
Interface engineering in transition metal carbides for electrocatalytic hydrogen generation and nitrogen fixation
Min
Kuang†
a,
Wenjing
Huang†
a,
Chidanand
Hegde
b,
Wei
Fang
 a,
Xianyi
Tan
a,
Chuntai
Liu
c,
Jianming
Ma
*cd and
Qingyu
Yan
*a
a,
Xianyi
Tan
a,
Chuntai
Liu
c,
Jianming
Ma
*cd and
Qingyu
Yan
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: alexyan@ntu.edu.sg
bSingapore Centre for 3D Printing, Department of Mechanical and Aerospace Engineering, Nanyang Technological University, Singapore 639798, Singapore
cKey Laboratory of Materials Processing and Mold, Ministry of Education, Zhengzhou University, Zhengzhou 450002, China
dSchool of Physics and Electronics, Hunan University, Changsha 138634, China. E-mail: nanoelechem@hnu.edu.cn
First published on 17th September 2019
Abstract
With an increasing energy consumption rate and rising global population, constructing sustainable energy technologies has become one of the major scientific challenges. Therefore, the development of electrocatalytic conversion technologies that can convert renewable resources, such as water and nitrogen, into value-added chemicals or fuels (e.g., hydrogen and ammonia) can be crucial. A number of transition metal carbides (TMCs) have been investigated over the past few years as effective electrocatalysts for various reactions. This is mainly owing to their unique electronic structures, which leads to high electrical conductivity and chemical stability. Moreover, the reactivity of TMC-based electrocatalysts is highly dependent on their surface and interfacial properties. This review focuses on tuning nanostructures and interfaces to enhance the electrocatalytic activity of TMC-based materials for hydrogen production and nitrogen fixation. The mechanisms behind the surface and interface engineering are discussed, including the synergy effects, facet binding energy, active defects, and low-coordinated sites. In particular, studies on activity enhancement through design of the interfacial phase, composition, and structure in TMC-based electrocatalysts are highlighted. The effective tuning strategies might pave the way for future development of highly active TMC-based electrocatalysts for sustainable energy-related conversion.
 Min Kuang | Min Kuang received her PhD degree (2018) in the Laboratory of Advanced Materials at Fudan University in China, under the supervision of Prof. Gengfeng Zheng. She is currently a postdoctoral fellow in School of Materials Science and Engineering at Nanyang Technological University in Singapore, under the supervision of Prof. Qingyu Yan. Her research focuses on the development of hybrid earth-abundant nanostructured electrocatalysts for clean and renewable energy-related reactions, including water splitting, fuel cells, electrochemical CO2 reduction and nitrogen fixation. |
 Wenjing Huang | Wenjing Huang received her PhD Chemistry from Soochow University in 2018. Now she works as a research fellow under the supervision of Prof. Qingyu Yan in MSE at Nanyang Technological University. Her current research focuses on application of nanostructured materials in electrocatalysis. |
 Jianming Ma | Jianmin Ma is an Associate Professor in Hunan University, China. He received his BSc degree in Chemistry from Shanxi Normal University in 2003 and his PhD degree in Materials Physics and Chemistry from Nankai University in 2011. During 2011–2015, he also conducted research in several overseas universities as a postdoctoral research associate. His research interests are focused on energy storage and conversion, green catalytic technologies, and theoretic calculations. |
 Qingyu Yan | Qingyu Yan is currently a professor in School of Materials Science and Engineering in Nanyang Technology University. He obtained his BS in Materials Science and Engineering, Nanjing University. He finished his PhD from Materials Science and Engineering Department of State University of New York at Stony Brook. After that, He joined the Materials Science and Engineering Department of Rensselaer Polytechnic Institute as a postdoctoral research associate. He joined School of Materials Science and Engineering of Nanyang Technological University as an assistant professor in early 2008 and became a Professor in 2018. |
1. Introduction
Electrochemical conversion of renewable resources to valuable chemicals and fuels is regarded as one of the sustainable strategies to meet the rising global energy demands in an environmentally benign approach.1–4 Hydrogen (H2) is one of the prime clean energy carriers and ammonia (NH3) is drawing increasing attention as a promising chemical intermediate.5,6 While hydrogen can be produced by the electrolysis of water, electrochemical nitrogen fixation is a promising route to ammonia synthesis.7,8 However, the electrochemical generation of H2 and NH3 using renewable energy resources (solar, hydropower or wind energy) is still not in industrial practice, owing in part to the high cost of these electrochemically generated products relative to their conventionally produced counterparts.9,10 Most importantly, these electrocatalytic reactions highly dependent on the development of new electrocatalysts with excellent reactivity and stability.11–14 Furthermore, the majority of the active and stable catalysts for these processes are well known to be precious metals, whose low-abundance and high cost limit their large scale industrial application.15 Therefore, this presents an urgent demand for scalable synthesis of earth-abundant electrocatalytic materials that are highly active and stable.Transition metal carbides (TMCs), with structures consisting of carbon (C) atoms occupying the interstitial sites of their parent metals, have sparked increasing attention as efficient electrocatalysts toward several electrochemical reactions, such as the hydrogen evolution reaction (HER),16 oxygen evolution reaction (OER),17 nitrogen reduction reaction (NRR),18 and oxygen reduction reaction (ORR).19 TMC-based electrocatalysts, as a class of special “interstitial alloys”, are interesting because of their advantageous physical and chemical properties, including their precious-metal-like electronic structures, high electrical conductivities, excellent stabilities, and good corrosion resistance.20 The precious-metal-like electronic structures render TMCs effective to adsorb and activate hydrogen, thus exhibiting intrinsic activities like Pt-based electrocatalysts.21 In addition, most transition metals, with the Pt-group metals as exceptional examples, are intensively capable of forming TMCs with significant variations in their crystal structures. Fig. 1a exhibits the Group 4–10 metals and their stable carbides.22 As we can see from Fig. 1a, the transition metals on the left and center can alloy with the C atoms to form carbides generally with the formulas of MC and M2C respectively. On the other hand, for the transition metals on the right, metal-rich stoichiometries of M3C and M4C are more prevalent. It is worth noting that the bonding arises from the interaction of 2s and 2p orbitals of the C atoms with the d orbital of the transition metals. Thus, with rising sp electrons, the parent metal structure progressively transitions from body-centered cubic (bcc) to hexagonal close-packed (hcp) to face-centered cubic (fcc) (Fig. 1b).23,24 Likewise, elements in Group 4 and 5 prefer the bcc structure owing to their partially-filled bands capable of accommodating large amounts of sp-C orbitals, while Group 6 elements adopt the hcp structure and other elements in Groups 7 and 8 favor the fcc structure.23,25 Moreover, it has been demonstrated that the broadening of the parent metal d-band, due to the introduction of C atoms, would raise the density of states (DOS) at the Fermi level, hence resulting in high electrocatalytic activities.26
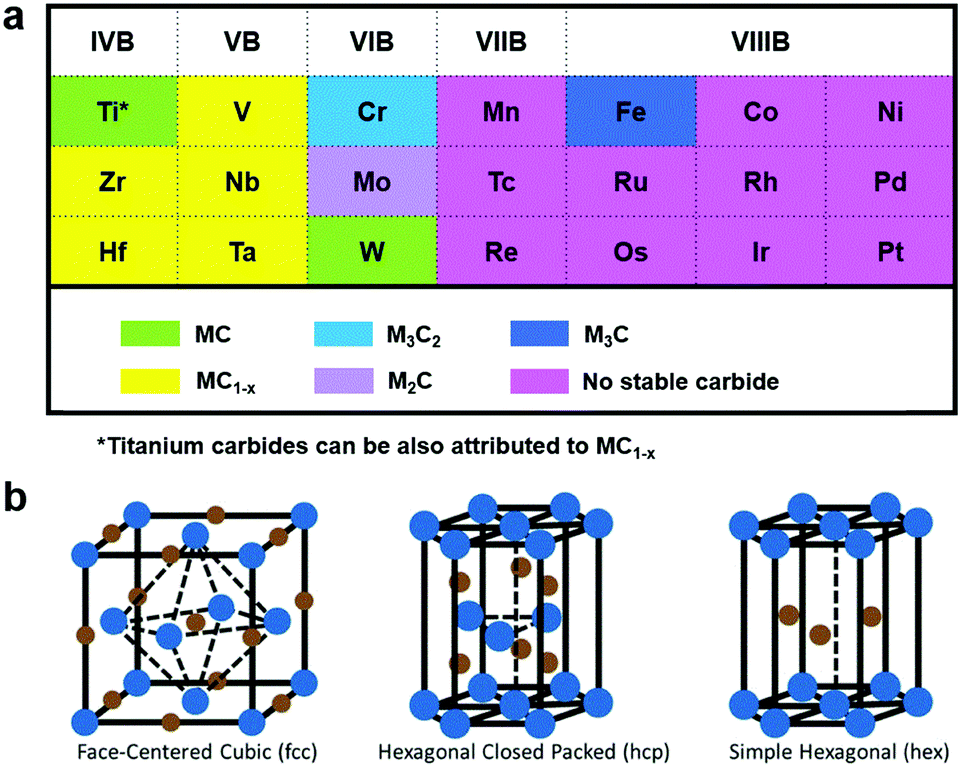 | ||
