
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe anomaly in bioactive sol–gel borate glasses†
William C.
Lepry
 and
Showan N.
Nazhat
*
and
Showan N.
Nazhat
*
Department of Mining and Materials Engineering, McGill University, Montreal, QC, Canada. E-mail: showan.nazhat@mcgill.ca
First published on 7th July 2020
Abstract
Borate glasses differ from silicates as they do not always exhibit linear property trends with the addition of modifying elements. Rather, various property maxima, notably boron coordination, are observed at different modifier contents, a phenomenon termed as the borate anomaly. Furthermore, the higher dissolution rates of bioactive borate glasses have led to their consideration in not only hard tissue regeneration, but also in soft tissue repair. Yet, many of these borate compositions are based on traditional bioactive silicate glasses, which may not be optimized for their chemistries. Here, by exploiting the sol–gel process, we have for the first time extended the range of amorphous borate glasses to (x)CaO–(100−x)B2O3, where x = 20, 30, 40, 50, 60, 70 (mol%) to investigate the effect of alkaline-earth content on glass structure, texture, and bioactivity. Magic angle spinning nuclear magnetic resonance, infrared spectroscopy, and surface area and pore volume values confirmed that modifier content affected both the structural and textural properties of the glass in accordance with the borate anomaly. Modifier content also dictated the rate and conversion of glass to either calcite or hydroxycarbonated apatite in simulated body fluid, in vitro, according to X-ray diffraction, infrared spectroscopy, and scanning electron microscopy. We believe this is the first report demonstrating the borate anomaly in bioactive sol–gel glasses – over the widest compositional range for a binary alkaline-earth borate system – in terms of both structural and textural properties. The results aim to help provide a basis for designing borate glasses for targeted biomedical applications.
Introduction
Adding modifiers, such as alkali (M2O) or alkaline-earth (MO) cations, to silicate glasses disrupts the glass network by forming non-bridging oxygens, which help charge balance the modifier cations and reduce the overall glass network connectivity.1 This typically results in a linear decrease in the coordination number of the glass forming unit and decreases the glass transition temperature (Tg)2 and hardness3 as well as surface area and pore volume in sol–gel derived silicate glasses.4 In contrast to silicate glasses, where the silicon tetrahedron forms the main structural unit, vitreous borate glasses are based on planar, trigonally coordinated BO3 groups, which can also form larger structural units, such as boroxol rings.5 Here, modifier addition initially increases the glass network connectivity by forming 4-coordinated BO4− units up to a relative maxima, which then decreases upon further modifier additions, and giving rise to the borate anomaly.6–8 This maxima of tetrahedrally coordinated boron units (N4) occurs around 35–40 mol% depending on the type and amount of M2O content, according to nuclear magnetic resonance1 with the range being generally higher for MOs.9 However, this range is beyond the composition where the reversal of other physical properties, such as Tg (≈27 mol% M2O) and coefficient of thermal expansion (≈20 mol% M2O) making it inadequate to use 4-coordinated boron values alone to explain this phenomenon.10The borate anomaly can be further explained by the equilibrium reactions for the two isomers of metaborate or orthoborate units that are either 3- or 4-coordinated, and their equilibrium, which occurs at different modifier concentrations.11,12 The type and amount of alkali or alkaline-earth modifiers relative to B2O3 (M2O/B2O3 or MO/B2O3, respectively, and typically termed “R”), also determines whether they serve as charge compensating or modifying roles in the glass structure; greatly impacting its properties.9
Although the inherently lower network connectivity of borate glasses generally limits their commercial applications,13 they have demonstrated great promise in biological applications attributable to their more rapid dissolution and full conversion to bone mineral (hydroxycarbonated apatite; HCA) when compared to silicate-based glasses.14–16 Their ability to quickly release ions has also propelled their use in soft tissue repair, such as wound healing.17–21 Nevertheless, questions still remain about using mineralizing glasses in soft tissue sites.22
The bioactive properties of borate glasses can be further enhanced by using the sol–gel process, which produces glasses with increased textural properties.23 Previously, we have reported on a wide compositional range of 4-component sol–gel derived borate glasses (SGBGs; 36–61 mol% B2O3) that have demonstrated specific surface areas and porosities at least two orders of magnitude greater than the melt-quench equivalent, leading to a 25-fold increase in HCA conversion rate.23 We have also shown that SGBGs can be produced using a variety of sol–gel precursors and processing parameters while still maintaining their high bioactivity, by converting to HCA within 2 hours,24 with ternary, sodium-free compositions also exhibiting the same level of bioactivity.25 SGBGs have also been incorporated into polymer matrices to enhance composite bioactivity,26 while compositions doped with silver, have demonstrated antibacterial properties.27
Interest in bioactive borate glasses has led to many ‘unique’ compositions27–43 such as “1605”.44–46 However, despite fundamental differences between borate and silicate glass structure, many borate compositions, including our previous work,23–26,47 have been based on “traditional” silicate glass compositions like Bioglass® “45S5”,15,23–26,47–50 “13–93”,14,16,17,51–69 and/or “S53P4”70–73 where SiO2 is either partially or fully substituted by B2O3.74 The tendency to use these compositions likely stems from the fact that many of these silicate glass compositions are commercially and clinically available. Yet, it is well known that the addition or full substitution of borate in silicate-based compositions will drastically change glass structure and properties. While many of these borate-substituted glass compositions do improve bioactivity,15,49,54 there is still an opportunity to optimize borate glass compositions for targeted tissue engineering applications,19 by using knowledge of their unique chemistries and structure.11,74,75 For example, although calcium is a critical component of bioactive glasses, it is difficult to process amorphous high-calcium containing compositions through traditional melt-quench methods.9,76,77 Furthermore, compared to alkali-borate glasses, there is distinct lack of studies on alkaline-earth-borates,76,78,79 in particular on their bioactivity.
Early investigated borate sol–gel glass systems were based on binary alkali–borate glasses ranging from 10–40 mol% modifier addition,80–83 along with doping of other elements such as Si84 and Ti.85 In terms of alkaline earth sol–gel borate glasses, a wide range binary79 and some ternary86 MgO containing compositins have also been examined. Overall, little has been explored regarding borate-based sol–gel glasses,87,88 though borate modifier additions are common in other sol–gel glasses, mostly to increase the degradation rate, thus improving the bioactivity.89–91 Many of these compositional ranges, however, were not wide enough to properly examine the borate anomaly.
In this study, by exploiting our sol–gel process,23 we have extended the range of six binary formulations: (x)CaO–(100 − x)B2O3 where x = 20, 30, 40, 50, 60, and 70 (mol%) to investigate the anomaly effect of alkaline-earth addition on borate glass properties. It was determined that both the structural and textural properties of the sol–gel glasses corresponded with the borate anomaly. Calcium content also impacted glass bioactivity by dictating both, the rate and conversion to either calcite or HCA in simulated body fluid (SBF). We anticipate this unprecedented extension of SGBG compositional range will not only advance our knowledge to better optimize borate glass compositions for targeted tissue engineering, but also their design in other specialty borate applications.
Experimental section
Sol–gel processing and compositional determination
Table 1 gives an overview of the SGBG compositions investigated in this study. The glasses were fabricated based on a modified, previously described method.23 For example, to make 1 g of “B60”, 1.16 g of boric acid (≥99.5%) and 11 mL anhydrous ethanol (Sigma Aldrich, Canada) were mixed and magnetically stirred in a watch glass-covered Teflon beaker at 40 ± 3 °C to aid dissolution. Once the solution became clear, 5.93 g of calcium methoxyethoxide (20% in methoxyethanol, Gelest, USA), was added and the sol was mixed for an additional 30 min and then aged in a sealed polypropylene vial at 37 °C for 10 days. The sol was then transferred to crystallization dishes and air dried at room temperature (RT) for 2 days, forming a gel, followed by oven drying at 120 °C for a further 2 days. Finally, all glasses underwent the same calcination step at 400 °C in air at a rate of 3 °C min−1, with a 2 h-dwell period, followed by furnace cooling. The calcined glasses were then ground to a particle size fraction of 25–75 μm and stored in a desiccator until analysis (Fig. S1, ESI†).| Ca20 | Ca30 | Ca40 | Ca50 | Ca60 | Ca70 | |
|---|---|---|---|---|---|---|
| a Each glass was measured in triplicate and the standard deviation was less than 1%. | ||||||
| CaO | 20 (26.1) | 30 (33.1) | 40 (42.1) | 50 (51.9) | 60 (62.6) | 70 (73.3) |
| B2O3 | 80 (73.9) | 70 (66.9) | 60 (57.9) | 50 (48.1) | 40 (37.4) | 30 (26.7) |
The compositions of the SGBGs were quantified using an inductively coupled plasma–optical emission spectrophotometer (Thermo Scientific iCAP 6500, USA) where 0.01 g of glass particles (n = 3) was dissolved in pure nitric acid for 2 h at 95 °C followed by dilution. Serially diluted solutions of boron (0.5, 5, 50 ppm) and calcium (0.2, 2, 20 ppm), were used as standards.
