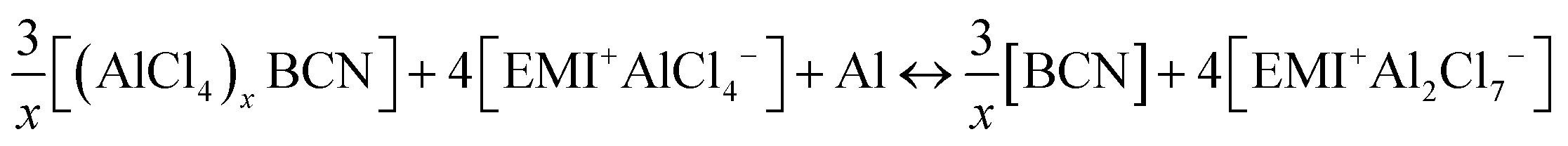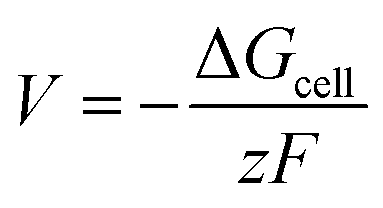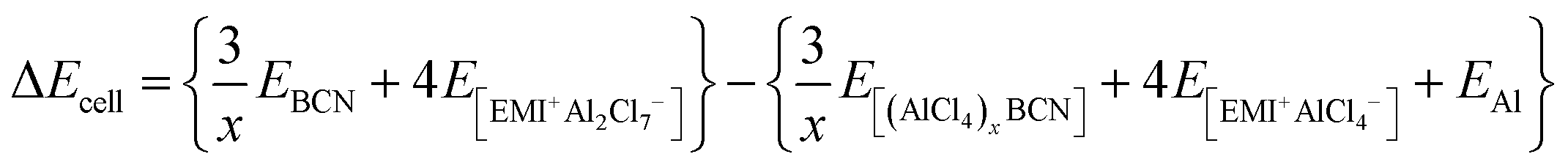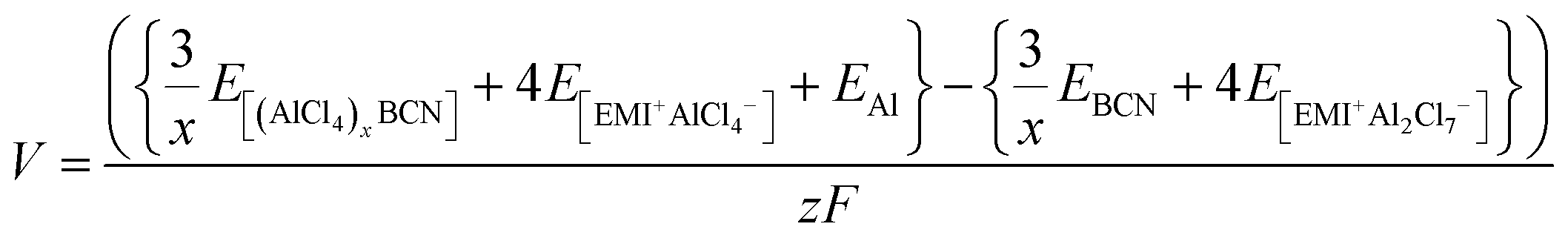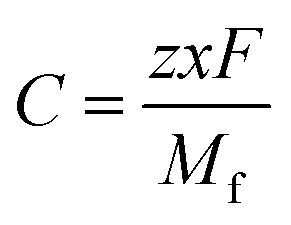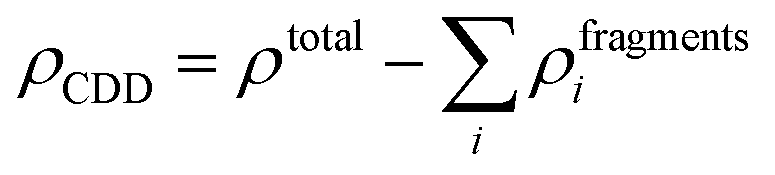DOI:
10.1039/D0MA00501K
(Paper)
Mater. Adv., 2020,
1, 2418-2425
BCN monolayer for high capacity Al-based dual-ion batteries†
Received
12th July 2020
, Accepted 6th September 2020
First published on 7th September 2020
Abstract
Recent advances in the field of Al dual-ion batteries have put forward a major challenge of developing a novel and advanced cathode material that can provide high specific capacity, besides maintaining a constant high voltage. In this context, significant research has been carried out to identify new electrochemical energy storage materials, which suggest the applicability of low dimensional materials as an excellent choice due to their high surface-to-volume ratio. Herein, we perform first principles calculations to investigate the BCN monolayer as a suitable cathode material for Al dual-ion batteries. AlCl4 has been found to reversibly adsorb on the BCN monolayer with a significant charge transfer of 0.9 |e| from BCN to AlCl4, indicating the oxidation of BCN monolayer during the charging process. Moreover, the BCN monolayer shows excellent structural stability on systematically increasing the concentration of adsorbed AlCl4, and could provide an open circuit voltage of 1.8 V (average voltage is 1.43 V) with a high specific capacity of 316.9 mA h gBCN−1 at the maximum concentration of AlCl4 adsorption. The adsorption of AlCl4 also enables good electronic conductivity. Diffusion energy calculations reveal a minimum energy barrier of 0.12 eV for the migration of AlCl4 within BCN, ensuring a fast charge/discharge rate. Consequently, these findings suggest that the BCN monolayer could be a potential cathode material for Al dual-ion batteries, and motivates us to carry out further studies on similar low dimensional materials to improve the performance of Al dual-ion batteries.
1. Introduction
With the continuously increasing energy demand, secondary batteries have been widely used as energy conversion and storage units in devices, ranging from portable electronics to heavy electronic vehicles.1,2 Li-ion batteries and lead-acid batteries, being the most commercialized batteries, are quite efficient but have certain limitations in terms of safety, abundance and manufacturing cost.3–7 In this context, multivalent metal ion (Mg, Zn, Al) batteries can be a better choice due to their high abundance, low cost and high volumetric capacity.8–15 Amongst the recently developed batteries, Al batteries have garnered interest due to the higher abundance (third most abundant metal in earth's crust), high gravimetric (3 A h g−1) and volumetric capacity (8.04 A h cm−3) compared to other metal ions.12–15 However, these merits are due to the anodic part and there is an essential need to look out for materials to improve the cathodic part, which can provide suitable electrochemical properties. Lin et al. assembled an Al battery having Al metal as an anode and three dimensional graphitic foam as a cathode in an ionic liquid electrolyte of [EMIm]Cl/AlCl3 (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3) whose working mechanism involves the reversible intercalation/deintercalation of AlCl4 on the cathode side during the charging/discharging process.16,17 Simultaneously, the electrochemical deposition/dissolution of Al takes place at the anodic part during charging/discharging. This cell provides a good discharge voltage of 2 V with a specific capacity of 70 mA h g−1 and an ultrahigh charge–discharge rate. Since then, different forms of graphite and conducting polymers have been utilized as cathode materials for Al batteries.13,18–22 Similarly, theoretical studies have also been carried out for carbonaceous cathode materials, such as BC3, C3N and G/h-BN.23–25 Among various investigated cathode materials, low dimensional materials have shown improved results in terms of storage capacity, voltage, and cyclic stability.22,26–29 Importantly, two-dimensional materials have attracted special interest due to their high surface-volume ratio, mechanical flexibility, cycle life and thermal stability.30–33 Thus, for the improvement of the electrochemical properties of Al dual-ion batteries, there is a need to look out for other low-dimensional cathode materials. One such potential candidate is the 2D hexagonal graphenic BCN monolayer, recently synthesized by Beniwal and co-workers using bis-BN cyclohexane (B2C2N2H12) as a precursor for the epitaxial growth of the BCN monolayer on an Ir(111) substrate under ultrahigh vacuum.34 Graphene and hBN were also previously synthesized on Ir(111) and Rh(111) substrates.35–37 The authors further predict that due to strong intermolecular bonding between the atoms in the monolayer, free-standing monolayers can be obtained by exfoliation.34 In previous studies, we have observed that BC3 gives lower capacity with higher voltage for bulk systems, while C3N delivers higher capacity with lower voltage compared to graphite.23,24 BCN is isoelectronic in nature to graphene, but has both electron-deficient B and electron-rich N atoms, which may provide a unique adsorption environment for AlCl4. BCN is semiconducting in nature with a bandgap (1.50 eV) that is intermediate between graphene and h-BN.34 Apart from these features, BCN shows higher directional anisotropy than graphene, suggesting a lower barrier for ion diffusion.38 Moreover, BCN has a smaller Young's modulus value and higher flexibility than graphene, which justifies its mechanical stability.38 All of these properties suggest that the BCN monolayer may be a potential electrode material candidate.