| Fig. 1 (a) Typical transition metals in the TMC compounds. Reproduced with permission from ref. 22. Copyright 2013, Elsevier Ltd. (b) Common crystal structure in TMC compounds. The blue points represent the metal atoms and the brown points represent the carbon atoms. Reproduced with permission from ref. 23. Copyright 2016, John Wiley and Sons, Inc. | ||
Unfortunately, many of the reported TMCs suffer from limited specific surface areas, which hinders their widespread applications in the field of electrocatalysis.27 Furthermore, during the carbide synthesis process, the generation of surface contaminants blocks the active sites and cavities, resulting in the degradation of electrocatalytic activities.28 In general, carbides are typically prepared by solid–solid or gas–solid reactions to achieve high surface area, namely, direct pyrolysis of metal carbonyl compounds, or reaction of metal/metal oxides with a C source.22 Consequently, the aggregation and/or excessive growth of TMCs during pyrolysis at relatively high temperatures leads to a reduction in the active sites and electrocatalytic activities for electrochemical reactions.29 Up until now, significant efforts have been dedicated to enhancing electrocatalytic activities via the engineering of structures and interfaces, including nanostructuring,30 heteroatom doping,31 morphology control,32 and introduction of various carbon-based materials.33,34 Furthermore, the problem of char contamination has also been studied.35–37 One of the effective solutions is the strategy of carburizing the metal precursor using compounds such as g-C3N4,38 carbon nanotubes (CNT),39,40 graphene,16,41–43 and other inorganic carbon sources.44 Compared with their organic counterparts (like aniline, dopamine, and melamine), the diffusion speed of C atoms away from the inorganic compounds is slower, thereby avoiding the char contamination.45 More specially, oxygen (O2) plasma treatment is also demonstrated to be an effective method to eliminate the surface contamination with minimal effect on the carbide structure.46 Based on the discussion above, rational design and controllable preparation of TMCs with the utmost exposure of active sites is very important to realize efficient electrochemical reactions.
The Pt-like electrocatalytic behavior of TMCs can be traced back to the pioneering investigations by R. B. Levy and M. Boudart in 1973.47 They reported that carbon sp electrons enhanced the apparent electron to atom ratio, thereby forming a more Pt-like electronic configuration. A series of TMC-based electrocatalysts were subsequently reported, such as molybdenum carbides, tungsten carbides, titanium carbides, etc., which exhibited high activity in various electrocatalytic technologies.48,49 With the development of advanced characterization techniques, novel preparation strategies and theoretical methods, the mechanisms behind the electrocatalytic activities of TMCs have been widely explored. For instance, Jingguang Chen and co-workers48 reported that the d-band center of both Mo- and C-terminated β-Mo2C surfaces could be broadened via the hybridization between d-orbitals of Mo and s/p-orbitals of C, thereby resulting in the enhancement of electrocatalytic activities toward the HER. Shuit Tong Lee and co-workers45 reported the preparation of phase-pure and ultrasmall W2C nanoparticles via an improved carburization strategy. Kian Ping Loh and co-workers50 reported the synthesis of edge segregated Mo2C electrocatalysts consisting of conventional AA-stacked T-phase Mo2C and Bernal-stacked (AB-stacked) Mo2C crystals using the diffusion-mediated method. The density functional theory (DFT)-based calculations demonstrated that the AB-stacked Mo2C crystals possessed a d-band closer to the Fermi energy compared to the AA-stacked Mo2C crystals, resulting in the improvement of electrocatalytic activities toward the HER. R. R. Adzic and co-workers40 demonstrated that the inlaid structure coupled with the electronic modification could facilitate the electrocatalytic activity of carbides, as exemplified by Mo2C and CNT (covalent binding). The X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) results suggested that this unique anchored structure induced a charge-transfer from Mo to C, which could further downshift the d-band center of Mo, and thus formed a moderate hydrogen binding energy. Most recently, by utilizing first-principles DFT, Q. Jiang et al.51 investigated cobweb-like MoC6 electrocatalysts and they claimed that the d orbitals of Mo atoms could be strongly hybridized with the orbitals of graphyne, leading to strong interactions between the Mo atom and graphyne. The as-prepared MoC6 exhibited high activity and selectivity toward the NRR at low potential and ambient conditions, which was mainly ascribed to the selective stabilization of N2H* species and destabilization of the NH2* species.
Research on TMC-based materials is an emerging field. Further developments can expand the widespread application of TMCs in electrochemical technologies (involving hydrogen generation and nitrogen fixation) to potentially substitute the noble-metal catalysts. In addition, TMCs can be surface decorated with active materials or chemically doped with metal/non-metal atoms or heterostructures to direct the desired electrocatalysis of energy-related reactions, and such surface/interface modifications can be used to tune multiple electrocatalytic steps. In this review, we summarize recent advances in hydrogen generation and nitrogen fixation via the HER and NRR using TMC-based materials. Emphasis is given to discussions on the relationship between surface/interface engineering and electrocatalytic activity, and mechanisms for the increasing electrocatalytic performance. The important strategies of surface/interface engineering such as control of nanocrystal shape, size, crystallographic orientation, doping, chemical state, support material, and composition are described. Finally, an outlook of future prospects for hydrogen generation and nitrogen fixation by TMC-based electrocatalysts is described.
2. Fundamentals of electrochemical HER and NRR
2.1 HER
The HER is the cathodic half reaction of a water electrolyzer, which involves distinct mechanisms for acidic and basic media.52 The reaction proceeds through three possible sub-reaction steps (as illustrated by Table 1).5 For instance, in acidic media, the first step is the hydrogen adsorption step, namely, the as-called Volmer reaction. In this step, an electron combines with a proton to generate an adsorbed H atom (Hads) intermediate on the surface of the electrode. After the formation of Hads, the HER can either proceed by the Heyrovsky reaction or Tafel reaction or both (Fig. 2).53 From the perspective of HER mechanism, the generation of Hads and its binding energy play an essential role in the process of HER. Therefore, the free energy of hydrogen adsorption (ΔGH) is extensively used as an activity descriptor to rationally design active HER electrocatalysts. If the ΔGH is negative, the intermediate has a strong bonding with the surface of the electrode, favoring the Volmer reaction, while suppressing the Heyrovsky or Tafel reaction; in contrast, if the ΔGH is positive, the intermediate is bound weakly to the surface of the electrode, leading to a slower Volmer reaction that hinders the overall turnover rate.| Reactions | Reaction steps | Equations | Tafel slope (mV dec−1) |
|---|---|---|---|
| Acidic solution: 2H+ + 2e− → H2 | Volmer | H+ + e− → H* | b = 2.3RT/αF = 120 |
| Heyrovsky | H* + H+ + e− ↔ H2 | b = 2.3RT/2F = 30 | |
| Tafel | H* + H* ↔ H2 | b = 2.3RT/(1 + α)F = 40 | |
| Alkaline/neutral solution: 2H2O + 2e− → H2 + 2OH− | Volmer | H2O + e− ↔ H* + OH− | |
| Heyrovsky | H2O + e− ↔ H2 + OH− | ||
| Tafel | H* + H* ↔ H2 |
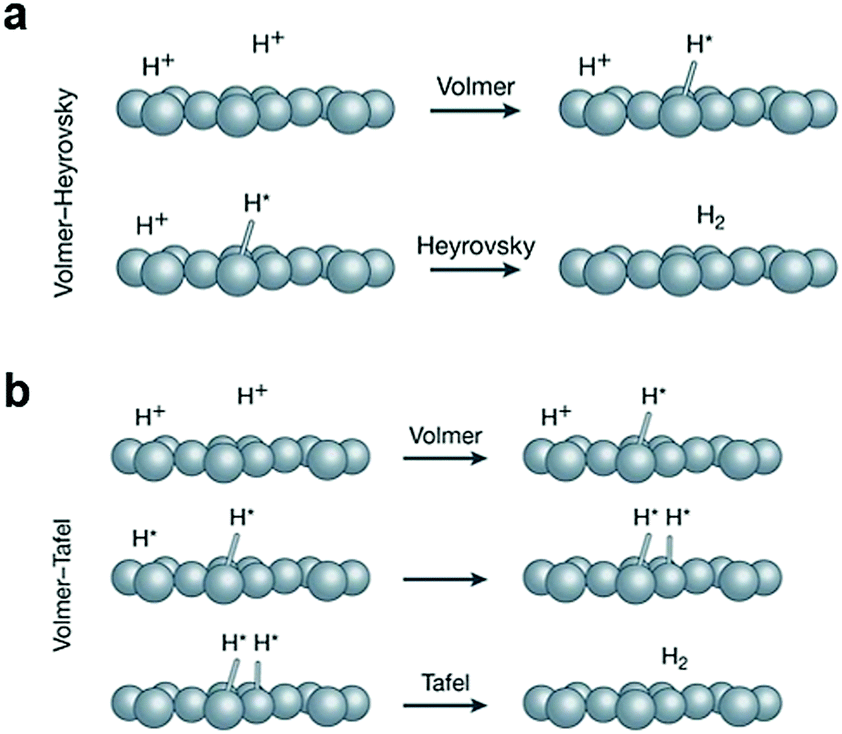 | ||
| Fig. 2 (a) The Volmer–Heyrovsky and (b) the Volmer–Tafel mechanism of the HER. Reproduced with permission from ref. 53. Copyright 2018, Nature Publishing Group. | ||
