
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest-conditioned multicolor writing on cellulose nanocrystal canvases†
Miguel A.
Soto‡
 a,
Dongjie
Zhang‡
ab,
Yitao
Xu
a,
Yihan
Shi
a,
Brian O.
Patrick
a,
Wadood Y.
Hamad
c and
Mark J.
MacLachlan
*ade
a,
Dongjie
Zhang‡
ab,
Yitao
Xu
a,
Yihan
Shi
a,
Brian O.
Patrick
a,
Wadood Y.
Hamad
c and
Mark J.
MacLachlan
*ade
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: mmaclach@chem.ubc.ca
bMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, P. R. China
cTransformation and Interfaces Group, Bioproducts, FPInnovations, 2665 East Mall, Vancouver, British Columbia V6T 1Z4, Canada
dCanada Quantum Matter Institute, University of British Columbia, 2355 East Mall, Vancouver, BC, V6T 1Z4 Canada
eWPI Nano Life Science Institute, Kanazawa University, Kanazawa, 920-1192, Japan
First published on 9th October 2020
Abstract
Attractive ion–ion interactions allow trapping the tetracationic host cyclobis(paraquat-p-phenylene) (CBPQT4+) within solid matrices composed of negatively charged scaffolds, namely cellulose nanocrystals (CNCs). Through this supramolecular approach, we have fabricated CNC-CBPQT4+ films that change color upon sequenced guest uptake-molecular recognition. The evolution of color is conditioned to the selection of a guest and the self-assembly of an in-film host–guest complex. Here, we have used films and guests as synergetic paper and ink materials to generate responsive text and patterns that transform (disappear, change color, etc.) upon stimulation (temperature, pH, or competitive guests). This concept could be utilized for anticounterfeiting purposes, responsive inkjet printing, data encryption/decryption, and time-evolving patterns.
Since ancient time, mankind has used pigments and dyes to communicate in pictographic and written forms. The localization of coloring agents on a canvas generates imprints or patterns that are relatively static (unresponsive) and long-lasting.1 Alternatively, the concept of dynamic (responsive) patterns and writing has emerged as an intriguing approach that allows access to rewritability,2 transformable information,3 and data encryption.4 These features originate from the dynamic components incorporated into the canvas and/or ink materials, which transform upon stimulation. An applied trigger, e.g. heat,5,6 light,7,8 pH,9 or chemical species10–12 changes the structure of the molecular components and hence their optical (or luminescent) properties, leading to the appearance, switching, or disappearance of color.13–18
Cellulose nanocrystals (CNCs) are spindle-shaped particles obtained by the acid (H2SO4) hydrolysis of bulk cellulose. This treatment renders nanoparticles that contain sulfate half-ester groups on their surface.19 When dispersed in water, CNCs form colloidal suspensions that are stabilized by (sulfate–sulfate) interparticle repulsions.20,21 CNCs are attracting a great deal of attention for photonics,22–24 mechanical support,25–27 stimuli-responsive gels,28–30 and other applications.31–33 We recently demonstrated that CNCs can be used to produce gel receptors that change color as a result of in-gel guest recognition processes.34 The addition of the polyionic host cyclobis(paraquat-p-phenylene) (CBPQT4+, Fig. 1a)35 to a dispersion of CNCs modifies the ionic strength of the medium, mitigates interparticle repulsions, and causes gel formation. The molecular host remains trapped and active within the gel network (CNC-CBPQT4+). The materials can uptake and recognize guests from solution, a process that results in a color change.