:1.3) whose working mechanism involves the reversible intercalation/deintercalation of AlCl4 on the cathode side during the charging/discharging process.16,17 Simultaneously, the electrochemical deposition/dissolution of Al takes place at the anodic part during charging/discharging. This cell provides a good discharge voltage of 2 V with a specific capacity of 70 mA h g−1 and an ultrahigh charge–discharge rate. Since then, different forms of graphite and conducting polymers have been utilized as cathode materials for Al batteries.13,18–22 Similarly, theoretical studies have also been carried out for carbonaceous cathode materials, such as BC3, C3N and G/h-BN.23–25 Among various investigated cathode materials, low dimensional materials have shown improved results in terms of storage capacity, voltage, and cyclic stability.22,26–29 Importantly, two-dimensional materials have attracted special interest due to their high surface-volume ratio, mechanical flexibility, cycle life and thermal stability.30–33 Thus, for the improvement of the electrochemical properties of Al dual-ion batteries, there is a need to look out for other low-dimensional cathode materials. One such potential candidate is the 2D hexagonal graphenic BCN monolayer, recently synthesized by Beniwal and co-workers using bis-BN cyclohexane (B2C2N2H12) as a precursor for the epitaxial growth of the BCN monolayer on an Ir(111) substrate under ultrahigh vacuum.34 Graphene and hBN were also previously synthesized on Ir(111) and Rh(111) substrates.35–37 The authors further predict that due to strong intermolecular bonding between the atoms in the monolayer, free-standing monolayers can be obtained by exfoliation.34 In previous studies, we have observed that BC3 gives lower capacity with higher voltage for bulk systems, while C3N delivers higher capacity with lower voltage compared to graphite.23,24 BCN is isoelectronic in nature to graphene, but has both electron-deficient B and electron-rich N atoms, which may provide a unique adsorption environment for AlCl4. BCN is semiconducting in nature with a bandgap (1.50 eV) that is intermediate between graphene and h-BN.34 Apart from these features, BCN shows higher directional anisotropy than graphene, suggesting a lower barrier for ion diffusion.38 Moreover, BCN has a smaller Young's modulus value and higher flexibility than graphene, which justifies its mechanical stability.38 All of these properties suggest that the BCN monolayer may be a potential electrode material candidate.
In this work, we have theoretically investigated the electrochemical properties of BCN as a cathode material for Al dual-ion batteries using density functional theory (DFT). We have considered a configuration of the BCN monolayer containing equal proportions of B, C and N atoms, and studied the adsorption behavior and nature of interaction of AlCl4 on the BCN monolayer. The electronic properties have been determined through density of states (DOS) calculation of pristine BCN and AlCl4-adsorbed BCN. The electrochemical properties have been examined by subjecting the monolayer to step-by-step AlCl4 adsorption. The diffusion characteristics were also carried out and overall, our results point towards the aptness of the BCN monolayer as an Al dual-ion battery cathode.
2. Computational details
The first principles calculations were carried out using the projector-augmented wave (PAW) method, as implemented in the Vienna Ab Initio Simulation Package (VASP).39–42 The generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE) was used for describing the exchange–correlation potential, along with a plane-wave cut-off energy of 470 eV.43 All structures were fully optimized until the total energy convergence criteria of 10−5 eV were met, and the Hellmann–Feynman forces on all atoms were less than 0.01 eV Å−1. A Γ-centered k-point grid of 9 × 17 × 1 was used for the unit cell optimization, while a 5 × 5 × 1 Γ-centered k-point grid was used for the 2 × 4 × 1 supercell optimization. To avoid all periodic interactions, a vacuum of 20 Å was considered along the z-direction. The DFT-D3 approach was used to include van der Waals interactions and dispersion energy corrections.44 To calculate the diffusion energy barrier for the AlCl4 molecule across the BCN monolayer, the climbing image nudged elastic band (CI-NEB) method was used by considering six intermediate images between the fully optimized initial and final structural geometry with the energy convergence criteria of 10−4 eV for each image.45 Bader charge analysis was performed by means of the Henkelman program, using the near grid algorithm refine edge method to understand the extent of charge transfer from BCN to AlCl4.46–49 DOS calculations were performed using a Γ-centered k-point grid of 15 × 15 × 3 for the BCN monolayer and AlCl4-adsorbed BCN monolayer. To examine the thermal stability of the 2 × 4 × 1 BCN monolayer, we performed ab initio molecular dynamics (AIMD) simulations using the Nosé thermostat model.50 A k-point grid of 5 × 5 × 1 was considered to perform the simulation at 300 and 400 K, as the batteries were expected to work using room temperature ionic liquid electrolyte for 10 picoseconds (ps) with a time step of 1 femtosecond (fs).
The overall cell reaction for the adsorption of x number of AlCl4 molecules on the BCN monolayer can be written as:
| | 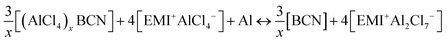 | (1) |
The cell voltage can be calculated by the Nernst equation,
| | 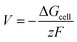 | (2) |
where Δ
Gcell is the change in Gibbs free energy of the cell reaction,
z is the total number of electrons involved in the cell reaction, and
F stands for the Faraday constant. Δ
G for a reaction is given as
| | | ΔGcell = ΔEcell + PΔVcell − TΔScell | (3) |
At 0 K temperature, the contribution of the volume effect (Vcell) and entropy (Scell) can be neglected. Hence, ΔGcell can be approximated to the change in internal energy (ΔEcell).51 This change in the internal energy for the overall cell can be calculated by eqn (4).
| | 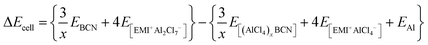 | (4) |
where
EBCN is the total energy of the BCN system, while
E[(AlCl4)xBCN],
E[EMI+Al2Cl7−], and
E[EMI+AlCl4−] are the total energies of the AlCl
4-absorbed BCN system, EMI
+Al
2Cl
7− and EMI
+AlCl
4−, respectively.