Glass particle characterization
The average size (Davg) and median diameter (D50) of the glass particles was determined using a Horiba LA-920 (ATS Scientific Inc., Canada). The textural properties were measured with nitrogen gas adsorption and desorption isotherms collected with a Micromeritics TriStar 3000 (Micromeritics Instrument Corporation, USA) gas sorption system (n = 3). Specific surface area (SSA) values were determined using the Brunauer–Emmett–Teller (BET) method92 while the average pore width and pore volume (PV) values were calculated using the Barrett–Joyner–Halenda (BJH) method.93Magic angle spinning nuclear magnetic resonance (MAS-NMR)
11B magic angle spinning (MAS) NMR analysis was carried out on a Bruker Advanced NMR spectrometer in the NMR-3 at Dalhousie University (Canada) with a 16.4 T magnet (224.67 MHz 11B Larmor frequency) using a probe head for rotors of 2.5 mm diameter. The samples were spun at 10 and 25 kHz to determine center bands and to identify spinning sidebands. In addition, a spectrum of an empty rotor was acquired under identical conditions. The NaBH4 resonance served as secondary chemical shift standard at −42.1 ppm relative to BF3·Et2O. The 11B NMR spectra were accumulated using a single pulse length of 0.56 μs corresponding to a 15 degree pulse angle in the nearly cubic environment of NaBH4. The small pulse angles were chosen to allow the comparison of sites with different quadrupole couplings. Rough spin lattice relaxation times were determined using a saturation recovery sequence. The pulse repetition times were chosen to be on the order of the longest relaxation time, which varied between 4 and 25 seconds. Between 80–160 scans were accumulated varying with the boron concentration. The substantial boron background was removed by subtracting the spectrum of an empty rotor acquired under similar conditions having accumulated 320 scans. The integral values are given between 23.0 to 6.0 ppm and from 6.0 to −5.0 ppm.Bioactivity in SBF
Kokubo's SBF (pH 7.4) was used to examine the in vitro mineralization of the glasses.94 Glass particles were added to sterile 50 mL falcon tubes containing SBF at a 1.5 mg mL−1 ratio and stored at 37 ± 1 °C. The vials were gently agitated twice per day to prevent agglomeration. The ability of the glasses to form HCA was examined at 0.5 h, 2 h, 6 h, 1 d, and 7 d time points, where the powders were gently rinsed with deionized water then twice with anhydrous ethanol, dried overnight at room temperature, and then dried in an oven at 60 °C for 1 day.Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR)
ATR-FTIR spectroscopy of the glass particles, as made and post immersion in SBF was carried out between 4000 and 650 cm−1 with a resolution of 4 cm−1 using 64 scans per sample using a Spectrum 400 (PerkinElmer, USA). The collected spectra were baseline corrected then normalized to the total area surface area under absorption bands using Spectrum software (PerkinElmer, USA).X-ray diffraction (XRD)
XRD diffractograms of the glass particles, as made and post immersion in SBF were analyzed using a Bruker D8 Discover X-ray diffractometer (Bruker AXSS Inc., USA) equipped with a CuKα (λ = 0.15406 nm) target set to a power level of 40 mV and 40 mA. Three frames were collected from 15–75 2 theta (°), using an area detector, and merged in post processing while phase identification was carried out using X’Pert Highscore Plus (PANalytical, Netherlands).Scanning electron microscopy (SEM)
SEM imaging of the glass particles, as made and post immersion in SBF was carried out on Pt sputter coated samples and analyzed with an Inspect F50 Field Emission Scanning Electron Microscope (FEI Corporation, U.S.A.) at 5 kV.Results and discussion
Compositional range and sol–gel processing
Table 1 gives an overview of the investigated SGBG compositions with the measured values used in this study. There was an average borate loss of 3.2 ± 1.4 mol%, which is similar to previous melt-quench95 and sol–gel79,80 borate glasses. Similar to our previously processed sodium-free SGBGs25 or compositions made with either tri-ethyl or -methyl borate as precursors,24 no gelation was observed after 10 days of ageing. However, upon drying in air, a gel-like solid structure was formed (“dried”, Fig. S1, ESI†), which is similar to previous mechanisms of gel formation observed in earlier alkali-borate sol–gel glasses.96 After calcination and grinding, most glasses were off-white in color except for Ca20 which became darker (Fig. S1, ESI†). This change in color can be attributed either to carbon deposits from the partially hydrolyzed alkoxide groups84 or to pyrolysis of residual organics80 as has been previously observed at higher calcination temperatures with other SGBGs.23,24Prior studies have reported that the glass forming region of calcium-borates was limited between about 20 to 60 mol% CaO since alkaline-earths tend to either crystalize at high content or exhibit liquid–liquid phase separation at low content.9,76,77 This may explain why there is far less literature on alkaline-earth borates compared to alkali-borate glasses.9,76,78,97–99 Although Manupriya et al. previously reported on a series of melt-quench compositions of similar range described in this study, there was insufficient characterization data regarding their amorphous nature.100 By using our low-temperature sol–gel process, to the best of our knowledge, this is the first report of an expanded compositional range up to 73.3 mol% CaO, as indicated by the two broad, amorphous humps in XRD diffractograms (Fig. S2, ESI†). While modifiers are typically not the main component of a glass composition, these ‘invert-glasses’101 have been explored with borates10,102 and, similar to the borate anomaly, they often exhibit relative minima and maxima in property trends.103
Structural analysis of calcined glasses
The SGBG bonding regions were examined using ATR-FTIR spectroscopy (Fig. 1a). Three main regions associated with borate-based glasses were identified: the B–O stretching of the BO4 units (850–1200 cm−1), the B–O stretching of the BO3 units (1200–1500 cm−1), and the B–O–B bending of the BO3 units, as indicated by the band at ∼720 cm−1.83,86,104 The B–O stretching of boroxol rings are characteristic of the shoulder peak at ∼863 cm−1, whereas the B–O linkages of BO4 are indicated by the broad band between ∼942 and ∼1000 cm−1.90,105–107 Ca60 and 70 showed a greater shoulder peak at around 862 cm−1 with a less-defined BO4 region. Fig. 1b shows the 11B MAS-NMR line spectra, which provide geometrical information on the borate unit. All SGBGs exhibited a large, sharp peak near 0 ppm, which can be attributed to 11B nuclei occupying a relatively symmetric site in the chemical structure (e.g., BO4 units). The stronger quadrupole interaction of BO3 produced a broader resonance between 20 and 10 ppm. These relative integral intensities shift according to modifier content over the range of SGBGs.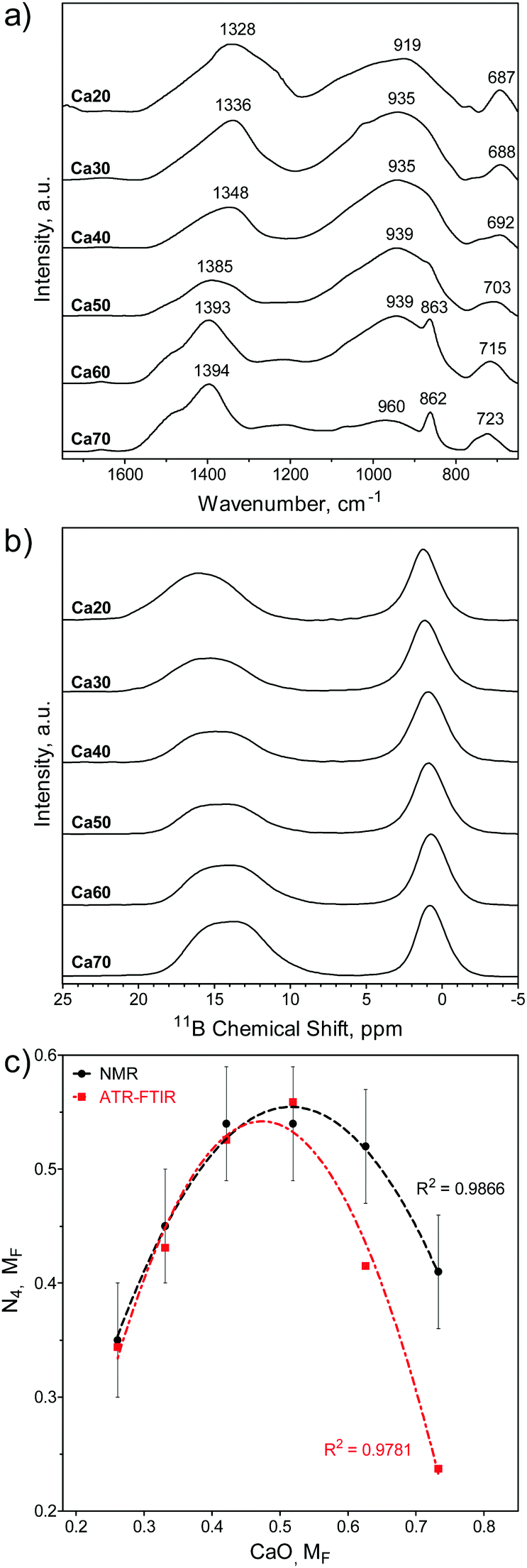 | ||
| Fig. 1 Structural properties of SGBGs. (a) ATR-FTIR and (b) 11B MAS-NMR line spectra of calcined SGBGs indicated typical peaks relating to 3- and 4-coordinated boron. (c) Plot of the relative 4-coordinated regions from ATR-FTIR (ESI†) and 11B MAS-NMR spectra demonstrated the borate anomaly as a relative maxima in N4 units was observed at approximately 0.5 MF CaO. Simple quadratic equations using a least squares fit are shown to help demonstrate the trend. Linear fits and “R” plots are shown in Fig. S3 (ESI†). | ||
The molecular fraction (MF) of 4-coordinated borate units (N4) can be visualized graphically over the compositional range in Fig. 1c using the ATR-FTIR (Table S1, ESI†) and the deconvolved 11B MAS-NMR spectra (Fig. 1b), which is also plotted in terms of ‘R’ (CaO/B2O3 mol%; Table S2 and Fig. S3, ESI†). The relative maxima in N4 fraction for 11B MAS-NMR spectra is the same at 42.1 and 51.9 mol% CaO (Fig. 1c) which is slightly higher than the 11B MAS-NMR N4 maxima of similar melt-quench glasses (around 40 mol% CaO).76 The relative N4 maxima using the relative maxima in absorption of the 3- and 4-coordinated bonding regions in ATR-FTIR (Table S1, ESI†) occurs at 51.9 mol% CaO. This increased N4 maxima using IR spectra has also been previously observed with binary CaO–B2O3 melt-quench glasses which was reported to being at around 45 mol% CaO.9 More compositional data are needed to better elucidate the exact maxima.