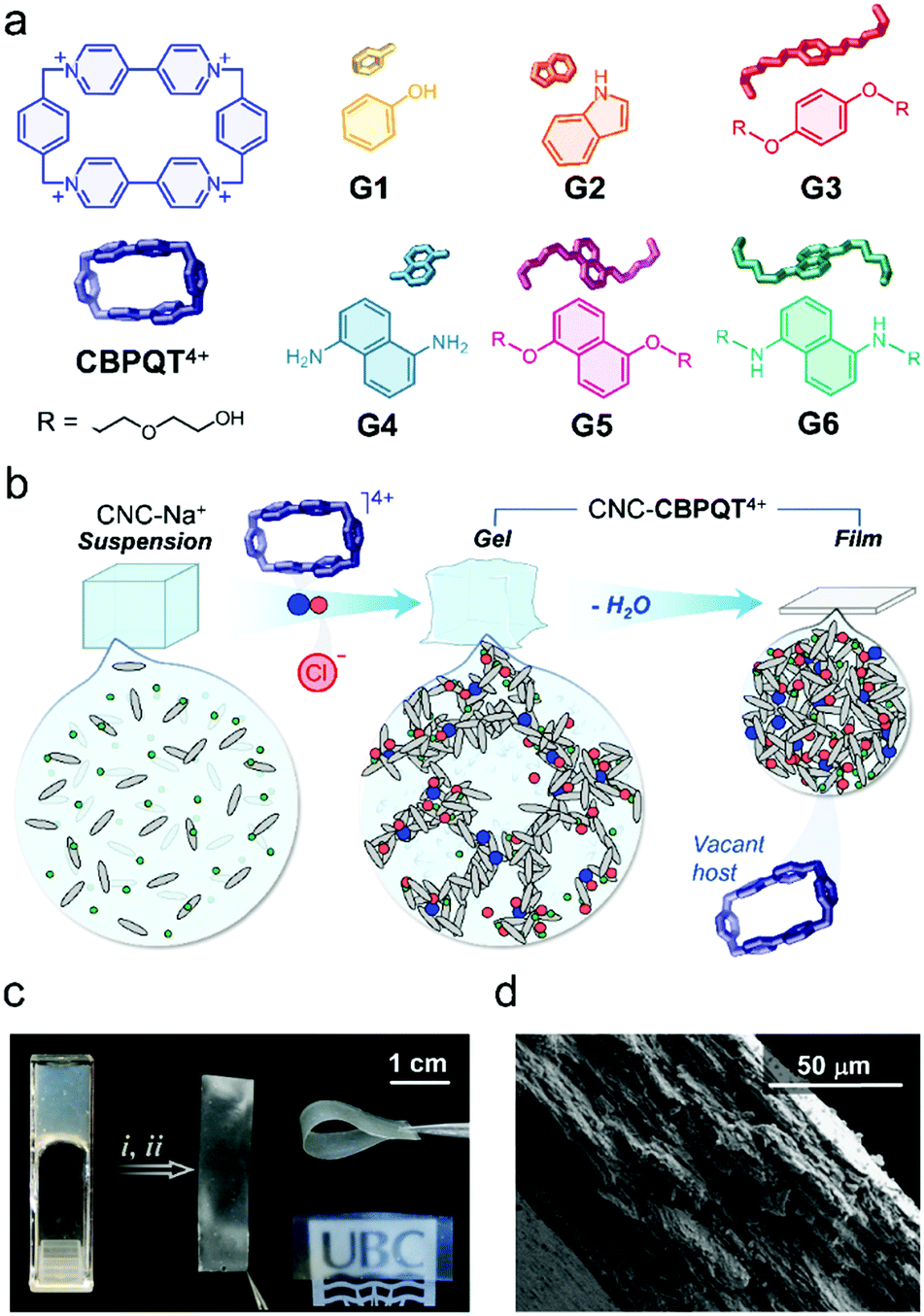 | ||
| Fig. 1 (a) Chemical structures and cartoons of the components utilized in the preparation of canvas (CBPQT4+) and ink materials (G1–G6). (b) Schematic representation for the fabrication of CNC-CBPQT4+ films. (c) Photographs of a CNC-CBPQT4+ gel (left) and films (right). Films obtained by (i) casting followed by (ii) evaporation (host ≈ 3 wt%). (d) SEM image featuring the cross section of a CNC-CBPQT4+ film (host ≈ 3 wt%). | ||
Here, we investigate the processing of CNC-CBPQT4+ gels into films (Fig. 1b) and their use as receptors that selectively respond to and recognize guest molecules. These materials have been used to develop new paper and ink products that produce stimuli-responsive imprints. The supramolecular methodology reported here offers two main advantages over previously designed cellulosic paper materials.36 First, the ion-induced fabrication of canvases is robust and readily accessible (e.g. no layering or covalent functionalization needed). Second, the coloration mechanism (via host–guest recognition) allows access to multichromatic outcomes, rendering patterns with clear contrast that are observable under ambient light.
Initially, we anticipated that (i) the optical properties of the in-film host–guest complexes could be adjusted by diversifying the structure of the guest species (G1–G6, Fig. 1a),37,38 which would be key to perform multicolor tinting/writing, as well as stimuli-responsive patterning; (ii) the macrocycle CBPQT4+ would remain embedded within the films due to its interaction (via electrostatic ion–ion attraction) with the negatively charged CNC particles; and (iii) CBPQT4+ would prevail as an active host as it has a non-collapsible shape-persistent cavity.
To prove our concept, we started with the preparation of CNC-CBPQT4+ gels by mixing sodium-neutralized cellulose nanocrystals (CNC-Na+, 6 wt%) with varied amounts of [CBPQT]Cl4. The final concentrations of host were fixed to 1.3, 2.5, 3.8, and 7.5 mM. Upon gel formation, the materials were cast onto polystyrene substrates and dried under ambient conditions (details in ESI†), yielding self-standing colorless films. We calculated that these materials contained 1–7 wt% of embedded macrocycle. Low amounts of host (1 and 2 wt%) produced transparent films, whereas higher ratios (3 and 7 wt%) afforded semitransparent materials (Fig. 1c), all ranging from 84 to 89% transmittance at 500 nm (Fig. S1, ESI†). The CNC-CBPQT4+ films did not show obvious sensitivity to air or moisture, and thermally decomposed above ca. 200 °C due to the carbonization of cellulose nanocrystals (Fig. S2, ESI†).
At the microscale, we identified the CNC spindles randomly oriented on the surface of the films (Fig. S3, ESI†) and confirmed a layered structure through the cross-section of the CNC-CBPQT4+ materials (Fig. 1d), which suggested that the films would be relatively permeable. When submerged in water, the materials remained cohesive. Storing a film (ca. 7 wt% host) for three months in D2O at room temperature did not have an obvious effect on the material's stability. In fact, in a recorded 1H NMR spectrum of the supernatant solution, we did not observe host leaching out of the film (Fig. S4, ESI†). Likewise, the films remained stable in organic media, including ethanol, methanol, acetonitrile, nitromethane, and acetone.
