, this process should lead to a non-uniform distribution of inorganic throughout the hybrid layer's thickness. To investigate the distribution of Zn within the polymer layer, SIMS was performed on a hybrid film that was preheated for 24 h prior to infiltration. The SIMS depth profile showing Zn+, C+, and Si+ signals is shown in Fig. 7. The Zn concentration drops precipitously at about 2/3 of the film's thickness, suggesting that for these conditions it is not evenly distributed across the ∼60 nm hybrid film thickness. Combined with the FTIR data, these results begin to suggest that the greater number of functional groups in the longer pre-heated films lead to a near-surface hybrid layer that significant inhibits diffusion and full infiltration.
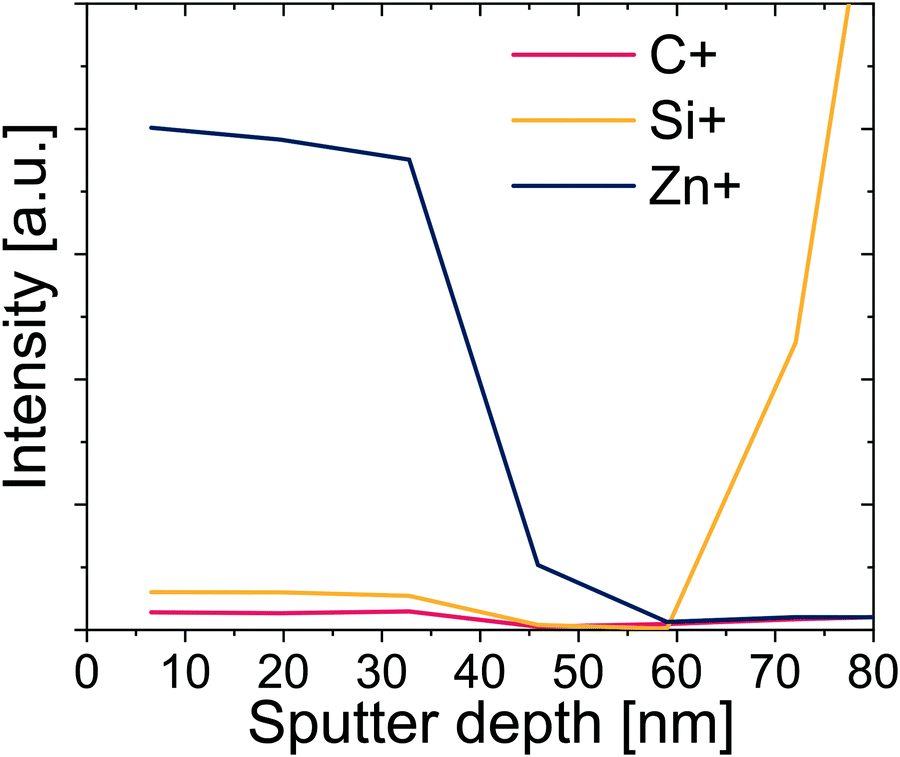 | ||
| Fig. 7 SIMS profiles for Zn+, Si+, and C+ as a function of sputter depth for an infiltrated polyisoprene films which was preheated for 24 h at 80 °C prior to infiltration. | ||
Fig. 8 plots the Zn loading for polyisoprene films pre-heated for different durations. Analyzing these data allows us to partially deconvolute the effects of polymer relaxation from the creation of reactive functional groups due to polymer oxidation. For this purpose, we divide the curve into three regions. In region I, a strong increase in the amount of inorganic loading is observed with increasing pre-heating time. The increase in loading can be related to the creation of functional groups, and the effect is greater than the loss of free volume due to relaxation. In Region II, Zn loading decreases with pre-heating time. As shown in Fig. 2(a), Region II also corresponds to a significant decrease in neat polymer film thickness with pre-heating. This decrease in film thickness is attributed to a loss of free volume, which appears to have a greater effect on the loading than the addition of more functional groups, leading to a decrease in Zn loading. For the longest pre-heating times (Region III), in which both the polymer relaxation and chemical oxidation reach completion (Fig. 2), Zn uptake becomes constant irrespective of pre-heating time. Based on FTIR (Fig. S6, ESI†) and SIMS (Fig. 7) data, it is likely that films within Region III are also not fully infiltrated. Instead, these films only have a saturated near-surface layer, likely of similar thickness dictated by the high concentration of reactive functional groups. It is also possible that the decrease in Zn loading in region II could be a combination of lower free volume and reduced infiltration due to the blocking layer. In fact, the fast and then slow diffusion regimes of Fig. 4b may be indicative of these different contributions to the inorganic loading process.