To evaluate the electrocatalytic activity of target HER electrocatalysts, several crucial parameters such as the total electrode activity, Tafel slope, exchange current density (j0), faradaic, efficiency (FE), stability and turnover frequency (TOF) are essential to be measured/calculated carefully.52,54,55 In general, linear sweep voltammetry (LSV) is performed to estimate the total electrode activity, in which low sweep rates (e.g., 2 or 5 mV s−1) are commonly applied to minimize the non-faradaic capacitive current. Based on the LSV curves, two important parameters for the HER can be evaluated; namely, the onset potential and the overpotential (η). Onset overpotential is the excess potential at which the current density starts to enhance significantly. Furthermore, the overpotential (η), which is the excess potential to reach the current density of 10 mA cm−2 is generally regarded as the benchmark to compare the HER activity of as-prepared electrocatalysts, corresponding to photoelectrochemical hydrogen generation efficiency of 12.3%.56–58 In addition, the Tafel slope and j0 are two essential parameters which can be determined from the Tafel equation (η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) log
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j), where j is the current density and b is the Tafel slope. Tafel slope is commonly correlated with the electrocatalytic mechanism of the HER (Table 1) and j0 reflects the rate of electron transfer under equilibrium conditions. A promising electrocatalyst should offer a low Tafel slope and high j0 simultaneously during the HER process. FE, also called coulombic efficiency, is the ratio of the experimental to the theoretical H2 generation. Thereinto, the theoretically produced H2 can be calculated by estimating the total charge transfer from potentiostatic or galvanostatic electrolysis plots and the experimental H2 production can be measured by gas chromatography (GC). In general, the FE is less than 100% owing to the existence of side reactions in the HER process. Furthermore, in order to realize the largescale application of the catalysts, the long-term stability is another important parameter, which can be measured by two methods: CV and galvanostatic/potentiostatic electrolysis. Finally, the TOF, which is a measure of the intrinsic activity of each electrocatalytic site, is defined as the number of reactants that an electrocatalyst can convert to a desired product at each electrocatalytic site per unit time. But, it is still a challenge to preciously measure the TOF due to the heterogeneous catalytic reaction often occurring on the electrode surface.52 Despite the fact that the measured TOF is relatively imprecise, it may still give insights into the comparative catalytic activity or efficiency of two or more materials if carefully executed. In conclusion, promising HER electrocatalysts should possess a low overpotential, small Tafel slope, high j0, large TOF, and long-term stability.
j), where j is the current density and b is the Tafel slope. Tafel slope is commonly correlated with the electrocatalytic mechanism of the HER (Table 1) and j0 reflects the rate of electron transfer under equilibrium conditions. A promising electrocatalyst should offer a low Tafel slope and high j0 simultaneously during the HER process. FE, also called coulombic efficiency, is the ratio of the experimental to the theoretical H2 generation. Thereinto, the theoretically produced H2 can be calculated by estimating the total charge transfer from potentiostatic or galvanostatic electrolysis plots and the experimental H2 production can be measured by gas chromatography (GC). In general, the FE is less than 100% owing to the existence of side reactions in the HER process. Furthermore, in order to realize the largescale application of the catalysts, the long-term stability is another important parameter, which can be measured by two methods: CV and galvanostatic/potentiostatic electrolysis. Finally, the TOF, which is a measure of the intrinsic activity of each electrocatalytic site, is defined as the number of reactants that an electrocatalyst can convert to a desired product at each electrocatalytic site per unit time. But, it is still a challenge to preciously measure the TOF due to the heterogeneous catalytic reaction often occurring on the electrode surface.52 Despite the fact that the measured TOF is relatively imprecise, it may still give insights into the comparative catalytic activity or efficiency of two or more materials if carefully executed. In conclusion, promising HER electrocatalysts should possess a low overpotential, small Tafel slope, high j0, large TOF, and long-term stability.
However, accurately measuring and evaluating the activity of various electrocatalysts is difficult owing to the various measurement methods/conditions, such as electrolyte compositions, deposited substrates, and pH values. Therefore, standard electrocatalytical measurements are of great value. As a successful example, Thomas F. Jaramillo and co-workers59 proposed a benchmark to explore various HER electrocatalysts (Fig. 3), which may be greatly significant for the development of standard measure techniques. Aiming toward higher accuracy of reported results, we need to pay special attention to the following points:60,61 First, the electrode setup should well define the overpotential of the working electrode. Second, if the researchers use the manual iR drop compensation at the benchmark current densities, they should exhibit a detailed iR compensated method, and especially the percentage of iR drop compensation should be cited in reports. Third, in order to ensure the accuracy of the activities, researchers should choose a suitable reference electrode and calibrate it experimentally.
 | ||
| Fig. 3 Suggested protocol for benchmarking the performance of electrocatalysts for the HER. Reproduced with permission from ref. 59. Copyright 2015, American Chemical Society. | ||
2.2 NRR
The electroreduction of N2 is another crucial energy conversion reaction to obtain a useful chemical intermediate, namely, NH3.10,62 It is worth noting that the HER is the major side reaction during the electrochemical NRR process, owing to its lower kinetic barrier and comparable thermodynamic potential. In contrast to the HER, the NRR involves a variety of proton–electron conversion processes in aqueous solution, including multiple surface-bound reaction intermediates.63,64 Until now, the reaction mechanisms of the electrochemical NRR on the heterogeneous materials have been classified into two types, viz. associative and dissociative mechanisms (Fig. 4a).65 In the dissociative pathway, the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds are firstly cleaved, and subsequently, the two N-atoms undergo hydrogenation processes independently. On the other hand, in the associative pathway, the N2 is firstly adsorbed on the electrocatalyst surface and subsequently undergoes a hydrogenation process with two N-atoms binding to each other. Moreover, the associative mechanism can be further classified into the alternating and distal pathway based on the different hydrogenation sequences. In the alternating pathway, the two N atoms are simultaneously hydrogenated, whereas, in the distal pathway, the N atom far away from the electrode surface is hydrogenated first and released as the NH3 molecule, while the adsorbed N atom is hydrogenated subsequently to form NH3.
N bonds are firstly cleaved, and subsequently, the two N-atoms undergo hydrogenation processes independently. On the other hand, in the associative pathway, the N2 is firstly adsorbed on the electrocatalyst surface and subsequently undergoes a hydrogenation process with two N-atoms binding to each other. Moreover, the associative mechanism can be further classified into the alternating and distal pathway based on the different hydrogenation sequences. In the alternating pathway, the two N atoms are simultaneously hydrogenated, whereas, in the distal pathway, the N atom far away from the electrode surface is hydrogenated first and released as the NH3 molecule, while the adsorbed N atom is hydrogenated subsequently to form NH3.