Previous FTIR data have shown that N4 units decreased with increasing M2O field strength, but over >45 mol%, the trend reversed in NMR data.76 Among binary M2O–B2O3 glasses (where M = Li, Na, K, Rb, Cs) the N4 unit fraction is consistent up to 33 mol% M2O (e.g., R = 0.5), but it was found that Li has the highest fraction of B4 units and that heavier alkali elements result in larger chemical shifts.108
Textural and physical properties
Fig. 2 shows a plot of the SGBG textural properties versus MF (summarized in Table S2, ESI†). All particles were ground to a similar median particle size (D50) range to directly compare their textural properties. There was an increase in SSA (Fig. 2a) and PV (Fig. 2b) values with an increase in MF, reaching a relative maximum at 42.1 mol% CaO, which was followed by a decrease with further CaO addition. There was also a correlation between glass textured surfaces and their SSA and PV values (Fig. S1 and Table S2, ESI†). Pore width did not follow a trend with increasing CaO content.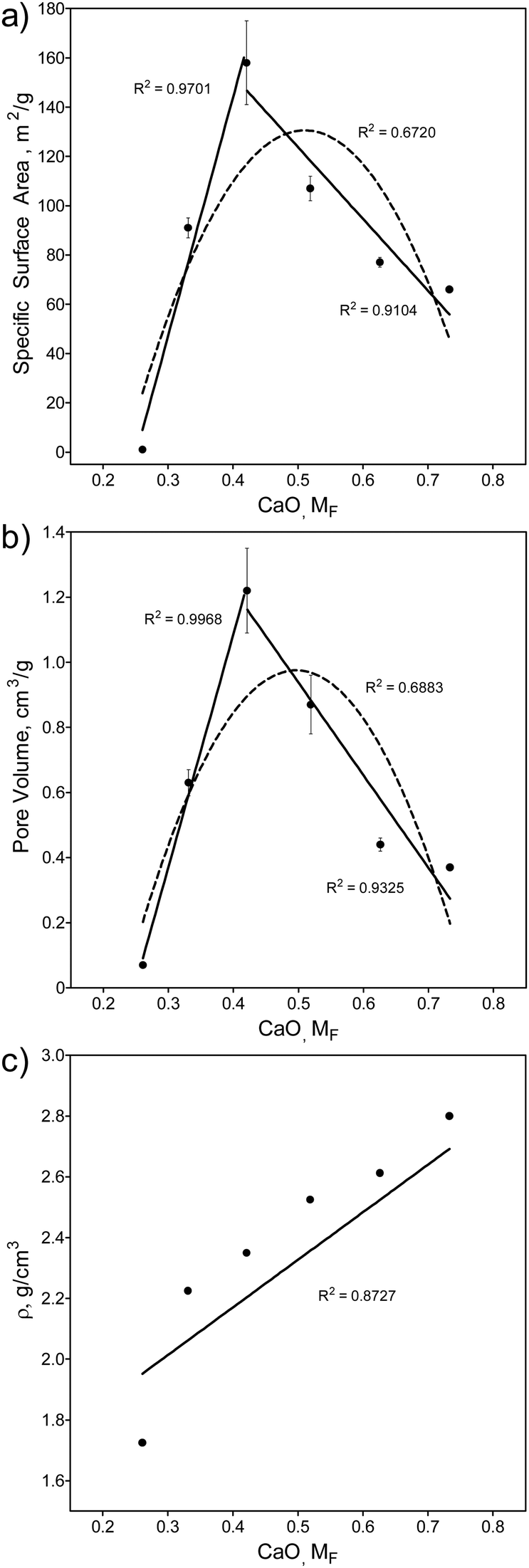 | ||
| Fig. 2 Textural and physical properties of SGBGs versus MF. (a) Specific surface area (SSA) and (b) pore volume (PV) change with CaO content according to the borate anomaly, by indicating relative maxima values. In contrast, (c) density increases linearly with CaO content. Simple linear (solid line) and quadratic (dashed line) equations using a least squares fit are shown to help demonstrate the trend. Fig S4 (ESI†) shows these values plotted in terms of “R”. | ||
The observed increase in surface area can be attributed to the higher glass network connectivity, as previously observed with SGBGs23 and other sol–gel systems.4 However, this trend is in direct contrast to binary CaO–SiO2 glasses,109,110 which demonstrated a linear decrease and increase in SSA and PV, respectively, with an increase in CaO content (Fig. S5 and S6, ESI†). For the borate compositional range in this study, SSA and PV values followed a similar trend to the structural ATR-FITR and NMR data (Fig. 1). This relative maxima in SSA and PV (42.1 mol% CaO) is similar to the relative maxima of 4-coordinated borate units in 11B MAS NMR, but lower compared to the maxima using ATR-FTIR (Fig. 1c). Similar to the original borate anomaly, where the maximum in N4 species does not necessarily correlate with the relative maxima in property trends,1,5,10 this perhaps is a feature of the borate anomaly in terms of textural properties, where the structure and textural properties do not precisely coincide. This might be due to the effect of calcination temperature, which is known to significantly influence SSA and PV of sol–gel derived glasses.23 On the other hand, the addition of CaO resulted in a linear increase in density (Fig. 2c) which has previously been shown in other borate melt-quench systems77,100 and follows an opposite trend compared to binary CaO–SiO2 glasses (Fig. S7, ESI†).109,110
Bioactivity in SBF