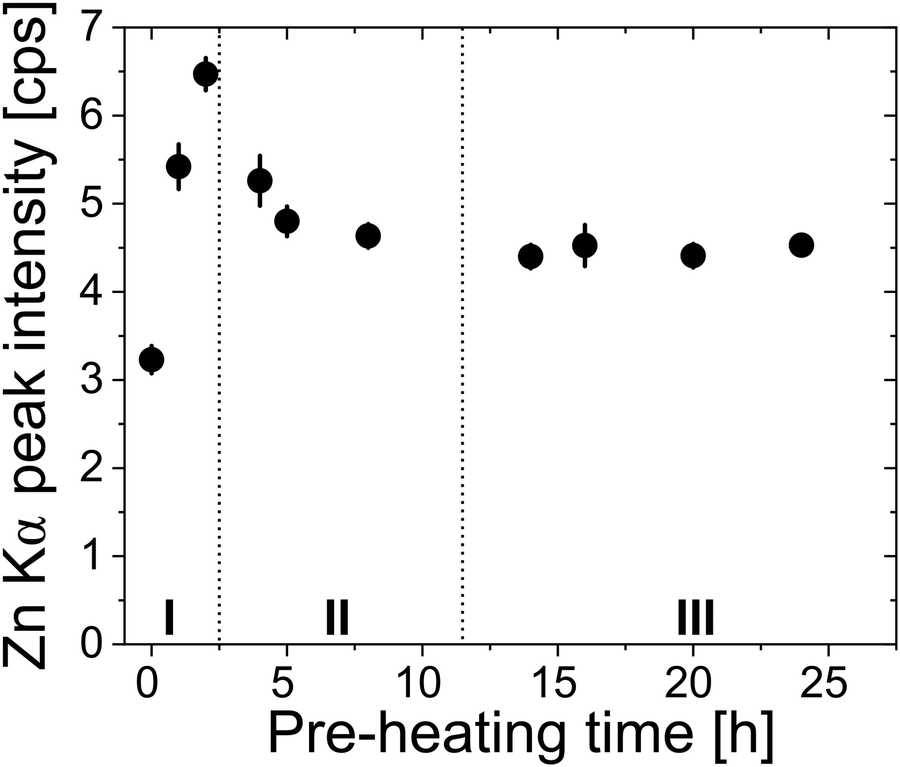 | ||
| Fig. 8 XRF measurements of zinc concentration in hybrid films as a function of cis-polyisoprene pre-heating time prior to infiltration. General sample size per data point is n = 2. Error bars represent the standard deviation. | ||
Fig. 9 provides a schematic sketch summarizing our current understanding of the mechanisms controlling DEZ/ZnO entrapment in polyisoprene polymers as a function of pre-heating time. The infiltration of unheated polyisoprene films results in a small to modest amount of loading. While the DEZ is permanently entrapped, the exact chemical bonding mechanism is not known. Increasing pre-heating times lead to relaxation of the polyisoprene film and increased loading due to creation of functional groups through oxidation. Further increase of pre-heating time leads to further relaxation and dense loading in the top of the film. This dense hybrid layer blocks further sorption and diffusion of the precursor, leaving uninfiltrated regions at the bottom of the film.
