
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hit-to-lead optimization of a benzene sulfonamide series for potential antileishmanial agents†
Paul J.
Koovits
a,
Marco A.
Dessoy
a,
An
Matheeussen
b,
Louis
Maes
b,
Guy
Caljon
b,
Leonardo L. G.
Ferreira
 c,
Rafael C.
Chelucci
c,
Simone
Michelan-Duarte
c,
Adriano D.
Andricopulo
c,
Simon
Campbell
d,
Jadel M.
Kratz
d,
Charles E.
Mowbray
d and
Luiz C.
Dias
*a
c,
Rafael C.
Chelucci
c,
Simone
Michelan-Duarte
c,
Adriano D.
Andricopulo
c,
Simon
Campbell
d,
Jadel M.
Kratz
d,
Charles E.
Mowbray
d and
Luiz C.
Dias
*a
aInstitute of Chemistry, University of Campinas (UNICAMP), Rua Josué de Castro, S/N, Cidade Universitária, Campinas, SP 13083-861, Brazil. E-mail: ldias@iqm.unicamp.br; Tel: +55 19 3521 3097
bLaboratory of Microbiology, Parasitology and Hygiene (LMPH), University of Antwerp, Universiteitsplein 1, 2610 Wilrijk, Belgium
cLaboratory of Medicinal and Computational Chemistry, Physics Institute of Sao Carlos, University of Sao Paulo, Av. Joao Dagnone 1100, 13563-120, Sao Carlos-SP, Brazil
dDrugs for Neglected Diseases initiative (DNDi), 15 Chemin Louis-Dunant, 1202 Geneva, Switzerland
First published on 25th August 2020
Abstract
A series of benzene sulphonamides with good potency and selectivity against Leishmania spp. intracellular amastigotes was identified by high-throughput screening. Approximately 200 compounds were synthesized as part of a hit-to-lead optimization program. The potency of the series appears to be strongly dependent on lipophilicity, making the identification of suitable orally available candidates challenging due to poor pharmacokinetics. Despite not identifying a clinical candidate, a likely solvent exposed area was found, best exemplified in compound 29. Ongoing detailed mode-of-action studies may provide an opportunity to use target-based medicinal chemistry to overcome the issues with the current series.
Leishmaniasis is a neglected parasitic protozoal disease that causes an estimated 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 deaths annually and infects up to 12 million people, leaving up to a billion at risk across Latin America, Africa, Asia and the Middle East.1,2 The disease is caused by the kinetoplastid Leishmania,3 of which there are over 20 species, and is transmitted by the female phlebotomine sand fly. There are several clinical forms, the most common being cutaneous leishmaniasis (CL) and visceral leishmaniasis (VL). Annually, there are an estimated 700000–1200000 new cases of CL,4 which presents itself as skin lesions on exposed body parts such as the arms, legs, and face, leaving permanent scars and severe disabilities. VL or kala-azar is the more serious form and is fatal without treatment. It is characterized by anaemia, enlarged organs (spleen and liver), fever and weight loss. VL is specifically caused by L. infantum (L. inf.) or L. donovani (L. don.). L. inf. is zoonotic in Latin America, Europe, and Central Asia, where canines act as the major reservoir. In South Asia and East Africa, L. don. is considered anthroponotic and is responsible for 95% of VL cases worldwide.5 Whilst the numbers of new infections has been decreasing annually and dropped below 20000 in 2018,6 it is estimated that only 20–45% of cases are reported to the WHO.7,8 Despite these large numbers, infections by L. inf. generally remain sub-clinical and VL only develops in cases where the patient is immunosuppressed, such as patients with HIV or severe malnourishment.9 The latter is one of the reasons why leishmaniasis particularly affects poor populations in rural areas.
000–30000 deaths annually and infects up to 12 million people, leaving up to a billion at risk across Latin America, Africa, Asia and the Middle East.1,2 The disease is caused by the kinetoplastid Leishmania,3 of which there are over 20 species, and is transmitted by the female phlebotomine sand fly. There are several clinical forms, the most common being cutaneous leishmaniasis (CL) and visceral leishmaniasis (VL). Annually, there are an estimated 700000–1200000 new cases of CL,4 which presents itself as skin lesions on exposed body parts such as the arms, legs, and face, leaving permanent scars and severe disabilities. VL or kala-azar is the more serious form and is fatal without treatment. It is characterized by anaemia, enlarged organs (spleen and liver), fever and weight loss. VL is specifically caused by L. infantum (L. inf.) or L. donovani (L. don.). L. inf. is zoonotic in Latin America, Europe, and Central Asia, where canines act as the major reservoir. In South Asia and East Africa, L. don. is considered anthroponotic and is responsible for 95% of VL cases worldwide.5 Whilst the numbers of new infections has been decreasing annually and dropped below 20000 in 2018,6 it is estimated that only 20–45% of cases are reported to the WHO.7,8 Despite these large numbers, infections by L. inf. generally remain sub-clinical and VL only develops in cases where the patient is immunosuppressed, such as patients with HIV or severe malnourishment.9 The latter is one of the reasons why leishmaniasis particularly affects poor populations in rural areas.
Today, there are insufficient treatment options (Fig. 1),5,10 all of which are associated with severe side-effects and/or other limitations, often requiring intravenous or painful intramuscular injections as well as monitoring in hospital. Miltefosine (MIL) is presently the only approved oral drug for VL and CL. Although MIL has good efficacy, it has a long treatment regimen (28 days); has dose-limiting side-effects (due to vomiting and diarrhoea); is teratogenic which limits use in pregnancy;11 and there are reports of resistance and variable efficacy in different geographical areas.12–14 Whilst the current pipeline looks more promising than in previous years, with three candidates in phase I and several more in pre-clinical phases,15–17 due to the pressing need for new medicines it is imperative that we continue to develop novel, affordable, effective and safe drugs for leishmaniasis.
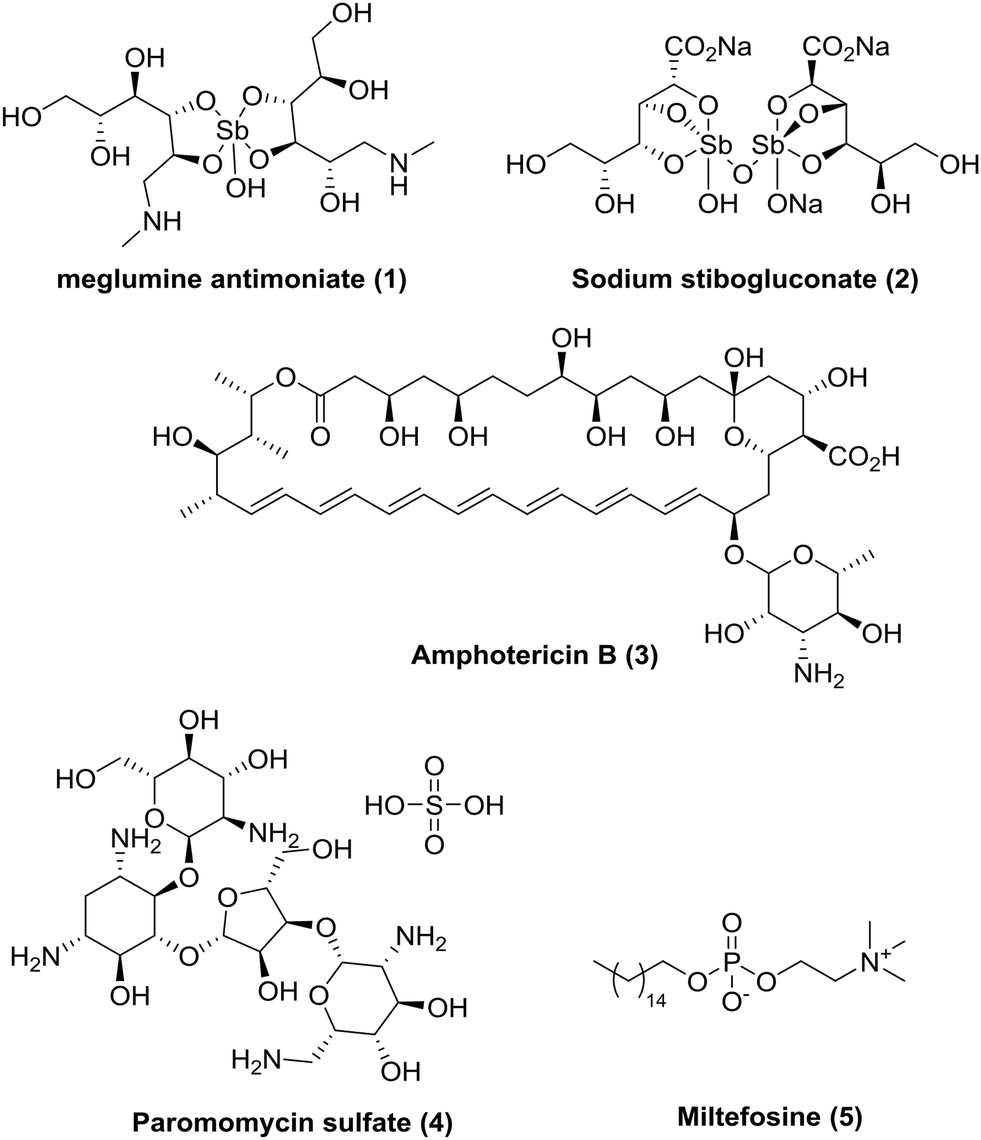 | ||
| Fig. 1 Current available drugs for visceral leishmaniasis. | ||
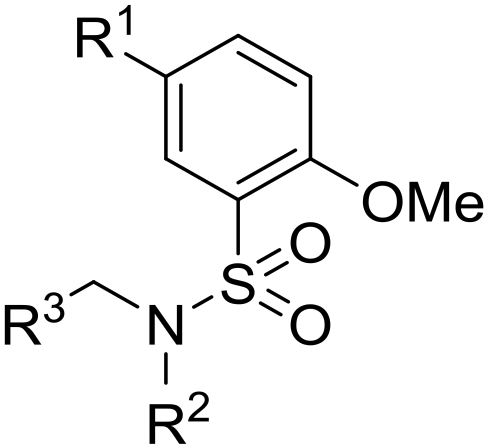
As part of our collaboration with the Drugs for Neglected Diseases initiative (DNDi), we have been examining new chemical series with anti-trypanosomal activity as potential leads for drug discovery programs.18 Sulphonamide hits 6–8 (Fig. 2)19 with moderate to good potency (pIC50 4.92–5.82) and selectivity were identified in a cidal high-throughput phenotypic screen of a commercial library against axenic L. don. amastigotes (MHOM/SD/62/1S-CL2, LdBOB), as previously described.20 Further profiling indicated that intracellular amastigotes of L. inf. were slightly more sensitive (pIC50 6.23–6.35). However, all these hits were highly lipophilic (clogP ≥ 5) and were rapidly cleared in vitro in the presence of both human and mouse microsomes (S9 fractions). To rapidly investigate their structure–activity relationship (SAR), a library of approximately 100 commercial analogues was purchased and tested in parallel against intracellular amastigotes of both L. inf. and L. don. (see ESI†). Active compounds could be clustered into two sub-sets, both of which contained a 2-alkoxybenzene-sulphonamide moiety (Fig. 2). The first sub-set consisted of a long alkyl chain (at least 4 carbons) on the ortho-substituted oxygen and possessed a secondary sulphonamide. The second sub-set consisted of a simple methoxy group at the ortho position and a tertiary sulphonamide. Of these sub-sets, no compounds offered a significant advantage over the initial hits in terms of potency or lower lipophilicity. The previous heightened potency against L. inf. over L. don. (3 to 5-fold) was consistently observed as well, although further investigation of this discrepancy was not pursued in the scope of this work. A hit-to-lead program was initiated focusing primarily on the tertiary sulphonamide sub-set (a) as these compounds offered slightly higher potency and lower lipophilicity, as well as ease of synthesis. It was also felt that improved metabolic stability would be more readily achieved without a large lipophilic alkyl chain in the molecule. The initial hits were divided into two different subsets: tertiary sulphonamides (hit 6) and secondary sulphonamides (hits 7 and 8). Attempts to reduce lipophilicity of the hit 6 by replacing the tertiary sulphonamide moiety with a secondary one led to complete loss of potency. The observed loss of potency most likely is related to the drop in logD that such modification implies. A mandatory requisite for the potency of secondary sulphonamides (hits 7 and 8) is the presence of a fatty alkoxy chain on the 2 position of the benzenesulfonyl core. Overall, this requirement tends to increase the logD for this class of analogues, leading to compounds with poor metabolic stability.
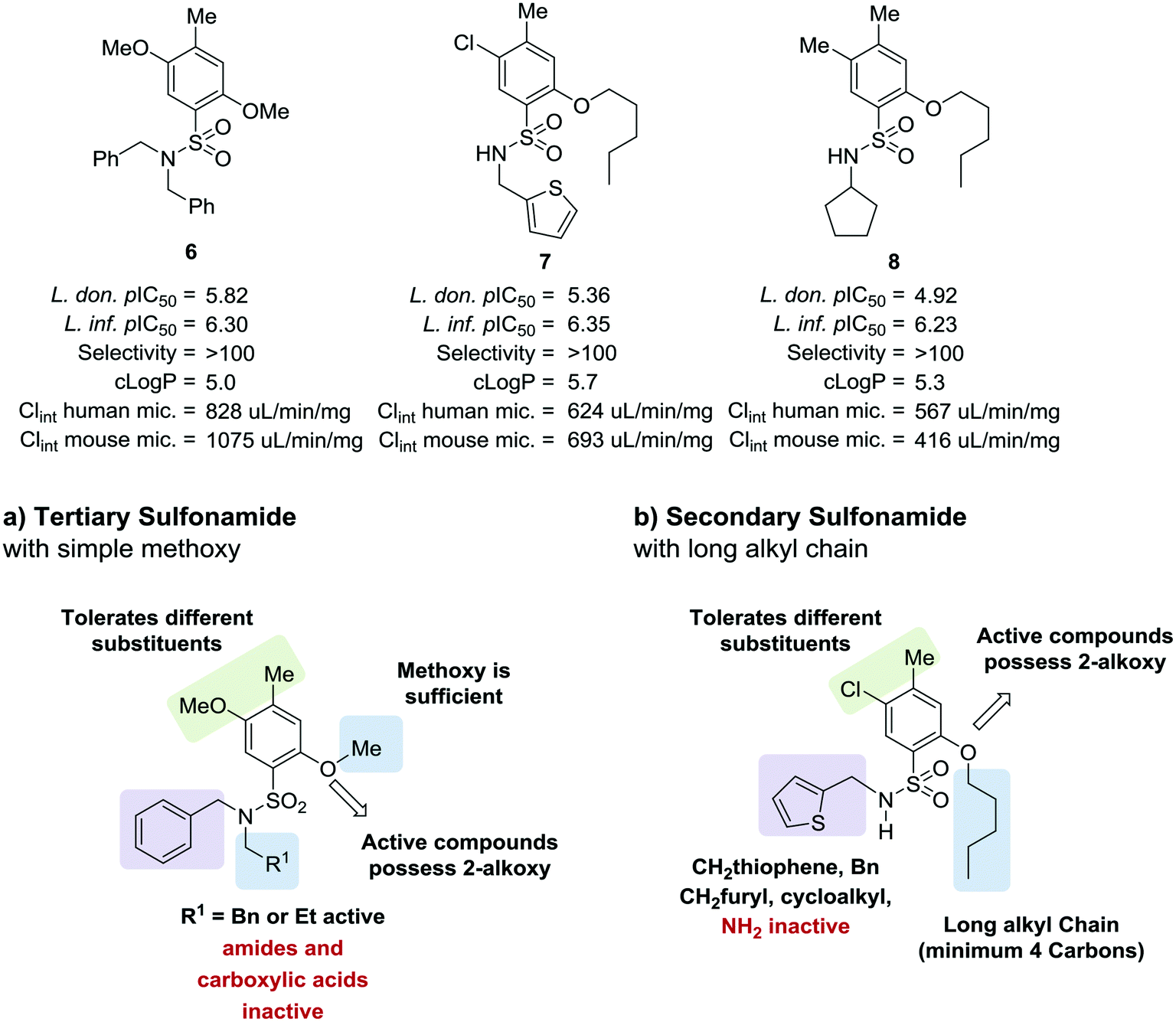 | ||
| Fig. 2 Initial hits and preliminary structure activity relationship (SAR). | ||
Initially, a variety of substituents in the parent aromatic ring was explored in combination with the dibenzylsulphonamide portion (Table 1). It was found that the 4-methyl group from the original hit 6 was not necessary for activity (compound 9), allowing a reduction in lipophilicity as well as removing a possible metabolic soft spot. As observed in the initial hits 7 and 8, the 5-methoxy substituent was also not essential for potency (compound 10), allowing removal of the p-dimethoxyaryl moiety, which has been associated with inherent toxicity risks. The 2-alkoxy group, however, was confirmed to be crucial, since removal resulted in a loss of potency (compounds 11–14). The 2-trifluoromethoxy analogue 15 retained some potency (pIC50 = 5.96), but with a large increase in lipophilicity (elogD = 5.1) and therefore was not further pursued. Removal of the alkyl ether group to give a phenol was investigated, and 5-cyano analogue 16 was chosen because of ease of synthesis,21 but resulted in loss of potency (pIC50 = 4.51). Re-installing a methoxy group (compound 17) restored potency (pIC50 = 6.36), confirming that the phenol group is not tolerated.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | L. inf. pIC50a | PMM pIC50b | Microsome Clintc (μL min−1 mg−1) | elogDd |
LLE | |
| Human | Mouse | |||||||
|
a Geometric mean of a minimum of two experiments ± standard error. Assay was run according to experimental description (see ESI†) using miltefosine as a positive control.
b Geometric mean of a minimum of two experiments ± standard error. Assay was run according to experimental description (see ESI†) using tamoxifen as a positive control.
c Intrinsic in vitro clearance calculated using liver microsomes.
d HPLC measured logD.
e Analogues with two p-fluorobenzyl groups on sulphonamide nitrogen instead of simple benzyl groups.
|
||||||||
| 9 | 2-OMe | OMe | 6.43 ± 0.24 | <4.19 | 242 | 1300 | 4.6 | 2.86 |
| 10 | 2-OMe | H | 6.48 ± 0.00 | <4.19 | n.a. | n.a. | n.a | 2.68 |
| 11 | 4-OMe | H | 4.34 ± 0.15 | <4.19 | 184 | 2412 | 4.7 | 0.70 |
| 12 | 2-F | F | <4.19 | <4.19 | n.a. | n.a. | n.a. | 0.07 |
| 13 | 2-CN | H | 4.95 ± 0.00 | <4.19 | 699 | n.a. | 4.3 | 1.40 |
| 14 | 2-SO2Me | H | 4.49 ± 0.06 | <4.19 | 417 | 2880 | 4.0 | 1.77 |
| 15 | 2-OCF3 | H | 5.96 ± 0.21 | <4.19 | 92.9 | 689 | 5.1 | 1.15 |
| 16 | 2-OH | CN | 4.51 ± 0.05 | 4.34 ± 0.15 | 27.4 | 1100 | 3.7 | 1.10 |
| 17 | 2-OMe | CN | 6.36 ± 0.04 | <4.19 | 304 | n.a. | 4.3 | 2.88 |
| 18 | 2-OMe | F | 5.86 ± 0.04 | <4.19 | 955 | n.a. | 4.7 | 2.11 |
| 19 | 2-OMe | Cl | 5.86 ± 0.04 | <4.19 | 407 | 1352 | 5.0 | 1.61 |
| 20 | 2-OMe | CO2H | 4.24 ± 0.05 | <4.19 | <23 | 94.0 | 2.5 | 1.11 |
| 21 | 2-OMe | CONH2 | 5.13 ± 0.04 | <4.19 | 108 | 1670 | 3.6 | 2.40 |
| 22 | 2-OMe |
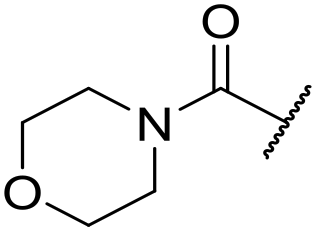
|
6.34 ± 0.04 | 4.34 ± 0.15 | 810 | 2025 | 3.8 | 3.02 |
| 23 | 2-OMe |
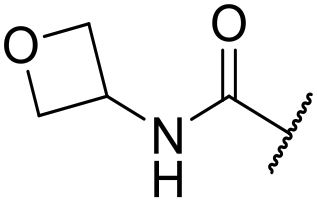
|
6.10 ± 0.00 | <4.19 | 178 | 395 | 4.0 | 3.11 |
| 24 | 2-OMe |
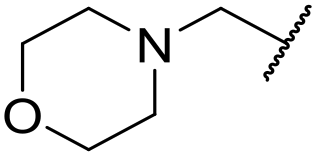
|
6.28 ± 0.02 | <4.19 | 49.4 | 3200 | 4.3 | 2.84 |
| 25 | 2-OMe |
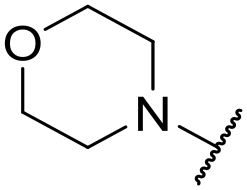
|
6.49 ± 0.00 | <4.19 | 421 | 1400 | 4.2 | 2.98 |
| 26 | 2-OMe | SO2Me | 5.89 ± 0.21 | <4.19 | 296 | n.a. | 3.9 | 3.04 |
| 27 | 2-OMe | SO2NH2 | 6.25 ± 0.02 | <4.19 | 94.1 | 884 | 3.6 | 3.84 |
| 28 | 2-OMe | SOMe | 6.23 ± 0.13 | <4.19 | 233 | 929 | 3.5 | 3.08 |
| 29 |
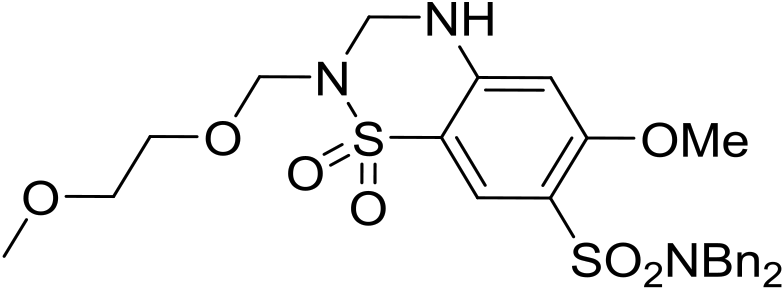
|
6.18 ± 0.08 | <4.19 | 518 | 1370 | 4.0 | 3.69 | |
| 30 |
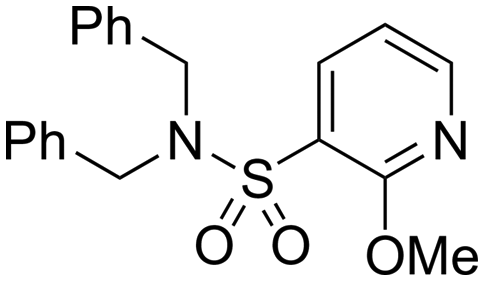
|
6.19 ± 0.09 | 4.19 | 849 | 2567 | 4.3 | 3.20 | |
| 31 |
|
4.68 ± 0.21 | 4.19 | 676 | 2450 | 3.7 | 2.42 | |
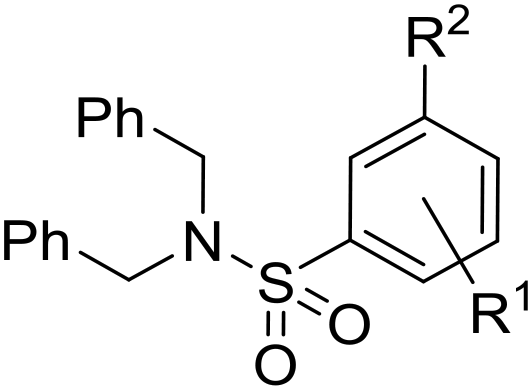
The substituent in the 5-position was further explored and found to be tolerant towards almost all changes (see Table 1, compounds 17–28), except for the carboxylic acid 20 (pIC50 = 4.24). Nitrile 17, amides 22 and 23, sulfone 26, sulphonamide 27, and sulfoxide 28 were the most promising analogues offering the best ligand-lipophilicity efficiency (LLE) and improved in vitro clearance (albeit still too high for in vivo studies). Morpholine 25 was the most potent of these analogues (pIC50 = 6.49) but was not further pursued due to perceived toxicity risks associated with anilines, and the likelihood of oxidation to a reactive iminoquinone-like intermediate.
It was hypothesized that the large degree of tolerability around the 5-position was consistent with these substituents pointing towards solvent. Compound 29 provides more support for this idea, as the capped methoxyethoxymethyl (MEM) derivative still possessed sub-micromolar potency, raising the exciting prospect of using polyethyleneglycol (PEG) based linkers as tool compounds to explore the mechanism of action.
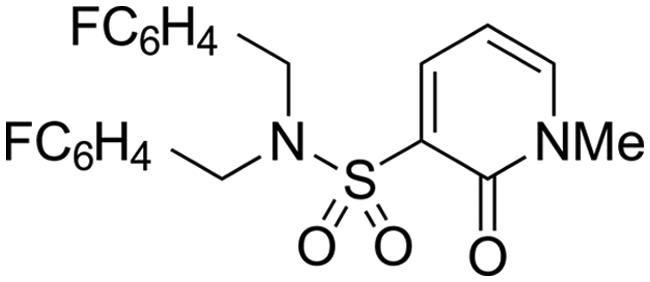
Finally, we investigated some heterocyclic aromatic sulphonamides, such as compound 30, which retained potency but led to poor metabolic stability. Pyridinone analogues unfortunately showed a significant loss of potency (see Table 1, compound 31 and ESI†).
The lack of improvement in in vitro clearance from changes to the upper portion of the molecule suggested that metabolic soft spots are likely to be present in the substituents on the sulphonamide nitrogen. It was hoped that changes here could result in improved metabolic stability. The two suspected spots for metabolic oxidation were the para-positions of the benzene ring and the benzyl positions. Firstly, fluorination at the para-position was tested and offered similar potency (pIC50 = 6.27 for 32versus 6.43 for 9), although clearance data could not be obtained for this analogue (failed in automatic detection high-throughput assay). Replacement of the 4-fluorobenzyl group with a benzene ring (or pyridine – see ESI†) reduced potency by more than 1.5log units (Table 2, compound 33, pIC50 = 4.94). We next examined smaller or more polar alternatives (see Table 2, compounds 33–42 and ESI†). At best, these analogues led to a 0.75log unit reduction in potency versus the parent dibenzyl analogue 9, and in some cases even complete loss of activity. The most promising analogue was the methoxyethane derivative 38 which still possessed moderate potency (pIC50 = 5.68) as well as lower lipophilicity (elogD = 3.5) and slightly improved metabolic stability compared to previous analogues. Replacing the 2,5-dimethoxybenzene group of 38 with other substituents from Table 1, such as the cyano and morpholine amide moieties, further improved the in vitro clearance as exemplified by compounds 41 and 42, but with a loss of potency (pIC50 = 5.05 and 4.36 respectively). It is possible that the elogD of this series has a lower limit of about 3.5, as no compounds have been observed with good potency (pIC50 ≥ 6.0) below this value.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | L. inf. pIC50a | PMM pIC50b | Microsome Clint,uc (μL min−1 mg−1) | elogDd |
LLE | |
| Human | Mouse | ||||||||
|
a Geometric mean of a minimum of two experiments ± standard error. Assay was run according to experimental description (see ESI†) using miltefosine as a positive control.
b Geometric mean of a minimum of two experiments ± standard error. Assay was run according to experimental description (see ESI†) using tamoxifen as a positive control.
c Intrinsic in vitro clearance calculated using liver microsomes.
d HPLC measured logD.
e Expected degradation product would be carboxylic acid 20 which was inactive.
|
|||||||||
| 32 | OMe |
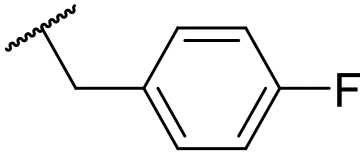
|
C6H4F | 6.27 ± 0.00 | <4.19 | n.a. | n.a. | 4.7 | 2.49 |
| 33 | OMe |
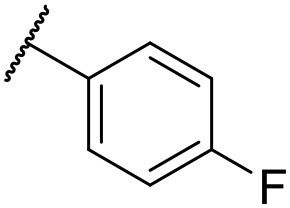
|
C6H4F | 4.94 ± 0.25 | <4.19 | 482 | 1560 | 4.3 | 1.60 |
| 34 | OMe |
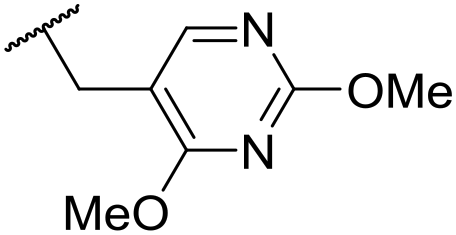
|
C6H4F | 4.27 ± 0.08 | <4.19 | 498 | 3148 | 3.8 | 1.58 |
| 35 | OMe |
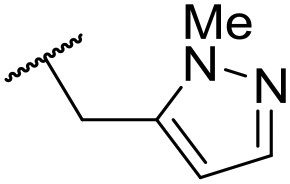
|
C6H4F | 5.28 ± 0.26 | 4.34 ± 0.15 | 651 | 5480 | 3.5 | 3.49 |
| 36 | OMe |
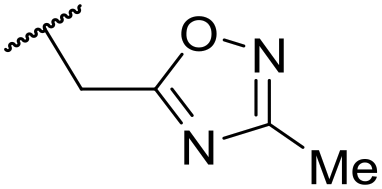
|
C6H4F | 5.11 ± 0.05 | <4.19 | 350 | 3054 | 3.5 | 3.03 |
| 37 | OMe | Me | C6H4F | 4.93 ± 0.04 | <4.19 | 414 | n.a. | 3.7 | 2.67 |
| 38 | OMe | Et | Ph | 5.60 ± 0.03 | <4.19 | 534 | 3497 | 3.8 | 3.04 |
| 39 | OMe |
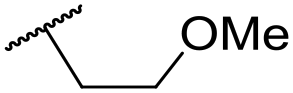
|
Ph | 5.68 ± 0.00 | <4.19 | 253 | n.a. | 3.5 | 3.62 |
| 40 | OCF3 |
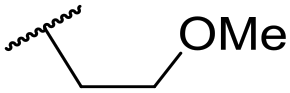
|
C6H4F | 5.08 ± 0.11 | <4.19 | 194 | 222 | 3.8 | 1.78 |
| 41 | CN |
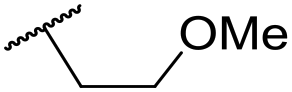
|
C6H5F | 5.05 ± 0.03 | <4.19 | <23 | 585 | 3.4 | 2.98 |
| 42 |
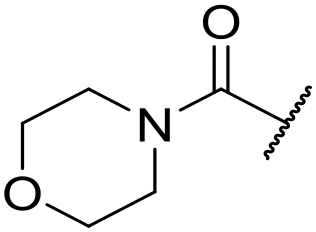
|
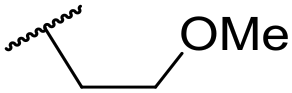
|
C6H5F | 4.36 ± 0.06 | <4.19 | 28 | 60 | 2.9 | 2.74 |
| 43 | CN |
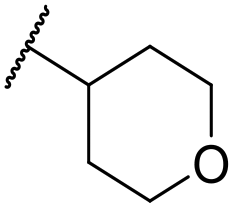
|
C6H5F | <4.19 | <4.19 | 194 | 253 | 3.4 | 1.64 |
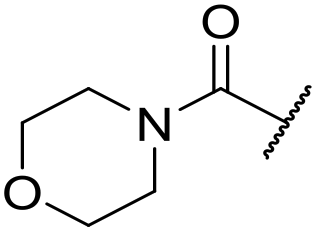
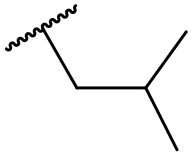
| 44 |
|
|
C6H5F | 5.16 ± 0.13 | <4.19 | 1050 | 1400 | 3.6 | 2.08 |
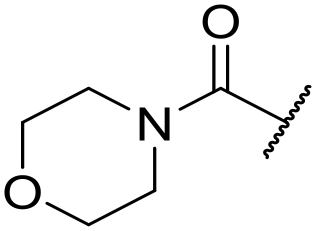
| 45 |
|
|
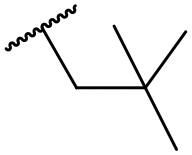
Ph | 6.00 ± 0.10 | <4.19 | 1600 | 2580 | 3.8 | 2.45 |
| 46 |
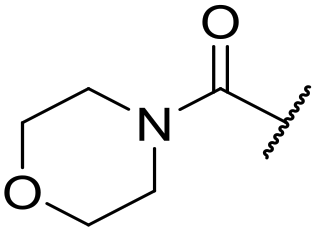
|
|
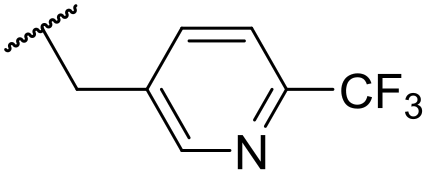
C6H5F | 5.76 ± 0.06 | <4.19 | 265 | 534 | 3.7 | 2.44 |
| 47 |
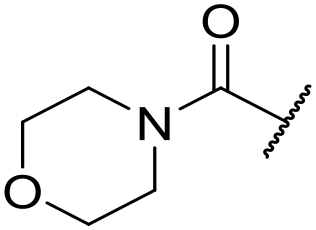
|
|
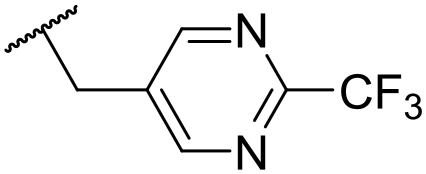
Ph | 5.35 ± 0.25 | <4.19 | 73 | 152 | 3.5 | 2.86 |
| 48 |
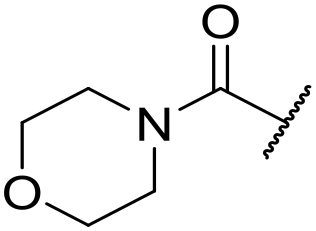
|
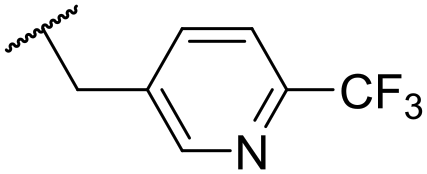
|
t Bu | 4.34 ± 0.15 | <4.19 | 143 | 379 | 3.6 | 0.89 |
| 49 | CN |
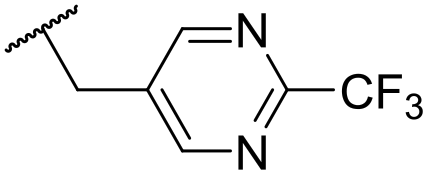
|
Ph | 5.41 ± 0.16 | <4.19 | 100 | n.a. | 3.8 | 2.76 |
| 50 | CN |
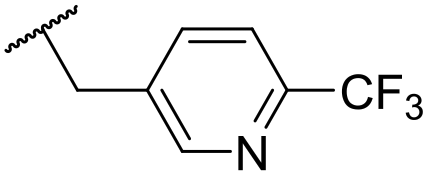
|
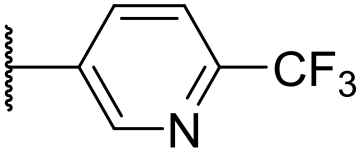
|
4.41 ± 0.23 | <4.19 | <23 | 95 | 3.8 | 1.13 |
At this point, results from metabolite identification (MetID) experiments (see ESI† Tables S2 and S3) suggested that the benzyl CH2 was indeed the major metabolic soft spot as previously postulated. Several analogues containing sterically hindered aliphatic groups as lipophilic alternatives to the benzyl group were synthesised to reduce clearance whilst maintaining potency (Table 2, compounds 43–45). The more polar 4-tetrahydropyranyl 43 was inactive and the isobutyl analogue 44 only possessed moderate potency against L. inf. (pIC50 = 5.16). The bulkier neopentyl analogue 45 showed good potency (pIC50 = 6.00) albeit without much improvement in metabolic stability. The substituents which led to the largest improvement in clearance without completely compromising potency were trifluoro-pyridine and trifluoro-pyrimidine analogues (Table 2, compounds 46–47). Replacement of a phenyl group with a trifluoro-pyridine, as in 46, gave a 4-fold improvement in the in vitro mouse clearance but suffered from a decrease in potency (pIC50 = 5.76). The trifluoro-pyrimidine analogue 47 gave a further 4-fold improvement in clearance but once more with a drop in potency (pIC50 = 5.35). Attempts to switch the morpholine amide for a nitrile group (compound 49) did not lead to any improvement, and the replacement of the second benzyl group with a neopentyl (compound 48) or another trifluoropyridyl (compound 50) resulted in inactive compounds, albeit with further improvements in metabolic stability.
Despite not having identified a likely lead, the compounds with the best compromise between potency and clearance, trifluoro-pyridine 46 and trifluoro-pyrimidine 47, were examined in more detail (Table 3). In addition to the poor in vitro clearance it was found that these compounds had extremely low aqueous solubility, which is likely to be due to their high lipophilicity. Furthermore, they possessed surprisingly poor stability with only 22% and 42% (for 46 and 47, respectively) remaining after 6 h incubation in mouse plasma. Amide hydrolysis may be responsible for poor stability, although this has not been observed in microsomes, but further analogues would need to be tested to confirm this hypothesis.
| Compound | 46 | 47 |
|---|---|---|
| L. inf. pIC50 | 5.76 | 5.35 |
| In vitro Clint (human) | 265 μL min−1 mg−1 | 73 μL min−1 mg−1 |
| In vitro Clint (mouse) | 534 μL min−1 mg−1 | 152 μL min−1 mg−1 |
| Plasma protein binding (PPB) | 95.9% | 88.3% |
| Mouse plasma stability (6 h) | 22% remaining | 42% remaining |
| Kinetic solubility (pH 2.0) | 3.8 μg mL−1 | 11.3 μg mL−1 |
| Kinetic solubility (pH 7.4) | 3.2 μg mL−1 | 6.2 μg mL−1 |
Despite evaluating almost 200 compounds, no suitable candidate with an acceptable balance of potency and physicochemical properties could be identified, and it was decided to halt further work on this series. In particular, the potency of these sulphonamides appears to be dependent on lipophilicity with almost all compounds with elogD < 3.5 losing activity. As can be seen in Fig. 3, it proved difficult to synthesise compounds with LLE > 4. It should be noted that attempts to return to the initially identified subset of secondary sulphonamides (Fig. 2) and improve lipophilicity also led to significant reduction in potency (see ESI†).
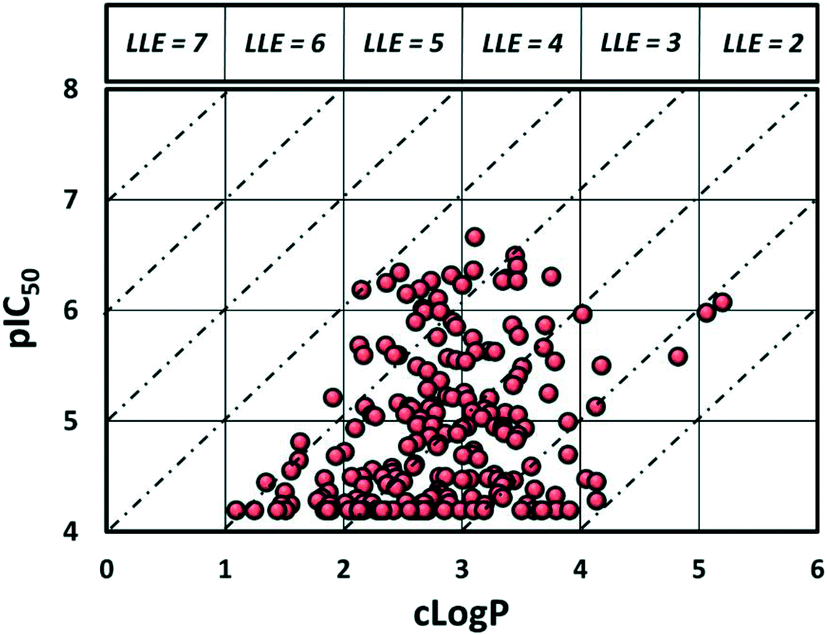 | ||
| Fig. 3 Plot of lipophilicity versus potency. | ||
One of the reasons why drug discovery is notoriously challenging in leishmaniasis is the difficulty of reaching the parasites. The amastigotes reside in parasitophorous vacuoles inside the hosts' macrophages,22,23 requiring molecules to cross three cell membranes with differing pH gradients.10 There are several examples of target-based approaches for leishmaniasis failing to translate into cell-based assays due to this difficulty in permeating the parasite.24–26 This may be responsible for the loss of potency in the benzenesulphonamide compounds with lower lipophilicities. Additionally, given the species difference in drug sensitivity observed during initial SAR exploration, we also examined the activity of several analogues against L. inf. in a different, immortalized, cell line (THP-1). We noticed significant variability in potency (see ESI† Table S1), suggesting different host cell characteristics and host–parasite interactions may play an important role in the antileishmanial activity of this series and ultimately impact hit discovery campaigns.27–29 This may be of relevance when considering host cell selection for future screening efforts against leishmaniasis, particularly given the ethical and logistical constraints of using primary versus immortalized cells.
Despite the difficulties in optimizing these compounds, this series showed good potency and selectivity against intracellular forms of Leishmania spp. covering several species of CL and VL (see ESI†). The possibility of using these tool compounds in detailed mode of action studies is currently being explored. Such studies may provide an opportunity to use target-based medicinal chemistry to overcome the current issues with this series.
Material and methods
Parasite and cell cultures
Leishmania infantum MHOM/MA(BE)/67/ITMAP263 and L. donovani MHOM/ET/67/L82 amastigotes were collected from the spleen of an infected donor Golden hamster and used to infect primary peritoneal mouse macrophages (PMM). PMM were collected 2 days after peritoneal stimulation of Swiss mice with a 2% potato starch suspension and maintained in RPMI-1640 medium supplemented with 200 mM L-glutamine and 5% FCS. Cultures and assays were maintained at 37 °C under an atmosphere of 5% CO2.Compound solutions/dilutions
Compound stock solutions were prepared in 100% DMSO at 20 mM. The compounds were serially pre-diluted (2-fold) in 100% DMSO to maintain maximal solubility, followed by a one-step further dilution in demineralized water to achieve a final in-test DMSO concentration of <1%. The compounds were tested at 2-fold compound dilutions covering a range of 64 to 0.00024 μM.Susceptibility assay
For the intracellular amastigote susceptibility assay, 3 × 104 PMM were seeded in a 96-well plate. After 24 h, 5 × 105 amastigotes per well were added and incubated for 2 h at 37 °C. The compound dilutions were added next and the plates were further incubated for 5 days at 37 °C and 5% CO2. Total parasite burdens were microscopically assessed after Giemsa staining. In treated wells with high amastigote burdens, an overall estimate of the total burden per well was made without discrimination between the number of infected macrophages and the number of amastigotes per infected cell. In treated wells with low burdens, exact counting was performed. The results were expressed as % reduction in parasite burdens compared to control wells and an IC50 was calculated. Miltefosine was included as the reference drug (IC50 ∼5–10 μM).Cytotoxicity assays
The evaluation of toxicity to PMM was part of the in vitro susceptibility assay, determined by microscopic evaluation of cell detachment, lysis, and granulation. Evaluation was done by semi-quantitative scoring (no exact counting was performed) of at least 500 cells distributed over adjacent microscopic fields. The results were expressed as % reduction in normal cells compared to untreated control wells and an CC50 (50% cytotoxic concentration) was determined. Tamoxifen was used as reference for cytotoxicity (CC50 ∼ 5–10 μM).ADME assays
For experimental determination of logD, test compounds were prepared at a physiologically relevant pH of 7.4 at 200 nM and 2% DMSO in a 50/50 mix of mobile phase A (5% methanol in 10 mM ammonium acetate and adjusted to pH 7.4) and B (100% methanol adjusted to pH 7.4) with an appropriate internal standard at 4 nM, and injected onto an Ascentis Express RP Amide column. Retention times were compared to a standard curve of nine commercial drugs covering a logD range of −1.86 to 6.1. The retention times of each of the standards was plotted against the published logD values. The resulting equation for this line (y = mx + b) was used to calculate the logD values for the test compounds where ‘x’ was the retention time in minutes of the test compound and the resultant “y” was the experimental logD value.
For the experimental stability determination of test compounds in liver microsomes in the presence of NADPH, a clearance rate was determined. Assay conditions were 0.25 mg mL−1 liver microsomal protein from the species of interest (mouse and human), 0.5 μM test compound, pH 7.4 and 37 °C. The reagent was purchased commercially, and the work did not involve the use of animals or humans. Samples were taken at 0, 5, 10, 15, 20 and 30 min. The reaction was started after T0 was taken, with the addition of NADPH at 0.5 μM. The reaction was stopped by addition of 95% acetonitrile/5% methanol containing an internal standard. Time point samples were combined in compound groups of six that had been pre-sorted by mono molecular weight and analyzed by LC/MS/MS. Peak area ratios (analyte peak area/internal standard peak area) were converted to % remaining using the area ratio at time 0 as 100%. Half-life (t1/2 = ln(2)/k) and intrinsic clearance (Clint = k × 1000/(0.25)) in μL min−1 mg−1 were calculated from % remaining versus incubation time. From this plot, the slope (k) was determined. Data was qualified if t1/2 > 4X the last time point.
Author contribution
P. J. K. took the lead in writing the manuscript; P. J. K. and M. A. D. carried out design and synthetic chemistry efforts; A. M., L. L. G. F., R. C. C. and S. M.-D. carried out biological experiments; L. M., G. C., A. D. A., J. M. K., C. E. M. and L. C. D. conceived experiments, provided guidance about data interpretation and design of compounds; S. C. contributed to compound design, interpretation of results and to writing the manuscript; J. M. K, C. E. M. and L. C. D. conceived and planned the project.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We would like to thank FAPESP (Grants 2015/19495-5, 2015/50655-9 and 2013/07600-3) as well as DNDi for funding; UNICAMP NMR and technical staff for assistance; The Thomson Mass Spectrometry Lab at UNICAMP for use of HRMS equipment; the High-Throughput ADME team at AbbVie for ADME testing and Dr. Dale Kempf (AbbVie) and Dr. Michael Schrimpf (AbbVie) for helpful discussions; the Translational Parasitology Group at the Drug Discovery Unit, University of Dundee, for carrying out the primary screen and initial analogue testing; DNDi is grateful to its donors, public and private, who have provided funding for all DNDi activities since its inception in 2003. A full list of DNDi's donors can be found at http://www.dndi.org/donors/donors/.References
- DNDi, Leishmaniasis - Disease Factsheet, https://www.dndi.org/wp-content/uploads/2019/09/Factsheet2019_Leishmaniasis.pdf, (accessed 4 February 2020).
- World Health Organization, Unveiling the neglect of leishmaniasis - WHO infographic, https://www.who.int/leishmaniasis/Unveiling_the_neglect_of_leishmaniasis_infographic.pdf?ua=1, (accessed 4 February 2020).
- K. Stuart, R. Brun, S. Croft, A. Fairlamb, R. E. Gürtler, J. McKerrow, S. Reed and R. Tarleton, J. Clin. Invest., 2008, 118, 1301–1310, DOI:10.1172/JCI33945.
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. de Boer, PLoS One, 2012, 7, e35671, DOI:10.1371/journal.pone.0035671.
- F. Alves, G. Bilbe, S. Blesson, V. Goyal, S. Monnerat, C. Mowbray, G. Muthoni Ouattara, B. Pécoul, S. Rijal, J. Rode, A. Solomos, N. Strub-Wourgaft, M. Wasunna, S. Wells, E. E. Zijlstra, B. Arana and J. Alvara, Clin. Microbiol. Rev., 2018, 31, 1–30, DOI:10.1128/CMR.00048-18.
- World Health Organization, Global Health Observatory data repository - Number of cases of visceral leishmaniasis reported Data by country, http://apps.who.int/gho/data/node.main.NTDLEISHVNUM?lang=en, (accessed 4 February 2020).
- World Health Organization (WHO), Wkly. Epidemiol. Rec., 2016, vol. 91, pp. 287–296 Search PubMed.
- World Health Organization (WHO), Wkly. Epidemiol. Rec., 2017, vol. 92, pp. 557–565 Search PubMed.
- J. Alvar, S. Yactayo and C. Bern, Trends Parasitol., 2006, 22, 552–557, DOI:10.1016/j.pt.2006.09.004.
- M. C. Field, D. Horn, A. H. Fairlamb, M. A. J. Ferguson, D. W. Gray, K. D. Read, M. De Rycker, L. S. Torrie, P. G. Wyatt, S. Wyllie and I. H. Gilbert, Nat. Rev. Microbiol., 2017, 15, 217–231, DOI:10.1038/nrmicro.2016.193.
- T. P. C. Dorlo, M. Balasegaram, J. H. Beijnen and P. J. de Vries, J. Antimicrob. Chemother., 2012, 67, 2576–2597, DOI:10.1093/jac/dks275.
- S. Rijal, B. Ostyn, S. Uranw, K. Rai, N. R. Bhattarai, T. P. C. Dorlo, J. H. Beijnen, M. Vanaerschot, S. Decuypere, S. S. Dhakal, M. L. Das, P. Karki, R. Singh, M. Boelaert and J. C. Dujardin, Clin. Infect. Dis., 2013, 56, 1530–1538, DOI:10.1093/cid/cit102.
- B. Ostyn, E. Hasker, T. P. C. Dorlo, S. Rijal, S. Sundar, J. C. Dujardin and M. Boelaert, PLoS One, 2014, 9, e100220, DOI:10.1371/journal.pone.0100220.
- S. Srivastava, J. Mishra, A. K. Gupta, A. Singh, P. Shankar and S. Singh, Parasites Vectors, 2017, 10, 1–11, DOI:10.1186/s13071-017-1969-z.
- S. Khare, A. S. Nagle, A. Biggart, Y. H. Lai, F. Liang, L. C. Davis, S. W. Barnes, C. J. N. Mathison, E. Myburgh, M.-Y. Gao, J. R. Gillespie, X. Liu, J. L. Tan, M. Stinson, I. C. Rivera, J. Ballard, V. Yeh, T. Groessl, G. Federe, H. X. Y. Koh, J. D. Venable, B. Bursulaya, M. Shapiro, P. K. Mishra, G. Spraggon, A. Brock, J. C. Mottram, F. S. Buckner, S. P. S. Rao, B. G. Wen, J. R. Walker, T. Tuntland, V. Molteni, R. J. Glynne and F. Supek, Nature, 2016, 537, 229–233, DOI:10.1038/nature19339.
- S. Wyllie, S. Brand, M. Thomas, M. De Rycker, C.-W. Chung, I. Pena, R. P. Bingham, J. A. Bueren-Calabuig, J. Cantizani, D. Cebrian, P. D. Craggs, L. Ferguson, P. Goswami, J. Hobrath, J. Howe, L. Jeacock, E.-J. Ko, J. Korczynska, L. MacLean, S. Manthri, M. S. Martinez, L. Mata-Cantero, S. Moniz, A. Nühs, M. Osuna-Cabello, E. Pinto, J. Riley, S. Robinson, P. Rowland, F. R. C. Simeons, Y. Shishikura, D. Spinks, L. Stojanovski, J. Thomas, S. Thompson, E. Viayna Gaza, R. J. Wall, F. Zuccotto, D. Horn, M. A. J. Ferguson, A. H. Fairlamb, J. M. Fiandor, J. Martin, D. W. Gray, T. J. Miles, I. H. Gilbert, K. D. Read, M. Marco and P. G. Wyatt, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9318–9323, DOI:10.1073/pnas.1820175116.
- Drugs for Neglected Diseases initiative, DNDi R&D Portfolio December 2019, https://www.dndi.org/diseases-projects/portfolio/, (accessed 24 February 2020).
- P. J. Koovits, M. A. Dessoy, A. Matheeussen, L. Maes, G. Caljon, C. E. Mowbray, J. M. Kratz and L. C. Dias, Bioorg. Med. Chem. Lett., 2020, 30, 126779, DOI:10.1016/j.bmcl.2019.126779.
- For another example of sulphonamide based scaffolds with anti-leishmanial activity see: L. C. S. Pinheiro, M. D. L. G. Ferreira, F. F. Silveira, L. M. Feitosa and N. Boechat, Synthetic compounds with sulphonamide moiety against Leishmaniasis: an overview, Med. Chem. Res., 2019, 28, 1807–1817, DOI:10.1007/s00044-019-02432-3.
- A. Nühs, M. De Rycker, S. Manthri, E. Comer, C. A. Scherer, S. L. Schreiber, J. R. Ioset and D. W. Gray, PLoS Neglected Trop. Dis., 2015, 9(9), e0004094, DOI:10.1371/journal.pntd.0004094.
- A. F. Kornahrens, A. B. Cognetta, D. M. Brody, M. L. Matthews, B. F. Cravatt and D. L. Boger, J. Am. Chem. Soc., 2017, 139, 7052–7061, DOI:10.1021/jacs.7b02985.
- J. C. Antoine, E. Prina, T. Lang and N. Courret, Trends Microbiol., 1998, 6, 392–401, DOI:10.1016/S0966-842X(98)01324-9.
- N. M. Novozhilova and N. V. Bovin, Biochemistry, 2010, 75, 686–694, DOI:10.1134/S0006297910060027.
- L. S. Torrie, S. Brand, D. A. Robinson, E. J. Ko, L. Stojanovski, F. R. C. Simeons, S. Wyllie, J. Thomas, L. Ellis, M. Osuna-Cabello, O. Epemolu, A. Nühs, J. Riley, L. Maclean, S. Manthri, K. D. Read, I. H. Gilbert, A. H. Fairlamb and M. De Rycker, ACS Infect. Dis., 2017, 3, 718–727, DOI:10.1021/acsinfecdis.7b00047.
- V. Corpas-Lopez, S. Moniz, M. Thomas, R. J. Wall, L. S. Torrie, D. Zander-Dinse, M. Tinti, S. Brand, L. Stojanovski, S. Manthri, I. Hallyburton, F. Zuccotto, P. G. Wyatt, M. De Rycker, D. Horn, M. A. J. Ferguson, J. Clos, K. D. Read, A. H. Fairlamb, I. H. Gilbert and S. Wyllie, ACS Infect. Dis., 2019, 5, 111–122, DOI:10.1021/acsinfecdis.8b00226.
- J. A. Hutton, V. Goncalves, J. A. Brannigan, D. Paape, M. H. Wright, T. M. Waugh, S. M. Roberts, A. S. Bell, A. J. Wilkinson, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, J. Med. Chem., 2014, 57, 8664–8670, DOI:10.1021/jm5011397.
- S. Hendrickx, L. Van Bockstal, G. Caljon and L. Maes, PLoS Neglected Trop. Dis., 2019, 13, e0007885, DOI:10.1371/journal.pntd.0007885.
- C. H. Franco, L. M. Alcântara, E. Chatelain, L. Freitas-Junior and C. B. Moraes, Trop. Med. Infect. Dis., 2019, 4, 82, DOI:10.3390/tropicalmed4020082.
- G. Yang, N. Lee, J.-R. Ioset and J. H. No, SLAS Discovery, 2017, 22, 125–134, DOI:10.1177/1087057116673796.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00165a |
| This journal is © The Royal Society of Chemistry 2020 |