With the films fabricated, we focused on the self-assembly of the inclusion complexes in solution. Most of the selected guests (G1–G6) are known to interact with CBPQT4+ in polar organic solvents (e.g. acetone and acetonitrile),35,39–42 while complex formation has been scarcely explored in water.43
We proved that each individual guest self-assembles with the host in aqueous media to render colorful inclusion complexes via charge-transfer (CT) and π-stacking (see ESI†). As an example, adding one equivalent of G4 to a solution of [CBPQT]Cl4 (2.5 mM) in D2O (Fig. 2a), resulted in an immediate color change from colorless to blue, which indicated intercomponent self-assembly. In the recorded 1H NMR spectrum (Fig. 2b), we observed a dramatic low frequency shift for all G4 resonances (a, b, and c); e.g. Δδa = −5.2 ppm. The α and β host signals displayed a similar trend (e.g. Δδα = −0.3 ppm, see Fig. S5, ESI†). The shielding of host and guest resonances implied strong intercomponent π–π stacking within the formed inclusion complex G4 ⊂ CBPQT4+. A UV-vis spectrum of this system showed a broad, unstructured CT band at 678 nm (Fig. S6, ESI†), which is ascribed to the interaction of the electron-rich guest G4 with the electron-deficient host. The geometry of the host–guest complex was further confirmed by single-crystal X-ray diffraction. In the solid state, G4 resides inside the cavity of CBPQT4+ (Fig. 2c), displaying a mean plane-to-plane (bipyridinium-naphthalene) distance of 3.35 Å, which reasserts strong intercomponent π-stacking (see full data in ESI†).
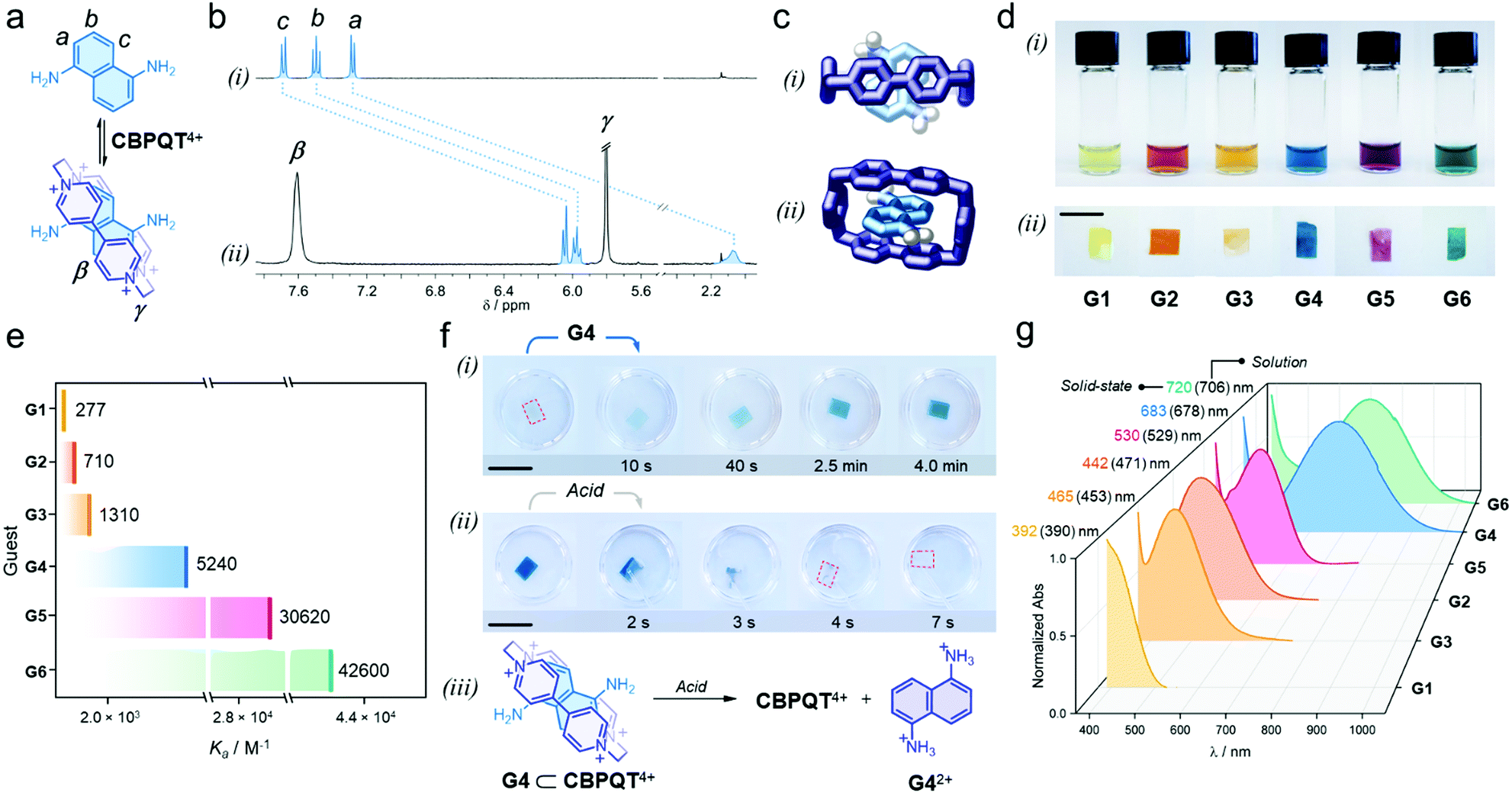 | ||
| Fig. 2 (a) Self-assembly of the host–guest complex G4 ⊂ CBPQT4+; proton labeling is included in the chemical structures. (b) Partial 1H NMR spectra (400 MHz, D2O) of (i) G4 and (ii) an equimolar solution of G4 and [CBPQT]Cl4 (G4 = 2.5 mM). (c) Solid-state structure of the inclusion complex G4 ⊂ CBPQT4+; (i) top and (ii) side views. Chloride ions and solvent molecules are omitted for clarity. Only amine H-atoms are shown. (d) Photographs showing the series of host–guest complexes assembled (i) in solution and (ii) within CNC films (3 wt% host). (e) Association constants (Ka) measured in solution via spectrophotometric titrations (EtOH:H2O, 3/1, v/v, 25 °C). Shown values correspond to the average of three independent measurements (see Table S1, ESI†). (f) Snapshots of (i) the uptake-recognition of G4 and (ii) its acid-triggered release; the red outline highlights the colorless CNC films. (iii) Scheme of the acid-promoted exclusion of G42+. (g) UV-vis spectra of the assembled in-film host–guest complexes (3 wt% host); this plot includes λmax detected in the solution and solid state. Scale bars = 1 cm. | ||
When comparing all formed complexes between CBPQT4+ and G1–G6, two main differences are noteworthy: their apparent color and stability. The assembled complexes cover a wide region of the visible spectrum: yellow for G1; orange for G2 and G3; blue for G4; purple for G5; and green for G6 (Fig. 2d(i)), and have affinities (Ka) that range from 277 ± 10 M−1 for G1 to 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 ± 2300 M−1 for G6 (see Fig. 2e and Table S1, ESI†).44 These variations arise from the electronic properties of the guests, which contain different (i) cores (phenylene vs. naphthalene), (ii) electron-donating substituents (O–R vs. N–R), and (iii) may or may not bear lateral chains – which contribute to complex stabilization via H-bonding.45,46
600 ± 2300 M−1 for G6 (see Fig. 2e and Table S1, ESI†).44 These variations arise from the electronic properties of the guests, which contain different (i) cores (phenylene vs. naphthalene), (ii) electron-donating substituents (O–R vs. N–R), and (iii) may or may not bear lateral chains – which contribute to complex stabilization via H-bonding.45,46