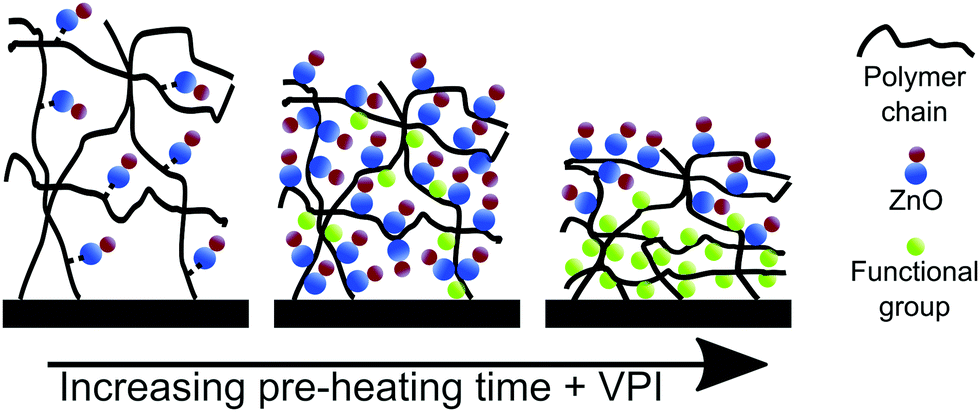 | ||
| Fig. 9 Schematic of ZnO loading into polyisoprene as a function of polymer pre-heating time. Pre-heating is believed to affect both the concentration of functional groups reactive to DEZ and the free volume in the polymer. | ||
F. Dissolution of ZnO-polyisoprene hybrid films
Dissolution experiments are performed to better understand the physicochemical structure of the ZnO-polyisoprene hybrid materials. Hybrids are tested against toluene because it is a good solvent for the neat cis-polyisoprene polymer. Fig. 10 plots the normalized thickness as a function of immersion time for (a) pure polyisoprene polymers pre-heated for 0 h, 5, h and 24 h and (b) infiltrated ZnO-polyisoprene hybrids formed from these various preheated conditions.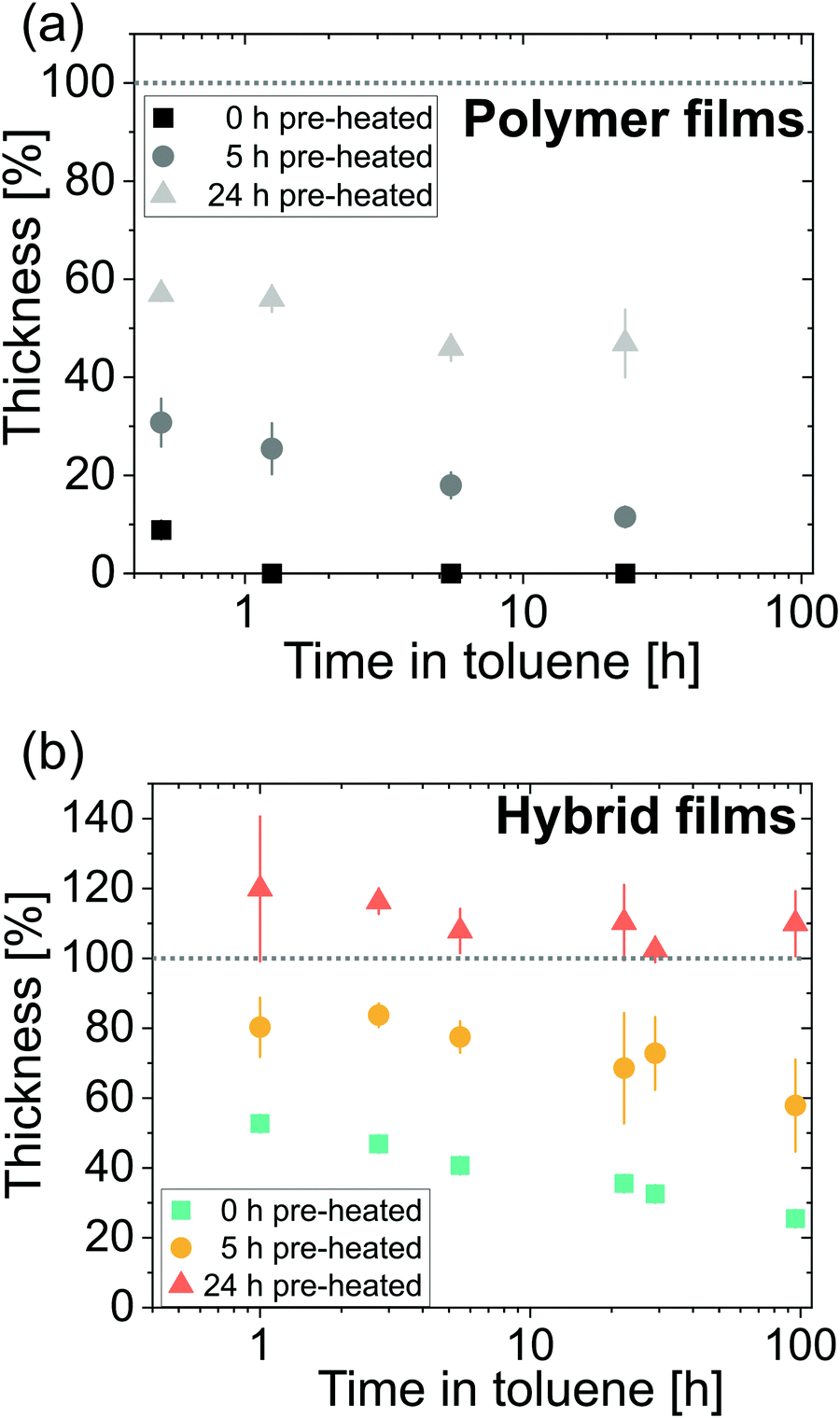 | ||
| Fig. 10 Normalized film thickness of (a) pure and (b) infiltrated polyisoprene films as a function of time in toluene. Polyisoprene films were pre-heated for either 0 h, 5 h, or 24 h prior to (a) dissolution or (b) infiltration and dissolution. Dotted lines indicate normalized film thickness (100%) prior to immersion in toluene. Sample size per data point n = 2. Error bars represent the standard deviation from measurements on multiple films. | ||
Polyisoprene polymers (Fig. 10(a)) rapidly lose thickness upon immersion in toluene. All films show a loss of at least 40% in thickness within 30 min of immersion. The unheated neat polymer dissolves most rapidly and is completely dissolved within 1 h. Neat polymers that are pre-heated prior to immersion in toluene show increased stability. The 5 h heated film loses around 90% of thickness within 25 h, but at least 50% of the 24 h heated film survives for 25 h of immersion. This stability is likely the result of oxidative cross-linking.22