The ability of binary SGBGs to form biologically relevant mineral was examined up to 7 days in SBF. Ionic release rates through glass dissolution and change in pH in SBF were compositionally dependent over this range (Fig. S8a, ESI†). With the exception of Ca20, which showed a slower rate of ionic release, the majority of boron and calcium ions in the SGBGs were rapidly released within 0.5 h (Fig. S8b and c, ESI†). ATR-FTIR spectra (Fig. 3) of the Ca30–60 SGBG range indicated the formation of a phosphate peak within 0.5 h in SBF, as shown by the strong band at ∼1020 cm−1 and its shoulder region at ∼961 cm−1, which are characteristic of the bending modes ν1 of PO43−.111 Carbonate peaks at ∼1470 and ∼1421 cm−1 represent the stretching modes ν1 and ν3 of CO32− respectively, and a sharp peak at around 870 cm−1 indicates the bending mode ν2 of CO32−.112,113 This latter band may also be a combination of the B–O stretching of boroxol rings found in the calcined glasses as well as the bending mode ν2 of CO32−. The ν2 bending mode of water can also be seen by the broad band at ∼1640 cm−1.114,115 At longer immersion times in SBF, more defined peaks were observed, indicating the formation of carbonated apatite, which were not observed in Ca20 until day 7, suggesting a slower conversion rate. Although Ca70 demonstrated phosphate peak formation within 0.5 h, the peaks at ∼870 and ∼1404 cm−1 as well as the ν4 CO32− in plane bending at ∼707 cm−1, were also indicative of calcite formation.116,117 The peaks at ∼871 and 1404 cm−1 decrease in intensity at longer immersion times and the latter appears similar to the bending mode ν2 of CO32−.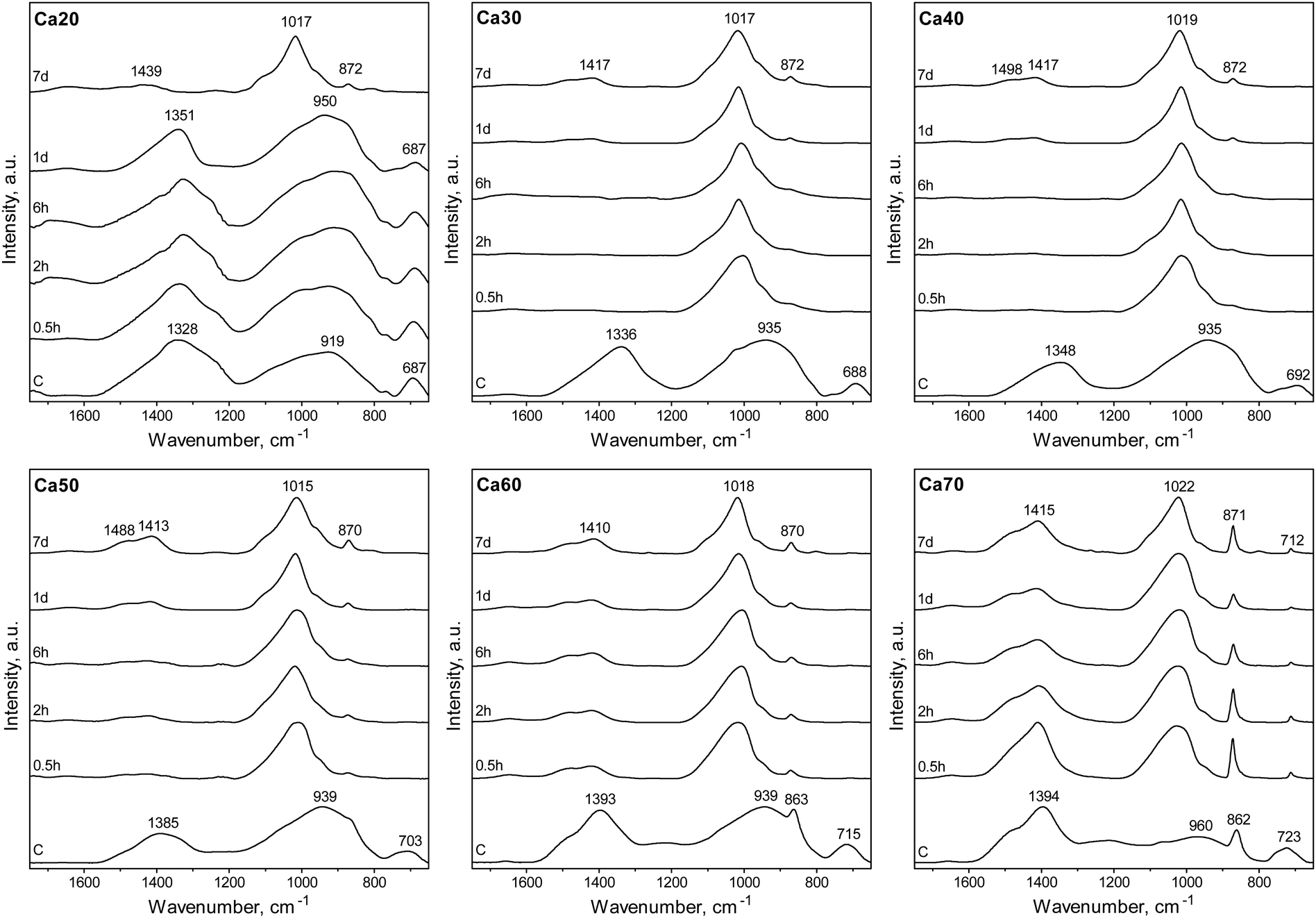 | ||
| Fig. 3 ATR-FTIR spectra of SGBGs as a function of immersion time in SBF. Characteristic apatite-like peak formation initiated within 0.5 h in SBF for all glasses except Ca20 which took 7 days and Ca70 which indicated calcite formation. | ||
Mineral conversion was confirmed by XRD diffractograms in which characteristic HCA peaks, at ∼25 and ∼32° 2θ (“●”, JCPDS 19-272), were observed for Ca30-50 SGBG range after 2 h in SBF (Fig. 4). These broad apatite peaks, which became sharper at longer times in SBF, suggested nanometer-sized or partially crystallized HCA.15 In contrast, peak formation was not observed until day 7 for Ca20. Furthermore, Ca60 and Ca70 did not initially convert to HCA, but calcite (“■”, JCPDS 5-586), which may be attributed to the higher calcium content in these compositions.118 However, at day 7, the prominent HCA peak (∼32° 2θ) began to form, suggesting a combination of mineral formation, and corroborating the ATR-FTIR data (Fig. 3).
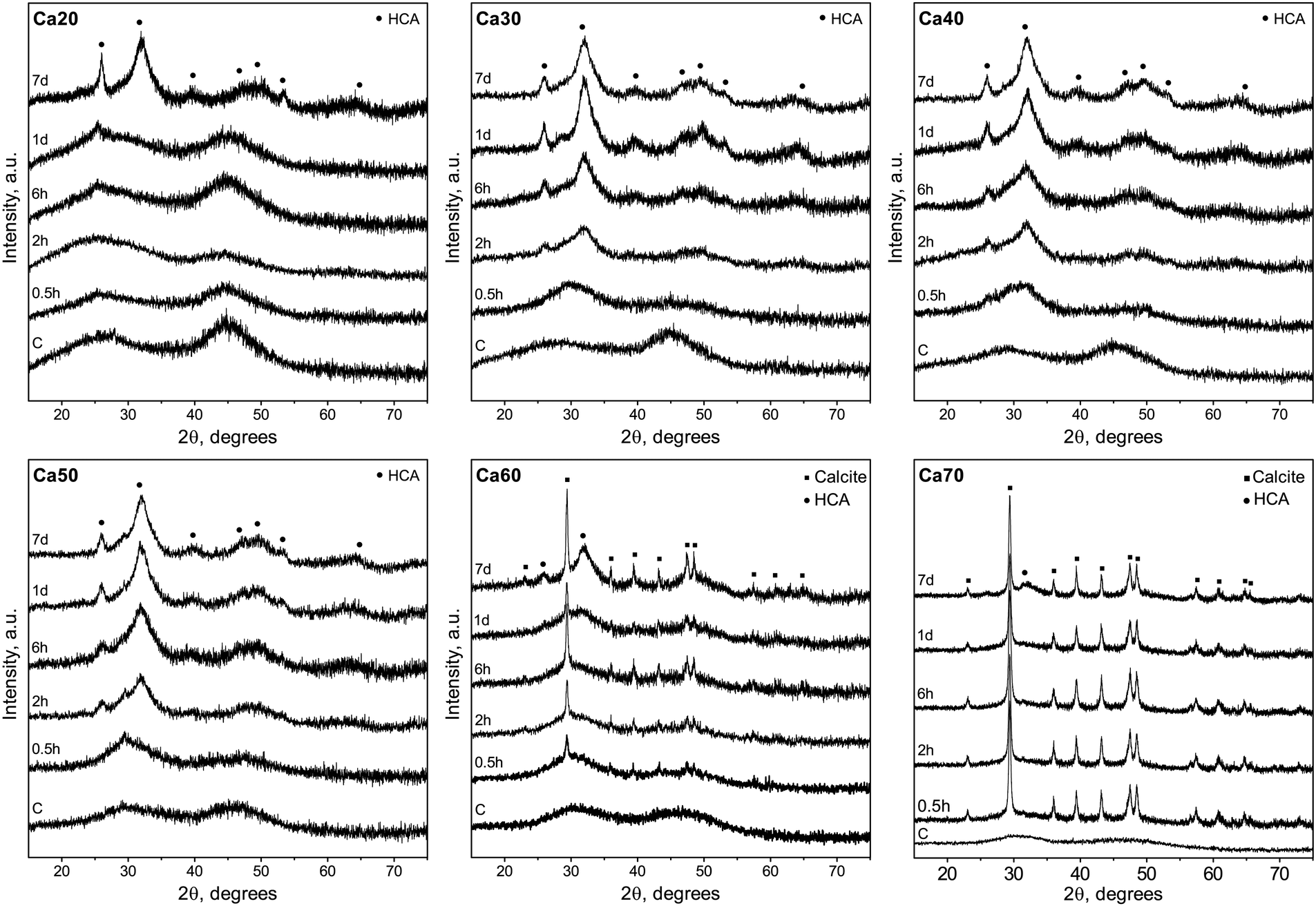 | ||
| Fig. 4 XRD diffractograms of SGBGs as a function of immersion time in SBF. Higher calcium containing glasses initially converted to calcite (“■”, JCPDS 5-586) but showed signs of HCA conversion (“●”, JCPDS 19-272) with longer immersion times. Glasses with less than 0.5 MF CaO favored HCA formation. | ||
SEM micrographs (Fig. 5) confirmed the findings in ATR-FTIR and XRD. For Ca30-50, spherulitic-like HCA crystals formed at 6 h and became more defined by day 7, which is similar to our previously studied SGBG formulations.23,24 The slower conversion of Ca20 was also confirmed by the absence of mineral formation at 6 h and 1 d, whereas at 7 d typical HCA crystals were observed. Ca60 and 70 showed the formation of typical geometrically shaped calcite crystals starting at 6 h. At longer immersion times, a combination of these and spherulitic-like HCA crystals were observed.