With the assembly of all host–guest systems proved, we investigated the performance of the CNC-CBPQT4+ films as guest receptors using a mixture of ethanol and water (3/1, v/v) as the medium.47 A solution of G4 (5 mL, 50 mM) was added to a vessel containing a CNC-CBPQT4+ film (15 mg, 3 wt% host). After a few seconds, we noticed that the film turned pale blue, getting more intense over time (Fig. 2f(i)). After ca. 4 min, the film became homogeneously colored (Video S1, ESI†). This suggests that as the guest permeates into the film, G4 reaches the confined host and undergoes molecular recognition to form G4 ⊂ CBPQT4+, revealed by its distinctive blue color. A UV-vis spectrum of the obtained film displayed a CT band centered at 683 nm, which compares well with the band identified for the same complex in solution (678 nm, Fig. S7, ESI†). Interestingly, the limiting guest uptake, and thus the maximum content of host–guest complex in a film, could be adjusted by varying the amount of embedded CBPQT4+ (1–7 wt%, see Fig. S8, ESI†).
We confirmed that all in-film complexes (using G1–G6) assemble over similar time scales upon the exposure of the CNC-CBPQT4+ films to a guest solution (Fig. 2d(ii)). The CT bands detected in the solid state resemble those observed in solution (Fig. 2g). Of note, molecules that did not feature the stereoelectronic properties of prototypical guests (G1–G6), were also absorbed by CNC-CBPQT4+ films but did not self-assemble with the host. Their uptake was optically undistinguishable (Fig. S9, ESI†).
In principle, all in-film complexes could be disassembled by disrupting the intercomponent interactions through controlled stimuli. We found that the pH-responsive guests (G4 and G6) could be readily released from the films by an acid treatment. For instance, the addition of a HCl solution (50 mM, ethanol/water (3/1, v/v)) to a film composed of G4 ⊂ CBPQT4+, quickly caused the disappearance of the blue color (Fig. 2f(ii), Video S2, ESI†). This is explained by the protonation of G4; the tetracationic host repels the formed dication (G42+, Fig. 2f(iii)) and this species is expelled from the host cavity. After removing the film from the HCl solution, the material did not recuperate its blue color, indicating complete guest protonation. By 1H NMR spectroscopy we detected G42+ in the acidic solution, which confirmed guest release (Fig. S10, ESI†). Similarly, guests G1–G3 and G5 were ejected from the films using temperature as the trigger (Fig. S11, ESI†). Heating the films at 70 °C while submerged in ethanol/water (3/1, v/v), caused guest dissociation and release. In either case, using heat or acid, the stimuli-controlled disassembly allowed for the recovery of vacant CNC-CBPQT4+ films.
As an additional means to disassemble an in-film complex, we evaluated the addition of competitive species. Given the different stability of the host–guest complexes (Fig. 2e), we anticipated that an in-film guest exchange process would be feasible. To prove it, we first submerged a CNC-CBPQT4+ film (3 wt% host) in a solution of G1 (50 mM, ethanol/water (3/1, v/v)) for 30 min; this turned the material from colorless to pale-yellow, implying self-assembly of G1 ⊂ CBPQT4+ within the CNC matrix (Fig. 3a). Under the same conditions, the produced film was subsequently immersed in a solution of G2 (50 mM) and left to equilibrate. This resulted in a color change from yellow to orange. As evidenced by the solution Ka values, CBPQT4+ prefers G2 over G1. Thus, G2 can displace G1 from the host cavity (and eventually from the CNC matrix), switching the in-film complex from G1 ⊂ CBPQT4+ (yellow) to G2 ⊂ CBPQT4+ (orange). Similarly, G2 was expelled from the host cavity by G3, this guest by G4, G4 by G5, and finally G5 by G6. All these exchange events were confirmed by color changes (Fig. 3a).