Infiltration of the films leads to increased stability for all pre-heating conditions. Unheated film and films pre-heated for 5 h do show some dissolution but at a much slower rate than the pure polymer films. Films which are pre-heated for 24 h prior to infiltration swell upon immersion (normalized thickness exceeds 100%). This result suggests that these hybrid materials are likely chemically cross-linked. For 5 h pre-heating, this cross-linking is probably incomplete. For unheated films, precursors may not be bound to the polymer at all, and rather act to simply inhibit polymer reptation and simply slow the dissolution rate.
The ability to increase chemical stability of polymers in solvents by VPI has been reported for different systems.8,15 In a recent study by McGuinness et al.,8 the stability of AlOx-poly(methyl methacrylate) hybrid films was tested in various solvents. The authors found that the infiltration temperature controlled the physicochemical structure of the final hybrid material and its resultant chemical dissolution performance. Low infiltration temperatures led to unbound inorganic in the polymer matrix whereas high temperatures led to chemical bonds between the precursor and the moieties of the polymer as well as cross-linking. Films infiltrated at high temperatures were found to be stable in all investigated solvents whereas films infiltrated at low temperature showed dissolution in some solvents, including dissolution of the inorganic in water. While the effect of infiltration temperature on dissolution behavior was not part of this study, we observe similar trends via the introduction of chemically reactive functional groups through polymer oxidation. Longer pre-heating times lead to the creation of more functional groups in the polymer with which the precursor can chemically react. These chemical reactions lead to more cross-linking that increase chemical stability.