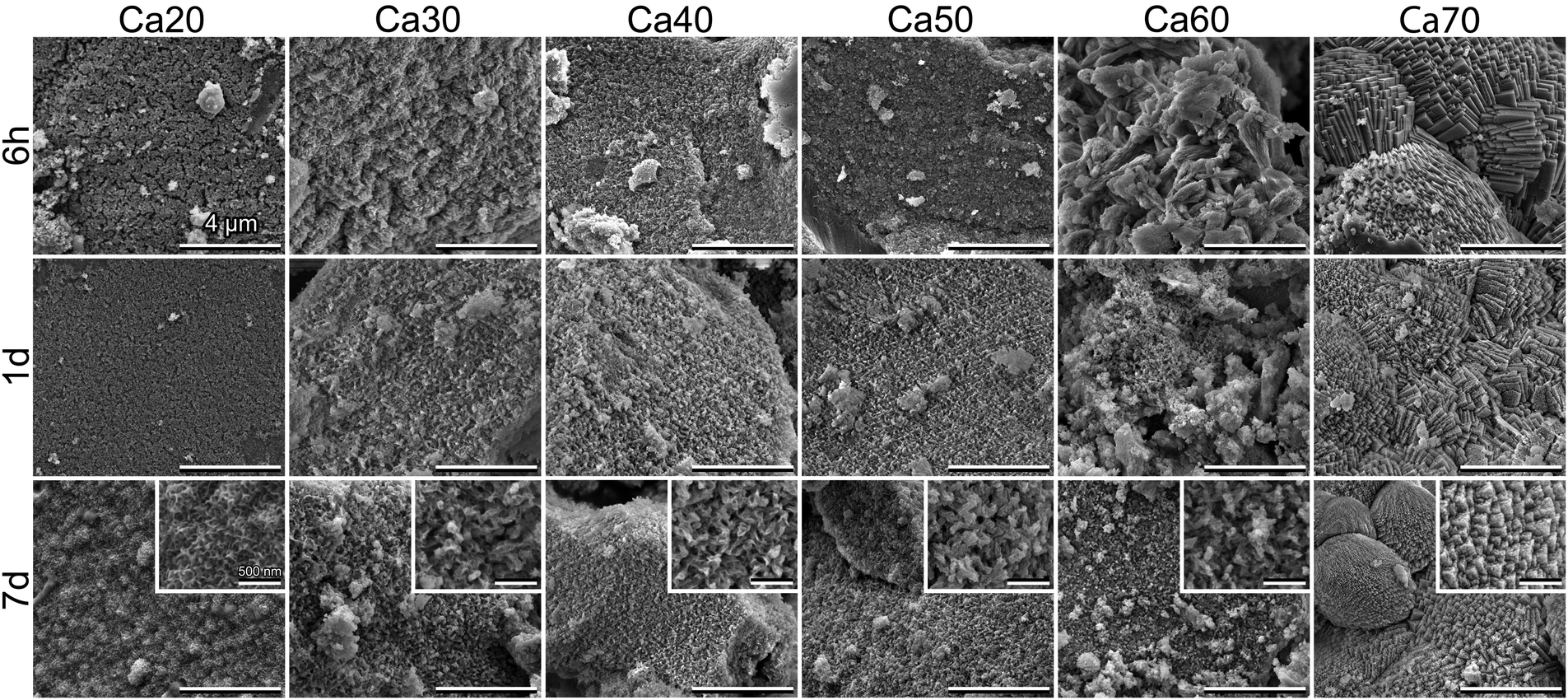 | ||
| Fig. 5 Scanning electron microscopy of SGBGs as a function of immersion time in SBF. Low CaO content glasses led to conversion to HCA as indicated by the spherulitic-like crystals whereas higher CaO content glasses initially converted to calcite as indicated by the typical geometric crystal patterns. At longer times, Ca60 and 70 showed signs of both calcite and HCA mineral formations. | ||
Although less common than calcium–phosphates, calcite is a bone graft substitute that can bond to bone without the formation of a surface apatite layer.119,120 Yet, as also highlighted by a recent study, it is often omitted during bioactivity assessments,118 which might be due to the fact that HCA formation has been generally recognized as a key indicator of standardized bioactivity testing.121 Nonetheless, silicate bioactive glasses122,123 as well as nano “45S5” glass compositions124 have shown conversion to calcite in SBF. In fact, factors such as glass particle size, choice of media, and their ratio will all influence if either HCA or calcite form. For example, finer particle size fractions of 13-93B3 borate glass have demonstrated calcite formation125 as has been observed with other bioactive ceramics and glasses.126 We have also recently shown that the bioactivity medium can dictate the conversion of a 4-component SGBG into either HCA or calcite.47
The interplay between glass composition and textural properties
The extended range of glasses enabled by the sol–gel process highlights the importance of the borate anomaly in both composition and texture during the design of bioactive borate glasses, as summarized in Fig. 6. Previously, it has been shown that by changing the form of a single bioactive glass composition (e.g., particles vs. fibers) through different processing methods (e.g., melt-quench vs. sol–gel), it is possible to extend its use from mineralized tissue repair to ‘non-traditional’ applications, such as wound healing.19,22 However, glass composition is still paramount in determining the rate and type of mineral conversion in a given environment.75,127 From the range investigated in this study, it is clear that glasses with more than ≈52 mol% CaO predominately convert to calcite (Fig. 4), despite their relatively high SSA and PV values (Fig. 2 and Table S3, ESI†). Yet, although ionic release rates and pH (Fig. S8, ESI†) were compositionally dependent, the importance textural properties cannot be ignored. While there was no direct comparison between sol–gel and melt-quench glass compositions in this study – partly due to difficulties in processing amorphous, high CaO containing glasses in bulk through melt-quenching9,76,128 – a previous study has reported that phosphate peak formation in the IR spectra of melt-quench (x)CaO(1 − x)B2O3 (x = 0.2 to 0.5, MF) glasses only appeared after 15 days in SBF, with the dissolution rate reaching a relative minima at x = 0.5, correlating with the maximum number of N4 units.129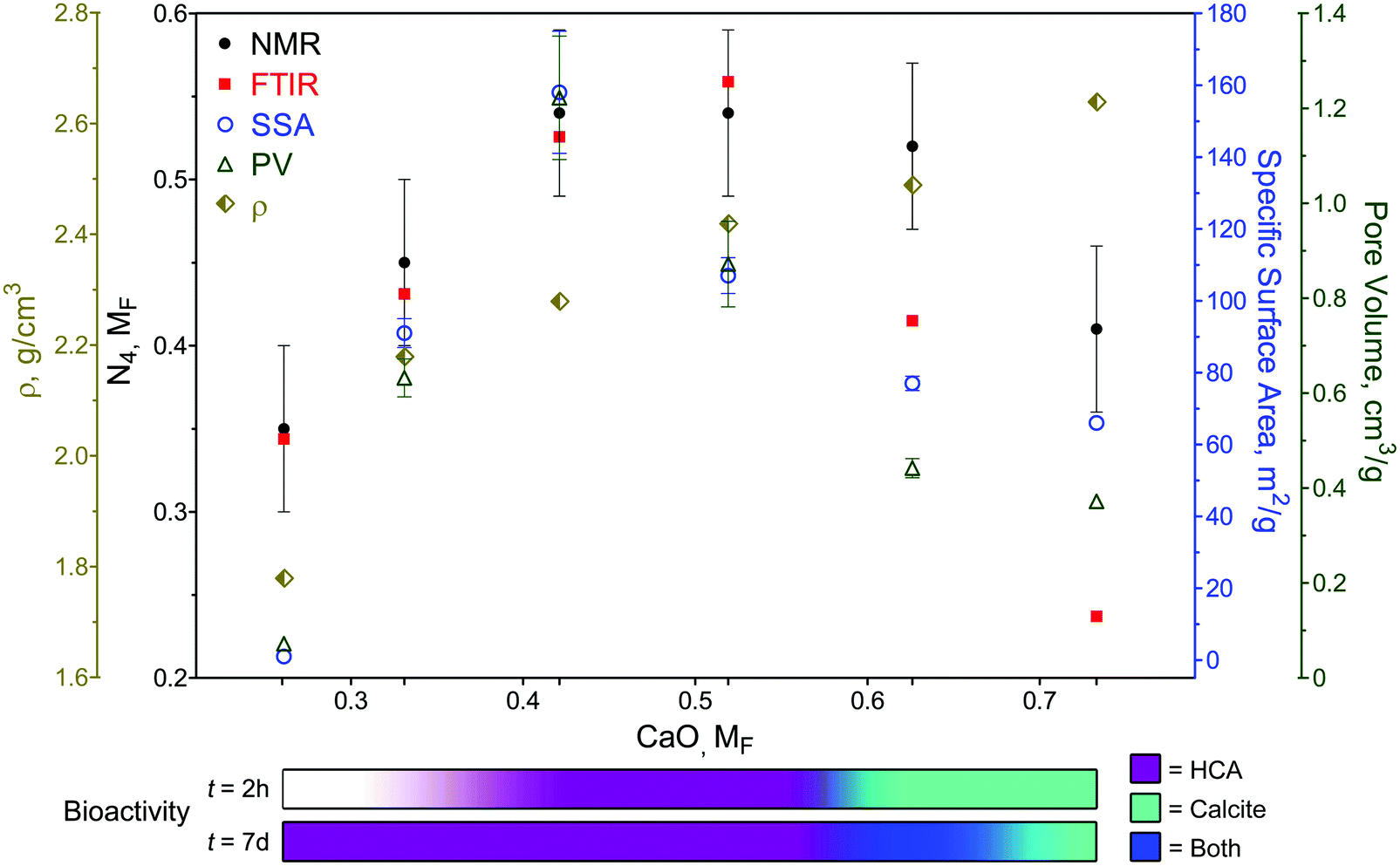 | ||
| Fig. 6 Overview of SGBG structural, physical, textural, and bioactive properties versus MF. Structural, textural, and bioactive properties were in accordance with the borate anomaly, whereas glass density (ρ) increased with MF. Bioactivity assessment in SBF is qualitatively displayed by the colored bars and is depended on CaO MF and immersion time. | ||
In traditional melt-quench glasses, an increase in N4 units creates a more chemically durable glass, whereas in sol–gel glasses, this maxima also coincides with higher SSA and PV values, which in turn impact bioactivity.4 Indeed, this was observed in this study, as IR spectra indicated phosphate peak formation within 0.5 h in SBF in Ca30-50 SGBGs (Fig. 3). This finding is also in line with our previous report, in which a 4-component borate glass composition was significantly more bioactive when processed through sol–gel.23 The effect of glass textural properties can also be observed on the rate of mineral formation, since glasses of higher SSA and PV values (i.e., Ca30-50) more rapidly converted to HCA compared to Ca20, which did not show mineral formation until day 7. However, the slow HCA conversion of Ca20 may also be attributed to its low CaO content and ionic release rates (Fig. S8, ESI†).