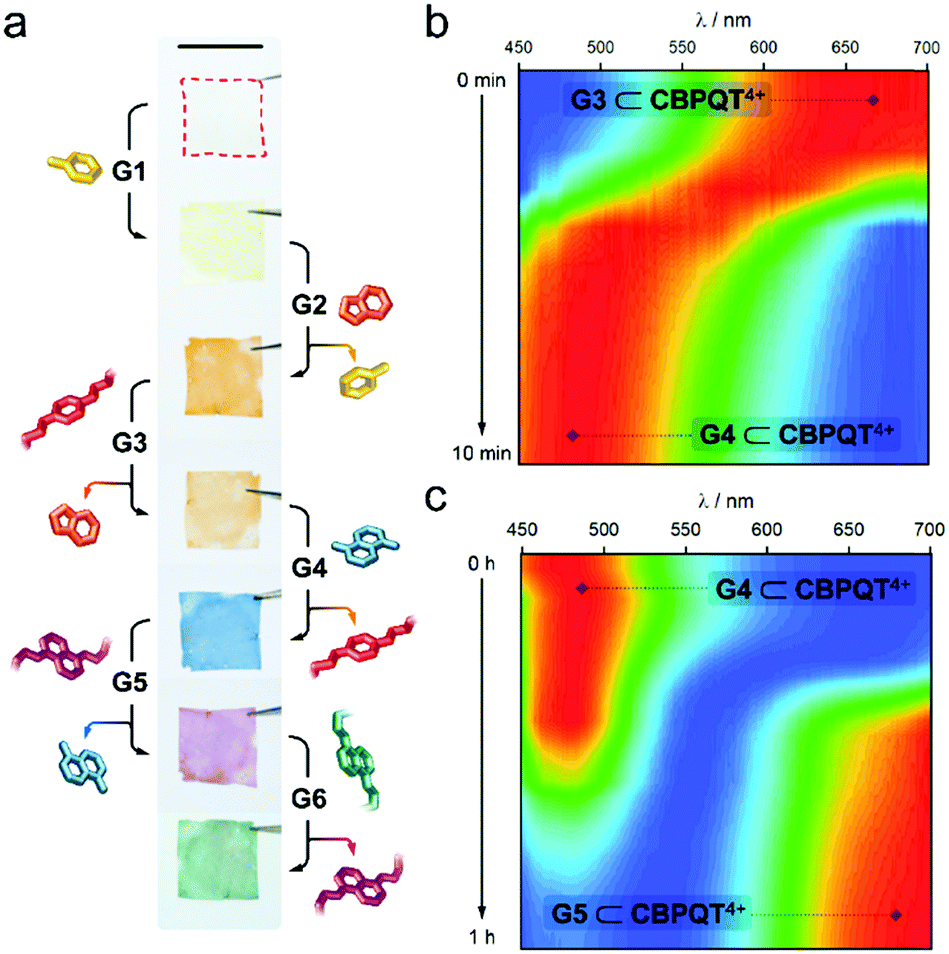 | ||
| Fig. 3 (a) Photographs of the in-film guest exchange process. The red outline highlights the colorless CNC film (3 wt% host). Scale bar = 1 cm. Time-resolved reflectance spectra for the in-film transformation of (b) G3 ⊂ CBPQT4+ into G4 ⊂ CBPQT4+ and (c) G4 ⊂ CBPQT4+ into G5 ⊂ CBPQT4+. The color scale goes from blue (minimum) to red (maximum); each 1-D spectrum was individually normalized prior to building the 2-D plots to emphasize the shift in reflectance maximum. | ||
The exchange process was monitored by reflectance spectroscopy, which gave evidence of exchange completeness and allowed for a relative analysis of the trading rates. Two examples are shown in Fig. 3b and c (see Fig. S12 for full data set, ESI†). The in-film complex G3 ⊂ CBPQT4+ appears orange in color and the corresponding reflectance spectrum features a band from 580 to 700 nm. Upon the addition of a solution of G4 (50 mM, ethanol/water (3/1, v/v)), a new band with a maximum between 450–540 nm (assigned to G4 ⊂ CBPQT4+) rapidly evolved, reaching saturation in ca. 10 min (Fig. 3b). The initial band was no longer evident. The obtained in-film complex was then transformed into G5 ⊂ CBPQT4+ by the addition of G5 in solution (50 mM, ethanol/water (3/1, v/v)). This caused the gradual disappearance of the 450–540 nm band, accompanied by the emergence of a new band at 625–700 nm (G5 ⊂ CBPQT4+), which reached a maximum in ca. 1 h (Fig. 3c). The difference in exchange rates can be explained by the diffusion of the guests within the film, which depends on the bulkiness of the guests; trading G3 by G4 takes only 10 min, while exchanging G4 with a bulkier molecule (G5) takes ca. six times longer. An inverse trading process from G6 to G2 was tested, however, the exchange was not fully completed, and, in some cases, the expected colors were not observed (Fig. S13, ESI†).
Writing on the surface of a CNC-CBPQT4+ film (Fig. 4a) would be accessible by localizing a guest (ink) on specific areas of the film receptor (canvas); the apparent color and (chemical/physical) susceptibility of the writing would be defined by the chosen ink. To prove this, a pen cartridge was loaded with a solution of G4 (50 mM, ethanol/water (3/1, v/v). The ink was then applied on a CNC-CBPQT4+ canvas (3 wt% host). After just a few seconds, the writing was revealed in blue color as a result of the in-film self-assembly of G4 ⊂ CBPQT4+ (Fig. 4b). Overwriting the pattern allowed increasing the color intensity, which suggested that free host remained on the initially written region (Video S3, ESI†). As expected, the writing was fully erased within seconds upon immersion of the film in an alcoholic HCl solution (50 mM).