IV. Conclusions
This study demonstrates the ability to use single-exposure vapor phase infiltration of DEZ + H2O on cis-polyisoprene polymers to create ZnO-polyisoprene hybrid materials. Unlike other VPI chemistries, volume changes (i.e., film thickness changes) with infiltration are not well correlated to inorganic mass loading. Volume changes appear to be a convolution of polymer relaxation processes (reduced volume) and mass uptake (volume expansion processes) that are often similar in magnitude. XRF spectroscopy is introduced as an alternative non-destructive method for quantifying the amount of inorganic loading in these hybrid films. Several complicating factors are identified in controlling the infiltration process of polyisoprene rubbers. Both changes in the free volume of the polymer and the potential to oxidize the polymer and form reactive carbonyl and hydroxide groups at modest temperatures (80 °C) contribute to the DEZ sorption, diffusion, and entrapment processes. With modest heating and oxidation of the polymer, it is possible to significantly increase the zinc oxide loading by more than 2×. IR spectroscopy confirms reaction with these oxidized functional groups, revealing both a decrease in the carbonyl intensity and the emergence of a C–O–Zn–R functional group. However, continued heating of the polymer decreases Zn loading. This decrease is attributed to either a decrease in the polymer's free volume or a highly cross-linked near surface layer that prevents further sorption and diffusion, the latter appearing to be more likely. Interestingly, even without these oxide functional groups, the DEZ appears to form a stable adduct within the polymer that is highly resistant to desorption, even without dosing the water co-reactant. However, the resulting hybrid films show significantly different dissolution behavior in toluene, a good solvent for the pure polymer. Infiltrated polyisoprene with negligible functional groups dissolves, albeit slowly, while infiltrated polyisoprene with many reactive functional groups shows much more limited dissolution or even swelling as would be expected for a cross-linked gel. These observations suggest distinct differences in the chemical bonding structure of these organic–inorganic hybrid materials. Further computational studies or in situ process characterization will be necessary to fully understand this behavior, however, significant opportunity exists in exploring these infiltrated hybrid materials as new accelerators for rubber vulcanization or modifiers to alter the physical properties of polyisoprene rubbers.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received partial funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 715403). This material is also based upon work partially supported by the National Science Foundation under Grant # 1921873. J. P. acknowledges personal funding from the Austrian Marshall Plan Foundation for funding his research stay at Georgia Tech. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542174). The authors acknowledge Yi Li for performing XPS measurements, Jamie P. Wooding for performing SIMS measurements, and Yulian Yao for AFM measurements. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of these funding agencies.References
- M. Morton, Rubber Technology, Van Nostrand Reinhold Company Inc, New York, 3rd edn, 1987 Search PubMed.
- K. Roy, S. C. Debnath and P. Potiyaraj, J. Elastomers Plast., 2020, 52, 167–193 CrossRef CAS.
- S. Mostoni, P. Milana, B. Di Credico, M. D’Arienzo and R. Scotti, Catalysts, 2019, 9, 664 CrossRef.
- G. Heideman, R. N. Datta, J. W. M. Noordermeer and B. van Baarle, J. Appl. Polym. Sci., 2005, 95, 1388–1404 CrossRef CAS.
- M. Knite, V. Teteris, A. Kiploka and J. Kaupuzs, Sens. Actuators, A, 2004, 110, 142–149 CrossRef CAS.
- Y. Ikeda and S. Kohjiya, Polymer, 1997, 38, 4417–4423 CrossRef CAS.
- V. S. Raman, A. Das, K. W. Stöckelhuber, S. B. Eshwaran, J. Chanda, M. Malanin, U. Reuter, A. Leuteritz, R. Boldt, S. Wießner and G. Heinrich, RSC Adv., 2016, 6, 33643–33655 RSC.
- E. K. McGuinness, C. Z. Leng and M. D. Losego, ACS Appl. Polym. Mater., 2020, 2, 1335–1344 CrossRef CAS.
- R. P. Padbury and J. S. Jur, Langmuir, 2014, 30, 9228–9238 CrossRef CAS PubMed.
- H. I. Akyildiz, R. P. Padbury, G. N. Parsons and J. S. Jur, Langmuir, 2012, 28, 15697–15704 CrossRef CAS.