EAl is the energy per Al atom in its bulk.
E[EMI+Al2Cl7−] and
E[EMI+Al2Cl7−] were calculated by optimizing EMI
+Al
2Cl
7− and EMI
+AlCl
4− as ion pairs in a box due to the unavailability of their crystal structures. The solvent effect was not considered in our calculation, as it has been reported that the difference between the solvation energies of AlCl
4− and Al
2Cl
7− is very small (∼0.1 eV) in such cases.
52
Therefore, the voltage (V) for the system can be calculated by using eqn (5),
| | 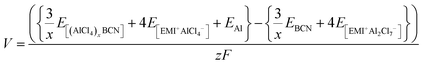 | (5) |
where
z is the electronic charge. However, the specific capacity (
C) can be calculated by using
eqn (6),
| | 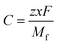 | (6) |
where
z is the number of electrons transferred per formula unit,
x is the number of AlCl
4− adsorbed on the BCN monolayer,
F is the Faraday constant and
Mf is the mass of the BCN formula unit.
3. Results and discussion
3.1. Structural stability of BCN
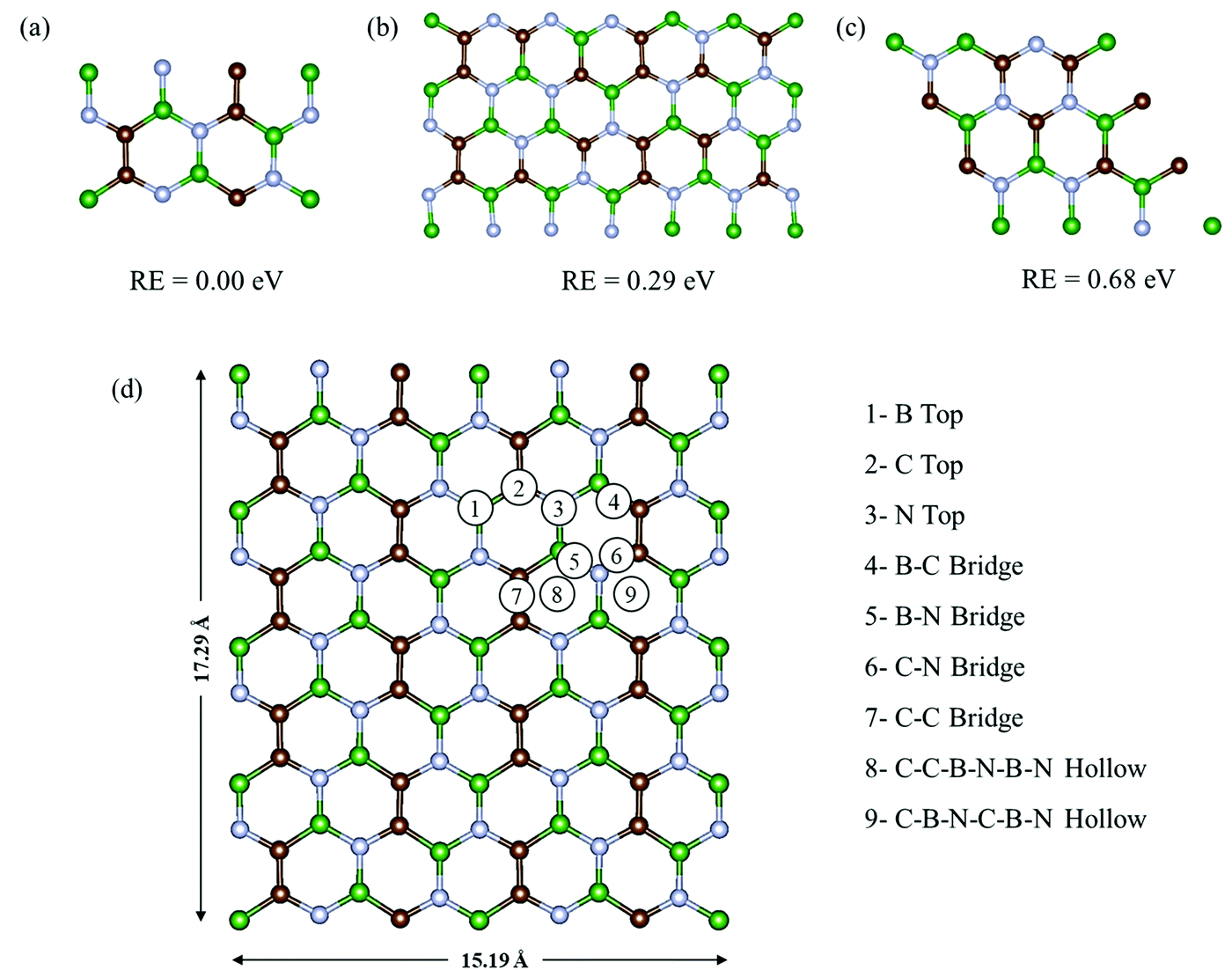
Fig. 1a–c show the three most commonly studied configurations of BCN, which contain B, C and N atoms in the same stoichiometric ratio of 1:1:1, with the arrangement of atoms and the connectivity between them being different. They are made up of different hexagonal units, as configuration (a) consists of a B2C2N2 unit, while (b) is composed of B2C2N2, B2C3N, BC3N2, and B3N3 units, and (c) is made up of B2C2N2, B3N3, B3C3, and C3N3 units. Among all three configurations, (a) was found to be relatively most stable energetically, followed by (b) and (c). The monolayer configurations (a) and (b) have B–C, B–N, C–N and C–C connecting bonds, while (c) does not have any C–C bonds. Upon comparing the bond lengths, we found that the C–C bonds were shortest with a bond length of 1.38 Å, followed by C–N, B–N and B–C with bond lengths of 1.40 Å, 1.45 Å, and 1.54 Å, respectively. This explains the lower stability of monolayer (c) with respect to (a) and (b), as it does not have strong C–C bonds. In contrast, monolayer (a) has the maximum number of C–C bonds, and is thus found to be the most stable. This is consistent with a previous theoretical study stating that the most stable BCN structures tend to maximize the number of B–N and C–C bonds.35 The higher stability of configuration (a) was also previously reported on the basis of the computed electronic energy and relative cohesive energy.34,38 Our cohesive energy calculations yielded a value of −8.38 eV per atom for the BCN unit cell of configuration (a), which is comparable to our calculated value of graphene (−8.9 eV). The reported value for graphene is around −8.1 eV.53 Thus, the monolayer once formed will not spontaneously decompose into individual atoms.
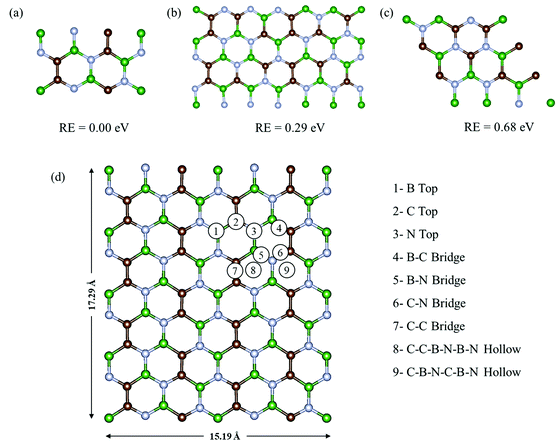 |
| | Fig. 1 Three plausible configurations (a)–(c) of the BCN monolayer with their relative energies, and (d) the top view of the possible adsorption sites on the 2 × 4 × 1 supercell of BCN. Here, the green, brown, and blue colors represent the B, C and N atoms, respectively. | |
Henceforth, we have adopted configuration (a) of the BCN monolayer to construct a 2 × 4 × 1 supercell, as presented in Fig. 1d for investigating the suitability of BCN as a cathode material. To start with, we have examined the thermal stability of the 2 × 4 × 1 monolayer. The effect of temperature on the phase stability was analyzed at 300 and 400 K. There is no appreciable energy change throughout the simulation for both cases, as shown in Fig. S1a and b (ESI†). Furthermore, we present the snapshots of the final structure after simulation in Fig. S1d and e (ESI†), which do not show much deviation from the initial structure. The graphene-like lattice structure is also retained at the end of simulation. Hence, we can consider our BCN monolayer supercell to be thermally stable.
3.2. Adsorption properties of AlCl4
During the charging of the Al dual-ion battery, the AlCl4 intercalates/adsorbs into the cathode material. Hence, in order to study the adsorption properties of AlCl4, both planar and tetrahedral configurations of AlCl4 have been considered for adsorption on the BCN monolayer. However, AlCl4 prefers the tetrahedral geometry after the adsorption on the BCN monolayer, as reported earlier for other carbonaceous cathodes.17,23–25 To identify the most stable site, all available possibilities were considered, as shown in Fig. 1d for a 2 × 4 × 1 supercell. Nine stable adsorption sites could be identified, which comprise the three top sites (B top, C top and N top), four bridge sites (B–C bridge, B–N bridge, C–N bridge and C–C bridge) and two hollow sites (C–B–N–C–B–N and C–C–B–N–B–N rings). The optimized structures of AlCl4 adsorption at these sites are also represented in Fig. S2 (ESI†). The relative adsorption energies of these sites are given in Table S1 (ESI†). The adsorption energies (Eadsorption) corresponding to these sites have been calculated by using eqn (7).| | | Eadsorption = (EBCN+AlCl4 − EBCN − EAlCl4) | (7) |
where EBCN+AlCl4 and EBCN are the total energies of the BCN system with and without AlCl4, respectively, while EAlCl4 is the total energy of one AlCl4 molecule. Hence, a more negative value of adsorption energy signifies the more stable adsorption of AlCl4 on that site. As a result, the AlCl4 adsorption is found to be most stable (−2.36 eV) at Site 8, which corresponds to the hollow site surrounded by the C–C–B–N–B–N ring, as illustrated in Fig. 1d. The average distance of Al of AlCl4 from BCN monolayer on its adsorption at the hollow site surrounded by C–C–B–N–B–N ring is also least (3.85 Å) among all the sites, as represented in Table S1 (ESI†). The distance between AlCl4 and the monolayer can be justified by the absence of any overlap between the electron clouds of the monolayer and AlCl4 anion, as the interaction can be thought of as predominantly anion-π type. This has been the case in the previously reported adsorption of AlCl4 in few-layered graphene, where the distance between C and Cl was also found to be more than 3 Å, as the Cl atoms have an ionic radii of 1.67 Å and the overall size of AlCl4 is estimated to be 6.09 Å.54 The adsorption of AlCl4 occurs in such a way that the three Cl atoms facing towards the monolayer tend to stabilize closer to the B atoms of the monolayer. This may be due to the possibility of interaction between the lone pairs of Cl and vacant p orbital of the B atoms. As a result, the AlCl4 molecule undergoes a slight deformation as the three Al–Cl bonds facing towards the BCN monolayer get elongated to 2.17 Å (from 2.15 Å), while the one facing upward gets shortened to 2.13 Å.
The adsorption of Al2Cl7 on the BCN monolayer was also investigated and the adsorption energy was calculated to be −2.77 eV, which is much more favourable than the AlCl4 adsorption (−2.36 eV). This is expected due to the presence of a higher number of electronegative Cl atoms in the Al2Cl7 anion, which can induce a higher charge transfer from the monolayer. However, despite being energetically favourable, there is less probability of Al2Cl7 adsorption during the charge/discharge cycle of the battery. Firstly, it is reported that for a 1:1 ratio of [EMIm]Cl:AlCl3, only AlCl4 exists and the ratio must be 1:1.3, indicating that the AlCl3 concentration should be higher to generate Al2Cl7 in the electrolyte.55 Thus, the concentration of the Al2Cl7 ions is much less compared to AlCl4. Second, during the charging of the battery by application of an external potential through the circuit, when AlCl4 intercalation takes place on the cathodic part, Al2Cl7 must undergo simultaneous reduction to generate Al and AlCl4.
| 4Al2Cl7− + 3e− ⇌ Al + 7AlCl4− (Anode) |
| Cn + AlCl4− ⇌ Cn[AlCl4] + e− (Cathode) |
As a result, there is less chance that low concentrations of Al2Cl7 achieved by keeping the ratio greater than 1:1, can engage in binding with the cathode material. Based on the very low probability of Al2Cl7 being available for binding at the cathodic part, the voltage equation (eqn (5)) was derived, considering only AlCl4 intercalation.
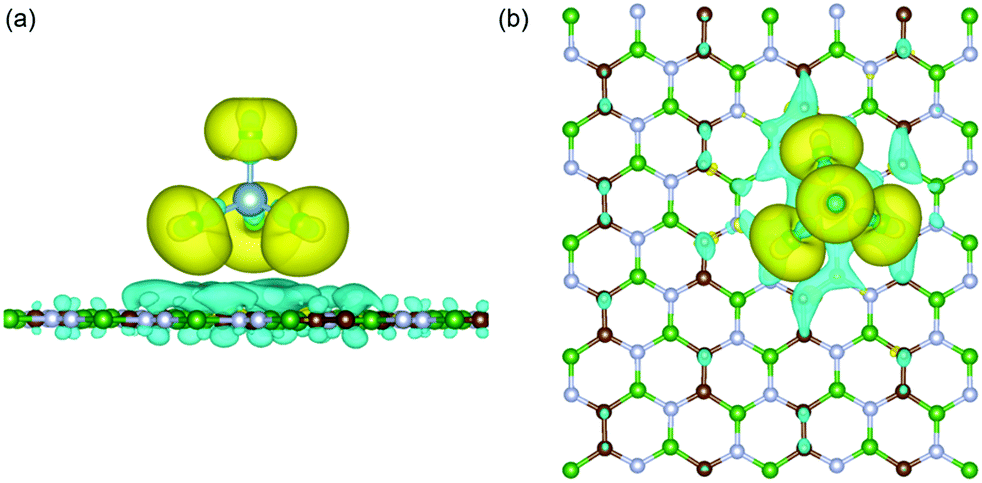
Next, to identify the nature of interaction between the adsorbed AlCl4 and BCN monolayer, the charge density difference (CDD) was evaluated using eqn (8):
| | 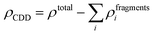 | (8) |
where
ρtotal is the total charge density of the AlCl
4-absorbed BCN system, and
ρfragmentsi is the charge density of the individual fragments from the system. The charge density of the individual fragments (
ρfragmentsi) was calculated using the pseudo structure in which the individual fragment retained the same structure as that in the system, while the remaining fragments were deleted. The CDD plot of the AlCl
4-absorbed BCN system at the most stable site is depicted in
Fig. 2. The nature of interaction was found to be ionic. On adsorption, there is an accumulation of electronic charge on each chlorine atom of AlCl
4, while depletion of electronic charge can be detected on the BCN monolayer atoms close to the adsorbed AlCl
4. As a result, the oxidation of the BCN monolayer cathode (along with simultaneous reduction of AlCl
4) during the charging mechanism can be concluded. Moreover, the charge depletion can be seen to occur from the C and N atoms, and not B atoms among the atoms surrounding the adsorption site due to the difference in electron density among them. Bader charge analysis of the system also shows that AlCl
4 accepts a net electronic charge of 0.9 |
e| from the BCN monolayer upon adsorption, signifying the cathode oxidation during charging.
 |
| | Fig. 2 (a) Side and (b) top view of the charge density difference plot for AlCl4 adsorbed on the BCN monolayer. Here, the green, brown, and blue colors represent the B, C and N atoms, respectively. Here, isosurface = 0.0009 |e| Å−3 (yellow for Δρ > 0 and blue for Δρ < 0). | |
3.3. Electrochemical and electronic properties
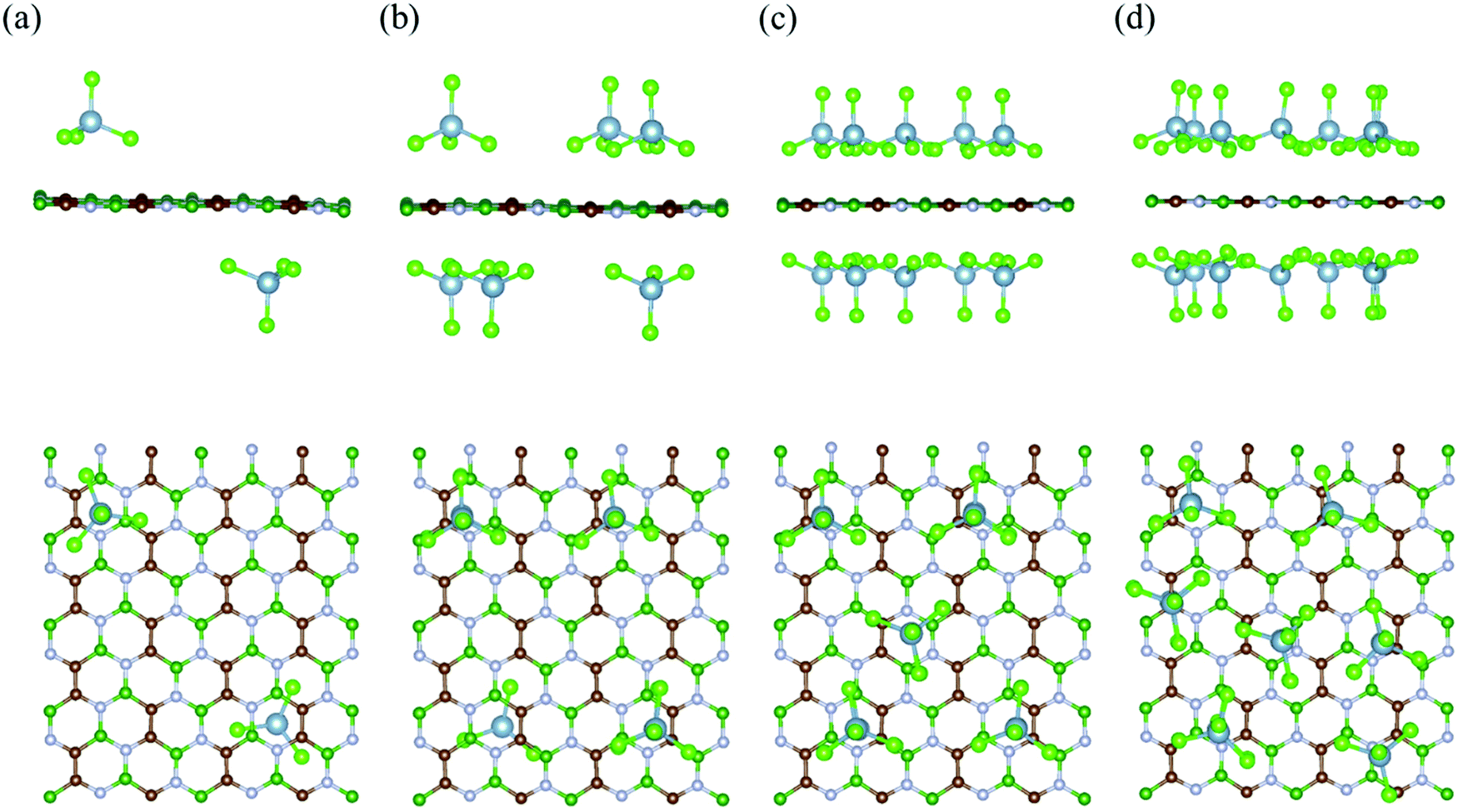
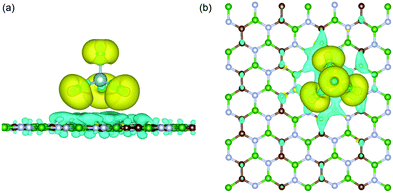
To calculate the electrochemical properties (like voltage and specific storage capacity) for the BCN cathode material, the BCN monolayer was systematically subjected to adsorb different concentrations of AlCl4. For the different concentrations of adsorbed AlCl4, all possible configurations were considered to find out the most stable configurations, as represented in Fig. 3 and Fig. S3 (ESI†). The maximum number of AlCl4 adsorbed on a 2 × 4 × 1 BCN monolayer was found to be 14.
 |
| | Fig. 3 Side and top views of the most stable configurations of the AlCl4-adsorbed BCN monolayer at different AlCl4 concentrations: (a) (AlCl4)2BCN, (b) (AlCl4)6BCN, (c) (AlCl4)10BCN, and (d) (AlCl4)14BCN. | |
To investigate the variation in adsorption behavior of the BCN monolayer with increasing concentration of AlCl4 adsorption, we calculated the adsorption energy for all cases, as presented in Table 1. As the concentration of AlCl4 adsorbed on BCN increases, the adsorption energy per AlCl4 molecule (as well as the charge transferred per AlCl4 molecule) was found to decrease. This decrease in adsorption energy might be due to the increase in repulsive forces, as the number of adsorbed AlCl4 molecule increases on the monolayer. The AlCl4 molecules occupy the most stable hollow site surrounded by the C–C–B–N–B–N ring in the monolayer up to 12 AlCl4 molecules. On further increasing the concentration, AlCl4 molecules do not necessarily occupy the most stable site and instead try to maintain distance among themselves, as shown in Fig. 3d. The maximum of 14 AlCl4 molecules are adsorbed reversibly on a 2 × 4 × 1 supercell of the BCN monolayer. Further adsorption of AlCl4 results in dissociation into AlCl3 and Cl, as shown in Fig. S4 (ESI†), which is not recommended for the reversible charging/discharging mechanism of a cathode material. However, the BCN monolayer was not found to show any kind of structural distortion on the adsorption of 14 AlCl4 molecules, and remained stable to be used as cathode material.
Table 1 The calculated adsorption energies, charge transfer to per AlCl4 from BCN and calculated theoretical voltage for different concentrations of AlCl4 adsorbed on the BCN monolayer
| Number of AlCl4 adsorbed |
Adsorption energy/AlCl4 (eV) |
Charge transfer to per AlCl4 molecule (|e|) |
Voltage (V) |
| 1 |
−2.36 |
−0.90 |
0.84 |
| 2 |
−2.21 |
−0.88 |
0.99 |
| 3 |
−2.05 |
−0.82 |
1.15 |
| 4 |
−1.95 |
−0.75 |
1.24 |
| 5 |
−1.85 |
−0.70 |
1.35 |
| 6 |
−1.79 |
−0.67 |
1.41 |
| 7 |
−1.74 |
−0.64 |
1.46 |
| 8 |
−1.70 |
−0.61 |
1.50 |
| 9 |
−1.63 |
−0.58 |
1.56 |
| 10 |
−1.59 |
−0.56 |
1.60 |
| 11 |
−1.55 |
−0.54 |
1.65 |
| 12 |
−1.52 |
−0.52 |
1.67 |
| 13 |
−1.46 |
−0.51 |
1.74 |
| 14 |
−1.38 |
−0.47 |
1.81 |
We calculated the theoretical charging voltage using eqn (5) for the systematic adsorption of AlCl4 molecules on the BCN monolayer. From Table 1, it is evident that the voltage for the adsorption of the first AlCl4 molecule is only 0.84 V, which continues to increase with increasing concentration of AlCl4. Hence, the average voltage obtained is 1.43 V. This seems to be inversely proportional with the change in adsorption energy per AlCl4, which is understandable from eqn (5). A more stable AlCl4-adsorbed system will require a lower charging voltage. As the AlCl4 concentration increases, the system gets destabilized due to the repulsion among the adsorbed anions, thus requiring a high charging voltage.24 The open circuit voltage for the maximum AlCl4-adsorbed system is 1.8 V, which is comparatively lower than that experimentally reported for the graphite cathode (2 V).16 However, the theoretical specific capacity calculated by eqn (6) for the BCN monolayer at the maximum AlCl4 adsorption is 316.9 mA h gBCN−1, which is much more than that experimentally reported for the graphite cathode (70 mA h g−1).16 Hence, the BCN monolayer can be considered an attractive high specific capacity cathode material that is suitable for Al dual-ion batteries.
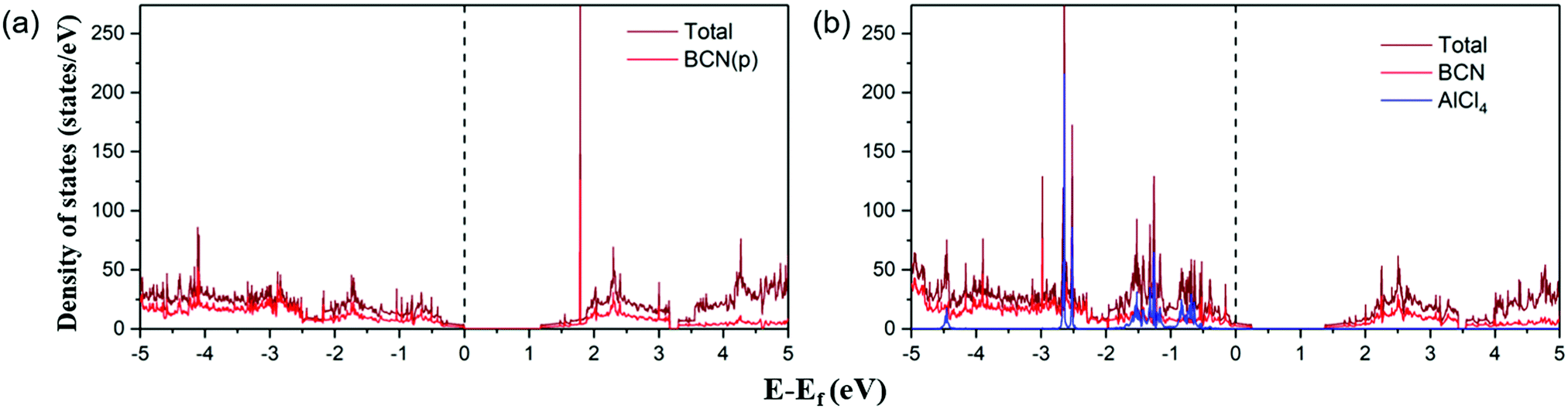
Furthermore, the electronic conductivity of a material also plays a crucial role in determining its suitability as a cathode material. We have plotted the total and partial density of states (DOS) of the pristine BCN and AlCl4-adsorbed BCN system, as represented in Fig. 4. In previous reports, configurations (b) and (a) of BCN have been found to be semiconducting in nature with a band gap of 1.5 eV and 1.18 eV using the HSE06 density functional and PBE functional, respectively.34,56 The DOS plots of pristine BCN of configuration (a) show the material to be semiconducting in nature with a small bandgap of 1.18 eV. The states near the Fermi level are mainly occupied by the BCN (2p) states. However, the DOS picture completely changes on adsorption of AlCl4 on the most stable site of the BCN monolayer. The Fermi level can be detected to shift towards the valence band compared to the pristine BCN DOS plot due to the charge transfer from BCN to AlCl4, which destabilizes the valence band. As a result, the states are introduced at the Fermi level, as there is interaction between the p states of AlCl4 and BCN. The band gap width was also observed to shrink with increasing concentration of AlCl4 adsorption (1.09 eV, 1.04 eV and 1.02 eV for 7, 10 and 14 AlCl4 adsorption, respectively), as represented in Fig. S5 (ESI†). Hence, the conductivity of the BCN monolayer may increase with AlCl4 adsorption, and satisfies the criteria for electronic conductivity of the cathode material.
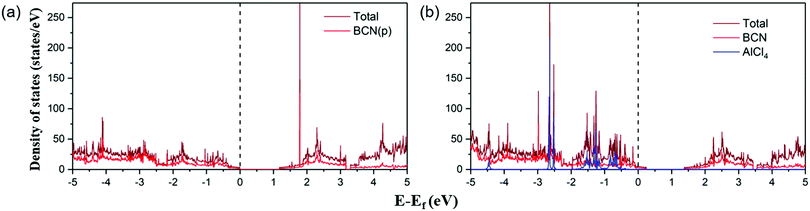 |
| | Fig. 4 Total and partial density of states of the (a) BCN monolayer and (b) AlCl4-adsorbed BCN monolayer. The Fermi level is set at zero. | |
3.4. Diffusion properties
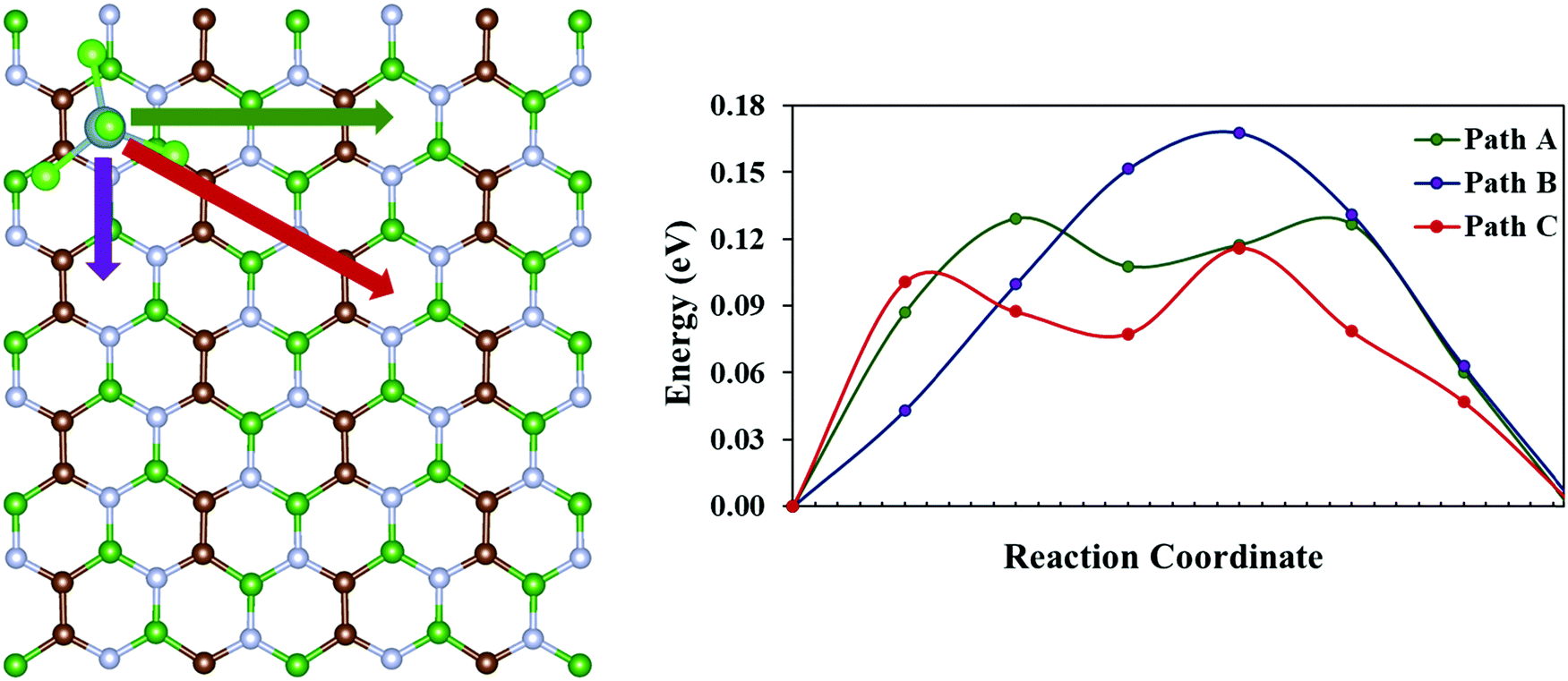
Another crucial factor that determines the performance of a battery is the charge/discharge rate of its cathode material. The charge/discharge rate depends on the migration of the adsorbed species within the cathode system. Thus, we have calculated the diffusion barrier for AlCl4 within similar adsorption sites of the BCN monolayer using the CI-NEB method. For this, a 2 × 4 × 1 supercell of the BCN monolayer has been considered. Site 8, which corresponds to the hollow site surrounded by C–C–B–N–B–N ring (as discussed earlier in Subsection 3.2), is taken as both the starting and the end point for AlCl4 diffusion because of its least adsorption energy. There are three possible pathways (path A, path B and path C) for AlCl4 diffusion, as illustrated in Fig. 5. The energy vs. reaction coordinate graph shows the anisotropic diffusion behavior, as path C is the most favorable path for AlCl4 diffusion with a smaller diffusion barrier (0.12 eV) compared to that of path A and path B, as represented in Table 2. Hence, we try to examine the stability of the highest energy transition state for path C compared to that of path A and path B. This relative stability of the highest energy transition states can be explained based on their orientations, as represented in Fig. S6 (ESI†). While trying to find the most stable orientations of AlCl4 adsorption, we saw the tendency of the three Cl atoms of the AlCl4 molecule in remaining closer to the B atoms of the BCN monolayer. Along the same line of thought, we find that the Cl atoms of AlCl4 for the most stable highest energy transition state (corresponding to path C) maintain a shorter average distance (3.47 Å) to their nearest B atoms of the monolayer compared to the corresponding transition states of path A and B, as given in Table 2. Overall, we find that the diffusion barrier of the minimum energy pathway (0.12 eV) is lower than the diffusion barrier calculated for the graphene bilayer as the cathode (0.19 eV) in a previous report.54 This demonstrates the ability of the BCN monolayer to allow trouble-free AlCl4 diffusion.
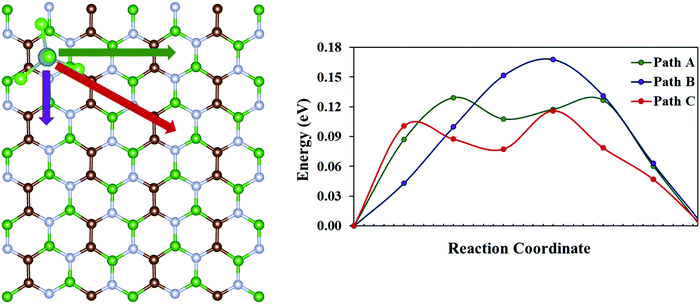 |
| | Fig. 5 Possible pathways for AlCl4 diffusion on the BCN monolayer and their corresponding diffusion energies. | |
Table 2 Diffusion energy barrier and average distance of Cl atoms of AlCl4 from the nearest B atoms in the BCN monolayer
| Diffusion path |
Diffusion energy barrier (eV) |
Average distance of Cl atoms of AlCl4 from the nearest B atoms in BCN (Å) |
| Path A |
0.13 |
3.50 |
| Path B |
0.17 |
3.68 |
| Path C |
0.12 |
3.47 |
4. Conclusion
In this work for the first time, the applicability of the BCN monolayer as a potential AlCl4 intercalating cathode material for Al dual-ion batteries has been discussed. The results show that AlCl4 adsorbs tetrahedrally with the BCN monolayer with reasonable adsorption energy. Furthermore, the Bader charge analysis and CDD show significant charge transfer of 0.9 |e| from the BCN monolayer to AlCl4, demonstrating the oxidation of BCN and reduction of AlCl4 during the charging mechanism. The systematic step-by-step adsorption of AlCl4 yields a maximum adsorption of 14 AlCl4 molecules on our considered BCN monolayer. As the number of adsorbed molecules increase, the adsorption energy per molecule decreases due to the repulsion among the anions. Also, the BCN monolayer retains its structure during the adsorption process, implying reversibility of the AlCl4 adsorption process. The voltage and capacity calculations reveal a significant open circuit voltage of 1.8 V (average voltage 1.43 V) with an ultrahigh specific capacity of 316.9 mA h gBCN−1. The DOS calculations predict the increase in conductivity of the BCN cathode on AlCl4 adsorption. Anisotropic properties are detected during the diffusion energy barrier calculations along different paths. The small energy barrier of 0.12 eV obtained for AlCl4 diffusion across the monolayer also predicts an unrestricted diffusion of the AlCl4 molecule. Overall, our study shows that the favorable AlCl4 adsorption, substantial voltage, ultra-high capacity, fast charge/discharge rate and the electrical conductivity of the BCN monolayer presents it as an alternative cathode material for Al dual-ion batteries.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank IIT Indore for lab and computing facilities. This work is supported by DST-SERB (Project Number CRG/2018/001131), and SPARC (Project Number SPARC/2018-2019/P116/SL) New Delhi. H. S. thanks IIT Indore for providing research facilities. S. D. thanks CSIR for a research fellowship.
References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 Search PubMed.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926–5940 Search PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 Search PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 223, 587–603 Search PubMed.
- S. K. Martha, B. Hariprakash, S. A. Gaffoor, D. C. Trivedi and A. K. Shukla, J. Chem. Sci., 2006, 118, 93–98 Search PubMed.
- G. J. May, A. Davidson and B. Monahov, J. Energy Storage, 2018, 15, 145–157 Search PubMed.
- R. H. Newman, J. Power Sources, 1994, 52, 149–153 Search PubMed.
- S. Rasul, S. Suzuki, S. Yamaguchi and M. Miyayama, Electrochim. Acta, 2012, 82, 243–249 Search PubMed.
- R. Zhang and C. Ling, MRS Energy Sustainability, 2016, 3, E1 Search PubMed.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 Search PubMed.
- B. Lee, H. R. Lee, H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, Chem. Commun., 2015, 51, 9265–9268 Search PubMed.
- Y. Ru, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 14391–14418 Search PubMed.
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203–5215 Search PubMed.
- Q. Li and N. J. Bjerrum, J. Power Sources, 2002, 110, 1–10 Search PubMed.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610–12612 Search PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 Search PubMed.
- P. Bhauriyal, A. Mahata and B. Pathak, Phys. Chem. Chem. Phys., 2017, 19, 7980–7989 Search PubMed.
- D. Y. Wang, C. Y. Wei, M. C. Lin, C. J. Pan, H. L. Chou, H. A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell and Y. J. Hsieh,
et al.
, Nat. Commun., 2017, 8, 14283 Search PubMed.
- Y. Wu, M. Gong, M.-C. Lin, C. Yuan, M. Angell, L. Huang, D.-Y. Wang, X. Zhang, J. Yang, B.-J. Hwang and H. Dai, Adv. Mater., 2016, 28, 9218–9222 Search PubMed.
- H. Sun, W. Wang, Z. Yu, Y. Yuan, S. Wang and S. Jiao, Chem. Commun., 2015, 51, 11892–11895 Search PubMed.
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554–2560 Search PubMed.
- X. Huang, Y. Liu, H. Zhang, J. Zhang, O. Noonana and C. Yu, J. Mater. Chem. A, 2017, 5, 19416–19421 Search PubMed.
- P. Bhauriyal, A. Mahata and B. Pathak, J. Phys. Chem. C, 2017, 121, 9748–9756 Search PubMed.
- P. Bhauriyal, P. Garg, M. Patel and B. Pathak, J. Mater. Chem. A, 2018, 6, 10776–10786 Search PubMed.
- P. Bhauriyal, G. Bhattacharyya, K. S. Rawat and B. Pathak, J. Phys. Chem. C, 2019, 123, 3959–3967 Search PubMed.
- L. Zhang, C. Chen, H. Luo, X. Zhou and Z. Liu, Adv. Energy Mater., 2017, 7, 1700034 Search PubMed.
- H. Chen, F. Guo, Y. Liu, T. Huang, B. Zheng, N. Ananth, Z. Xu, W. Gao and C. Gao, Adv. Mater., 2017, 29, 1605958 Search PubMed.
- J. Tu, H. Lei, Z. Yu and S. Jiao, Chem. Commun., 2018, 54, 1343–1346 Search PubMed.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 Search PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 Search PubMed.
- Q. Sun, Y. Dai, Y. Ma, T. Jing, W. Wei and B. Huang, J. Phys. Chem. Lett., 2016, 7, 937–943 Search PubMed.
- H. R. Jiang, Z. Lu, M. C. Wu, F. Ciucci and T. S. Zhao, Borophene: Nano Energy, 2016, 23, 97–104 Search PubMed.
- P. Bhauriyal, A. Mahata and B. Pathak, J. Phys. Chem. C, 2018, 122, 2481–2489 Search PubMed.
- S. Beniwal, J. Hooper, D. P. Miller, G. Costa, P. S. Chen, S. Y. Liu, P. A. Dowben, E. C. H. Sykes, E. Zurek and A. Enders, ACS Nano, 2017, 11, 2486–2493 Search PubMed.
- S. Lizzit and A. Baraldi, Catal. Today, 2010, 154, 68–74 Search PubMed.
- F. Orlando, R. Larciprete, P. Lacovig, I. Boscarato, A. Baraldi and S. Lizzit, J. Phys. Chem. C, 2012, 116, 157 Search PubMed.
- M. Corso, W. Auwärter, M. Muntwiler, A. Tamai, T. Greber and J. Osterwalder, Science, 2004, 303, 217 Search PubMed.
- S. Thomas and S. U. Lee, RSC Adv., 2019, 9, 1238–1246 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 Search PubMed.
-
G. Kresse and J. Furthmüller, J. Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 Search PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. J. Henkelman, J. Comput. Chem., 2007, 28, 899–908 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 Search PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 Search PubMed.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354 Search PubMed.
- M. L. Agiorgousis, Y.-Y. Sun and S. Zhang, ACS Energy Lett., 2017, 2, 689–693 Search PubMed.
- V. O. Özçelik and S. Ciraci, J. Phys. Chem. C, 2013, 117, 2175–2182 Search PubMed.
- S. C. Jung, Y. J. Kang, D. J. Yoo, J. W. Choi and Y. K. Han, J. Phys. Chem. C, 2016, 120, 13384–13389 Search PubMed.
- T. Jiang, M. J. Chollier Brym, G. Dubé, A. Lasia and G. M. Brisard, Surf. Coat. Technol., 2006, 201, 1–9 Search PubMed.
- S. Thomas, M. S. Manju, K. M. Ajith, S. U. Lee and M. Asle Zaeem, Phys. E, 2020, 123, 114180 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The total energy vs. time plot for AIMD simulation with optimized final structures, optimized structures of AlCl4 adsorption at all possible adsorption sites of the BCN monolayer, relative adsorption energy for all possible adsorption sites available for the adsorption of AlCl4 and the average distance of adsorbed AlCl4 from the BCN monolayer, side and top views of the most stable configuration of the AlCl4-adsorbed BCN monolayer at different concentrations, optimized configuration of 15 AlCl4 adsorbed system, DOS plots for 7, 10, 14 AlCl4 adsorbed systems, and the optimized highest energy transition states for different possible pathways of AlCl4 diffusion. See DOI: 10.1039/d0ma00501k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Sandeep
Das
,
Sandeep
Das