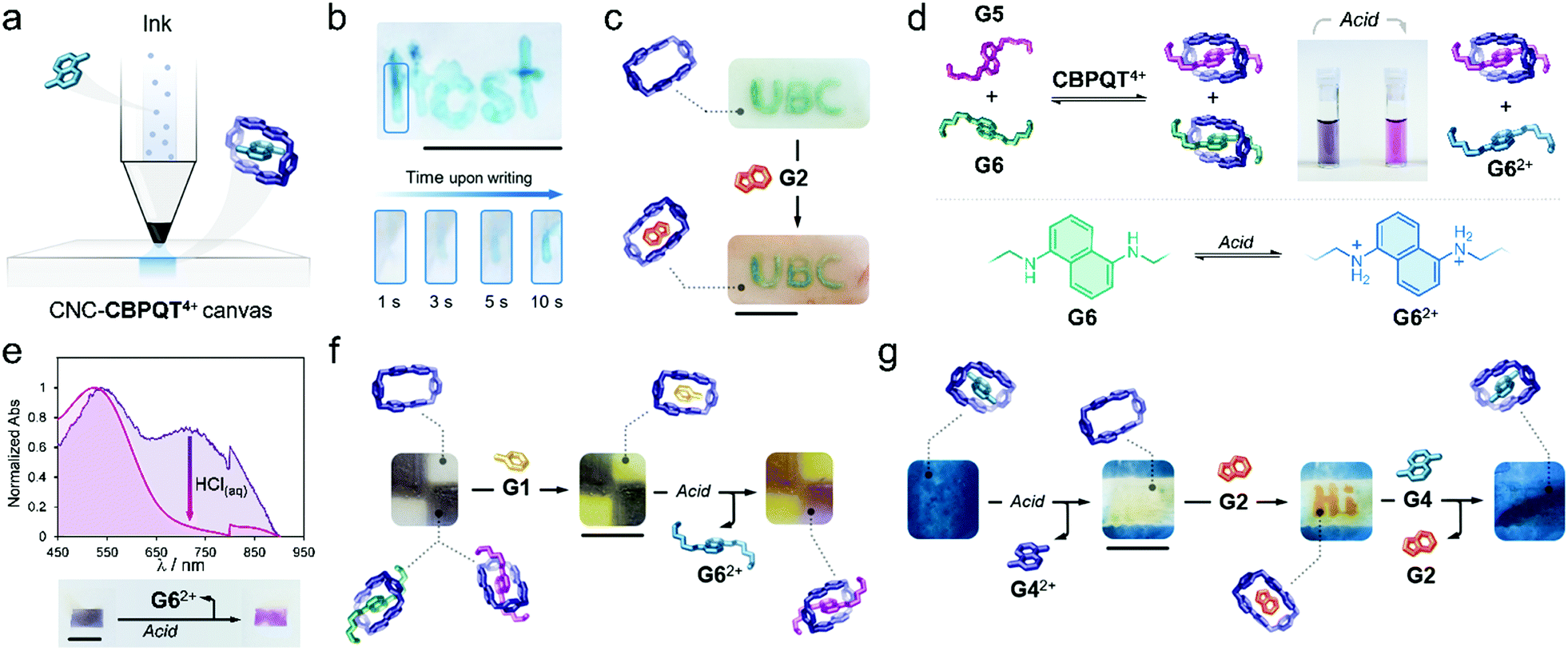 | ||
| Fig. 4 (a) Representation of the guest-conditioned writing approach. (b) Photograph of a CNC-CBPQT4+ film (3 wt% host) where an imprint (host) was incorporated using G4; the evolution of color upon writing is shown in the bottom of this figure. (c) Background tinting via the uptake and recognition of a non-competitive guest (G2). (d) Responsive system composed by coexisting host–guest complexes; the photograph shows a solution containing G5 ⊂ CBPQT4+ and G6 ⊂ CBPQT4+ (left) and its transformation into G5 ⊂ CBPQT4+ (right) through the protonation of G6 (bottom). (e) UV-vis spectra for the in-film selective disassembly of G6 ⊂ CBPQT4+ by acid treatment; the photograph at the bottom shows the macroscopic outcome. (f) pH-Responsive pattern composed by coexisting host–guest complexes; CNC-CBPQT4+ film (7 wt% host). (g) Tinting–erasing–writing–restoring in a single CNC-CBPQT4+ film (7 wt% host). Scale bars = 1 cm. | ||
Writing on a CNC-CBPQT4+ film leaves behind an uncolored, yet active, portion in the canvas: the background. We envisaged that the introduction of a scarcely competitive guest (relative to the ink) would allow for the selective tinting of the canvas background. Fig. 4c shows a film in which the acronym UBC was introduced with G6. Submerging this film in a solution of G2 (50 mM, ethanol/water (3/1, v/v), 1 min) quickly colored the background in orange (G2 ⊂ CBPQT4+) whereas the UBC pattern remained green (G6 ⊂ CBPQT4+). Because CBPQT4+ prefers G6 over G2 (solution Ka (G6) ⋙ Ka (G2)), G2 selectively assembles with the host that is available in the background, but G2 cannot compete for the occupied host located in the written portion.
Taking advantage of the guest-conditioned tinting and pH-responsiveness of some of the guests, we designed a dynamic ink for writing and patterning. Guests G5 and G6 were selected considering that: (i) in solution, the affinity constants for G5 and G6 with CBPQT4+ are comparable ((3.1 vs. 4.3) × 104 M−1, respectively), (ii) both guests offer contrasting colors in their complexed forms (green for G6 and purple for G5), and (iii) from the two species, only G6 is pH-sensitive.