- R. Z. Waldman, D. J. Mandia, A. Yanguas-Gil, A. B. F. Martinson, J. W. Elam and S. B. Darling, J. Chem. Phys., 2019, 151, 190901 CrossRef PubMed.
- A. Subramanian, N. Tiwale and C.-Y. Nam, JOM, 2019, 71, 185–196 CrossRef.
- S.-M. Lee, E. Pippel, U. Gosele, C. Dresbach, Y. Qin, C. V. Chandran, T. Brauniger, G. Hause and M. Knez, Science, 2009, 324, 488–492 CrossRef CAS.
- W. Wang, F. Yang, C. Chen, L. Zhang, Y. Qin and M. Knez, Adv. Mater. Interfaces, 2017, 4, 1600806 CrossRef.
- E. K. McGuinness, F. Zhang, Y. Ma, R. P. Lively and M. D. Losego, Chem. Mater., 2019, 31, 5509–5518 CrossRef CAS.
- D. H. Yi, C.-Y. Nam, G. Doerk, C. T. Black and R. B. Grubbs, ACS Appl. Polym. Mater., 2019, 1, 672–683 CrossRef CAS.
- C. Z. Leng and M. D. Losego, Mater. Horizons, 2017, 4, 747–771 RSC.
- D. R. Burfield and K. L. Lim, Macromolecules, 1983, 16, 1170–1175 CrossRef CAS.
- B. D. Piercy and M. D. Losego, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2015, 33, 043201 Search PubMed.
- K. A. M. dos Santos, P. A. Z. Suarez and J. C. Rubim, Polym. Degrad. Stab., 2005, 90, 34–43 CrossRef CAS.
- R. A. Saunders and D. C. Smith, J. Appl. Phys., 1949, 20, 953–965 CrossRef CAS.
- X. Colin, L. Audouin and J. Verdu, Polym. Degrad. Stab., 2007, 92, 886–897 CrossRef CAS.
- J. L. Morand, Rubber Chem. Technol., 1977, 50, 373–396 CrossRef CAS.
- M. Zaghdoudi, A. Kömmling, M. Jaunich and D. Wolff, Polymers, 2019, 11, 1280 CrossRef.
- T. Sato, K. Sakota, M. Egami, K. Shirano and T. Okaya, J. Appl. Polym. Sci., 1977, 21, 981–988 CrossRef CAS.
- G.-Y. Li and J. L. Koenig, Rubber Chem. Technol., 2005, 78, 355–390 CrossRef CAS.
- Y. Li, M. Kröger and W. K. Liu, Polymer, 2011, 52, 5867–5878 CrossRef CAS.
- C. Z. Leng and M. D. Losego, Phys. Chem. Chem. Phys., 2018, 20, 21506–21514 RSC.
- G. J. Van Amerongen, Rubber Chem. Technol., 1951, 24, 109–131 CrossRef CAS.
- A. R. Berens and H. B. Hopfenberg, J. Membr. Sci., 1982, 10, 283–303 CrossRef CAS.
- E. Cianci, D. Nazzari, G. Seguini and M. Perego, Adv. Mater. Interfaces, 2018, 5, 1801016 CrossRef.
- W. Xie, S. Khan, O. J. Rojas and G. N. Parsons, ACS Sustainable Chem. Eng., 2018, 6, 13844–13853 CrossRef CAS.
- G. T. Hill, D. T. Lee, P. S. Williams, C. D. Needham, E. C. Dandley, C. J. Oldham and G. N. Parsons, J. Phys. Chem. C, 2019, 123, 16146–16152 CrossRef CAS.
- E. C. Dandley, C. D. Needham, P. S. Williams, A. H. Brozena, C. J. Oldham and G. N. Parsons, J. Mater. Chem. C, 2014, 2, 9416–9424 RSC.
- X. Ye, J. Kestell, K. Kisslinger, M. Liu, R. B. Grubbs, J. A. Boscoboinik and C.-Y. Nam, Chem. Mater., 2017, 29, 4535–4545 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00304b |
| This journal is © The Royal Society of Chemistry 2020 |