Another interesting observation is that, much like the original borate anomaly, the relative maxima in textural property trends (42.1 mol% CaO) did not necessarily correlate with the relative maxima in N4 units, i.e., 42.1 and 51.9 mol% CaO in NMR, and 51.9 mol% CaO in ATR-FTIR (Fig. 6). This is likely due to the effect of calcination temperature, as it has been shown to not only influence the textural properties in sol–gel glasses,23,24 but also boron coordination in melt-quench glasses.130–132 Traditional melt-quench glasses exhibit a ‘fictive temperature’ that describes the glass in terms of an equivalent equilibrium liquid state at a different temperature.133 This temperature depends on the glass thermal history (e.g., processing parameters) and can influence N4 coordination.76 Sol–gel glasses also exhibit fictive temperatures134 which can be similar to equivalent melt-quench glass compositions despite their lower processing temperatures.135 Little is reported on borate glass fictive temperatures and, in this study, only one calcination temperature was used for all compositions. Yet, based on our previous work,23,24 we can speculate that different calcination temperatures will not only shift the relative N4 maxima, but also change the textural properties, which would influence the overall borate anomaly trends. Future studies will seek to elucidate the exact regions of relative property maxima, including thermal properties, in these sol–gel borate glass systems. Nevertheless, it must be emphasized that while the addition of modifiers often generates linear effects in traditional silicate glasses, special considerations are needed in the case of borate glasses to not only factor in the anomaly effect, but also the processing technique and resultant textural properties.
Conclusions
An extended range of amorphous, binary CaO–B2O3 glasses (26.1–73.3 mol% CaO) has been created through the sol–gel process. Calcium modifier content altered both the structural and textural properties of the glasses according to the borate anomaly. Furthermore, higher amounts of CaO lead to the formation of calcite rather than HCA in SBF, yet over longer submersion times, a combination of both minerals was observed. Textural properties influenced the rate of mineral conversion but did not impact ion release profiles, which were compositionally dependent. Therefore, the importance of both composition and processing route must be considered to optimize borate glass properties for targeted applications.Conflicts of interest
W. C. Lepry and S. N. Nazhat are named inventors on International Patent Applications: PCT/CA2015/000365 entitled “Borate-glass biomaterials” and PCT/CA2020/050421 entitled “Borate-glass compositions, methods of manufacture, and uses”.Acknowledgements
This study was supported by Canada NSERC, FRQNT, CFI, Aligo Innovation and McGill William and Rhea Seath for Innovation in Engineering Awards. W. C. L. is supported by the McGill Engineering Innovation Fellowship. Dr Ulrike Werner-Zwanziger, at the Nuclear Magnetic Resonance Research Resource (NMR-3) at Dalhousie University is thanked for the acquisition and evaluation of the NMR data. Ranjan Roy is thanked for assistance with the ICP Spectroscopy.References
- J. E. Shelby, Introduction to Glass Science and Technology, Royal Society of Chemistry, 2nd edn, 2005 Search PubMed.
- D. Brauer, M. Anjum, M. Mneimne, R. Wilson, H. Doweidar and R. Hill, J. Non-Cryst. Solids, 2012, 358, 1438–1442 CrossRef CAS.
- I. Farooq, M. Tylkowski, S. Müller, T. Janicki, D. S. Brauer and R. G. Hill, Biomed. Mater., 2013, 8, 065008 CrossRef PubMed.
- R. Li, A. Clark and L. Hench, J. Appl. Biomater., 1991, 2, 231–239 CrossRef CAS PubMed.
- A. C. Wright, G. Dalba, F. Rocca and N. M. Vedishcheva, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2010, 51, 233–265 CAS.
- A. C. Wright, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2010, 51, 1–39 CAS.
- J. Krogh-Moe, J. Non-Cryst. Solids, 1969, 1, 269–284 CrossRef CAS.
- D. R. Uhlmann and R. Shaw, J. Non-Cryst. Solids, 1969, 1, 347–359 CrossRef CAS.
- Y. D. Yiannopoulos, G. D. Chryssikos and E. I. Kamitsos, Phys. Chem. Glasses, 2001, 42, 164–172 CAS.
- A. C. Wright, Int. J. Appl. Glass Sci., 2015, 6, 45–63 CrossRef.
- D. S. Brauer and D. Moncke, Bioactive Glasses: Fundamentals, Technology and Applications, The Royal Society of Chemistry, 2017, pp. 61–88 10.1039/9781782622017-00061.
- D. Möncke, E. I. Kamitsos, D. Palles, R. Limbach, A. Winterstein-Beckmann, T. Honma, Z. Yao, T. Rouxel and L. Wondraczek, J. Chem. Phys., 2016, 145, 124501 CrossRef PubMed.
- M. Bengisu, J. Mater. Sci., 2016, 51, 2199–2242 CrossRef CAS.
- A. Yao, D. Wang, W. Huang, Q. Fu, M. N. Rahaman and D. E. Day, J. Am. Ceram. Soc., 2006, 90, 303–306 CrossRef.
- W. Huang, D. E. Day, K. Kittiratanapiboon and M. N. Rahaman, J. Mater. Sci.: Mater. Med., 2006, 17, 583–596 CrossRef CAS PubMed.
- Q. Fu, M. N. Rahaman, H. Fu and X. Liu, J. Biomed. Mater. Res., Part A, 2010, 95, 164–171 CrossRef PubMed.
- S. Zhao, L. Li, H. Wang, Y. Zhang, X. Cheng, N. Zhou, M. N. Rahaman, Z. Liu, W. Huang and C. Zhang, Biomaterials, 2015, 53, 379–391 CrossRef CAS PubMed.
- W. Jia, H. Hu, A. Li, H. Deng, C. L. Hogue, J. C. Mauro, C. Zhang and Q. Fu, Acta Biomater., 2019, 103, 306–317 CrossRef PubMed.
- V. Miguez-Pacheco, L. L. Hench and A. R. Boccaccini, Acta Biomater., 2015, 13, 1–15 CrossRef CAS PubMed.
- Y. Lin, R. F. Brown, S. B. Jung and D. E. Day, J. Biomed. Mater. Res., Part A, 2014, 102, 4491–4499 Search PubMed.
- X. Liu, M. N. Rahaman and D. E. Day, J. Mater. Sci.: Mater. Med., 2013, 24, 583–595 CrossRef CAS PubMed.
- S. Naseri, W. C. Lepry and S. N. Nazhat, J. Mater. Chem. B, 2017, 5, 6167–6174 RSC.
- W. C. Lepry and S. N. Nazhat, Chem. Mater., 2015, 27, 4821–4831 CrossRef CAS.
- W. C. Lepry, S. Naseri and S. N. Nazhat, J. Mater. Sci., 2017, 52, 8973–8985 CrossRef CAS.
- W. C. Lepry, S. Smith and S. N. Nazhat, J. Non-Cryst. Solids, 2018, 500, 141–148 CrossRef CAS.
- W. C. Lepry, S. Smith, L. Liverani, A. R. Boccaccini and S. N. Nazhat, Biomed. Glasses, 2016, 2, 88–98 Search PubMed.
- S. Naseri, W. C. Lepry, V. B. Maisuria, N. Tufenkji and S. N. Nazhat, J. Non-Cryst. Solids, 2019, 505, 438–446 CrossRef CAS.
- A. Abdelghany, H. ElBatal and F. EzzElDin, Ceram. Int., 2012, 38, 1105–1113 CrossRef CAS.
- D. E. Day, J. White, R. F. Brown and K. McMenamin, Glass Technol.: Eur. J. Glass Sci. Technol., Part A, 2003, 44, 75–81 CAS.
- X. Han and D. E. Day, J. Mater. Sci.: Mater. Med., 2007, 18, 1837–1847 CrossRef CAS PubMed.
- K. O’Connell, C. Pierlot, H. O’Shea, D. Beaudry, M. Chagnon, M. Assad and D. Boyd, J. Biomed. Mater. Res., Part B, 2017, 105, 1818–1827 CrossRef PubMed.
- A. H. Yao, D. P. Wang, W. H. Huang, Q. Fu, M. N. Rahaman and D. E. Day, J. Am. Ceram. Soc., 2007, 90, 303–306 CrossRef CAS.
- M. S. Hasan, U. Werner-Zwanziger and D. Boyd, J. Biomed. Mater. Res., Part A, 2015, 103, 2344–2354 CrossRef CAS PubMed.
- A. M. Abdelghany, Spectrochim. Acta, Part A, 2013, 100, 120–126 CrossRef CAS PubMed.