We found that the addition of one equivalent of a G5/G6 mixture (2/1 mol ratio) to a solution of CBPQT4+ (2.5 mM) in ethanol/water (3/1, v/v), produced a color change from colorless to deep violet. Two CT bands were evident (529 and 706 nm) in the UV-vis spectrum that corresponded to the coexistence of G5 ⊂ CBPQT4+ and G6 ⊂ CBPQT4 (Fig. S14, ESI†). Notably, the addition of acid (50 mM HCl, ethanol/water (3/1, v/v)) produced discoloration of the sample, going from dark violet to purple (Fig. 4d). This is explained by the protonation of G6, which leads to the dissociation of G6 ⊂ CBPQT4+, leaving in solution G62+ and complex G5 ⊂ CBPQT4+. We confirmed a similar behavior in the solid state (Fig. 4e). Immersing a CNC-CBPQT4+ canvas (3 wt% host) in a solution of G5/G6 (2/1 mol ratio, ethanol/water (3/1, v/v)) colored the film dark violet within minutes; in the recorded UV-vis spectrum, we observed two CT bands at 530 and 720 nm corresponding to assembled complexes G5 ⊂ CBPQT4+ and G6 ⊂ CBPQT4+, respectively. Treating the film with acid (50 mM HCl, ethanol/water (3/1, v/v)), caused the vanishing of the band centered at 720 nm, and produced a change in the color of the film from violet to purple (Fig. 4e) as an indication of the selective disassembly of G6 ⊂ CBPQT4+.
With the responsive ink prepared (G5/G6, 2/1 mol ratio), we colored two portions of a canvas (7 wt% host) to generate the film shown in Fig. 4f. Immersing this pattern in a solution of G1 (50 mM, ethanol/water (3/1, v/v), 1 min), switched the vacant portions of the film from colorless to yellow (G1 ⊂ CBPQT4+); the violet regions remained unaltered as G1 cannot displace G5 or G6 (solution Ka (G5/G6) ⋙ Ka (G1)). Further treatment of the pattern with HCl (50 mM, ethanol/water (3/1, v/v), 1 min) selectively switched the violet region into purple through the disassembly of G6 ⊂ CBPQT4+. Complexes G1 ⊂ CBPQT4+ and G5 ⊂ CBPQT4+ remained intact and confined in specific portions of the canvas.
As an additional approach, we explored a four-step sequential process consisting of tinting-erasing-writing-restoring, using a single CNC canvas. A film (7 wt% host) was submerged in a solution of G4 (50 mM, ethanol/water (3/1, v/v), 30 min), yielding the blue in-film complex G4 ⊂ CBPQT4 (Fig. 4g). A solution of HCl (50 mM, ethanol/water (3/1, v/v)) was then applied on the central portion of the canvas, producing localized protonation of G4 and subsequent exclusion of G42+ from the host cavity. Writing on the freshly erased region was then accessible. By using G2 (50 mM, ethanol/water (3/1, v/v)), orange writing was incorporated. The imprinted pattern was finally deleted by immersing the canvas in a solution of G4 (50 mM, ethanol/water (3/1, v/v)), 1 h), which filled up the vacant portion of the film and displaced G2 from the written region, ultimately restoring the homogeneous blue material.
Conclusions
In summary, we have used a supramolecular approach to produce guest-responsive CNC matrices composed of a tetracationic host (CBPQT4+) – which remains embedded, active, and vacant within the materials. In a sequenced process, these matrices can uptake and self-assemble with π-electron rich molecules (G1–G6), developing colored readouts that are fully reversible. Taking advantage of this, and the different stability of the assembled complexes, we have developed a new writing approach that is conditioned to the presence of host in the canvas and guest in the ink. The produced writing/patterns are responsive to competitive species (other guests), acid, and heat, which can cause color switching or disappearance. The features incorporated into these functional paper products might be valuable in anticounterfeiting, rewritability, and data transformation; concepts now under investigation by our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. S. thanks CONACYT for a postdoctoral fellowship. D. Z. thanks the Chinese Science Council (CSC) for a scholarship, and Professors Yuyan Liu and Zhongjun Cheng for their support. M. J. M. thanks NSERC for a Discovery grant and a CREATE NanoMat grant. Y. S. thanks UBC for a 4 year fellowship.Notes and references
- A. Abel, in Colour Design: Theories and Applications, ed. J. Best, Woodhead Publishing, Duxford, United Kingdom, 2nd edn, 2017, pp. 557–587 Search PubMed.
- J. Ge, J. Goebl, L. He, Z. Lu and Y. Yin, Adv. Mater., 2009, 21, 4259–4264 CrossRef CAS.
- Y.-D. Yang, X. Ji, Z.-H. Lu, J. Yang, C. Gao, H. Zhang, B. Z. Tang, J. L. Sessler and H.-Y. Gong, Nat. Commun., 2020, 11, 77 CrossRef CAS.
- Y. Ma, S. Liu, H. Yang, Y. Zeng, P. She, N. Zhu, C.-L. Ho, Q. Zhao, W. Huang and W.-Y. Wong, Inorg. Chem., 2017, 56, 2409–2416 CrossRef CAS.
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS.
- W. Wang, N. Xie, L. He and Y. Yin, Nat. Commun., 2014, 5, 5459 CrossRef CAS.
- A. Abdollahi, K. Sahandi-Zangabad and H. Roghani-Mamaqani, ACS Appl. Mater. Interfaces, 2018, 10, 39279–39292 CrossRef CAS.
- K. Isoda, M. Matsubara, A. Ikenaga, Y. Akiyama and Y. Mutoh, J. Mater. Chem. C, 2019, 7, 14075–14079 RSC.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef.
- Y. Ma, P. She, K. Y. Zhang, H. Yang, Y. Qin, Z. Xu, S. Liu, Q. Zhao and W. Huang, Nat. Commun., 2018, 9, 3 CrossRef.
- C. Sun, D. Zhu, H. Jia, C. Yang, Z. Zheng and X. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 26455–26463 CrossRef CAS.
