
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalt-mediated extraction of nanoscale Si building blocks: composite anode for Li-ion full battery with high energy density†
Jaegeon
Ryu
 a,
Minjun
Je
a,
Wooyeong
Choi
b and
Soojin
Park
*a
a,
Minjun
Je
a,
Wooyeong
Choi
b and
Soojin
Park
*a
aDeparment of Chemistry, Division of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea. E-mail: soojin.park@postech.ac.kr
bDepartment of Energy Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 23rd September 2020
Abstract
The agglomerating nature of nanomaterials during the preparation degrades the unique properties of nanostructured materials even in controlled metallothermic reactions. Here, we introduce a molten salt-mediated extraction of nanoscale (but high-quality) silicon building blocks from industrial wastes without unfavorable pretreatments. Selective reduction of silica using reactive complex reducing agents produces segregated nanosized silicon embedded in an inactive impurity matrix. Inert impurities in the unprocessed wastes are likely to dissolve in the mild etching condition after the reduction process. The resulting silicon has a defect-less structure with elemental high purity, which improves the initial electrochemical reversibility and cycle stability compared with those of commercial silicon nanoparticles even with smaller particle size. When incorporated with carbon layers and blended with traditional graphite at a high silicon content (~20 wt%), the formulated composite anodes showed high initial Coulombic efficiency and capacity retention over hundreds of cycles, thus enabling the practical demonstration of high energy full cells. The proposed cost-effective, efficient, and selective extraction of unavoidable industrial wastes can be extended to the preparation of segregated nanomaterials for various energy applications.
1. Introduction
Nanomaterials are becoming an essential platform to reshape the ever-changing electrochemical energy storage systems that call for material-level innovations with unique physicochemical properties.1 Lithium-ion batteries (LIBs), which predominate as a power source in the consumer electronics market over the past 30 years, have heavily exploited nanomaterials in various dimensional forms of conductive additives/scaffolds2,3 or electroactive elements,4 along with passive components (e.g., separators5,6 or current collectors7) when formulating the composite electrodes. Among them, electroactive parts (where the charge storage reactions practically occur during the battery operation) benefited from the nanomaterial designs, owing to the reduced diffusion path of the charge-carrier ions and structural flexibility to afford the fast-charging and large-volume-change environment.8 However, their large surface area will inevitably lead to the thickening of interfacial films (generally known as solid electrolyte interphase, SEI), lose their ionic conduction through the film, and avoid the formation of the dense electrode.9 In addition, their practical utilization has been plagued by costly production methods and severe agglomeration issues.Silicon (Si) has been a model subject for nanoscale designs since most lithium-alloying materials pose the chronic issue of large structural changes (∼370% in Li15Si4) upon lithium insertion/extraction, despite its high theoretical capacity of 3579 mA h g−1 and low operating voltage (<0.4 V versus Li/Li+).10 As Huang and co-workers disseminated the non-fracture size of Si anodes (ca. 150 nm) through in situ microscopy analysis and theoretical calculation,11 there are substantial amounts of reports about nanostructured Si anodes, which feature sizes in the safe range against the fracture via different synthetic approaches.12 Chemical vapor deposition (CVD) of monosilane (SiH4) has been employed onto the stacked silica beads,13 bundles of carbon nanotubes,14 and spherical graphite15 with a controllable thickness of conformal Si deposits, while serious potential hazards are inherent in the use of toxic and pyrophoric silane compounds. Unlike the above bottom-up synthesis, top-down methods (such as mechanical milling,16 metal-assisted catalytic etching,17 dealloying,18 and metallothermic reduction (MTR) of earth-abundant sources19 or industrial wastes20) provide relatively practical feasible solutions.
The typical MTR uses vaporized metals for reducing silica into a porous structure above the melting point of metals (>650 °C).21 However, without proper mediators, the generated exothermic heat aggressively destroys the initial morphology and incurs adverse agglomeration.22 The heat scavengers (e.g., NaCl, KCl) partly dissipate the thermal energy to retain the structure and relieve the risk of an explosion.23 However, the chemically inert molten salt will slow down the reduction kinetics or unexpectedly leave the undesirable byproducts (i.e., silicate derivatives), which degrade the battery performances, and high-reaction temperatures during the MTR with heat scavengers are not suitable for the mass production of nanoscale Si.24–26
Instead, the use of aluminum chloride (AlCl3) molten salt medium enabled a low-temperature operation (<250 °C) and production of crystalline nano-Si in broad size distribution, where some of the nanoparticles are vulnerable to fracture.27 Afterward, Song et al. revealed that bifunctional molten salts participate in the reduction reaction, as well as support the clustering of reduced seeds inside the molten salt medium after the fragmentation of the silica sources.28 The resulting hollow Si microparticles (regardless of silica feeding sources) consist of well-defined and segregated (not a single framework) crystalline Si building blocks (<200 nm), leading to highly stable lithium anodic performances in the LIBs. The strong reducing power of the metal–metal chloride complex was further demonstrated in the presence of magnesium sulfate, which produced sulfur-doped, quasi-metallic Si anodes.29 Thus, this modified MTR can greatly reduce the required thermal energy and safety risk to produce nanocrystalline Si compared with the conventional process. The reactive metal–metal chloride complexes are capable of selectively reducing the oxygen-containing species (metal oxides or hydroxides) into the well-defined and uniform crystals, and enables the efficient extraction of necessary components from the industrial wastes (i.e., slag), while excluding unnecessary processes.
Herein, we propose a salt-mediated extraction of nano-Si from the unprocessed slag waste (1–5 mm) containing undesirable impurities. The selective reduction of silica (or silicate) using the reducing complex yields a well-segregated form of nanosized Si (<150 nm). As embedded in the inactive matrix toward a reductive environment, severe agglomeration of Si nanomaterials can be avoided. The hardly etchable impurities in the unprocessed state that initially require a highly concentrated acidic environment (>5.0 M hydrochloric acid, HCl at high temperature) can be readily eliminated from the neutral water and relatively mild acid treatment (∼1.0 M HCl) during the subsequent leaching process. The refined nano-Si features a highly crystalline nature and a non-porous structure with a low surface area, which can avoid the excessive formation of SEI layers. This enables the high initial Coulombic efficiency (ICE) of 89.5% and more stable cycling stability compared with those of commercial counterparts with a size of 50 nm, even at a high capacity loading condition (>3 mA h cm−2). Incorporating the resulting high-quality Si from the slag waste with carbon passivation and traditional graphite further stabilizes the interfacial issues and achieves stable yet high energy density anodes for a practical Li-ion full battery.
2. Experimental
2.1 Material preparation
Slag waste (POSCO chemical), aluminum (1–3 μm, AVENTION), aluminum chloride (99.9%, Alfa Aesar), hydrochloric acid (35.0–37.0%, SAMCHUN CHEMICALS), and hydrofluoric acid (49%, J.T. Baker) were used as received without further treatment. For the preparation of nanoscale silicon building blocks (NSB), aluminum metal and aluminum chloride water were first evenly ground inside an Ar-filled glove box with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Then, unprocessed slag waste (1–5 mm) was homogenized through mild vortexing with the Al/AlCl3 mixture. The mixture was put into the home-made stainless steel reactor, which has an air-tight function and is already replaced with Ar gas in advance. After heating the reactor at 250 °C for 10 hours with a ramping rate of 3 °C min−1, it was naturally cooled down to ambient temperature to settle down and solidify any possible AlCl3 gas inside the reactor. The crude products were carefully rinsed with deionized water, 1 M hydrochloric acid, and 10% HF in series, and finally dried under vacuum at 80 °C for 12 hours before further analysis and electrode fabrication. Chemical-vapor-deposition (CVD) was employed for the carbon coating on the NSB samples at 900 °C for 5 min using toluene vapor (500 cc min−1) under Ar atmosphere. The estimated amounts of carbon were ∼10 wt%.
:5. Then, unprocessed slag waste (1–5 mm) was homogenized through mild vortexing with the Al/AlCl3 mixture. The mixture was put into the home-made stainless steel reactor, which has an air-tight function and is already replaced with Ar gas in advance. After heating the reactor at 250 °C for 10 hours with a ramping rate of 3 °C min−1, it was naturally cooled down to ambient temperature to settle down and solidify any possible AlCl3 gas inside the reactor. The crude products were carefully rinsed with deionized water, 1 M hydrochloric acid, and 10% HF in series, and finally dried under vacuum at 80 °C for 12 hours before further analysis and electrode fabrication. Chemical-vapor-deposition (CVD) was employed for the carbon coating on the NSB samples at 900 °C for 5 min using toluene vapor (500 cc min−1) under Ar atmosphere. The estimated amounts of carbon were ∼10 wt%.
2.2 Physical characterization
The long-range ordering and crystallinity of the prepared samples were analyzed through a powder X-ray diffractometer (SmartLab, Rigaku) and Micro Raman Spectrometer (inVia Raman microscope, Renishaw). Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) was used to determine the elemental composition of the products at each stage. The morphology and structure of the samples were observed through scanning electron microscopy (SEM, S-4800, Hitachi) and transmission electron microscopy (TEM, Tecnai G2 F20 X-Twin, FEI), along with energy-dispersive X-ray spectroscopy (EDS). The surface porosity of the samples was measured using an auto physisorption analyzer (ASAP2020, Micrometritics Instruments).2.3 Electrochemical evaluation
The prepared NSB-based samples were uniformly blended with the conductive nanocarbon (Super P, Timcal), polyacrylic acid (Sigma-Aldrich, Mw = ∼250000), and carboxymethyl cellulose sodium salt (Sigma-Aldrich, low viscosity) with a mass ratio of 70:15:7.5:7.5 using a planetary centrifugal mixer. The slurry was cast onto the copper foil, and dried under vacuum at 150 °C for 2 hours for the crosslinking. The mass loading of active materials was set to 0.7–2.0 mg cm−2. The electrodes were cut into disk shapes and assembled into the coin-type cell inside the Ar-filled glove box. Lithium metal was used as a reference, as well as the counter electrode, and the polyolefin microporous film was used as a separator. The organic electrolytes consisted of 1.3 M lithium hexafluorophosphate (LiPF6) in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) with a 10 wt% fluoroethylene carbonate (FEC) additive to promote stable SEI formation. The composite anode or NCA cathode were fabricated by mixing the active materials (C-NSB and natural graphite with a mass ratio of 20:80 or NCA), binder, and Super P with a mass ratio of 90:8:2 or 97:1.5:1.5, respectively, of which the discharge capacity (anode) corresponds to about 900 mA h g−1. After pressing the electrodes (packing density of ∼1.5 g cm−3 and ∼3.2 g cm−3 for blending the anode and cathode, respectively), the coin-type half and full cells were assembled, as described above. The assembled cells were aged at 40 °C for 2 hours before the measurements. Their electrochemical performances were measured through the galvanostatic battery cycler (WBCS3000Le, Wonatech). The cyclic voltammetry and electrochemical impedance spectroscopy were analyzed using a potentiostat (VMP3, Biologic) with a scan rate of 0.1 mV s−1 and 100 kHz–0.1 Hz with an amplitude of 10 mV, respectively. The voltage windows are 0.005–1.5 V for the anode, 3.0–4.3 V for the cathode, and 2.5–4.2 V for the full cell. The blending anodes, cathodes, and their assembly were charged in constant current and constant voltage (CCCV) mode, and discharged in constant current (CC) mode.
3. Results and discussion
3.1 Synthesis and characterization of Si building blocks
Ensuring the dimensional stability of the Si-based anodes has been realized by introducing multi-scale porosities into the Si structures, which can reduce the diffusion path of Li-ions and accommodate the volume expansion through the inward breathing of porous materials.30 However, this study offers an alternative way of preparing high-quality Si building blocks that can be potentially assembled into the Si–C composite anodes (commercial graphite-blended electrode), rather than the previous pore engineering on the Si anodes31–33 or other structural designs to prevent the particle fracture. This is because replacing the low-quality nanoscale Si materials that have been used in the industrial blending system can be considered a more viable solution for high-energy-density LIBs.The slag wastes are inevitable byproducts of smelting iron in hundreds-of-kilograms per ton of iron pig. They have been widely deployed on industrial purposes, such as a raw material for roadworthy cement, or civil engineering. The valuable components (i.e., SiO2) for the Si production are embedded in the slag wastes (∼30 wt%), and thus call for the efficient, selective, and scalable extraction methods into a pure Si, while minimizing the unnecessary processes. Recently, Chun et al. reported a series of synthetic procedures for preparing the mesoporous Si from the waste iron slag via magnesiothermic reduction aided by NaCl heat scavenger.34 However, it involves unfavorable ‘pre-treatments’, such as a ball-mill to grind the unprocessed slag particles and a ‘harsh acidic etching process (∼5 M HCl)’ at high temperature (∼80 °C), followed by the calcination for purification and further etching process after the reduction, which is not an efficient extraction of nano-Si from the industrial waste.
In this work, the proposed salt-mediated methodology can directly (but selectively) extract the Si building blocks from the unprocessed slag waste without any kind of ‘pre-treatment’ by strong reducing agents complexed by Al–AlCl3 (Fig. 1a). The unprocessed slag wastes look like sea sands with a rough surface and broad size distribution, showing the general characteristics of industrial wastes (Fig. 1b and Fig. S1a, ESI†). When the complex reductants react with the raw materials, it will preferentially adhere to the oxygen atoms of silica, which practically exist in the form of amorphous silicates, as corroborated by the smooth X-ray diffraction (XRD) pattern (Fig. S1b, ESI†). Through the ligand-promoted reduction mechanism, as revealed by our previous study,28 the activated aluminum monochloride (AlCl) will have a strong affinity toward the oxygen atoms of silicates and generates aluminum oxychloride (AlOCl) byproducts, while forming a new Si–Si bond. Importantly, the salt-mediated reduction process is readily scalable using a home-made stainless-steel reactor (Fig. S1c, ESI†).
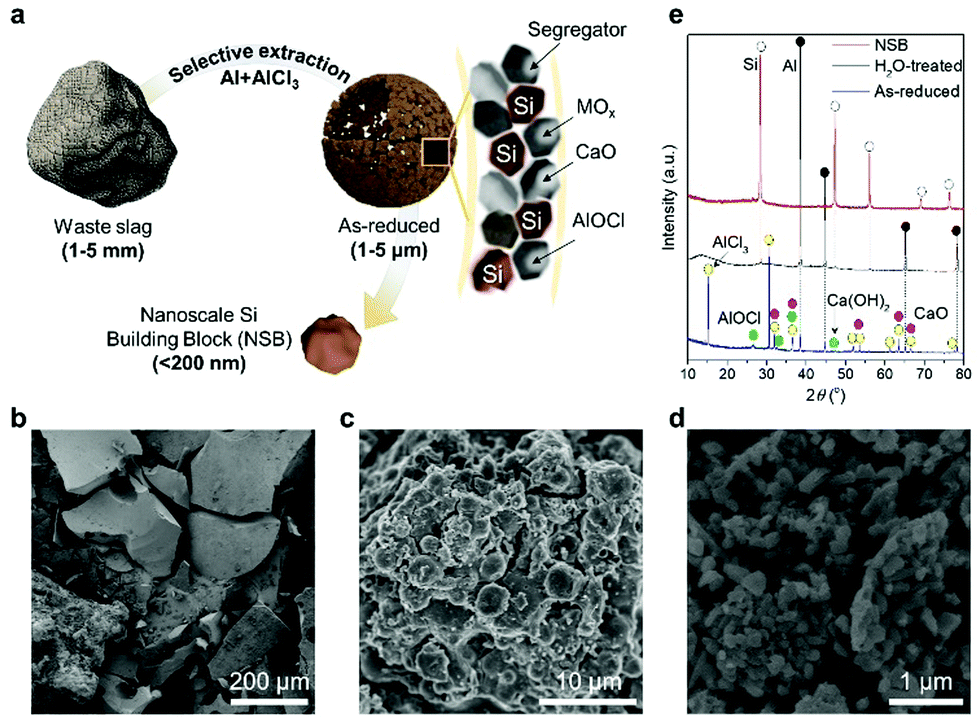 | ||
| Fig. 1 Schematic of preparation for refined Si building blocks. (a) Schematic illustration of a step-by-step preparation process from micrometer-sized waste slag to nanometer-sized NSB. SEM images of (b) bare slag, (c) as-reduced products after reduction process, and (d) NSB particles. (e) XRD pattern intermediates and products during the preparation of NSB. | ||
The successive reduction process fragments the millimeter-scale slag waste without the external mechanical milling process (Fig. 1c). The impurity oxides tightly anchored to Si–O bonds in the bare slag become liberated from the raw materials due to the selective extraction of Si. This chemically accelerated separation significantly lowers the activation energy to break the inherent covalent bonds inside the industrial wastes. Both liberated impurities and Si seeds are stabilized inside the molten salt, and clustered to yield the porous composite microspheres. After leaching out all the byproducts by neutral water and relatively mild acid, non-porous Si nanoparticles (which have a size of less than 150 nm) are evenly produced, as shown in Fig. 1d. The XRD patterns of the samples in each step reveal that the crude products (denoted as as-reduced) from the salt-mediated reduction have AlOCl, as well as the dominant AlCl3 and Al with a trace of Si and calcium hydroxide (Ca(OH)2) (Fig. 1e). The Ca-containing compounds are readily removed by the water dissolution and accordingly leave only Al metals, AlOCl, and Si, which can be refined through mild acid treatment. Thus, nanometer-sized Si building blocks (NSB) has a clear XRD peak assigned to the cubic crystal structure of Si (JCPDS No. 27-1402).
The fragmentation of the massive slag waste can be further unraveled by transmission electron microscopy (TEM) analysis (Fig. 2). A highly reactive complex species (i.e., Al-3(AlCl3)) broke the stable Si–O–M (M denotes the impurity metal elements) and in this manner, the reductants were almost totally shattering the slag wastes into Si seeds and unstable impurity debris. Through the localized Ostwald ripening process, the mixture formed the micrometer-sized (∼2 μm) porous spherical structure, but in the absence of the hollow-core observed in the previous study (Fig. 2a). Compared with the salt-mediated reduction of pure silica (or silicate), the relative portion of generated Si seeds is insufficient due to the majority impurities. Thus, the inner voids derived from the migration of the crystal seeds toward the surface were not observed. The high magnification TEM image showed the randomly dispersed morphology of the Si crystals embedded in the AlOCl byproducts and Ca derivatives (Fig. 2b and c). The inactive debris distributed over the intermediate microspheres performed a crucial role to effectively segregate each Si building block without serious agglomeration, which usually occurred in the preparation of nanoscale materials (Fig. 2d). The oval structured NSB has a uniform size distribution (100–150 nm), thus potentially showing the non-fracture property during battery operation. Although the microspheres are initially assembled with the impurity debris and NSB, a single NSB particle features a non-porous structure and a well-defined lattice fringe, corresponding to a d-spacing value of 0.31 nm for the Si(111) plane, as shown in Fig. 2e.
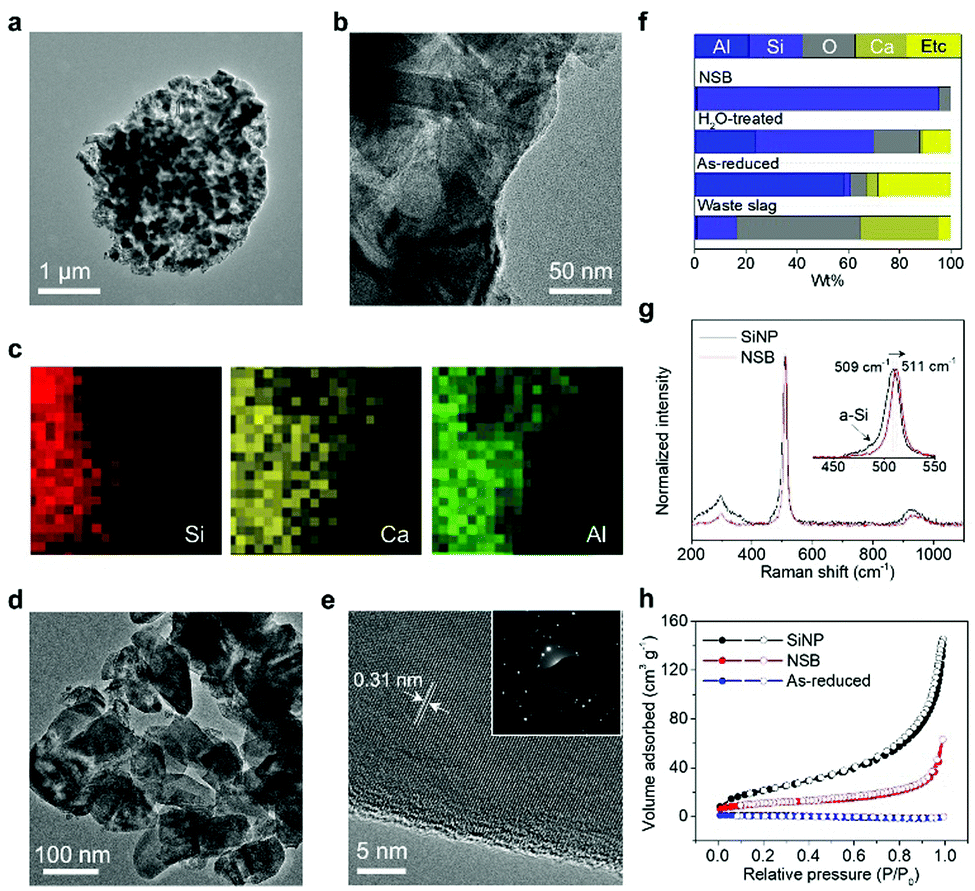 | ||
| Fig. 2 Structural characterizations of NSB. (a and b) TEM images and (c) energy-dispersive spectroscopy (EDS) elemental maps for as-reduced NSB. (d and e) TEM images of NSB (inset: selected area electron diffraction pattern). (f) Elemental composition of NSB in different stages. (g) Raman spectra of NSB and SiNPs. (h) Nitrogen adsorption–desorption isotherms of as-reduced NSB, NSB, and SiNPs. | ||
Interestingly, the native oxide layer of the NSB is much thinner (∼0.5 nm) than that of commercially available Si nanoparticles (SiNPs) (up to 1.5 nm), even after storage for more than 1 week in the ambient environment (Fig. S2 and S3a, ESI†). This observation is consistent with the low oxygen content in the NSB particles after the treatment (Fig. 2f). The thick oxide layers present in the commercial SiNPs will have detrimental effects on the electrochemical properties, such as low reversibility at the first cycle, high interfacial impedance, and electrical isolation of Si anodes inside the electrode, which are discussed later in detail.35 Despite recent progress in the synthesis of nanoparticles in the gas-phase, most vapor-synthesis incurs severe agglomeration of nanoparticles and the formation of dumbbell-like morphology, as in the case of commercial SiNPs.
SiNPs inherently have mixed amorphous/crystalline properties, as evidenced by the Raman spectra of SiNP and NSB samples (Fig. 2g). The crystalline Si band of SiNPs is located at a lower frequency shift (509 cm−1) than that of the NSB samples (511 cm−1), owing to the smaller particle size.36 However, the shoulder peak corresponding to the amorphous silicon band is observed at about 480 cm−1 in even the normalized spectrum for SiNP, which concurs with the TEM observation regarding their poor crystallinity. The relative comparison of their crystallinity was also confirmed by calculating the full-width-half-maximum (FWHM) values in the XRD patterns (Fig. S3b, ESI†). Such poor crystallinity might cause irregular solid-state Li-ion diffusion across the particles and consequently be unfavorable for battery-grade material preparation since the Si anodes exhibited anisotropic lithiation behavior.37 By contrast, the fully crystalline NSB particles with thin oxide layers and non-porous structures could be the high-performance electrode building blocks.
One important factor to determine the initial reversibility of most anodes relates to the surface porosity of the nanoparticles. This is because most nanostructured materials suffer from low ICE due to the substantial consumption of liquid electrolytes at the surface,38 thus posing a trade-off issue between the nanoscale designs for dimensional stability and low surface area for high ICE in general. Fig. 2h shows the nitrogen sorption isotherm plots of the crude product, NSB, and SiNPs. Unlike non-porous, as-reduced NSB coalesced by excessive amounts of byproducts, SiNPs have a macroporous characteristic that is ascribed to the small size of the particles and agglomerated structure, thereby having a large Brunauer–Emmett–Teller (BET) surface area of 76 m2 g−1. However, the isotherm plot for NSB shows no specific adsorption sites, except the inter-particle voids, which lowers the BET surface area to 39 m2 g−1. Thus, the pore volume of the NSB sample is much lower than that of the SiNPs, while both do not have a characteristic pore diameter, as corroborated by the pore size distribution curves and TEM images (Fig. S3c, ESI†).
3.2 Electrochemical test of Si building blocks
The electrochemical evaluation of the prepared NSB was conducted in a coin-type half-cell. Cyclic voltammogram (CV) curves show the typical alloying reaction of crystalline Si with Li at ∼0.1 V vs. Li/Li+, which entails amorphization of Si in the first cycle and a subsequent electrochemical evaluation of the prepared NSB was conducted in a coin-type half-cell. Cyclic voltammogram (CV) curves show the typical alloying reaction of crystalline Si with Li at ∼0.1 V vs. Li/Li+, which entails amorphization of Si in the first cycle and subsequent lithiation of amorphous Si into a-LixSi (where x is greater than 3.5), as shown in Fig. S4 (ESI†). Broad cathodic currents account for the irreversible electrolyte decomposition in both SiNP and NSB electrodes, which correspond to the SEI formation at the voltages of 0.7–1.5 V vs. Li/Li+.39Fig. 3a shows the initial galvanostatic voltage profiles of the two electrodes at 0.05C, and their discharge (lithiation) capacities are 3440 and 3520 mA h g−1 for the SiNP and NSB electrodes, respectively. Compared with the reported ICE elsewhere using the commercial SiNPs,40 the NSB electrode shows high ICE of 89.5%, although the NSB particles fall within the range of typical nanoscales below 200 nm, while the SiNP electrode shows ICE of 83.3%. The well-defined structure of the NSB, which has fewer defects without agglomerates, is attributed to the ICE improvement, suggesting that the prepared particle size is favorable to reducing the surface area in a way that does not fracture as well. | ||
| Fig. 3 Electrochemical properties of SiNP and NSB electrodes. (a) First voltage–capacity profiles at 0.05C, (b) Capacity retention at 0.5C, (c) rate capability results at various C-rates, and (d) Nyquist plots of SiNP and NSB electrodes. (e) Summary of electrode swelling and (f) volumetric capacities of the SiNP and NSB electrodes, respectively. | ||
During the subsequent electrochemical cycling, the SiNP electrode rapidly degraded in the first 20 cycles at 0.5C, despite its sufficiently small particle size (∼50 nm) (Fig. 3b). Surprisingly, the NSB electrode sustains the cycle life of the battery for 100 cycles at an areal capacity loading of 3.2 mA h cm−2, along with the stable capacity retention of 92.5% at 0.2C without protective coating layers or graphite blending, as shown in Fig. S5a (ESI†). Also, the NSB electrode demonstrates an improved rate capability, where the reversible capacities of 1320, 873, and 555 mA h g−1 are delivered at 3.0, 6.0, and 9.0 A g−1, respectively (Fig. 3c and Fig. S5b, c, ESI†). The Nyquist plots obtained from electrochemical impedance analysis (EIS) found that the charge transfer resistances of the NSB electrode are ten times lower than those of the SiNP electrode (Fig. 3d). The highly crystalline and nearly defect-free nature of NSB contributes to the reduced resistance of the charge-carrier ions.41
The dimensional stability of Si-based electrodes is of great importance to check its practical feasibility, and whether the volumetric parameter is compromised during the cycles. The fully lithiated NSB electrode in the first cycle expands only 12.6%, and it maintains as low as 12.6% after 20 cycles. This suggests that the NSB samples hold great potential as an efficient building block to formulate the high volumetric energy density electrode (Fig. 3e). Thus, the volumetric capacity of the NSB electrode reaches about 1680 mA h cm−3 without any calendaring process (Fig. 3f). On the contrary, the SiNP electrode substantially swelled at the initial cycle, and its expansion in the electrode level already exceeds the acceptable volume margin inside the commercial cell (∼40%). The SiNP electrode dramatically lost the designed volumetric capacity, and could only deliver 293 mA h cm−3 after 20 cycles. This was corroborated by the observed deep and wide cracks in the SiNP electrode from the ex situ SEM analysis (Fig. S6 and S7, ESI†). The commercial SiNP becomes severely agglomerated and coalesced larger than 200 nm, whereas the NSB particles show the well-dispersed electrode structure without microcracks and disintegration.
Despite the ensured dimensional stability, the intrinsically poor electrical conductivity of the Si anodes caused a large polarization at the increased current density (Fig. 3b). Also, the unavoidable crack formation on the Si surface during prolonged cycles triggered the accumulation of SEI layers in an uncontrolled way. Thus, moderate surface passivation with conductive carbon was introduced by the facile chemical vapor deposition method (Fig. S8, ESI†), thereby greatly improving the long-term stability of the electrodes at 1.0C. The carbon-coated NSB (C-NSB) electrodes showed 80% and 64% of capacity retention after 400 cycles with a capacity loading of ∼1.5 mA h cm−2 and ∼3.4 mA h cm−2, respectively (Fig. 4a). Importantly, the Coulombic efficiency (CE) of the C-NSB electrodes was quickly saturated over the initial few cycles. The average CE then reached 99.5%, indicating the feasibility of the graphite blending and full cell applications.
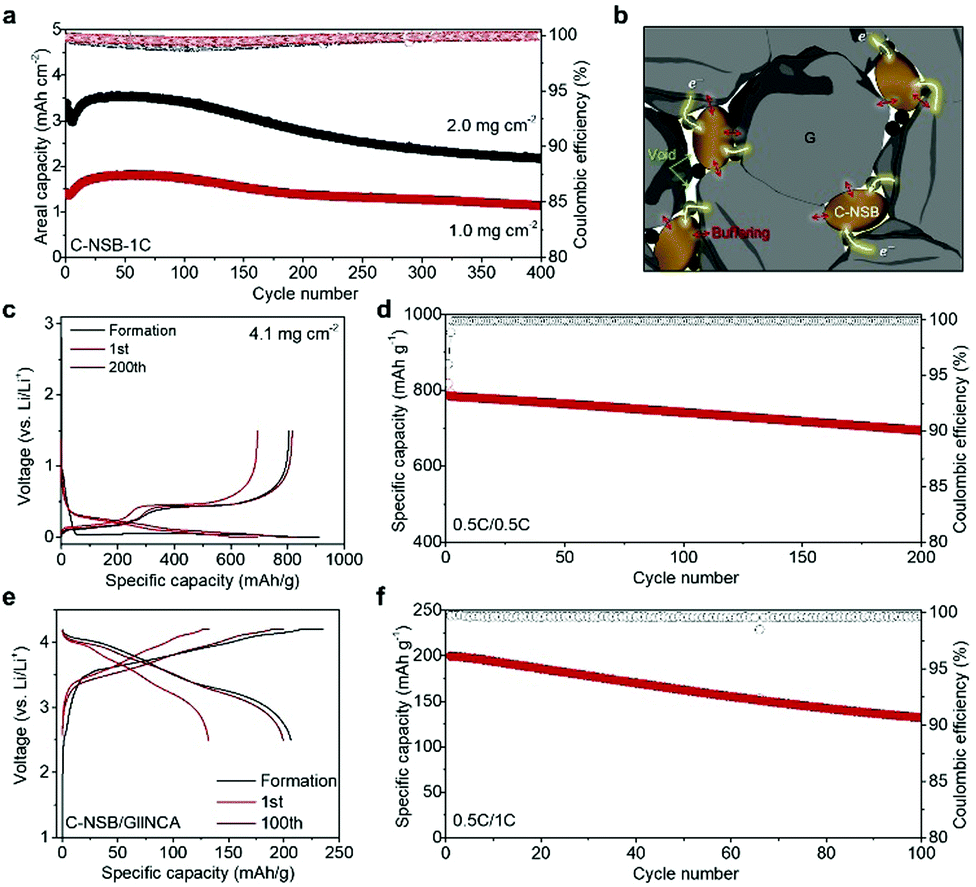 | ||
| Fig. 4 Battery performances of blending anodes and full cell. (a) Capacity retention of the C-NSB electrode at 1.0C. (b) Schematic illustration of the C-NSB/G electrode showing a buffer effect between the particle and native voids with a continuous conduction pathway. Voltage profiles (c and e) and capacity retention (d and f) of C-NSB/G anodes and C-NSB/G||NCA full cell, respectively. | ||
Compositing the Si-based anodes (nano-Si or SiOx) with carbon buffers has been investigated in multiple ways, such as microemulsion42 and spray-drying,43 while buffering and conductive carbon layers are mostly electrochemically inactive to lower the energy density of batteries. Instead, practical approaches rather blend a certain amount (<15%) of Si-based materials with stable graphite anodes, according to the designed specific capacity that falls into the range of 450–600 mA h g−1.44 The graphite blending electrodes with C-NSB (denoted as C-NSB/G) were formulated by uniform mixing of commercially available graphite and C-NSB particles (20%). Inside the C-NSB/G electrode, native void spaces between the C-NSB and bulk graphite particles are beneficial for relieving the stress induced by the volume expansion of C-NSB, and the intimate contact of the carbon layers and graphite will provide a continuous electron pathway over the thick electrode (Fig. 4b).
The C-NSB/G electrodes showed a high reversible capacity of 805 mA h g−1 and the ICE of 88.5% with a mass loading of 4.1 mg cm−2. This is well-matched with the designed parameters without any loss of active materials (Fig. 4c). During the repeated 200 cycles at 0.5C, the blending electrode showed a limited capacity loss of 0.075% per cycle, and retained 85.0% of the initial capacity (Fig. 4d). However, the Si contents and corresponding specific capacity are much higher than those of previous studies.45,46 Low-volume-change graphite and high-quality Si building block enable the high CE for the subsequent cycles as reaching above 99.9% without any fluctuation, which is a good indicator for full cell assembly. The C-NSB/G anode was paired with the recent emerging high-capacity lithium nickel cobalt aluminum oxide (NCA) cathode with N/P of 1.1 (capacity ratio of the anode to cathode), as shown in Fig. 4e. The C-NSB/G||NCA full cell delivered the initial discharge capacity of 206 mA h g−1 at 0.1C in the voltage window of 2.5–4.2 V (vs. Li/Li+). After the stabilization at 0.2C, the capacity retention was monitored for the subsequent cycles at 0.5C (Fig. 4f). The reversible capacity of the C-NSB/G||NCA full cell reaches 132 mA h g−1 after 100 cycles. The observed cycling stability can be attributed to the high-quality Si building blocks with appropriate feature size, defect-less structure, and surface carbon layers, which demonstrated the potential of practical applications.
4. Conclusion
In summary, an efficient, scalable, and eco-friendly method of salt-mediated extraction from the industrial wastes, enabled the production of high-quality nanoscale Si building blocks for the battery application. A metal–metal chloride complex with a high reducing power selectively reduced the reactive oxygen-containing species without additional pretreatments, such as the mechanical mill and harsh acidic leaching process, while other impurities remained intact. The intermediate structure of the hollow and porous spheres randomly contains Si building blocks and impurity debris to avoid the typical agglomeration issues raised in nanoparticle preparation. After the mild condition of subsequent byproducts dissolution, the resulting Si showed a well-defined structure with a narrow size distribution, while its feature size is smaller than the critical fracture size of the Si anodes. The electrode involving the NSB-based particles manifested dimensional stability with a high initial Coulombic efficiency, and their composite electrode with commercial graphite was demonstrated in half and full cells under the near practical testing conditions. The proposed salt-mediated selective extraction of valuable components from the industrial wastes can be extended to other practical applications with an inherently dispersed nature of nanoparticles in a battery grade.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) grants funded by the Korean government (Ministry of Science and ICT) (No. 2019 R1A2C2084149).References
- E. Pomerantseva, F. Bonaccorso, X. L. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, 969 CrossRef.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 RSC.
- F. Y. Su, C. H. You, Y. B. He, W. Lv, W. Cui, F. M. Jin, B. H. Li, Q. H. Yang and F. Y. Kang, J. Mater. Chem., 2010, 20, 9644–9650 RSC.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS.
- T. H. Cho, M. Tanaka, H. Ohnishi, Y. Kondo, M. Yoshikazu, T. Nakamura and T. Sakai, J. Power Sources, 2010, 195, 4272–4277 CrossRef CAS.
- S. Yoo, J. H. Kim, M. Shin, H. Park, J. H. Kim, S. Y. Lee and S. Park, Sci. Adv., 2015, 1, e1500101 CrossRef.
- Y. Yue and H. Liang, Small Methods, 2018, 2, 1800056 CrossRef.
- Y. Y. Liu, Y. Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- Y. M. Sun, N. A. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- U. Kasavajjula, C. S. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS.
- J. Ryu, T. Bok, S. Kim and S. Park, ChemNanoMat, 2018, 4, 319–337 CrossRef CAS.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. A. Liu, L. B. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS.
- W. Wang and P. N. Kumta, ACS Nano, 2010, 4, 2233–2241 CrossRef CAS.
- M. Ko, S. Chae, J. Ma, N. Kim, H. W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 16113 CrossRef CAS.
- M. Gauthier, D. Mazouzi, D. Reyter, B. Lestriez, P. Moreau, D. Guyomard and L. Roue, Energy Environ. Sci., 2013, 6, 2145–2155 RSC.
- B. M. Bang, H. Kim, H. K. Song, J. Cho and S. Park, Energy Environ. Sci., 2011, 4, 5013–5019 RSC.
- Y. T. Yuan, W. Xiao, Z. Y. Wang, D. J. Fray and X. B. Jin, Angew. Chem., Int. Ed., 2018, 57, 15743–15748 CrossRef CAS.
- D. S. Jung, M. H. Ryou, Y. J. Sung, S. B. Park and J. W. Choi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12229–12234 CrossRef CAS.
- M. Choi, J. C. Kim and D. W. Kim, Sci. Rep., 2018, 8, 960 CrossRef.
- Z. Xing, J. Lu and X. Ji, Small Methods, 2018, 2, 1800062 CrossRef.
- J. Ryu, S. Choi, T. Bok and S. Park, Nanoscale, 2015, 7, 6126–6135 RSC.
- B. H. Meekins, Y. C. Lin, J. S. Manser, K. Manukyan, A. S. Mukasyan, P. V. Kamat and P. J. McGinn, ACS Appl. Mater. Interfaces, 2013, 5, 2943–2951 CrossRef CAS.
- W. Luo, X. F. Wang, C. Meyers, N. Wannenmacher, W. Sirisaksoontorn, M. M. Lerner and X. L. Ji, Sci. Rep., 2013, 3, 2222 CrossRef.
- J. Ryu, D. Hong, S. Choi and S. Park, ACS Nano, 2016, 10, 2843–2851 CrossRef CAS.
- J. Ryu, D. Hong, M. Shin and S. Park, ACS Nano, 2016, 10, 10589–10597 CrossRef CAS.
- N. Lin, Y. Han, J. Zhou, K. Zhang, T. Xu, Y. Zhu and Y. Qian, Energy Environ. Sci., 2015, 8, 3187–3191 RSC.
- G. Song, J. Ryu, J. C. Kim, J. H. Lee, S. Kim, C. Wang, S. K. Kwak and S. Park, Commun. Chem., 2018, 1, 42 CrossRef.
- J. Ryu, J. H. Seo, G. Song, K. Choi, D. Hong, C. Wang, H. Lee, J. H. Lee and S. Park, Nat. Commun., 2019, 10, 2351 CrossRef.
- Q. F. Xiao, M. Gu, H. Yang, B. Li, C. M. Zhang, Y. Liu, F. Liu, F. Dai, L. Yang, Z. Y. Liu, X. C. Xiao, G. Liu, P. Zhao, S. L. Zhang, C. M. Wang, Y. F. Lu and M. Cai, Nat. Commun., 2015, 6, 8844 CrossRef CAS.
- X. L. Li, M. Gu, S. Y. Hu, R. Kennard, P. F. Yan, X. L. Chen, C. M. Wang, M. J. Sailor, J. G. Zhang and J. Liu, Nat. Commun., 2014, 5, 4105 CrossRef CAS.
- W. L. An, B. A. Gao, S. X. Mei, B. Xiang, J. J. Fu, L. Wang, Q. B. Zhang, P. K. Chu and K. F. Huo, Nat. Commun., 2019, 10, 1447 CrossRef.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151–10154 CrossRef CAS.
- J. Chun, S. An and J. Lee, J. Mater. Chem. A, 2015, 3, 21899–21906 RSC.
- S. Xun, X. Song, L. Wang, M. E. Grass, Z. Liu, V. S. Battaglia and G. Liu, J. Electrochem. Soc., 2011, 158, A1260–A1266 CrossRef CAS.
- J. H. Parker, D. W. Feldman and M. Ashkin, Phys. Rev., 1967, 155, 712–714 CrossRef CAS.
- S. W. Lee, M. T. McDowell, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 3034–3039 CrossRef CAS.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- N. Lin, Y. Han, J. Zhou, K. L. Zhang, T. J. Xu, Y. C. Zhu and Y. T. Qian, Energy Environ. Sci., 2015, 8, 3187–3191 RSC.
- T. T. Truong, Y. Qin, Y. Ren, Z. Chen, M. K. Chan, J. P. Greeley, K. Amine and Y. Sun, Adv. Mater., 2011, 23, 4947–4952 CrossRef CAS.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS.
- D. S. Jung, T. H. Hwang, S. B. Park and J. W. Choi, Nano Lett., 2013, 13, 2092–2097 CrossRef CAS.
- S. Chae, N. Kim, J. Ma, J. Cho and M. Ko, Adv. Energy Mater., 2017, 7, 1700071 CrossRef.
- S.-B. Son, L. Cao, T. Yoon, A. Cresce, S. E. Hafner, J. Liu, M. Groner, K. Xu and C. Ban, Adv. Sci., 2019, 6, 1801007 CrossRef.
- J. Ryu, D. Hong, H.-W. Lee and S. Park, Nano Res., 2017, 10, 3970–4002 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Sample characterizations, electrochemical performance results (CV, voltage profiles, and cycling stability), electrode analysis. See DOI: 10.1039/d0ma00640h |
| This journal is © The Royal Society of Chemistry 2020 |