- J. Doucet, E. Tonkopi, A. Nuschke, M. L. Tremblay, K. Brewer, S. Beyea, M. Filiaggi, R. Abraham, U. Werner-Zwanziger and D. Boyd, J. Non-Cryst. Solids, 2019, 510, 26–35 CrossRef CAS.
- K. L. Goetschius, M. A. Beuerlein, C. M. Bischoff and R. K. Brow, J. Non-Cryst. Solids, 2018, 487, 12–18 CrossRef CAS.
- K. Zhang, A. Alaohali, N. Sawangboon, P. T. Sharpe, D. S. Brauer and E. Gentleman, Dent. Mater., 2019, 35, 919–927 CrossRef CAS PubMed.
- K. MacDonald, K. O’Connell and D. Boyd, Mater. Lett., 2018, 231, 142–145 CrossRef CAS.
- M. A. Marzouk and H. A. ElBatal, Process. Appl. Ceram., 2014, 8, 167–177 CrossRef.
- Z. M. Al-Rashidy, A. E. Omar, T. H. Abd El-Aziz and M. M. Farag.
- O. Rodriguez, D. J. Curran, M. Papini, L. M. Placek, A. W. Wren, E. H. Schemitsch, P. Zalzal and M. R. Towler, J. Non-Cryst. Solids, 2016, 433, 95–102 CrossRef CAS.
- R. Shafaghi, O. Rodriguez, S. Phull, E. H. Schemitsch, P. Zalzal, S. D. Waldman, M. Papini and M. R. Towler, Mater. Sci. Eng., C, 2020, 107, 110351 CrossRef CAS PubMed.
- J. L. George and R. K. Brow, J. Non-Cryst. Solids, 2015, 426, 116–124 CrossRef CAS.
- Q. Yang, S. Chen, H. Shi, H. Xiao and Y. Ma, Mater. Sci. Eng., C, 2015, 55, 105–117 CrossRef CAS PubMed.
- S. Chen, Q. Yang, R. K. Brow, K. Liu, K. A. Brow, Y. Ma and H. Shi, Mater. Sci. Eng., C, 2017, 73, 447–455 CrossRef CAS PubMed.
- S. Jung, T. Day, T. Boone, B. Buziak and A. Omar, Biomed. Glasses, 2019, 5, 67–75 Search PubMed.
- W. C. Lepry, E. Rezabeigi, S. Smith and S. N. Nazhat, Biomed. Glasses, 2019, 5, 98–111 Search PubMed.
- J. Ning, A. Yao, D. P. Wang, W. H. Huang, H. L. Fu, X. Liu, X. Q. Jiang and X. L. Zhang, Mater. Lett., 2007, 61, 5223–5226 CrossRef CAS.
- R. F. Brown, M. N. Rahaman, A. B. Dwilewicz, W. Huang, D. E. Day, Y. Li and B. S. Bal, J. Biomed. Mater. Res., Part A, 2008, 88, 392–400 Search PubMed.
- Manupriya, K. S. Thind, G. Sharma, K. Singh, V. Rajendran and S. Aravindan, J. Am. Ceram. Soc., 2007, 90, 467–471 CrossRef CAS.
- J. Watters Richard, F. Brown Roger and E. Day Delbert, Biomed. Glasses, 2015, 1, 173–184 Search PubMed.
- K. C. Kolan, M. C. Leu, G. Hilmas and T. Comte, Proceedings of the 24th Annual International Solid Freeform Fabrication Symposium, 2013, pp. 816–826.
- H. Fu, Q. Fu, N. Zhou, W. Huang, M. N. Rahaman, D. Wang and X. Liu, Mater. Sci. Eng., C, 2009, 29, 2275–2281 CrossRef CAS.
- K. Schuhladen, X. Wang, L. Hupa and A. R. Boccaccini, J. Non-Cryst. Solids, 2018, 502, 22–34 CrossRef CAS.
- S. B. Jung, D. E. Day, R. F. Brown and L. Bonewald, Advances in Bioceramics and Porous Ceramics V, 2012, pp. 65–74 Search PubMed.
- L. Bi, M. N. Rahaman, D. E. Day, Z. Brown, C. Samujh, X. Liu, A. Mohammadkhah, V. Dusevich, J. D. Eick and L. F. Bonewald, Acta Biomater., 2013, 9, 8015–8026 CrossRef CAS PubMed.
- L. Bi, B. Zobell, X. Liu, M. N. Rahaman and L. F. Bonewald, Mater. Sci. Eng., C, 2014, 42, 816–824 CrossRef CAS PubMed.
- G. A. Funk, J. C. Burkes, K. A. Cole, M. N. Rahaman and T. E. McIff, J Bone Jt. Infect., 2018, 3, 187–196 CrossRef PubMed.
- X. Cui, Y. Gu, L. Li, H. Wang, Z. Xie, S. Luo, N. Zhou, W. Huang and M. N. Rahaman, J. Mater. Sci.: Mater. Med., 2013, 24, 2391–2403 CrossRef CAS PubMed.
- H. Yin, C. Yang, Y. Gao, C. Wang, M. Li, H. Guo and Q. Tong, J. Alloys Compd., 2018, 743, 564–569 CrossRef CAS.
- H. Ding, C.-J. Zhao, X. Cui, Y.-F. Gu, W.-T. Jia, M. N. Rahaman, Y. Wang, W.-H. Huang and C.-Q. Zhang, PLoS One, 2014, 9, e85472 CrossRef PubMed.
- X. Cui, C. Zhao, Y. Gu, L. Li, H. Wang, W. Huang, N. Zhou, D. Wang, Y. Zhu and J. Xu, J. Mater. Sci.: Mater. Med., 2014, 25, 733–745 CrossRef CAS PubMed.
- X. Zhang, W. Jia, Y. Gu, W. Xiao, X. Liu, D. Wang, C. Zhang, W. Huang, M. N. Rahaman, D. E. Day and N. Zhou, Biomaterials, 2010, 31, 5865–5874 CrossRef CAS PubMed.
- H. Wang, S. Zhao, X. Cui, Y. Pan, W. Huang, S. Ye, S. Luo, M. N. Rahaman, C. Zhang and D. Wang, J. Mater. Res., 2015, 30, 2722–2735 CrossRef CAS.
- M. Ottomeyer, A. Mohammadkah, D. Day and D. J. Westenberg, 2016.
- Y. Gu, W. Huang, M. N. Rahaman and D. E. Day, Acta Biomater., 2013, 9, 9126–9136 CrossRef CAS PubMed.
- K. Schuhladen, L. Stich, J. Schmidt, A. Steinkasserer, A. R. Boccaccini and E. Zinser, Biomater. Sci., 2020, 8, 2143–2155 RSC.
- N. J. Thyparambil, L. C. Gutgesell, C. C. Hurley, L. E. Flowers, D. E. Day and J. A. Semon, J. Mater. Sci.: Mater. Med., 2020, 31, 13 CrossRef CAS PubMed.
- X. Liu, Z. Xie, C. Zhang, H. Pan, M. N. Rahaman, X. Zhang, Q. Fu and W. Huang, J. Mater. Sci.: Mater. Med., 2010, 21, 575–582 CrossRef CAS PubMed.
- M. Ojansivu, A. Mishra, S. Vanhatupa, M. Juntunen, A. Larionova, J. Massera and S. Miettinen, PLoS One, 2018, 13, e0202740 CrossRef PubMed.
- M. Fabert, N. Ojha, E. Erasmus, M. Hannula, M. Hokka, J. Hyttinen, J. Rocherullé, I. Sigalas and J. Massera, J. Mater. Chem. B, 2017, 5, 4514–4525 RSC.
- S. Prasad, S. Datta, T. Adarsh, P. Diwan, K. Annapurna, B. Kundu and K. Biswas, J. Non-Cryst. Solids, 2018, 498, 204–215 CrossRef.
- X. Lu, L. Deng, P.-H. Kuo, M. Ren, I. Buterbaugh and J. Du, J. Mater. Sci., 2017, 52, 8793–8811 CrossRef CAS.
- P. Balasubramanian, T. Büttner, V. Miguez Pacheco and A. R. Boccaccini, J. Eur. Ceram. Soc., 2018, 38, 855–869 CrossRef CAS.
- D. S. Brauer, Angew. Chem., Int. Ed., 2015, 54, 4160–4181 CrossRef CAS PubMed.
- J. Wu and J. F. Stebbins, J. Am. Ceram. Soc., 2014, 97, 2794–2801 CrossRef CAS.
- N. P. Lower, J. L. McRae, H. A. Feller, A. R. Betzen, S. Kapoor, M. Affatigato and S. A. Feller, J. Non-Cryst. Solids, 2001, 293-295, 669–675 CrossRef CAS.
- T. Jin, G. M. Bernard, M. Miskolzie, V. V. Terskikh and V. K. Michaelis, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2018, 59, 174–180 Search PubMed.
- M. A. Karakassides, D. Petridis, G. Mousdis, C. Trapalis and G. Kordas, J. Non-Cryst. Solids, 1996, 202(1), 198–202 CrossRef CAS.
- N. Tohge and J. Mackenzie, J. Non-Cryst. Solids, 1984, 68, 411–418 CrossRef CAS.
- M. C. Weinberg, G. F. Neilson, G. L. Smith, B. Dunn, G. Moore and J. Mackenzie, J. Mater. Sci., 1985, 20, 1501–1508 CrossRef CAS.
- C. Brinker, K. Ward, K. Keefer, E. Holupka, P. Bray and R. Pearson, Aerogels, Springer, 1986, pp. 57–67 Search PubMed.
- E. I. Kamitsos, M. A. Karakassides and G. D. Chryssikos, Phys. Chem. Glasses, 1987, 28, 203–209 CAS.
- A. Irwin, J. Holmgren, T. Zerda and J. Jonas, J. Non-Cryst. Solids, 1987, 89, 191–205 CrossRef CAS.
- R. Ota, N. Asagi, J. Fukunaga, N. Yoshida and T. Fujii, J. Mater. Sci., 1990, 25, 4259–4265 CrossRef CAS.
- E. Kamitsos, M. Karakassides and G. D. Chryssikos, J. Phys. Chem., 1987, 91, 1073–1079 CrossRef CAS.
- C. Lu, S. Dimov and R. Lipson, Chem. Mater., 2008, 20, 5296–5300 CrossRef CAS.
- M. Bengisu, E. Yilmaz, H. Farzad and S. T. Reis, J. Sol-Gel Sci. Technol., 2008, 45, 237–243 CrossRef CAS.
- R. L. Ciceo, D.-L. Trandafir, T. Radu, O. Ponta and V. Simon, Ceram. Int., 2014, 40, 9517–9524 CrossRef.
- D. Carta, D. Qiu, P. Guerry, I. Ahmed, E. A. Abou Neel, J. C. Knowles, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 2008, 354, 3671–3677 CrossRef CAS.
- D. Carta, J. C. Knowles, P. Guerry, M. E. Smith and R. J. Newport, J. Mater. Chem., 2009, 19, 150–158 RSC.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- L. G. Joyner, E. P. Barrett and R. Skold, J. Am. Chem. Soc., 1951, 73, 3155–3158 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- K. MacDonald, M. A. Hanson and D. Boyd, J. Non-Cryst. Solids, 2016, 443, 184–191 CrossRef CAS.
- J. Hemry, M. Weinberg and D. R. Uhlmann, J. Mater. Sci., 1998, 33, 3853–3858 CrossRef CAS.
- J. F. Stebbins, P. Zhao and S. Kroeker, Solid State Nucl. Magn. Reson., 2000, 16, 9–19 CrossRef CAS PubMed.
- A. C. Wright, R. N. Sinclair, C. E. Stone, J. L. Shaw, S. A. Feller, T. Kiczenski, R. B. Williams, H. A. Berger, H. E. Fischer and N. M. Vedishcheva, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2012, 53, 191–204 CAS.
- N. Ohtori, K. Takase, I. Akiyama, Y. Suzuki, K. Handa, I. Sakai, Y. Iwadate, T. Fukunaga and N. Umesaki, J. Non-Cryst. Solids, 2001, 293, 136–145 CrossRef.
- Manupriya, K. S. Thind, G. Sharma, V. Rajendran, K. Singh, A. Gayathri Devi and S. Aravindan, Phys. Status Solidi A, 2006, 203, 2356–2364 CrossRef CAS.
- H. Trapp and J. Stevels, Phys. Chem. Glasses, 1960, 1, 107–118 Search PubMed.
- J. Banerjee, G. Ongie, J. Harder, T. Edwards, C. Larson, S. Sutton, A. Moeller, A. Basu, M. Affatigato, S. Feller, M. Kodama, P. M. Aguiar and S. Kroeker, J. Non-Cryst. Solids, 2006, 352, 674–678 CrossRef CAS.
- C. R. Kurkjian and W. R. Prindle, J. Am. Ceram. Soc., 1998, 81, 795–813 CrossRef CAS.
- A. M. Deliormanlı, Ceram. Int., 2012, 38, 6435–6444 CrossRef.
- C. Gautam, A. K. Yadav and A. K. Singh, ISRN Ceram., 2012, 2012, 428497 Search PubMed.
- S. Agathopoulos, D. U. Tulyaganov, J. M. Ventura, S. Kannan, M. A. Karakassides and J. M. Ferreira, Biomaterials, 2006, 27, 1832–1840 CrossRef CAS PubMed.
- L. Balachandera, G. Ramadevudub, M. Shareefuddina, R. Sayannac and Y. Venudharc, ScienceAsia, 2013, 39, 278–283 Search PubMed.
- V. K. Michaelis, P. M. Aguiar and S. Kroeker, J. Non-Cryst. Solids, 2007, 353, 2582–2590 CrossRef CAS.
- A. Martinez, I. Izquierdo-Barba and M. Vallet-Regi, Chem. Mater., 2000, 12, 3080–3088 CrossRef CAS.
- P. Saravanapavan and L. L. Hench, J. Biomed. Mater. Res., 2001, 54, 608–618 CrossRef CAS PubMed.
- C. Rey, M. Shimizu, B. Collins and M. J. Glimcher, Calcif. Tissue Int., 1991, 49, 383–388 CrossRef CAS PubMed.
- D. Nelson and J. Featherstone, Calcif. Tissue Int., 1981, 34, S69–S81 Search PubMed.
- N. S. Chickerur, M. S. Tung and W. E. Brown, Calcif. Tissue Int., 1980, 32, 55–62 CrossRef CAS PubMed.
- R. Z. LeGeros, Arch. Oral Biol., 1975, 20, 63–71 CrossRef CAS PubMed.
- B. O. Fowler, E. C. Moreno and W. E. Brown, Arch. Oral Biol., 1966, 11, 477–492 CrossRef CAS PubMed.
- R. Chester and H. Elderfield, Sedimentology, 1967, 9, 5–21 CrossRef CAS.
- F. A. Andersen and L. Brecevic, Acta Chem. Scand., 1991, 45, 1018–1024 CrossRef CAS.
- M. Mozafari, S. Banijamali, F. Baino, S. Kargozar and R. G. Hill, Acta Biomater., 2019, 91, 35–47 CrossRef CAS PubMed.
- K. Tsuru, A. Otsu, M. Maruta, A. Valanezhad, G. Kawachi, A. Takeuchi, S. Matsuya and I. Kunio, 2012.
- Y. Fujita, T. Yamamuro, T. Nakamura, S. Kotani, C. Ohtsuki and T. Kokubo, J. Biomed. Mater. Res., 1991, 25, 991–1003 CrossRef CAS PubMed.
- A. L. Macon, T. B. Kim, E. M. Valliant, K. Goetschius, R. K. Brow, D. E. Day, A. Hoppe, A. R. Boccaccini, I. Y. Kim, C. Ohtsuki, T. Kokubo, A. Osaka, M. Vallet-Regi, D. Arcos, L. Fraile, A. J. Salinas, A. V. Teixeira, Y. Vueva, R. M. Almeida, M. Miola, C. Vitale-Brovarone, E. Verne, W. Holand and J. R. Jones, J. Mater. Sci.: Mater. Med., 2015, 26, 115 CrossRef PubMed.
- J. R. Jones, P. Sepulveda and L. L. Hench, J. Biomed. Mater. Res., 2001, 58, 720–726 CrossRef CAS PubMed.
- V. Cannillo, F. Chiellini, P. Fabbri and A. Sola, Compos. Struct., 2010, 92, 1823–1832 CrossRef.
- M. Mackovic, A. Hoppe, R. Detsch, D. Mohn, W. J. Stark, E. Spiecker and A. R. Boccaccini, J. Nanopart. Res., 2012, 14, 966 CrossRef.
- A. M. Deliormanlı, Ceram. Int., 2013, 39, 8087–8095 CrossRef.
- L. L. Hench and J. M. Polak, Science, 2002, 295, 1014–1017 CrossRef CAS PubMed.
- H. R. Fernandes, A. Gaddam, A. Rebelo, D. Brazete, G. E. Stan and J. M. F. Ferreira, Materials, 2018, 11, 2530 CrossRef CAS PubMed.
- Y. Yiannopoulos, G. D. Chryssikos and E. Kamitsos, Phys. Chem. Glasses, 2001, 42, 164–172 CAS.
- Manupriya, K. S. Thind, K. Singh, V. Kumar, G. Sharma, D. P. Singh and D. Singh, J. Phys. Chem. Solids, 2009, 70, 1137–1141 CrossRef CAS.
- J. Wu and J. F. Stebbins, J. Non-Cryst. Solids, 2010, 356, 2097–2108 CrossRef CAS.
- J. Wu and J. F. Stebbins, J. Non-Cryst. Solids, 2013, 362, 73–81 CrossRef CAS.
- P. K. Gupta, M. L. Lur and P. J. Bray, J. Am. Ceram. Soc., 1985, 68, C-82 CrossRef CAS.
- J. C. Mauro, R. J. Loucks and P. K. Gupta, J. Am. Ceram. Soc., 2009, 92, 75–86 CrossRef CAS.
- M. Yamane, S. Aso, S. Okano and T. Sakaino, J. Mater. Sci., 1979, 14, 607–611 CrossRef CAS.
- R. Puyané, P. F. James and H. Rawson, J. Non-Cryst. Solids, 1980, 41, 105–115 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further data and discussion involving glass structure including XRD and ATR-FTIR as well as pH and inductively coupled plasma-optical emission spectrometry. In addition, property comparisons to related work are also shown. See DOI: 10.1039/d0ma00360c |
| This journal is © The Royal Society of Chemistry 2020 |