- L. Sheng, M. Li, S. Zhu, H. Li, G. Xi, Y.-G. Li, Y. Wang, Q. Li, S. Liang, K. Zhong and S. X.-A. Zhang, Nat. Commun., 2014, 5, 3044 CrossRef.
- H. Sun, N. Gao, J. Ren and X. Qu, Chem. Mater., 2015, 27, 7573–7576 CrossRef CAS.
- X. Du, T. Li, L. Li, Z. Zhang and T. Wu, J. Mater. Chem. C, 2015, 3, 3542–3546 RSC.
- W. Jeong, M. I. Khazi, D.-H. Park, Y.-S. Jung and J.-M. Kim, Adv. Funct. Mater., 2016, 26, 5230–5238 CrossRef CAS.
- A. S. Terpstra, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2017, 27, 1703346 CrossRef.
- C. Sun, S. Su, Z. Gao, H. Liu, H. Wu, X. Shen and W. Bi, ACS Appl. Mater. Interfaces, 2019, 11, 8210–8216 CrossRef CAS.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- S. Shafeiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, Rheol. Acta, 2013, 52, 741–751 CrossRef CAS.
- X. M. Dong, J.-F. Revol and D. G. Gray, Cellulose, 1998, 5, 19–32 CrossRef CAS.
- Y. Cao, L. Lewis, W. Y. Hamad and M. J. MacLachlan, Adv. Mater., 2019, 31, 1808186 CrossRef.
- Y. Liu and P. Wu, Adv. Funct. Mater., 2020, 30, 2002193 CrossRef CAS.
- Y. Cao, P.-X. Wang, F. D’Acierno, W. Y. Hamad, C. A. Michal and M. J. MacLachlan, Adv. Mater., 2020, 32, 1907376 CrossRef CAS.
- J. Yang, C.-R. Han, X.-M. Zhang, F. Xu and R.-C. Sun, Macromolecules, 2014, 47, 4077–4086 CrossRef CAS.
- A. Sheikhi and T. G. M. van de Ven, ACS Appl. Nano Mater., 2018, 1, 1841–1852 CrossRef CAS.
- S. Zhang, D. Huang, H. Lin, Y. Xiao and X. Zhang, Biomacromolecules, 2020, 21, 2400–2408 CrossRef CAS.
- T. Hiratani, O. Kose, W. Y. Hamad and M. J. MacLachlan, Mater. Horiz., 2018, 5, 1076–1081 RSC.
- A.-L. Oechsle, L. Lewis, W. Y. Hamad, S. G. Hatzikiriakos and M. J. MacLachlan, Chem. Mater., 2018, 30, 376–385 CrossRef CAS.
- L. Lewis, S. G. Hatzikiriakos, W. Y. Hamad and M. J. MacLachlan, ACS Macro Lett., 2019, 8, 486–491 CrossRef CAS.
- K. De France, Z. Zeng, T. Wu and G. Nyström, Adv. Mater., 2020, 2000657, DOI:10.1002/adma.202000657.
- B. L. Tardy, J. J. Richardson, L. G. Greca, J. Guo, H. Ejima and O. J. Rojas, Adv. Mater., 2020, 32, 1906886 CrossRef CAS.
- Z. Gao, Z. Lou, W. Han and G. Shen, ACS Appl. Mater. Interfaces, 2020, 12, 24339–24347 CrossRef CAS.
- D. Zhang, M. A. Soto, L. Lewis, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2020, 59, 4705–4710 CrossRef CAS.
- B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1988, 27, 1547–1550 CrossRef.
- Z. Li, X. Yang, W. Li and H. Liu, Carbohydr. Polym., 2019, 210, 350–363 CrossRef CAS.
- W.-Q. Deng, A. H. Flood, J. F. Stoddart and W. A. Goddard, J. Am. Chem. Soc., 2005, 127, 15994–15995 CrossRef CAS.
- T. Ikeda, I. Aprahamian and J. F. Stoddart, Org. Lett., 2007, 9, 1481–1484 CrossRef CAS.
- J. W. Choi, A. H. Flood, D. W. Steuerman, S. Nygaard, A. B. Braunschweig, N. N. P. Moonen, B. W. Laursen, Y. Luo, E. DeIonno, A. J. Peters, J. O. Jeppesen, K. Xu, J. F. Stoddart and J. R. Heath, Chem. – Eur. J., 2005, 12, 261–279 CrossRef.
- A. Mirzoian and A. E. Kaifer, J. Org. Chem., 1995, 60, 8093–8095 CrossRef CAS.
- M. Asakawa, W. Dehaen, G. L’abbé, S. Menzer, J. Nouwen, F. M. Raymo, J. F. Stoddart and D. J. Williams, J. Org. Chem., 1996, 61, 9591–9595 CrossRef CAS.
- C. H. Sue, S. Basu, A. C. Fahrenbach, A. K. Shveyd, S. K. Dey, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2010, 1, 119–125 RSC.
- M. Bria, G. Cooke, A. Cooper, J. F. Garety, S. G. Hewage, M. Nutley, G. Rabani and P. Woisel, Tetrahedron Lett., 2007, 48, 301–304 CrossRef CAS.
- K a values measured in a mixture of ethanol/water (3/1, v/v).
- R. Castro, K. R. Nixon, J. D. Evanseck and A. E. Kaifer, J. Org. Chem., 1996, 61, 7298–7303 CrossRef CAS.
- P. E. Mason, W. S. Bryant and H. W. Gibson, Macromolecules, 1999, 32, 1559–1569 CrossRef CAS.
- Ethanol/water (3/1, v/v) was chosen as all examined guests (G1–G6) are soluble in this mixture.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2016097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00606h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |