
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnchoring and packing of self-assembled monolayers of semithio-bambusurils on Au(111)†
Pramod Patil
Kunturu
a,
Özlem
Kap
b,
Kai
Sotthewes
 b,
Pierre
Cazade
c,
Harold J. W.
Zandvliet
*b,
Damien
Thompson
*c,
Ofer
Reany
*d and
Jurriaan
Huskens
*a
b,
Pierre
Cazade
c,
Harold J. W.
Zandvliet
*b,
Damien
Thompson
*c,
Ofer
Reany
*d and
Jurriaan
Huskens
*a
aMolecular Nanofabrication Group, MESA + Institute for Nanotechnology, Faculty of Science and Technology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: j.huskens@utwente.nl
bPhysics of Interfaces and Nanomaterials, MESA+ Institute for Nanotechnology, University of Twente, P. O. Box, 217, 7500AE Enschede, The Netherlands. E-mail: h.j.w.zandvliet@utwente.nl
cDepartment of Physics, Bernal Institute, University of Limerick, Limerick V94 T9PX, Ireland. E-mail: damien.thompson@ul.ie
dDepartment of Natural Sciences, The Open University of Israel, 1 University Road, Ra'anana 43107, Israel. E-mail: oferre@openu.ac.il
First published on 17th December 2019
Abstract
Semithio-bambusurils are a unique family of anion-binding host macrocycles that form self-assembled monolayers (SAMs) on Au(111). SAMs of semithio-bambus[n]uril homologs with different cage sizes (1: n = 4; 2: n = 6) have been investigated using electrochemistry, X-ray photoelectron spectroscopy (XPS), scanning tunneling microscopy (STM), and molecular dynamics (MD) simulations. Electrochemical measurements showed that electron transfer occurs via tunneling through the SAMs, and the low resistivity indicated an open layer architecture. XPS confirmed that thiocarbonyl sulfur atoms are chemisorbed to the Au(111) surface, and STM revealed the formation of ordered domains in a rectangular lattice for 1 and a highly ordered triangular/hexagonal lattice for 2. MD simulations substantiated the STM data by quantifying the balance between molecule–surface bonding, molecular conformations, and supramolecular packing that drive the formation of SAMs that maximize their surface coverage within the limits of conformational strain.
Design, System, ApplicationThe attachment of receptors to a transducer surface provides a general strategy to design sensor devices. Yet, these receptor molecules may undergo large conformational changes that may remain obscure when not inspecting the molecular structure of the receptor in its surface-immobilized state. The current manuscript provides an example of a class of receptor molecules with thiocarbonyl groups purposefully integrated in the molecular design in order to provide anion recognition capabilities. Upon binding of the receptors to a gold surface, which can be used as an electrode for electrochemical sensing, the molecules appear to undergo large conformational changes that optimize their layer formation properties but at the expense of their anion binding. The lessons learned in this detailed surface-analytical and molecular dynamics study may provide insights into the effect of surface binding to the molecular conformations of the receptors and may assist in developing new strategies to promote receptor binding with retention of recognition. |
Introduction
Self-assembled monolayers (SAMs) have promising applications in biotechnology, materials science, nano and molecular electronics, electrochemistry and sensor devices. Due to their remarkable properties, they can, among others, control surface wetting, electron transport, adhesion, and molecular recognition, including the detection of biomolecules.1–8 A high degree of order and dense packing of molecules in the film is a prerequisite for the use of SAM-based sensor devices.9–11 Furthermore, the complexation sites must show high binding affinity and specificity to maximize the electrical response and minimize fouling.12 One of the most useful SAM-based sensing devices involves immobilization of highly selective receptors to bind charged molecules and ions in electrochemical sensors and biosensing applications.13–16 In this context, synthetic receptors based on host macrocycles such as cyclodextrins, calixarenes cucurbiturils, pillarenes, and bambusurils exhibit strong binding affinities and a rich host–guest chemistry.17–23Bambus[n]urils (BUs, n = 4,6) are a family of cavitands composed of 2,4-disubstituted glycoluril subunits interconnected by methylene bridges in an alternate arrangement.24,25 The glycoluril subunits in the BU structure present their convex side towards the interior with the methine hydrogens pointing inward, forming a highly electron-deficient cavity with both polarization and electrostatic interactions contributing to anion binding. Following the predictions that replacement of oxygen atoms in bambusurils by other heteroatoms could significantly modify their anion binding properties, the calculated molecular electrostatic potentials of hetero-BUs were found to follow a general trend of anion-affinity: S > O > NH.26 These predictions were also confirmed experimentally indicating that, although all analogs are excellent anion binders, semithio-BU[6] is an effective transmembrane transporter capable of polarizing lipid membranes through the uniport mechanism of chloride anions.27 Similarly, semiaza-BU[6]s exhibit the simultaneous accommodation of three anions, linearly positioned along the main symmetry axis, which is reminiscent of natural chloride channels in E. coli.28
Here, we report the SAM formation ability of both semithio-BU homologs (n = 4, 6), 1 and 2, respectively (Fig. 1, Fig. S1†)29 on a gold surface. The semithio-BU SAMs were characterized by their contact angles, X-ray photoelectron spectroscopy (XPS), and electrochemistry. Anion binding was studied using electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). We used scanning tunneling microscopy (STM) combined with molecular dynamics (MD) simulations to determine the geometry of the semithio-BU SAMs in order to understand their anchoring on gold, intermolecular packing and anion recognition properties.
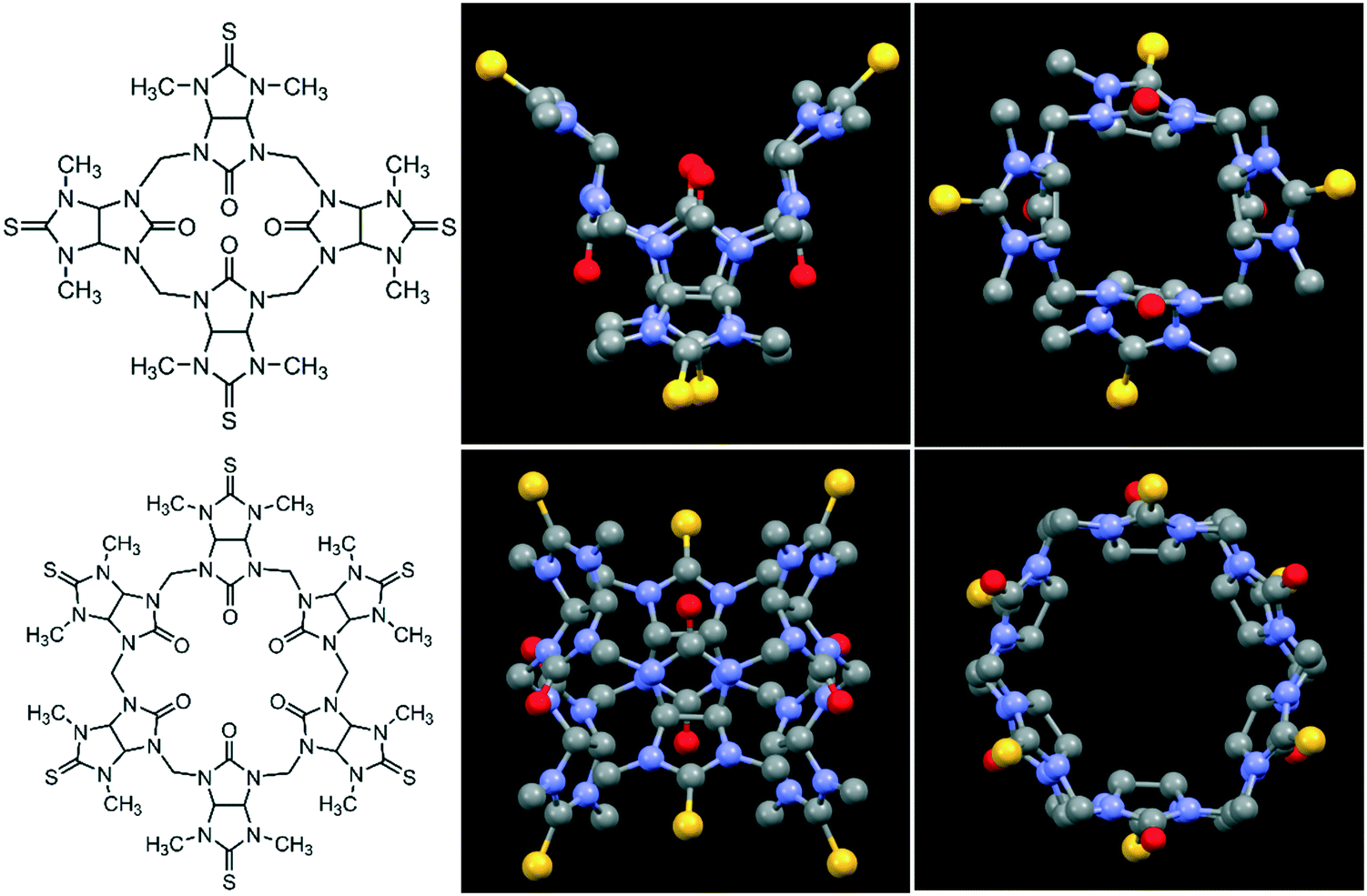 | ||
| Fig. 1 Molecular structures (left) of semithio-BU[4], 1 (top) and semithio-BU[6], 2 (bottom), and the corresponding side and top views of the X-ray structures.29 Hydrogen atoms and solvent molecules were omitted for clarity (unit cells with solvent shown in Fig. S1†). Color code: oxygen, red; sulfur, yellow; nitrogen, blue; carbon, grey. | ||
Results and discussion
SAMs were prepared by immersing Au substrates into DMSO solutions of 1 and 2 at different temperatures as described in Methods. The contact angle (CA) values summarized in Table 1 at ambient temperature show clearly that both 1 and 2 monolayers exhibit a similar hydrophilicity.The contact angles show almost no dependence on preparation temperature in the range of 25 to 80 °C (Table S1†), indicating that the temperature has no noticeable effect on the SAM formation. The cleaned bare gold layer is more hydrophobic than the SAMs, which indicates that both semithio-BU homologs have been successfully adsorbed onto the Au surface. The thiocarbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S) moieties within both portals of 1 and 2 are expected to have a high affinity for Au, which drives SAM formation on the Au surface. Regarding SAM stability, monitoring the contact angle of the SAMs over the course of a week (58° and 62° for 1 and 2, respectively) did not show significant changes.
S) moieties within both portals of 1 and 2 are expected to have a high affinity for Au, which drives SAM formation on the Au surface. Regarding SAM stability, monitoring the contact angle of the SAMs over the course of a week (58° and 62° for 1 and 2, respectively) did not show significant changes.
Infrared reflection absorption spectroscopy (IRRAS) showed three characteristic peaks of 1 and 2 (Fig. S2,† top and bottom, respectively): at 1260 cm−1, 1060 cm−1 and 950 cm−1 corresponding to (C)–(N), (C)(S) and (C)–(N–CS) stretching vibrations, respectively.
XPS measurements (Fig. 2a and b) clearly identified carbon, sulfur, nitrogen, and oxygen in the SAMs (Fig. 2 and S3†). The C1s signal can be deconvoluted into three components at 284.7, 286.8 and 289.1 eV, which are assigned to C–C, CS and CO bonds, respectively (Fig. 2c and d). There is qualitative agreement between the XPS spectra and elemental composition for the SAM and powder samples of 1 and 2 (Table S2†), but a more detailed comparison is complicated by the presence of solvent and/or salts in the powders (see Fig. S4† and the discussion following it). XPS was also used to determine the average number of sulfur atoms that are involved in binding the macrocycles to the gold surface. For sulfur-containing SAMs, e.g., thiols and sulfides, it is known that adsorption on gold results in a negative shift of about 1.5 eV in the XPS signal of the S2p electrons. Hence, with bound sulfur 2p3/2 to the gold surface the XPS signal is observed at 161.9 eV compared to unbound sulfur 2p3/2 observed at 163.4 eV.10,12 Both XPS spectra of SAMs of 1 and 2 in the S2p region showed broad peaks with a binding energy (BE) of sulfur (S2p3/2) located around ∼162 eV (Fig. 2e and f). An additional set of signals, with much lower intensity, is visible at around 168 eV as well, indicative of oxidized S. In particular, no detectable intensity is present in the BE region at ∼164 eV for any of these two SAMs, which means that unbound sulfur atoms (S2p1/2 BE of ca. 165 eV) are not detected by XPS. The most intense doublet at 162 eV is attributed to surface-bound Au–S species. The smaller doublets, at 168 eV, are tentatively attributed to oxidized sulfur in 1, while we note that SAMs of 2 are fully unoxidized. Potentially, some of the sulfur atoms may get oxidized by Au oxide, present after piranha treatment, or by ambient oxygen,30 but evidence of oxidation is also seen in the powder spectra (Fig. S4†). The peak separation in the doublets is about 1.2 eV and the intensity ratio is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, corresponding to the S 2p3/2 and S 2p1/2 lines of semithio-BUs. These data confirm that the majority or all of the sulfur atoms are connected to the gold surface upon SAM formation, with a minor fraction (approximately 15%) in SAMs of 1 oxidized which prohibits their precise assignment as free or bound species.
:1, corresponding to the S 2p3/2 and S 2p1/2 lines of semithio-BUs. These data confirm that the majority or all of the sulfur atoms are connected to the gold surface upon SAM formation, with a minor fraction (approximately 15%) in SAMs of 1 oxidized which prohibits their precise assignment as free or bound species.
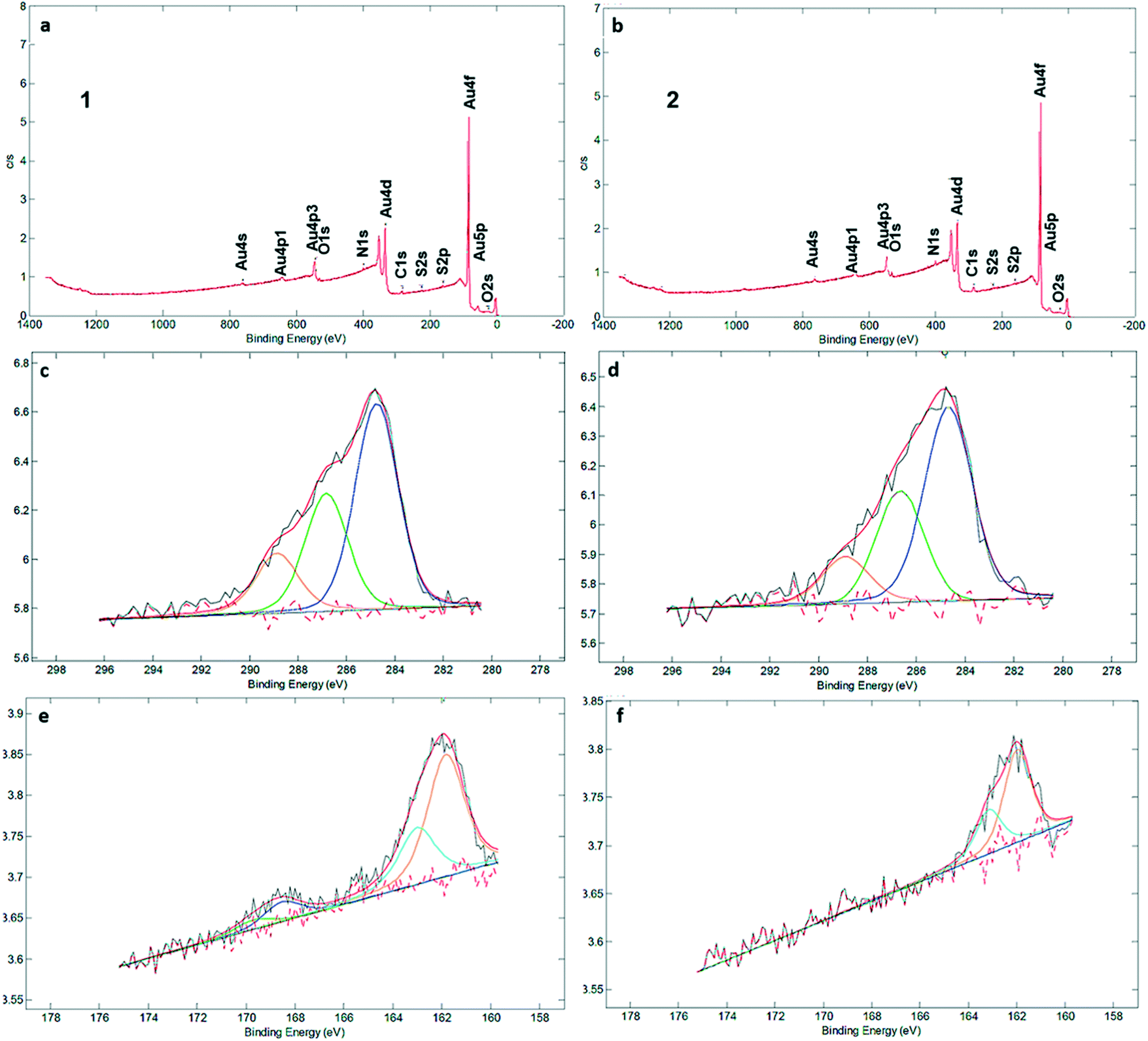 | ||
| Fig. 2 XPS spectra for monolayers of 1 (a/c/e) and 2 (b/d/f): full scans (a, b) C 1s (c, d), and S2p (e and f). All spectra were calibrated relative to the C1s peak located at 284.50 eV. In the element scans, raw data are in black, the overall fit in red, and the separate fitted peaks that sum up to the overall fit in the other colors. | ||
Electrochemical measurements, employing the redox behavior of the redox couple Fe(CN)63−/4– supplied to solution, are commonly used to probe the thickness and compactness of a monolayer on gold. Previous studies have shown that the diffusion-limited current, measured during cyclic voltammetry experiments at a monolayer-covered electrode, depends on the presence of pinholes in the monolayer.32,33 The efficiency of the monolayer in blocking redox reactions can be estimated by the kinetics of the redox probe, as expressed by the peak-to-peak separation ΔEp. CV in a 0.5 M K2SO4 electrolyte was used to determine the capacitance C of the monolayers (Table 1). The electrode adsorbs ions, resulting in an electrochemical double layer, which provides a capacitance to the surface. The capacitance of the monolayer electrical double layer (Cdl) is related to the effective thickness and order of the SAM on the gold substrate10 and can be calculated from the measured current density.
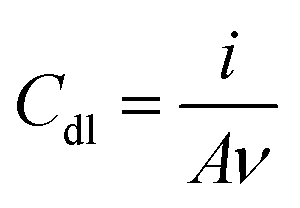
CV measurements (Fig. 3a) showed that the capacitance decreases upon SAM formation, with capacitances reduced from 11.6 μF cm−2 for bare gold to 7.67 μF cm−2 for 1 and 6.07 μF cm−2 for 2 (Table 1). All tested samples (1 and 2) showed a slightly enhanced resistance compared to bare gold samples. Overall, the relatively low resistance (Fig. S5†) of the SAMs of 1 and 2 and the concomitantly relatively high capacitance in the presence of redox couple (Fig. 3b) indicate a rather thin and open SAM structure, possibly with a high number of pinhole defects present in the monolayer. This electrochemically open structure is expected from the macrocyclic structure of these hosts and the absence of well-packing long alkyl chains in these molecules.
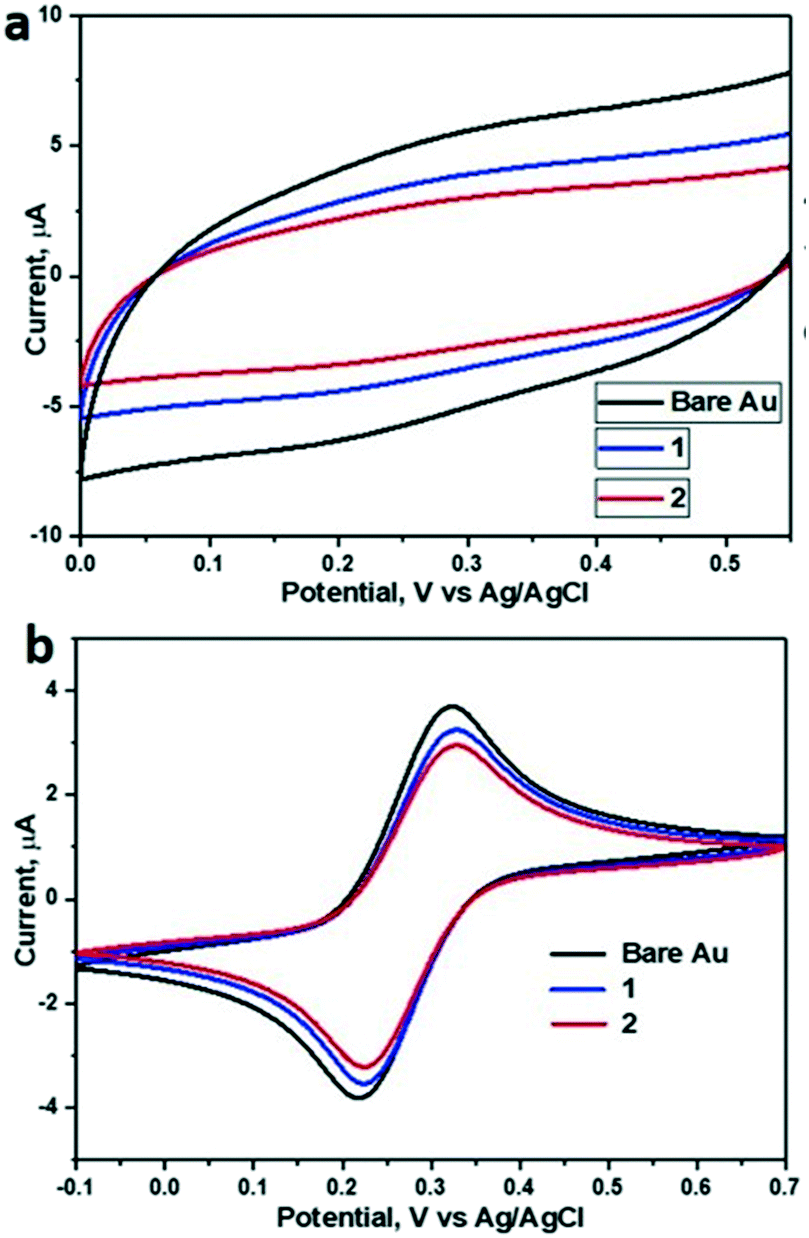 | ||
| Fig. 3 Cyclic voltammograms of the bare gold (black), 1 (blue) and 2 (red) SAM covered gold electrodes. Supporting electrolytes was (a) 0.5 M K2SO4 and (b) 0.5 M K2SO4 + 2 mM Fe(CN)63−/4–, scan rate 100 mV s−1. | ||
In line with earlier data obtained in solution,26,29 we attempted to use the SAMs as anion binders, and therefore we tried to probe the electrochemical performance at various concentrations of KBr solutions (Fig. S6†). Surprisingly, although semithio-BU[6] is a known strong bromide anion binder, addition of KBr solution (in the range of 1 mM to 30 mM) did not lead to detectable changes in the electrochemical parameters of the SAM of 2. Therefore, we assumed, also based on the XPS results which indicate involvement of most of the sulfur atoms in binding, that the attachment and assembly of 1 and 2 on gold was accompanied by large changes in their conformations relative to their conformational state in solution. Recently, Reany and co-workers reported that perthio-BUs exhibit a yet unknown compact conformation, which explains their diminished anion binding properties but still bind to thiophilic metals.31 Hence, we investigated the changes in the conformation of the current semithio-BUs on gold by scanning tunneling microscopy (STM) and molecular modeling.
Fig. 4a depicts an STM image of a SAM of 1 on Au(111). Several vacancy islands are present at the Au(111) surface. The vacancy islands have a depth of 2.5 Å, i.e., one Au atomic layer, indicating that these vacancy islands are vacancies in the Au surface and not in the semithio-bambusuril SAM. The formation of vacancy islands in the Au(111) surface upon the adsorption of S-containing molecules is a well-known phenomenon that relieves surface stress caused by the formation of the strong Au–S bonds.33,34
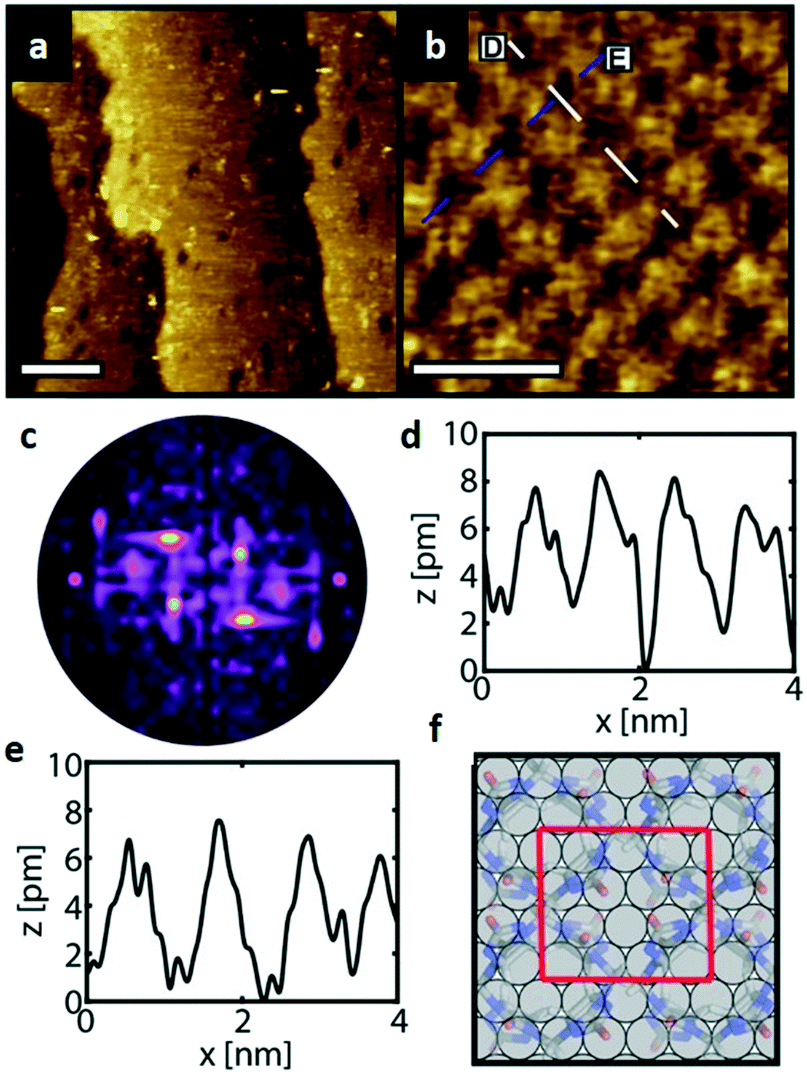 | ||
Fig. 4 STM images of a SAM of 1 on Au(111). (a) Large scale topographic image (100 × 100 nm2, scale bar is 20 nm), taken at 250 pA and 700 mV. (b) STM image of a rectangular ordered domain (5.2 × 5.2 nm2, scale bar 2 nm). (c) The corresponding FFT of (b) showing rectangular symmetry. (d and e) STM height profile of the white and blue lines in (b). (f) Proposed 4 × 2 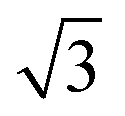 unit cell (red rectangle, 1.15 × 1 nm2) of 1 on the unreconstructed Au(111) surface. unit cell (red rectangle, 1.15 × 1 nm2) of 1 on the unreconstructed Au(111) surface. | ||
Fig. 4b shows a zoomed image of the ordered arrangement of the SAM of 1. A fast Fourier transform (FFT) (Fig. 4c) of the image displayed in Fig. 4b reveals a rectangular symmetry in line with the four-fold symmetry of the semithio-bambus[4]uril 1 (Fig. 1a, top). The two periodicities extracted from the apparent height profiles (Fig. 4d–e) correspond to 1 and 1.15 nm, respectively. Based on these findings we propose that the molecules self-assemble in a (4 × 2 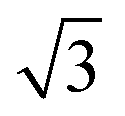 ) unit cell (red box in Fig. 4f) where (1 × 1) refers to the unreconstructed Au(111) surface with a nearest neighbor distance of 0.288 nm. The dark depressions are most likely the cavities present within the semithio-bambusuril monolayers,17,35 whereas the brighter areas are the sulfur bonding groups.36,37 From the XPS data (see Fig. 2 and Table 1) it is clear that the sulfur atoms in 1 form strong bonds with the Au(111) surface. Unfortunately, the resolution of the images is insufficient to draw any firm conclusions regarding the exact number of binding sites and geometry of the molecules.
) unit cell (red box in Fig. 4f) where (1 × 1) refers to the unreconstructed Au(111) surface with a nearest neighbor distance of 0.288 nm. The dark depressions are most likely the cavities present within the semithio-bambusuril monolayers,17,35 whereas the brighter areas are the sulfur bonding groups.36,37 From the XPS data (see Fig. 2 and Table 1) it is clear that the sulfur atoms in 1 form strong bonds with the Au(111) surface. Unfortunately, the resolution of the images is insufficient to draw any firm conclusions regarding the exact number of binding sites and geometry of the molecules.
Similarly, Fig. 5a shows an STM image of a SAM of 2 on Au(111). Again, vacancy islands with a depth of 2.5 Å were observed indicating that the islands are in the Au (111) layer, albeit more and larger vacancy islands were found as compared to 1. Fig. 5b shows a zoomed image of 2 monolayer. The FFT of the image displayed in Fig. 5b (Fig. 5c) reveals a hexagonal symmetry, which is in line with the six-fold symmetry of the semithio-BU[6], 2, molecule (Fig. 1a, bottom).The observed periodicity is 0.86 nm, which corresponds to the distance between the sulfur atoms of 2 and thus, the bright spots are most likely the sulfur atoms.36,38 This is also supported by the relatively larger fluctuations observed in the apparent height profiles (Fig. 5d and e) of the monolayer of 2 on gold in comparison to that of 1.
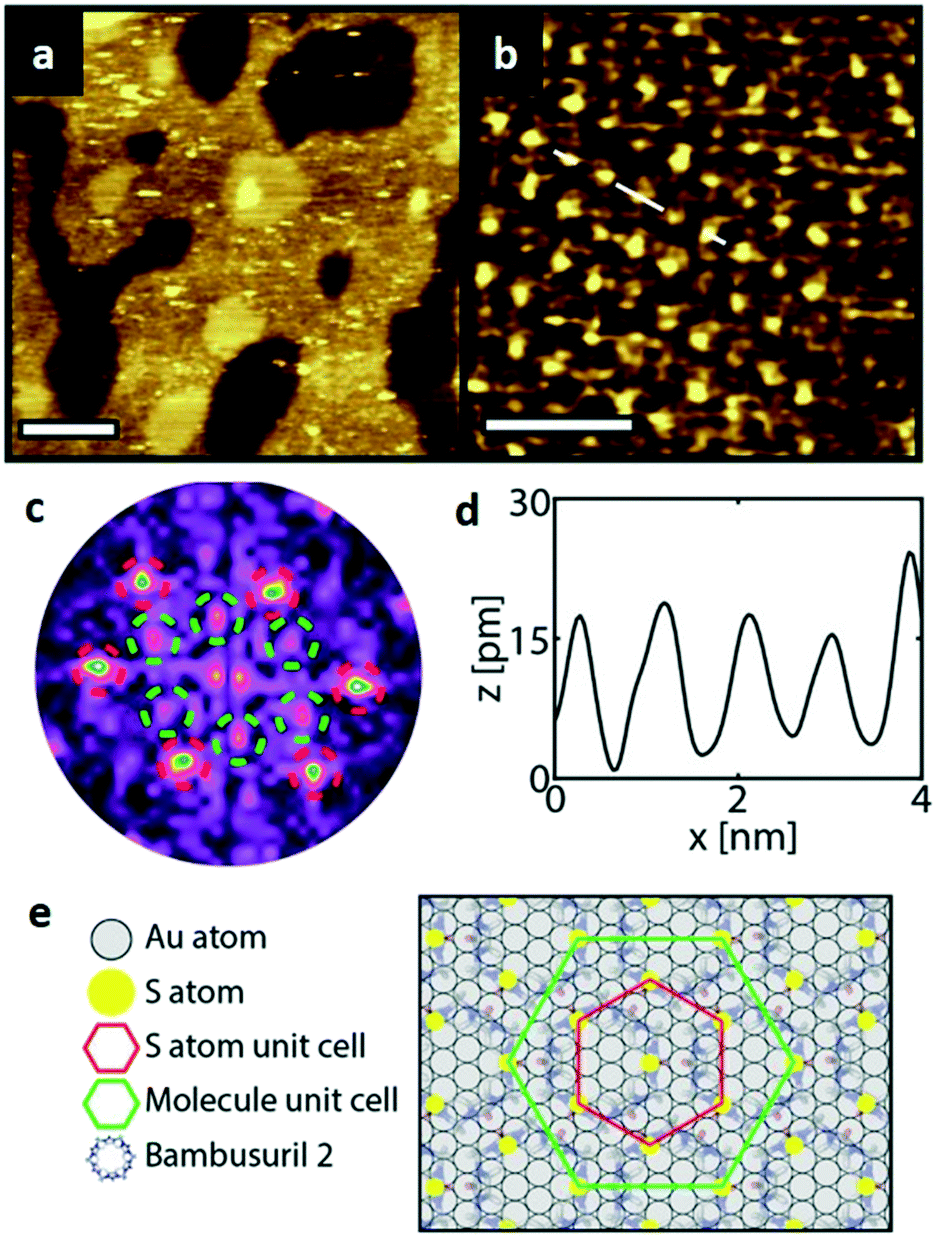 | ||
Fig. 5 STM images of a SAM of 2. (a) Large scale topographic image (100 × 100 nm2, scale bar is 20 nm), 190 pA and 600 mV. (b) STM image of a hexagonal ordered domain (9.2 × 9.2 nm2, scale bar 3 nm). (c) the corresponding FFT of (b) showing the same hexagonal symmetry with a 0.86 nm periodicity (red) and hexagonal symmetry with a periodicity of approximately 1.5 nm (green). (d) STM height profile of the white line in (b) confirming a 0.86 nm distance between the sulfur atoms. (e) Proposed (3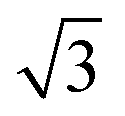 × 3 × 3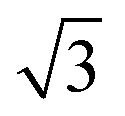 )R30° unit cell (green hexagon) of 2 on Au(111) with the (3 × 3) sulfur hexagon marked in red (0.86 × 0.86 nm2). )R30° unit cell (green hexagon) of 2 on Au(111) with the (3 × 3) sulfur hexagon marked in red (0.86 × 0.86 nm2). | ||
A closer look at the FFT in Fig. 5c reveals a second, but substantially weaker, (3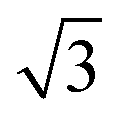 × 3
× 3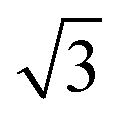 )R30° periodicity in a hexagonal symmetry (green circles). This larger periodicity is 30° rotated with respect to the sulfur (3 × 3) unit cell and corresponds to the arrangement of the semithio-BU[6], 2, molecules shown in Fig. 5e. The unit cell is a (3
)R30° periodicity in a hexagonal symmetry (green circles). This larger periodicity is 30° rotated with respect to the sulfur (3 × 3) unit cell and corresponds to the arrangement of the semithio-BU[6], 2, molecules shown in Fig. 5e. The unit cell is a (3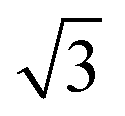 × 3
× 3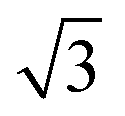 ) hexagon and contains 9 sulfur atoms and three molecules. Based on these findings we propose that all six thiocarbonyl sulfur atoms in the semithio-BU[6], 2, interact with the Au(111) surface. XPS results suggest that all 6 of the sulfur atoms bind to the gold (Table 1 and Fig. 2f) while MD simulations (vide infra, Fig. 8c) predict that 5 to 6 sulfur atoms interact with the surface. We propose that all six sulfur atoms point down towards the surface, however, three of the sulfur atoms make a direct and strong covalent bond with the gold surface, whereas the other three sulfur atoms interact differently with the gold surface, i.e. via a Au adatom.39 This type of bonding could be a result of an oxidized sulfur atom37 of high conformational strain, which leads to weakening of the Au–S bond.40 When Au adatom bonding occurs, the Au–Au contact is weakened, which makes the Au–S complex more mobile and leads to a reduced intensity in the STM images. However, more research is needed to find the exact origin of the different bonding configurations. The latter falls outside scope of our current work. To further resolve the atomic scale features of the exact binding modes we supplemented the STM data with MD computer simulations (Fig. 6).
) hexagon and contains 9 sulfur atoms and three molecules. Based on these findings we propose that all six thiocarbonyl sulfur atoms in the semithio-BU[6], 2, interact with the Au(111) surface. XPS results suggest that all 6 of the sulfur atoms bind to the gold (Table 1 and Fig. 2f) while MD simulations (vide infra, Fig. 8c) predict that 5 to 6 sulfur atoms interact with the surface. We propose that all six sulfur atoms point down towards the surface, however, three of the sulfur atoms make a direct and strong covalent bond with the gold surface, whereas the other three sulfur atoms interact differently with the gold surface, i.e. via a Au adatom.39 This type of bonding could be a result of an oxidized sulfur atom37 of high conformational strain, which leads to weakening of the Au–S bond.40 When Au adatom bonding occurs, the Au–Au contact is weakened, which makes the Au–S complex more mobile and leads to a reduced intensity in the STM images. However, more research is needed to find the exact origin of the different bonding configurations. The latter falls outside scope of our current work. To further resolve the atomic scale features of the exact binding modes we supplemented the STM data with MD computer simulations (Fig. 6).
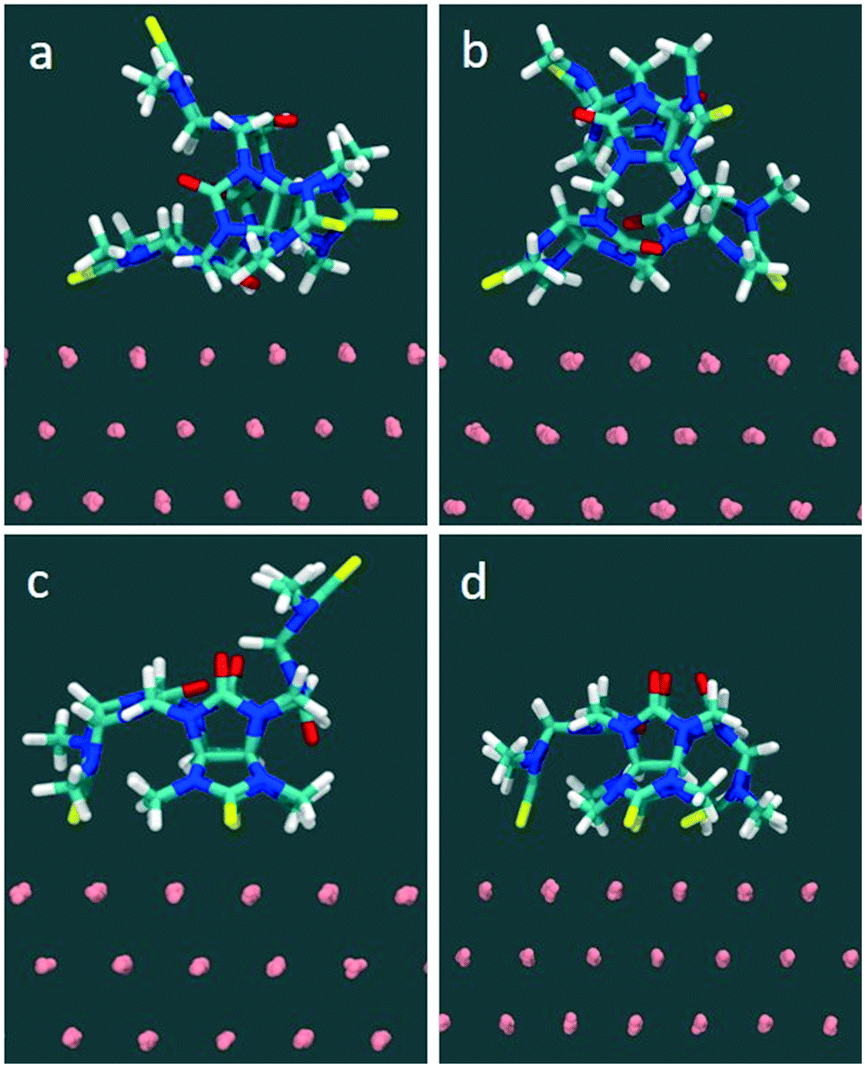 | ||
| Fig. 6 Molecule-surface adsorption modes of compound 1 generated from naturally occurring sulfur–gold interactions, (a) single-site binding complex (b) two-site binding complex, (c) three-site binding complex and (d) four-site binding complex. | ||
In the MD simulations (full details in Methods), compound 1 was initially placed in a DMSO solution exhibiting two major conformations. In one conformation, the glycoluril subunits are arranged in alternate conformation with a pair of thiocarbonyl groups pointing to one direction and another pair of thiocarbonyls groups pointing to the opposite direction (2/2). Another conformation includes three of the four thiocarbonyl groups pointing to one direction and the remaining thiocarbonyl group is flipped to the opposite direction (3/1) (see Fig. 6). In this fully solvated phase, the molecule did not spontaneously sample orientations with all four thiocarbonyl groups pointing in the same direction (4/0) during 100 nanoseconds of free, unconstrained molecular dynamics in DMSO, indicating that a putative binding mode with all sulfurs pointing down involves a significant penalty due to intramolecular conformational strain.
Molecule-surface binding modes (for one molecule adhering to a gold substrate) were generated from naturally occurring sulfur–gold interactions: for example, the monovalent complex was prepared from the solution conformation in which a sulfur spontaneously approached within bonding distance to a gold atom on the surface. The two-site binding complex was prepared from a suitably oriented single-site binding mode, and so on. The starting conformation and final conformations of 1 in water are shown in Fig. S7 and S8.†
Fig. 7 shows the calculated changes in energy for all four possibilities of binding states: single site via Au–S bond (1), and so on, compared to free 1 (0) with no Au–S bonds. All binding modes from mono- to tetravalent are stabilized relative to the unbound state (Fig. 7a), with increasing steric strain offset by stabilisation of −164 kJ mol−1 per additional Au–S bond.41 The conformational energy is quite similar for all binding modes up to trivalent (Fig. 7b) with the penalty for tetravalent due to the arched 4/0 conformation that does not occur spontaneously in simulations of 1 in solution. Small solvation penalties also come with each additional Au–S contact (Fig. 7c). The computed energies indicate that formation of the last Au–S bond creates significant strain in 1; the required high-energy conformation does not form spontaneously in the more loosely bound states. XPS (Table 1 and Fig. 2) showed that about 15% of the sulfur atoms are oxidized which corresponds to one sulfur atom per 1 molecule. The presence of an oxygen atom to bridge the fourth sulfur atom could potentially reduce the conformational strain by increasing the effective length of the molecule and reduce the steric penalty for formation of the fourth bond, as calculated previously for multivalent binding of PAMAM vs. PPI dendrimers.42
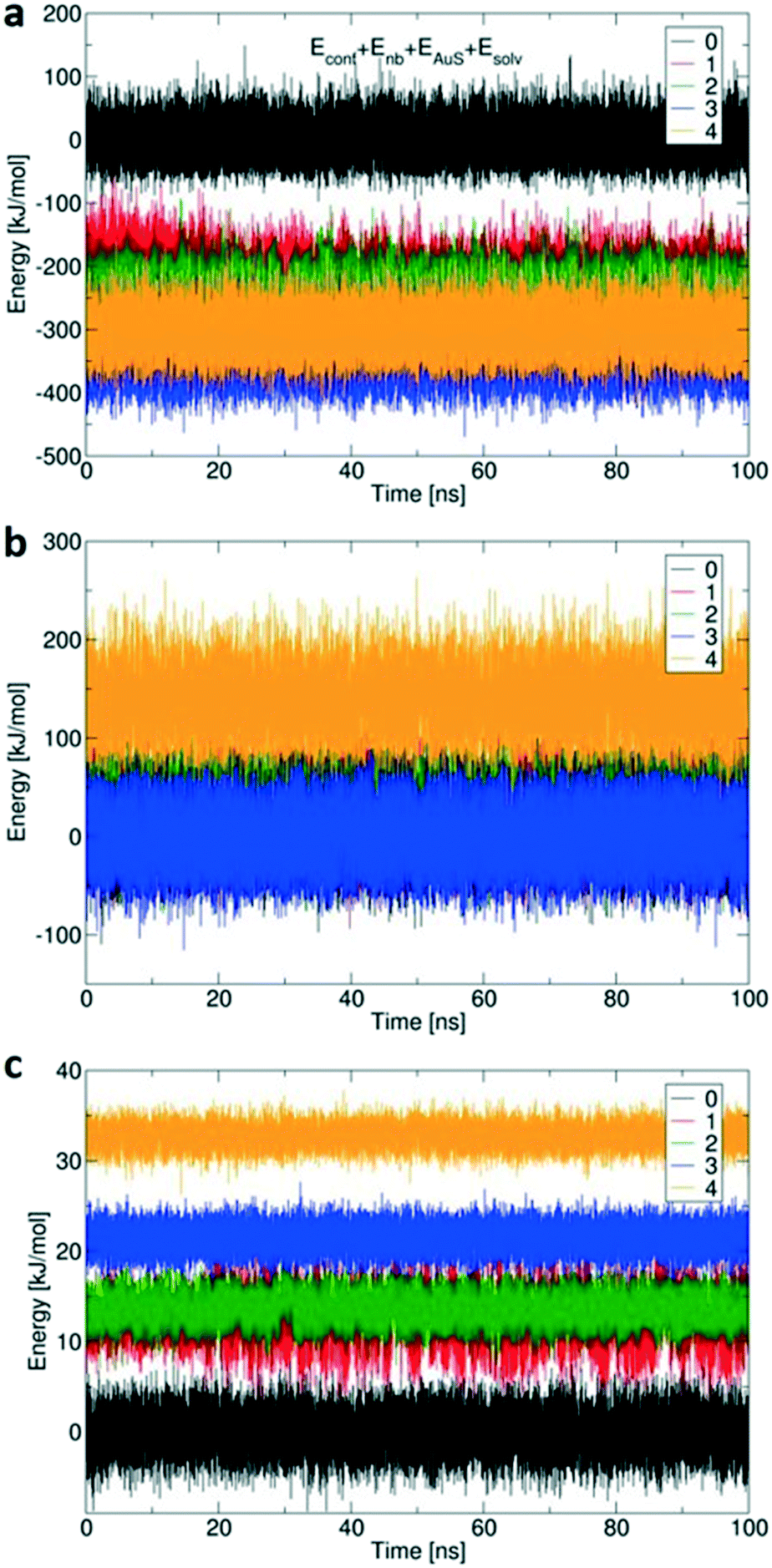 | ||
| Fig. 7 Calculated (a) total energy (summed over conformational energy Econf, nonbonded molecule-surface van der Waals interactions Enb, sulfur–gold covalent bonds EAuS, and solvation in DMSO Esolv), (b) conformational energy and (c) solvation energy terms, for all five states of 1: free with no Au–S bonding (0, black), with a single binding site of Au–S bond (1, red), with two binding sites (2, green), with three binding sites (3, blue) and all four binding sites (4, yellow). To aid comparison between binding modes, the energy of the unbound molecule is set to zero in each plot so energies reflect changes in conformational energy and in solvation energy when 1 binds to Au (111). | ||
Computed binding modes of compound 2 to Au(111) are given in Fig. 8. For the two three-binding modes, regardless of the exact order of thiocarbonyl binding pattern, we observe that, after 100 ns of simulation, the remaining unbound thiocarbonyl groups also point towards the gold surface. This observation clearly suggests that the flexibility of 2 allows larger conformational changes in comparison with 1. Regardless of the initial sulfur bonding pattern, 5 to 6 sulfur atoms were found to interact with the surface within 10 ns, reflecting the greater flexibility of 2 compared with 1 which reduces the steric constraints of the maximally bound conformation. Despite the fact that these calculations were performed for individual molecules, we do not expect SAM packing to have a large effect on the structures given the shortness of the anchoring groups.
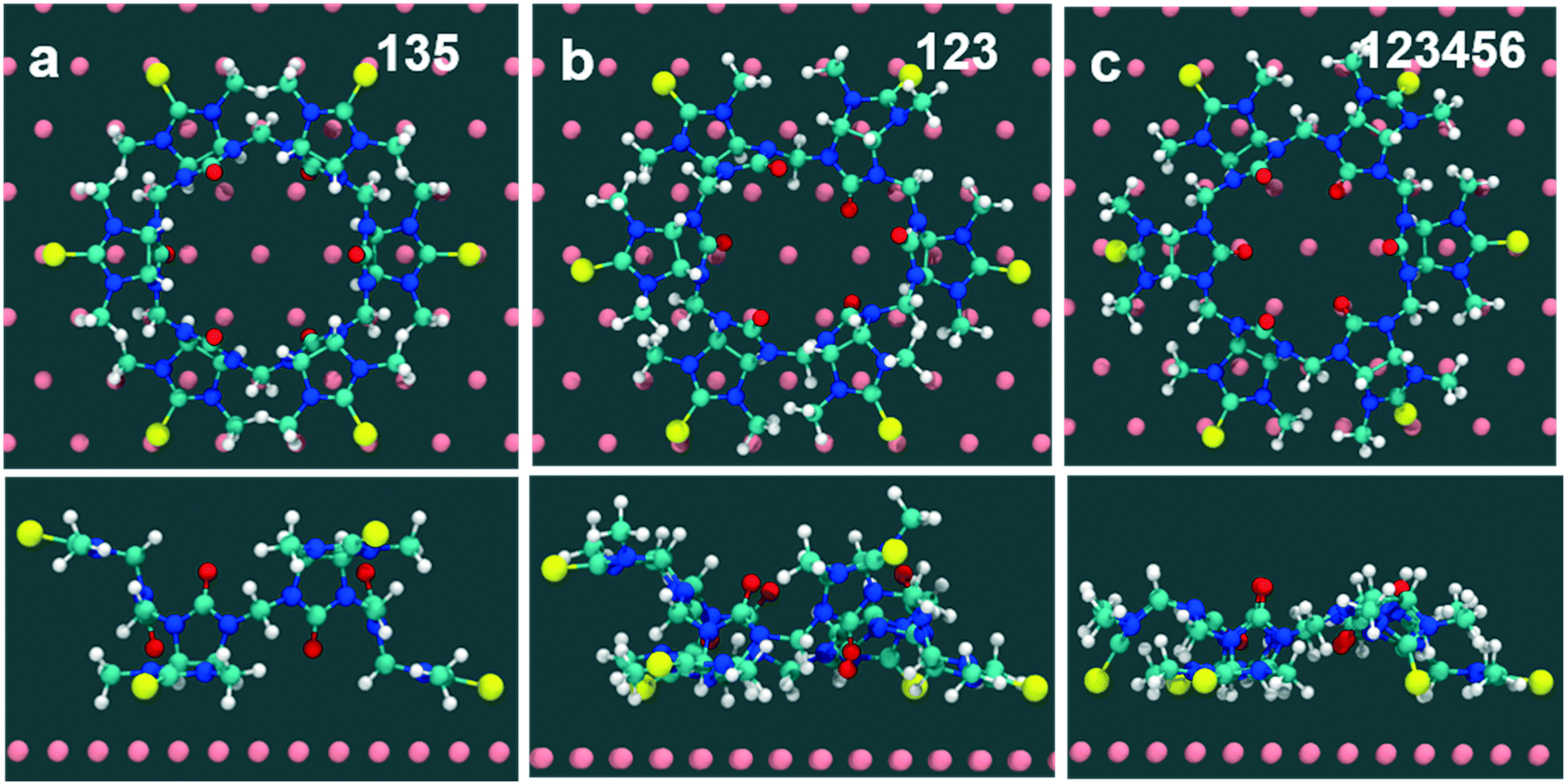 | ||
| Fig. 8 Molecule-surface adsorption complexes for compound 2 generated from naturally occurring sulfur–gold interactions, (a) with three alternate binding sites (135 mode) to the gold surface, (b) with three in-group binding sites (123 mode) to the gold surface, (c) with all six in-group binding sites (123456 mode) to the gold surface. | ||
Conclusions
The semithio-bambusurils 1 and 2 with 4 and 6 thiocarbonyl end groups, respectively, were used to prepare stable and ordered monolayers on gold. A range of physicochemical characterization techniques, combined with STM and MD simulations showed that the molecules undergo significant conformational rearrangements upon attachment to a gold surface. Moreover, these data suggest that the SAMs do not exhibit polymorphism. Apparently, these conformational changes reduce their anion-complexing ability. The molecules attach using most if not all of the S atoms, with the energetic benefit of Au–S binding compensating for any steric strain as the molecule is forced to adopt a more confined/pinned conformation on the surface. The energy balance can be tuned by using larger and more flexible macrocycles, here compound 2vs.1, by changing the anchoring group, and also by the possibility of oxidation of one of the S atoms in 1. In the future, SAMs of similar macrocycles, but with retained anion binding, could be computationally designed using larger-area molecular dynamics models and free energy calculations of anion discrimination. These SAM could be further engineered by use of electrodes on which S may bind more weakly, e.g. Pt or Cu,43 and by changing from thiocarbonyl to thioether attachment groups.44Experimental details
Materials
Semithio-bambusuril derivatives (1 and 2) were synthesized as described before.29 Dimethyl sulfoxide (DMSO, 99%, Sigma-Aldrich), potassium sulfate [(K2SO4), 99% Sigma-Aldrich], potassium ferricyanide [(K3Fe(CN)6), 99%, ACROS Organics], potassium ferrocyanide (K4Fe(CN)6), 99 +%, ACROS Organics]. All chemicals were analytical grade and were used without further purification.Preparation of SAMs
All glassware used to prepare monolayers was immersed in freshly prepared piranha solution (3:1 H2SO4: aq. H2O2) at 70 °C for 1 h. Warning: piranha solution should be handled with caution; it can detonate unexpectedly. Next, the glassware was rinsed with large amounts of high purity water (Millipore). Cleaned gold substrates were immersed with minimal delay into a 0.5 mM adsorbate (1 or 2) solution in 15 ml DMSO solution for overnight. The semithio-bambusurils monolayers were prepared at different temperatures (25 °C, 50 °C and 80 °C) overnight. Subsequently, the substrates were removed from the solution and rinsed with DMSO, and water to remove any physisorbed material. The 80 °C incubated samples were used for all measurements (CV, XPS and STM).
Monolayer characterization and instruments
Static contact angles were measured with Milli-Q water droplets on a Krüss G10 Contact Angle Measuring Instrument equipped with a CCD camera. Contact angle values were determined automatically by a drop shape analysis software. Contact angles were measured immediately after preparation of the SAMs (we prepared five samples of each one: bare Au, 1 and 2). Data for at least three drops were obtained and averaged.For IRRAS measurements, the cryostat was inserted into the optical path of a PEM module of a Nicolet FT-IR spectrometer, making sure that the incidence of the IR beam on the shroud windows was normal. The angle of incidence on the gold surface was 82° with a resolution of 4 cm−1, and 200 scans were collected.
XPS measurements were performed on a Quantera SXM (scanning XPS microprobe) from Physical Electronics, equipped with a monochromatic Al Kα X-ray source monochromatic at 1486.6 eV, producing approximately 50 W of X-ray power. Spectra were referenced to the main C 1s peak set at 284.0 eV. A surface area of 600 × 300 μm2 was scanned with an X-ray beam size about 200 μm (mappings are often done with beam sizes down to 9 μm).
All electrochemical measurements were performed on a CH instruments bipotentiostat 760D using a three-electrode setup using the SAM-covered gold plate as the working electrode (area = 0.43 cm2), a platinum disc as the counter electrode, and a Ag/AgCl reference electrode (Eo Ag/AgCl) 0.197 V vs. reversible hydrogen electrode (RHE) in aqueous 0.1 M K2SO4. Electrochemical impedance measurements were carried out from 1 × 106 to 0.1 Hz with amplitude of 10 mV at the formal potential of the measured species after a preconditioning step of 10 s.
STM measurements
STM imaging was conducted for SAMs using a Nanosurf Easy Scan 2 system under ambient conditions and at room temperature. All the images were acquired using a constant current mode using currents between 190 mA and 260 mA and sample bias between 450 mV and 800 mV to obtain high resolution images. All experiments were performed a mechanically cut tips (Pt/Ir (90:10)) (Nanosurf System). Au(111) was purchased commercially from Phasis, Switzerland. The substrates cleaned by a copious amount of Ethanol (>99%, Aldrich), 2-propanol (Merck, for analysis) and pure water.
Molecular dynamics simulations
Atomic resolution molecular dynamics simulations were performed using the Gromacs software45 and with the CHARMM/CGenFF force field.46 New parameters were derived by mapping quantum mechanical47 bonded and non-bonded parameters of 1 and 2 onto the CGenFF CHARMM dataset using ParamChem software. Au(111) is modeled as an infinite slab in the (x,y) plane. The slab is made of three layers (in the z direction) of fixed gold atoms. A box of DMSO of 3.5 × 3.5 × 3 nm is added on top of the slab. The B molecule is solvated in the DMSO phase. Periodic boundary conditions (PBC) are applied and MD simulations are performed in the NPT (constant pressure and constant temperature) ensemble, at a temperature of 300 K and a pressure of 1 bar, using a semi-isotropic coupling to the barostat. The cut-off for nonbonding interactions is set at 1.2 nm and electrostatics are calculated using the Particle Mesh Ewald summation method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gerard Kip is acknowledged for XPS measurements. P. P. K. is grateful to the Karnataka state government for providing a scholarship under the D Devraj Urs Videshi Vyasanga Vetana scheme. D. T. acknowledges support by Science Foundation Ireland (SFI) under awards number 15/CDA/3491 and 12/RC/2275 (SSPC), and acknowledges computing resources at the SFI/Higher Education Authority Irish Center for High-End Computing (ICHEC). The Open University of Israel (OUI) is gratefully acknowledged for financial support (OR).References
- H. Radecka, J. Radecki, I. Grabowska and K. Kurzątkowska, Electrochemical Sensors and Biosensors Based on Self-Assembled Monolayers: Application of Nanoparticles for Analytical Signals Amplification. In Functional Nanoparticles for Bioanalysis, in Functional Nanoparticles for Bioanalysis, Nanomedicine, and Bioelectronic Devices, 1, ACS Symp. Ser., 2012, 1112, 293–312 CrossRef CAS.
- S. Casalini, C. A. Bortolotti, F. Leonardi and F. Biscarini, Self-assembled monolayers in organic electronics, Chem. Soc. Rev., 2017, 46, 40–71 RSC.
- M. J. Shin, Y. J. Shin and J. S. Shin, Cholesterol recognition system by molecular imprinting on self-assembled monolayer, Colloids Surf., A, 2018, 559, 365–371 CrossRef CAS.
- M. Baghbanzadeh, F. C. Simeone, C. M. Bowers, K.-C. Liao, M. Thuo, M. Baghbanzadeh, M. S. Miller, T. B. Carmichael and G. M. Whitesides, Odd–Even Effects in Charge Transport across n-Alkanethiolate-Based SAMs, J. Am. Chem. Soc., 2014, 136, 16919–16925 CrossRef CAS PubMed.
- E. T. Mack, P. W. Snyder, R. Perez-Castillejos and G. M. Whitesides, Using Covalent Dimers of Human Carbonic Anhydrase II To Model Bivalency in Immunoglobulins, J. Am. Chem. Soc., 2011, 133, 11701–11715 CrossRef CAS PubMed.
- K. Sakakibara, J. P. Hill and K. Ariga, Thin-Film-Based Nanoarchitectures for Soft Matter: Controlled Assemblies into Two-Dimensional Worlds, Small, 2011, 7, 1288–1308 CrossRef CAS PubMed.
- D. Wasserberg, J. Cabanas-Danés, J. Prangsma, S. O'Mahony, P.-A. Cazade, E. Tromp, C. Blum, D. Thompson, J. Huskens, V. Subramaniam and P. Jonkheijm, Controlling Protein Surface Orientation by Strategic Placement of Oligo-Histidine Tags, ACS Nano, 2017, 11, 9068–9083 CrossRef CAS PubMed.
- K. S. Kumar, R. R. Pasula, S. Lim and C. A. Nijhuis, Long-Range Tunneling Processes across Ferritin-Based Junctions, Adv. Mater., 2016, 28, 1824–1830 CrossRef CAS PubMed.
- F. Ishiwari, G. Nascimbeni, E. Sauter, H. Tago, Y. Shoji, S. Fujii, M. Kiguchi, T. Tada, M. Zharnikov, E. Zojer and T. Fukushima, Triptycene Tripods for the Formation of Highly Uniform and Densely Packed Self-Assembled Monolayers with Controlled Molecular Orientation, J. Am. Chem. Soc., 2019, 141, 5995–6005 CrossRef CAS PubMed.
- M. W. J. Beulen, J. Bügler, M. R. de Jong, B. Lammerink, J. Huskens, H. Schönherr, G. J. Vancso, B. A. Boukamp, H. Wieder, A. Offenhäuser, W. Knoll, F. C. J. M. van Veggel and D. N. Reinhoudt, Host-Guest Interactions at Self-Assembled Monolayers of Cyclodextrins on Gold, Chem. – Eur. J., 2000, 6, 1176–1183 CrossRef CAS.
- J. J. Gooding and S. Ciampi, The molecular level modification of surfaces: from self-assembled monolayers to complex molecular assemblies, Chem. Soc. Rev., 2011, 40, 2704–2718 RSC.
- A. Perl, L. Kumprecht, T. Kraus, D. Armspach, D. Matt, D. N. Reinhoudt and J. Huskens, Self-Assembled Monolayers of α-Cyclodextrin Derivatives on Gold and Their Host−Guest Behavior, Langmuir, 2009, 25, 1534–1539 CrossRef CAS PubMed.
- D. Burshtain and D. Mandler, Studying the binding of Cd2+ by ω-mercaptoalkanoic acid self assembled monolayers by cyclic voltammetry and scanning electrochemical microscopy (SECM), J. Electroanal. Chem., 2005, 581, 310–319 CrossRef CAS.
- S. Flink, F. C. J. M. van Veggel and D. N. Reinhoudt, Sensor Functionalities in Self-Assembled Monolayers, Adv. Mater., 2000, 12, 1315–1328 CrossRef CAS.
- J. Movilli, A. Rozzi, R. Ricciardi, R. Corradini and J. Huskens, Control of Probe Density at DNA Biosensor Surfaces Using Poly(l-lysine) with Appended Reactive Groups, Bioconjugate Chem., 2018, 29, 4110–4118 CrossRef CAS PubMed.
- T. F. G. G. Cova, S. C. C. Nunes, A. J. M. Valente, T. M. V. D. Pinho e Melo and A. A. C. C. Pais, Properties and patterns in anion-receptors: A closer look at bambusurils, J. Mol. Liq., 2017, 242, 640–652 CrossRef CAS.
- G. Crini, Review: A History of Cyclodextrins, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- C. D. Gutsche, Calixarenes Revisited, Monographs in Supramolecular Chemistry, The Royal Society of Chemistry, Cambridge, 1998 Search PubMed.
- (a) K. I. Assaf and W. M. Nau, Cucurbiturils: from synthesis to high-affinity binding and catalysis, Chem. Soc. Rev., 2015, 44, 394–418 RSC; (b) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Cucurbituril-based molecular recognition, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed; (c) W. Liu, S. K. Samanta, B. D. Smith and L. Isaacs, Synthetic mimics of biotin/(strept) avidin, Chem. Soc. Rev., 2017, 46, 2391–2403 RSC; (d) O. Reany, A. Li, M. Yefet, M. K. Gilson and E. Keinan, Attractive interactions between heteroallenes and the cucurbituril portal, J. Am. Chem. Soc., 2017, 139, 8138–8145 CrossRef CAS PubMed; (e) R. Aav, S. Kaabel and M. Fomitsenko, Cucurbiturils: synthesis, structure, formation mechanism and nomenclature, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, vol. 3, pp. 203–220 Search PubMed.
- (a) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Pillar-Shaped Macrocyclic Hosts Pillar[n]arenes: New Key Players for Supramolecular Chemistry, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed; (b) J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, The aqueous supramolecular chemistry of cucurbit[n]urils, pillar[n]arenes and deep-cavity cavitands, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC.
- L. Jasikova, M. Rodrigues, J. Lapesova, T. Lizal, V. Sindelar and J. Roithova, Bambusurils as a mechanistic tool for probing anion effects, Faraday Discuss., 2019, 220, 58–70 RSC.
- O. Reany, A. Mohite and E. Keinan, Hetero-Bambusurils, Isr. J. Chem., 2018, 58, 449–460 CrossRef CAS.
- R. Pinalli, A. Pedrini and E. Dalcanale, Biochemical sensing with macrocyclic receptors, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC.
- J. Rivollier, P. Thuery and M.-P. Heck, Extension of the Bambus[n]uril Family: Microwave Synthesis and Reactivity of Allylbambus[n]urils, Org. Lett., 2013, 15, 480–483 CrossRef CAS PubMed.
- T. Lizal and V. Sindelar, Bambusuril Anion Receptors, Isr. J. Chem., 2018, 58, 326–333 CrossRef CAS.
- E. Solel, M. Singh, O. Reany and E. Keinan, Enhanced anion binding by heteroatom replacement in bambusurils, Phys. Chem. Chem. Phys., 2016, 18, 13180–13185 RSC.
- C. Lang, A. Mohite, X. Deng, F. Yang, Z. Dong, J. Xu, J. Liu, E. Keinan and O. Reany, Semithiobambus[6]uril is a transmembrane anion transporter, Chem. Commun., 2017, 53, 7557–7560 RSC.
- M. Singh, E. Solel, E. Keinan and O. Reany, Aza-Bambusurils En Route to Anion Transporters, Chem. – Eur. J., 2016, 22, 8848–8854 CrossRef CAS PubMed.
- M. Singh, E. Solel, E. Keinan and O. Reany, Dual functional semithio-Bambusurils, Chem. – Eur. J., 2015, 21, 536–540 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Quantifying thiol–gold interactions towards the efficient strength control, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- P. Mondal, E. Solel, N. Fridman, E. Keinan and O. Reany, Intramolecular van der Waals Interactions Challenge Anion Binding in perthio-Bambusurils, Chem. – Eur. J., 2019, 25, 13336–13343 CrossRef CAS PubMed.
- C. Cannes, F. Kanoufi and A. J. Bard, Cyclic voltammetry and scanning electrochemical microscopy of ferrocenemethanol at monolayer and bilayer-modified gold electrodes, J. Electroanal. Chem., 2003, 547, 83–91 CrossRef CAS.
- Z. Peng and S. Dong, Formation of a Self-Assembled Monolayer of 2-Mercapto-3-n-octylthiophene on Gold, Langmuir, 2001, 17, 4904–4909 CrossRef CAS.
- M. M. Biener, J. Biener and C. M. Friend, Sulfur-induced mobilization of Au surface atoms on Au(111) studied by real-time STM, Surf. Sci., 2007, 601, 1659–1667 CrossRef CAS.
- M. Sundararajan, R. V. Solomon, S. K. Ghosh and P. Venuvanalingam, Elucidating the structures and binding of halide ions bound to cucurbit[6]uril, hemi-cucurbit[6]uril and bambus[6]uril using DFT calculations, RSC Adv., 2011, 1, 1333–1341 RSC.
- K. Sotthewes, H. Wu, A. Kumar, G. J. Vancso, P. M. Schön and H. J. W. Zandvliet, Molecular Dynamics and Energy Landscape of Decanethiolates in Self-Assembled Monolayers on Au(111) Studied by Scanning Tunneling Microscopy, Langmuir, 2013, 29, 3662–3667 CrossRef CAS PubMed.
- K. Sotthewes, Ö. Kap, H. Wu, D. Thompson, J. Huskens and H. J. W. Zandvliet, Ordering of Air-Oxidized Decanethiols on Au(111), J. Phys. Chem. C, 2018, 122, 8430–8436 CrossRef CAS PubMed.
- S. Kurokawa, Y. Miyawaki and A. Sakai, Scanning Tunneling Microscopy Observation of Sulfur Adsorbates on Au(111) at Liquid Nitrogen Temperature, Jpn. J. Appl. Phys., 2009, 48, 08JB12 Search PubMed.
- J. Gao, F. Li, G. Zhu, Z. Yang, H. Lu, H. Lin, Q. Li, Y. Li, M. Pan and Q. Guo, Spontaneous Breaking and Remaking of the RS–Au–SR Staple in Self-assembled Ethylthiolate/Au(111) Interface, J. Phys. Chem. C, 2018, 122, 19473–19480 CrossRef CAS.
- J. A. Rodriguez, J. Dvorak, T. Jirsak, G. Liu, J. Hrbek, Y. Aray and C. González, Coverage Effects and the Nature of the Metal−Sulfur Bond in S/Au(111): High-Resolution Photoemission and Density-Functional Studies, J. Am. Chem. Soc., 2003, 125, 276–285 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- D. Thompson, Free Energy Balance Predicates Dendrimer Binding Multivalency at Molecular Printboards, Langmuir, 2007, 23, 8441–8451 CrossRef CAS PubMed.
- A. H. Pakiari and Z. Jamshidi, Nature and Strength of M−S Bonds (M = Au, Ag, and Cu) in Binary Alloy Gold Clusters, J. Phys. Chem. A, 2010, 114, 9212–9221 CrossRef CAS PubMed.
- D. Thompson, J. P. Hermes, A. J. Quinn and M. Mayor, Scanning the Potential Energy Surface for Synthesis of Dendrimer-Wrapped Gold Clusters: Design Rules for True Single-Molecule Nanostructures, ACS Nano, 2012, 6, 3007–3017 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, vol. 1–2, pp. 19–25 Search PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell Jr, CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J. Comput. Chem., 2010, 31, 671–690 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00149b |
| This journal is © The Royal Society of Chemistry 2020 |