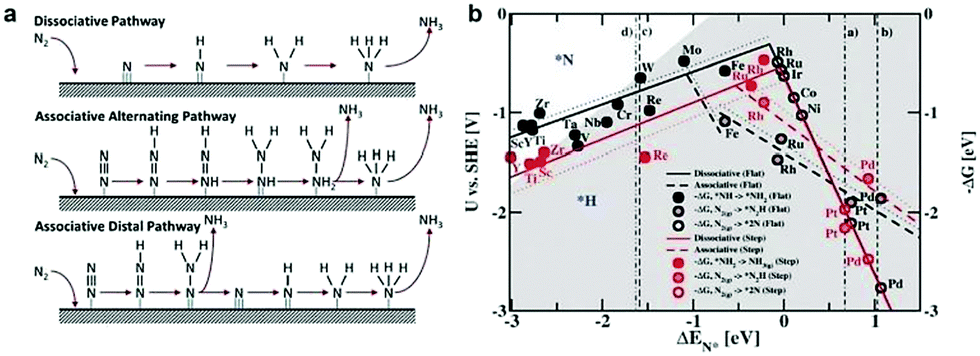 | ||
| Fig. 4 (a) Possible mechanisms for NRR on heterogeneous catalysts. Reproduced with permission from ref. 65. Copyright 2017, Elsevier Ltd. (b) Dissociative (solid lines) and associative (dashed lines) mechanisms, with (dotted lines) and without (solid lines) H-bonds effect on both stepped (red line) and flat (black line) metal surfaces. Reproduced with permission from Ref. 66. Copyright 2012, Royal Society of Chemistry. | ||
Similar to the electrochemical HER, the free energies of N2 admolecules and N adatoms are also regarded as an important descriptor to tune the activity and selectivity of an electroreduction nitrogen reaction. For instance, Nørskov and co-workers66 calculated the free energy profiles for the NH3 generation on both the flat and steeped surface using DFT, demonstrating the correlation between the adsorbed N intermediates and the selectivity of the NRR. Fig. 4b displays a volcano diagram for flat (black line) and stepped (red line) electrode surfaces for the NRR, containing associative (dotted lines) and dissociative (solid lines) mechanisms, with (dashed lines) and without (solid lines) H-bonds.66 As we can see from Fig. 4b, the most active metal surfaces for NH3 generation are Mo and Fe. However, these metal surfaces are evaluated to be more active for the HER instead of the NRR. In particular, most of the flat metal surfaces studied in this work, such as Co, Ni, Rh, Ir, and Pt, are predicted to be covered by H adatoms rather than N adatoms, resulting in low selectivity for the NRR. Note that several early transition metals (e.g., Zr, Ti, Y, and Sc) exhibit stronger N adatom binding energy compared to H adatoms. Thus higher selectivity to the NRR than the HER can be expected on these electrodes. Most recently, Nørskov and co-workers67 further reported a promising strategy to increase NRR selectivity by limiting the proton and electron supply, by numerous strategies such as increasing the barrier for proton transfer to the electrode surface, lowering the concentration of protons in the electrolyte, preventing the electron thermalization, and supplying a slow stream of electrons. Therefore, the theoretical investigations provide a good indicator to better understand the trend for electroreduction of N2 to NH3.
The NRR process severely suffers from poor reliability and reproducibility because of the low production rates of the NRR and ubiquitous contaminants.68 Similar to HER study, standard protocols for measuring the NRR are of fundamental importance for the NRR. In this regard, Chorkendorff and co-workers69 proposed a rigorous protocol to quantify the electrochemical reduction of N2 to ammonia (Fig. 5). A set of rigorous control experiments under an Ar, N2, and 15N2 atmosphere for each electrocatalyst should be performed to exclude ammonia contamination. Moreover, it is recommended that at least two testing methods be used to quantify the amount of ammonia, and an average of the two detection methods taken.70
 | ||
| Fig. 5 Protocol for the benchmarking of electrocatalysts for the NRR. Reproduced with permission from Ref 69. Copyright 2019, Nature Publishing Group. | ||
3. Surface structure engineering for the HER
3.1 Doping with heteroatoms
In the context of an electrocatalytic reaction, tuning the surface active sites and adsorption energy of reactants can be achieved via engineering the composition and nanostructure of electrocatalysts. Doping with heterogeneous atoms is a viable alternative to increase the number of active sites and optimize the kinetic process, and modulate the electronic structure and adsorption free energy of electrocatalysts synergistically.Apart from this, as demonstrated by previous studies, early-transition-metal carbides possess strong H-binding, which prejudices against gaseous H2 release.71 Taking Mo2C for example, there are high d-orbital vacancies around the EF which impede HER kinetics.71 TMC electrocatalyst doping with rich electrons elements reduces the vacancies effectively. The optimization of the H binding energy of TMC materials enhanced the kinetic reaction of the HER effectively.36,72,73 Rich electron dopants including metal atoms such as Fe, Ni, Co, Pt, and Pd,74–80 and non-metal atoms such as B, N, P, and S,30,81–85 show positive effects on modulating the surface composition and the internal electronic structure of the electrocatalysts. To cite N-doped Mo2C nanosheets as an example, the 1.0 nm ultrathin structures were constructed by transformation of the crystal phase and the surface atomic structure between MoO2 NSs and N-Mo2C NSs.30 Highly crystalline and porous structures were confirmed by HR-TEM (Fig. 6a). Besides, DFT calculations demonstrated that after N doping, new active sites associated with the N atom and Mo-3-T atom on the surface were introduced and with low |ΔGH*| values of 0.07 and 0.3 eV, respectively. The overpotential (η = 10 mA cm−2) of N-doped Mo2C nanosheets was only 99 mV owing to the robust sheet structure and N doping reduced the energy input for activating the HER (Fig. 6b). The morphology, phase transformation and composition of the N doped Mo2C NSs was affected by the amount of dicyandiamide and calcination temperature obviously. The authors also verified that the larger amount of dicyandiamide increased disordered carbon on the surface of the nanosheets during the calcining process. Besides, the more dicyandiamide led to phase transformation from Mo2C to MoC, and produced an excess amount of carbon coating the surface active sites of the Mo2C nanosheets resulting in poor HER activity (Fig. 6c). DFT calculations further verified that MoC possessed weaker HER activity than that of Mo2C (Fig. 6d). Hence, N doped Mo2C NS electrocatalysts with the appropriate amount of dicyandiamide possessed the best HER performance. Carbide electrocatalysts doped with transition metals which are rich in electrons such as group VIII metals can effectively reduce the unoccupied d orbitals of metal atoms in carbides.86,87 Wei and co-workers88 introduced a study about Ni-doped Mo2C supported on Ni foam, which showed excellent HER activity in alkaline solution. In another work, Yan and his colleagues reported a Fe-doped Ni3C electrocatalyst with remarkable water splitting performance in both acid and alkaline solutions.74 Thus, carbides doped with heterogeneous metal atoms (e.g. Fe, Ni, Co) exhibit remarkable enhancement in activity for the electrocatalytic hydrogen evolution reaction.80 To cite Co-doped Mo2C nanowires as an example, Gao and his co-workers75 reported a series of Co-doped Mo2C nanowire electrocatalysts, synthesized by means of a simple process of annealing Co-modified Mo3O10(C6H5NH3)2·2H2O precursors in an inert atmosphere. Nanowire structures provided abundant active sites in the radial direction for H2 generation and bubble release, while facilitating charge transfer in the axial dimension. After Co-doping into Mo2C, the electronic density around the Fermi level (EF) in Mo2C significantly increased which led to declination of the Mo–H bond toward optimized HER kinetics. XRD patterns undoubtedly confirmed that β-Mo2C was successfully prepared (Fig. 7a). On increasing the Co doping, the lattice parameter, a/b in β-Mo2C decreased resulting in a positive shift in the peaks of (100), (002) and (101) to higher angles (Fig. 7b and c). Analogous peak shifting was observed in electrocatalysts of Fe-doped Mo2C as reported by Leonard and his co-workers.89 Furthermore, the Mo2C unit cell shrunk on account of the replacement of some Mo atoms with the smaller Co in the lattice. On increasing the content of Co, the peak intensity of Co 2p3/2 and Co 2p1/2 in Co-Mo2C increased gradually. The peak position at 780.6 eV and 778.3 eV corresponded to metal Co and the peak position at 781.3 eV corresponded to oxidized Co, which was attributed to the interaction in the Mo2C lattice (Fig. 7d). The electronic feature of Mo in Mo2C varied after Co-doping, and there were three states (+2, +4 and +6) for Mo on the surface of the electrocatalysts. Mo4+ and Mo6+ were due to the inactive MoO2 and MoO3 as the carbides were exposed to air. Plenty of foregoing studies consider that Mo2+ species are the active centers for electrocatalytic HER. The Mo2+ signals were visibly red-shifted in Co-Mo2C (Fig. 7e), with increased Co-doping, which led to the lower binding energy. The shifting indicates that there are enriched electrons around Mo because of Co-doping. Besides, the Mo 3d peaks shifted to lower binding energy were reasonable on account of electron transfer from Co to Mo. The higher electronegativity of Mo (2.16) versus Co (1.88), and partially oxidized Co in Mo2C were the main contributing factors for electron transfer. Valence band (VB) study exhibited that a new peak at 3.45 eV arose with increasing amount of Co on account of more electrons around its Fermi level (Fig. 7f). The optimal Co-doping with the Co/Mo ratio of 0.020, guaranteed abundant Mo2+ species in Co doped β-Mo2C. Thus, Co-β-Mo2C nanowires acquired increased electron density by Co doping, which is in favor of its remarkable HER activity in both acid and alkaline solutions.
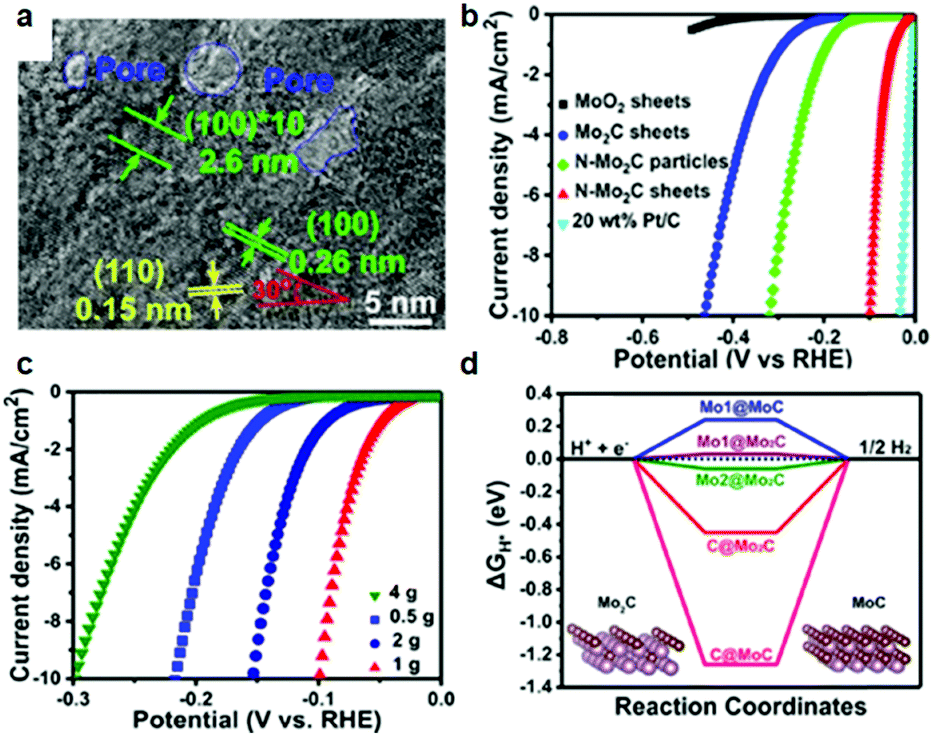 | ||
| Fig. 6 (a) HRTEM image of N-Mo2C NS. (b) Polarization curves of MoO2 NSs, Mo2C NSs, N-Mo2C nanoparticles, N-Mo2C NSs, and 20 wt% Pt/C on the GC electrode at 5 mV s−1 in 0.5 M H2SO4. (c) Polarization curves of N-Mo2C NSs with different content of N. (d) ΔGH* values of Mo and C atoms in MoC and Mo2C. Insets are the corresponding theoretical structural models. Reproduced with permission from ref. 30. Copyright 2017, American Chemical Society. | ||
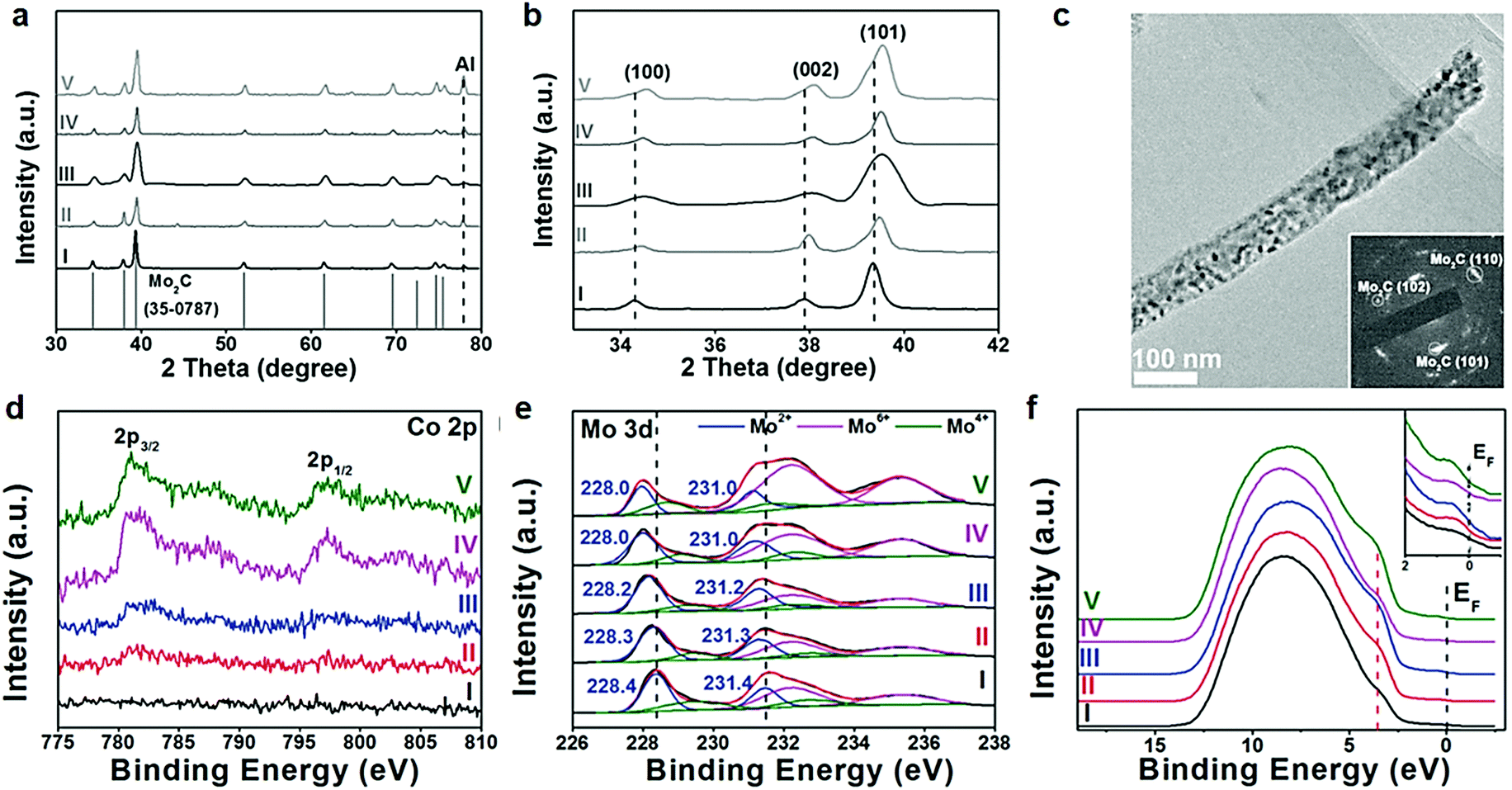 | ||
| Fig. 7 (a) XRD patterns of different phase Mo2C. (b) Corresponding zoomed-in regions showing evolution with Co-doping. (c) TEM image of Co-Mo2C-0.020. (d) XPS patterns of Co 2p and (e) Mo 3d in (I) Mo2C, (II) Co-Mo2C-0.012, (III) Co-Mo2C-0.020, (IV) Co-Mo2C-0.035, and (V) Co-Mo2C-0.049, and (f) their UPS spectra of the valence band. Ref. 75. Copyright 2016, John Wiley and Sons, Inc. | ||
Co-doping of organic–inorganic heteroatoms is recently reported as an effective way to increase the active sites of the catalyst. Recently, Lin and his co-workers90 reported porous MoC nanosheets co-doped with Zn, N metal–nonmetal atoms which demonstrated the advantages of metal/non-metallic heteroatom doping. A low melting point transition metal Zn acting as a dopant has attracted more attention in recent years due to the low electron negativity and divalent state of Zn.91–94 Besides, Chen and his co-workers91 confirmed that Zn was an efficient promoter for improving the HER catalytic activity of CoP. The bi-metal oxide precursor of ZnMoO4 acted as a template and a Zn doping source simultaneously. The porous Zn–N MoC nanosheets exhibited enhanced HER activity and remarkable long-time stability owing to the optimized Mo–H bonding as a result of the Zn, N co-doping and the large electrochemical surface area of the porous nanosheet structures. DFT calculations confirmed that the faster HER kinetics were achieved by Zn, N co-doping, on account of weakening the strong bond of Mo–H and facilitate the desorption of the absorbed hydrogen atom (Hads).
3.2 Creating surface porosity
As discussed above, electric conductivity, reactivity and the number of active sites are three key parameters, which affect the electrocatalytic activity of the catalysts. In terms of electrocatalysis process, exposure of more active sites is critical for the adsorption of intermediates in aqueous electrocatalytic reactions. Surface topography engineering is widely prevalent in the field of electrochemical energy conversion devices to enhance the adsorption of intermediates by the exposure of unique active sites. Transition metal carbide nanostructures with various dimensions and morphologies, such as nanodots, nanowires, nanobelts, nanorods, nanosheets, and 3D nanoframes,95–103 are theoretically and experimentally proved to exhibit marvelous HER and NRR electrocatalytic properties.71,95,104–106 Among them, carbides with porous structures were extensively studied because of excellent tunability of the pore size and shape in porous materials.104 It is understood that porous materials provide abundant active sites both on their surface and within their porous frameworks, which plays a pivotal role in the enhanced adsorption of reactants and intermediates.Metal–organic framework (MOF) materials to prepare mesoporous TMC materials have attracted much attention due to their unique structural characteristics in recent years.107,108 Continuous pore structures greatly facilitate electron transport and mass transfer during electrocatalytic reactions.109–112 MOF materials are extensively used in gas storage and separation, sensors, catalysts, and any other related fields.108–114 MOFs are materials of a porous coordination network formed by the interaction between metals and organic ligands, with a record surface area that exceeds that of activated carbon and zeolites. A large number of studies confirm that the introduction of MOFs into the catalyst plays an important role in promoting the electrocatalytic process. Huang et al.99 reported a porous Mo2C electrocatalyst with controlled compositions (doping) and morphologies (1D nanowires or 2D nanosheets) synthesized via carburizing cobalt or zinc-based zeolite-type MOF (ZIF-67 or ZIF-8) and cladding MoO3 nanosheets or nanowires under high temperature. A porous Mo2C electrocatalyst emerged with excellent electrocatalytic performance owing to unique pore structures. In this work, MOFs only acted as a precursor. However, the traditional method of preparing carbides relies on high temperature pyrolysis. Carbides with bulk structures form easily under high temperature conditions, resulting in loss of large number of active sites of the electrocatalysts.108 Recently, Lou et al.104 reported nano-octahedron MoCx electrocatalysts synthesized by a metal–organic framework (MOF)-assisted strategy, that exhibited excellent HER activity. Using the MOF-assisted strategy as a template to prepare porous transition metal carbides can effectively avoid diffusion/agglomeration during the pyrolysis process. And the porous structures remained intact even at higher temperatures. After preparing [Cu2(BTC)4/3(H2O)2]6[H3PMo12O40] (BTC = benzene-1,3,5-tricarboxylate) nano-octahedron structures, highly porous texture MoCx octahedral nanoparticles were finally obtained through a successive process of annealing and Fe3+ etching to remove Cu (Fig. 8a). The polycrystalline structure was confirmed by a selected-area electron diffraction (SAED) pattern (Fig. 8b). Some large particles formed at the corners of octahedral particles because of higher surface activity and stress, which was caused by collapse of MOFs, and MoCx nanocrystallites grew subsequently during high-temperature reaction. A number of MoCx clusters with a size of 5 nm anchored on the surface of amorphous carbon (Fig. 8c). The interplanar distance of 0.24 nm was consistent with (006) planes of η-MoC. Owing to the large electrochemical active surface of the unique nanostructures, porous MoCx nanooctahedrons showed excellent HER activity both in acid and alkaline media. The overpotentials at 10 mA cm−2 were 142 mV and 151 mV in acid and alkaline media respectively (Fig. 8d–f). And the electrocatalysts presented a small Tafel slope in both acid and alkaline media.
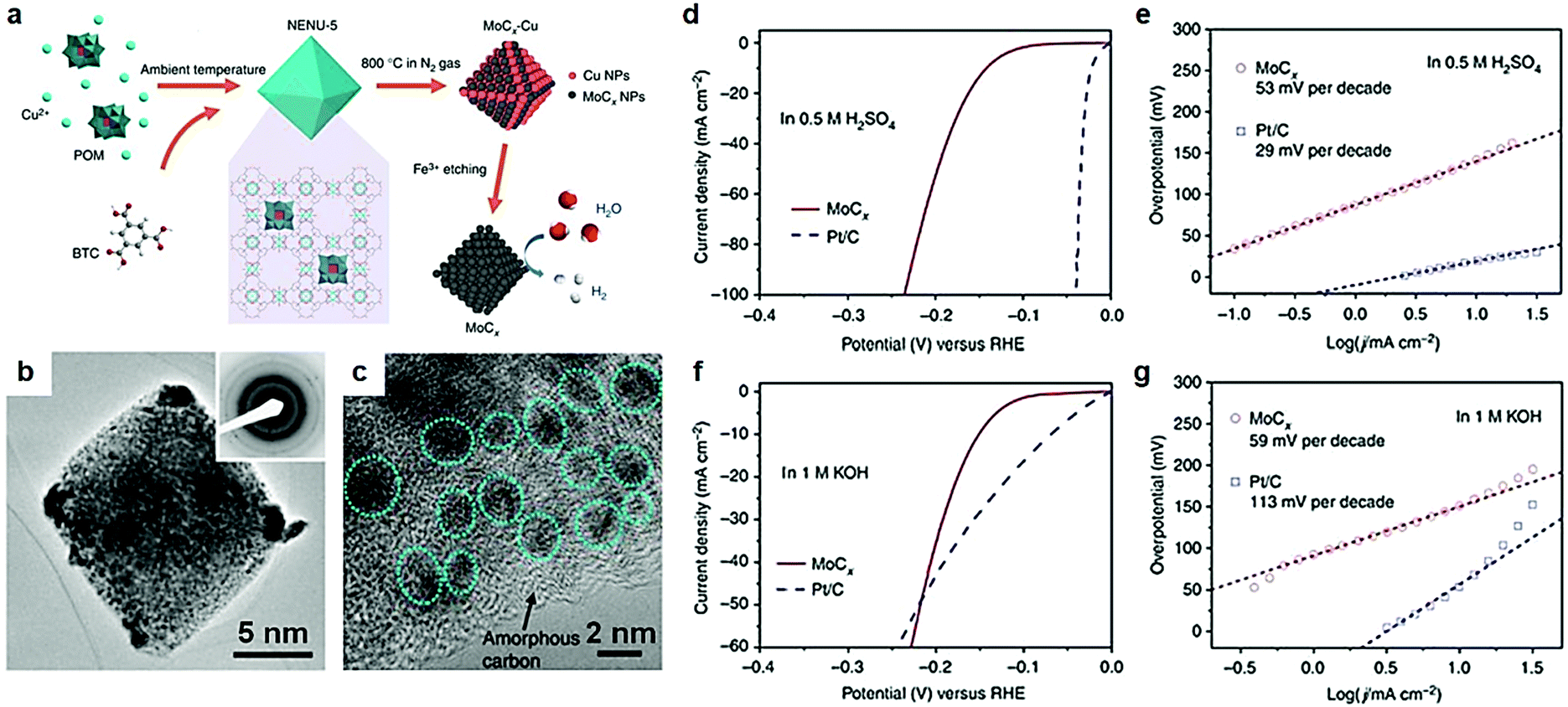 | ||
| Fig. 8 (a) Schematic illustration of the synthesis procedure for porous MoCx nano-octahedrons. (b) TEM images of MoCx nano-octahedrons; inset is the SAED pattern. (c) Magnified TEM image of MoCx nano-octahedrons. (d) Polarization curve at 2 mV s−1 and (e) Tafel plots in 0.5 M H2SO4. (f) Polarization curve at 2 mV s−1 and (g) Tafel plots in 1 M KOH. Ref. 104. Copyright 2015, Nature Publishing Group. | ||
3.3 Tailoring crystal facets
The electrochemical performance of nanostructures predominantly depends on the nature of their surface/interface properties.115 Tailoring high-index crystal facets is an effective method to enhance the surface energy of nanocrystals. Nanostructures with at least one miller index larger than 1 are considered high-index crystal faces.116,117 Surface structures with high surface energy tend to have high density of atoms with low-coordination numbers and kink atoms which greatly contribute to the catalyst performance of nanomaterials. Manufacturing high-index crystal facets to improve electrocatalytic activity of the catalysts is commonly found in the noble metal based electrocatalysts, such as Pt, Pd, and Au,118–123 which are usually applied for the reactions of fuel cells. However, structural instability of high index crystal facets for electrocatalytic reactions is the main problem to be solved. Mu and co-workers developed a subtle Cl2-assisted “micro-cutting-fragmentation” method to obtain carbon-cohered high-index (222) faceted tantalum carbide nanocrystals (TaC NCs@C) derived from TaC blocks.124 Low-index facets have a tendency to transform into high-index facets during high temperature processes.125 In this work, a series of experimental results demonstrated that the higher temperature favors fragmentation of the crystal into smaller nanocrystals by Cl2 gas.The chlorination temperature played a vital role in nanocrystal size and HER performance. Higher temperatures favored the formation of nanocrystals with a small size which was further confirmed by theoretical calculations based on the reactive kinetics of chlorination.126 Furthermore, carbide-derived carbon (CDC) growth on the surface of carbide blocks,127,128 can be understood as a diffusion-controlled mechanism based on linear-parabolic Deal-and Grove-like growth kinetics. The transition belts produced during the preparation process between high-index (222) facets of TaC NCs and in situ amorphous carbon layers prevented structural collapse during high temperature preparation and electrocatalytic processes. The challenge of structural instability of high crystal face index materials obtains a satisfactory solution.
Furthermore, DFT calculations uncovered the high-index (222) faceted TaC with no band gap as the crossover of the Dirac cone at the G point. In other words, a TaC electrocatalyst with high-index (222) had a fast electron transfer during an electrocatalytic hydrogen reduction process. Partial density of states verified that the Fermi level of all facets crosses the conduction band. Hence, the influence of electron mobility on different facets is insignificant, which is beneficial for the HER electrocatalysis. As we all know, Pt is the best electrocatalyst for the HER with a very low ΔGH* value of −0.09 eV.129–133 In comparison, the ΔGH* values of low-index (311), (220), (200) and (111) facets of the TaC electrocatalyst were −0.53 eV, −0.56 eV, −0.73 eV and −0.67 eV respectively. The TaC electrocatalyst with high-index (222) facets showed a least negative ΔGH* value of −0.23 eV. From the thermodynamic point of view, high-index (222) facets of TaC have the best electrocatalytic hydrogen evolution ability relative to other low-index crystal faces. The ultra-fine structure of carbon-cohered high-index (222) faceted tantalum carbide nanocrystals (TaC NCs@C) was obtained via using Cl2 to cut TaC blocks into TaC NCs at high temperature (Fig. 9a).124 The transition belts would be produced between high-index (222) facets of TaC NCs and in situ formed amorphous carbon layers which prevented conversion of high-index crystal facets. The high activity and stability of high-index (222) facets of TaC NC electrocatalysts was attributed to these unique structures. The authors maintained that Cl2 molecules preferentially reacted with Ta atoms located at small cracks existing in TaC blocks (Fig. 9b). This is the main reason for disorderly piled residue surface C atoms turning into amorphous carbon. The average size of TaC NCs was about 12.5 nm which uniformly encapsulated in the in situ formed carbon blocks during high-temperature reaction. The interplanar spacing of TaC NCs was 1.3 Å corresponding to high-index (222) facets (Fig. 9c). A weak carbon peak was exhibited in the XRD patterns (Fig. 9d). The authors considered that the cohered carbon was beneficial to maintain high-index faceted structures and exceptional dispersivity of nanostructured TaC NCs. The overpotential of high-index (222) facet carbon-cohered TaC NCs (146 mA cm−2) was substantially lower than that of the bulk TaC electrocatalyst (Fig. 9e). High-index facets of TaC NCs also showed an excellent Tafel slope of 143 mV dec−1, where the Tafel slope of bulk TaC was 232 mV dec−1 (Fig. 9f). The theoretical free energy of the hydrogen adsorption on different HER electrocatalysts indicated that the position of the j0 and ΔGH* values was closer to the peak (Fig. 9g), indicating a better HER activity.
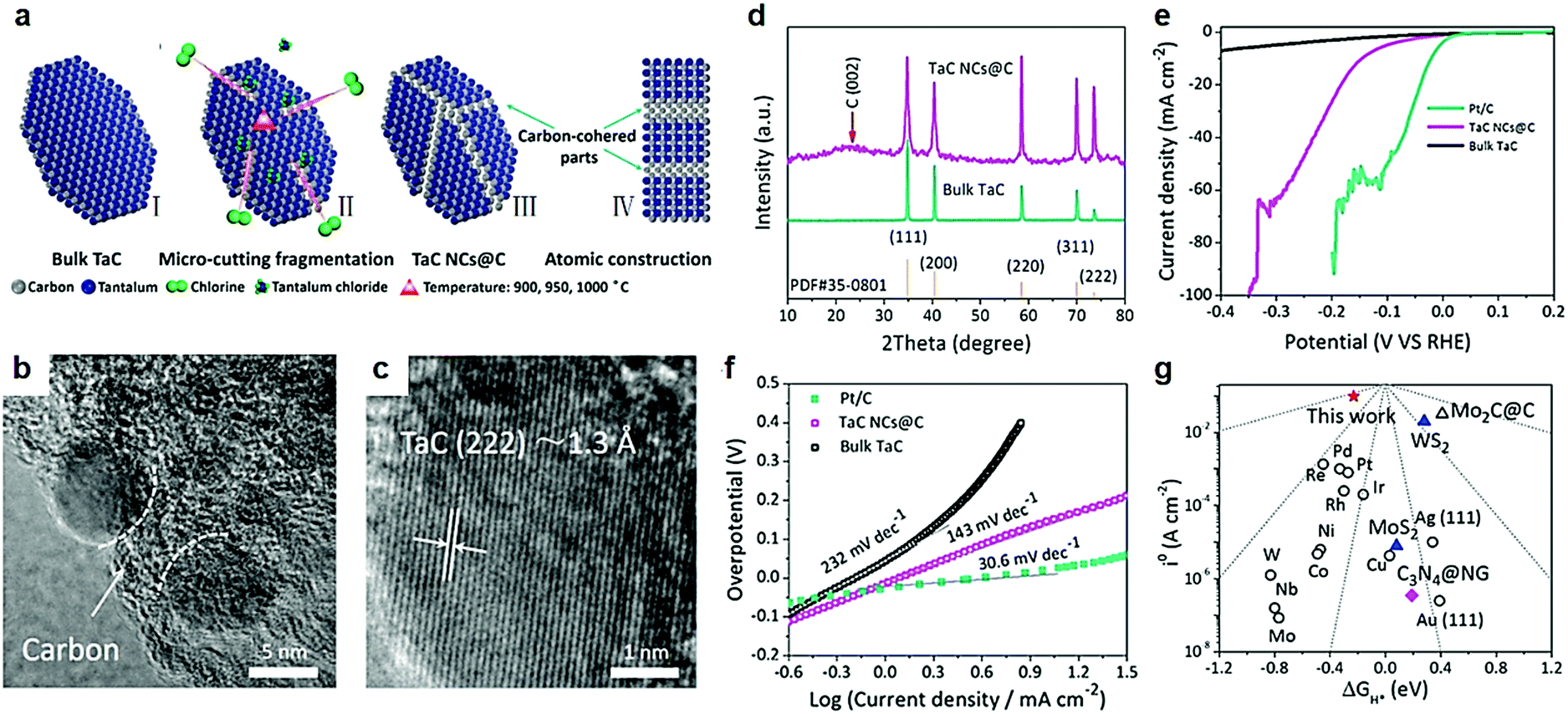 | ||
| Fig. 9 (a) Schematic formation of TaC NCs@C by a subtle chlorine-assisted micro-cutting fragmentation strategy. (I) Pristine TaC blocks; (II) intermediate state of the reaction; (III) final product TaC NCs@C; (IV) side-view model of the product. (b and c) High magnification TEM images of TaC NCs@C, presenting exposed high-index (222) facets on the surface of TaC NCs. (d) Powder XRD pattern of bulk TaC and TaC NCs@C. (e) Polarization curves and (f) Tafel plots (b) of commercial 20% Pt/C, TaC NCs@C and bulk TaC. (g) Volcano plots of i0 as a function of the ΔGH* for newly developed high-index (222) faceted TaC (red pentacle), common metal catalysts, molybdenum carbide and carbon hybrid catalyst, typical nanostructured MoS2 and WS2 catalyst as well as metal-free C3N4@N-doped graphene catalyst. Reproduced with permission from ref. 124. Copyright 2017, Elsevier Ltd. | ||
Another study on high-index facets was reported recently. Ma et al.134 prepared high-index {120} faceted dendritic hexagonal NiCx nanosheets on a Ni/CF substrate by a mild electrodeposition approach. The obtained dendritic NiC0.2NS/Ni/CF showed remarkable activity and stability of water splitting. The overpotential at 10 mA cm−2 was 121 mV, and the corresponding Tafel slope was 51 mV dec−1 in an alkaline medium. Furthermore, a dendritic NiC0.2NS/Ni/CF electrocatalyst offered superior catalytic stability of more than 100 h. Excellent electrocatalytic performance of NiC0.2NS/Ni/CF electrocatalysts was ascribed to several factors. The dendritic nanosheet morphology contributed abundant active sites for catalysis in NiC0.2NS/Ni/CF electrocatalysts. Besides, the enhanced electron transport characteristic between NiC0.2 nanosheets and the conductive substrate and the optimal carbon content also contributed to excellent HER performance. High-index crystal plane regulation improved the performance of electrocatalysts effectively. However, it is difficult to attain the best performance of electrocatalysts only by crystal plane regulation. Therefore, when considering the electrocatalyst design, it is quite essential to combine other techniques such as impurities doping, morphology modulation, electronic effects, interface engineering, etc.
3.4 Forming defects
Defects play a pivotal role in an electrocatalytic process because they can change the interaction between the surface and the target reactant molecule and reaction intermediate molecule. This can lower the reaction activation energy and consequently even change the reaction pathway. To improve the exposure of the surface active sites, defect engineering is regarded as a promising strategy and is often applied in the synthesis of nano-sized electrocatalysts. Defect-rich MoS2 was prepared through a reaction of a high concentration of precursors and excess thiourea.135 Thiourea performed the dual function as a reductant for conversion of Mo(VI) to Mo(IV) and as a morphological stabilizer.136 During the preparation of defective MoS2, the thiourea adsorbed on the surface of primary nanocrystallites hindered the oriented crystal growth. Owing to a defective structure, the MoS2 nanosheets have a small overpotential of 120 mV at a current density of 10 mA cm−2 and the Tafel slope of 50 mV dec−1. Thus, for transition metal carbides, defect engineering regulation plays an important role in the electrocatalytic process. Recently, Li et al.106 prepared macroporous inverse opal-like MoxC electrocatalysts with a prolific number of defects via a hard-template method. SiO2 spheres were mixed with dicyandiamide and Mo precursors before the carbonization process. In the following step, the SiO2 template was removed completely with HF (Fig. 10a).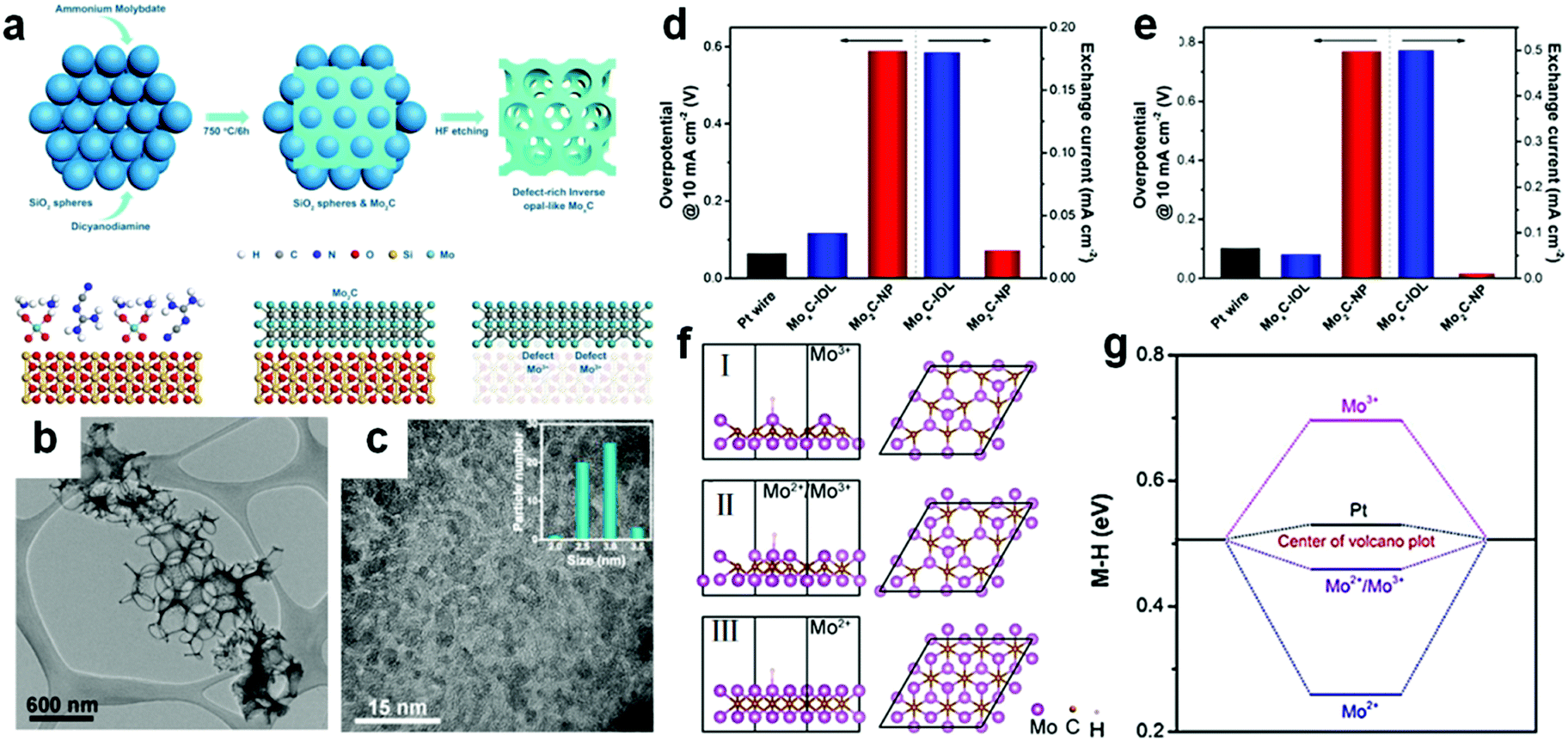 | ||
| Fig. 10 (a) Synthesis of inverse opal-like Mo2C without SiO2 spheres and formation of a rich density of defects. (b) Low-resolution TEM images of MoxC-IOL. (c) Magnified TEM image of MoxC-IOL. Inset is the corresponding particle size distribution of Mo2C in MoxC-IOL. (d) Overpotentials@10 mA cm−2 of Mo2C-NP, MoxC-IOL and Pt wire (left) and corresponding exchange current densities (right) in 0.5 M H2SO4. (e) Overpotentials@10 mA cm−2 of Mo2C-NP, MoxC-IOL, and Pt wire (left) and corresponding exchange current densities (right) in 1 M KOH, Scan rate: 5 mV s−1. (f) Hydrogen adsorption configuration on the Mo2C surfaces with Mo2+/Mo3+ = 0/1 (I), 0.4/0.6 (II), 1/0 (III). (g) Corresponding H binding energies. Ref 106. Copyright 2017, American Chemical Society. | ||
Macroporous inverse opal-like (IOL) MoxC structures with a number of defects were formed finally (Fig. 10b). MoxC IOL structures with different pore sizes were prepared for evaluating electrocatalytic performance. A pore size of 380 nm demonstrated the best hydrogen evolution performance among all pore sizes as the macroporous structures provided more convenient mass transport channels than mesoporous structures. Furthermore, MoxC nanoparticles existed in the IOL structure with a size of 2.5–3 nm (Fig. 10c). MoxC-IOL showed superior intrinsic electrocatalytic activity in both acid and alkaline media. The overpotentials at 10 mA cm−2 in acid and alkaline solution were 117 mV and 82 mV respectively (Fig. 10d and e). Besides, MoxC-IOL also exhibited large exchange current densities of 0.18 mA cm−2 and 0.5 mA cm −2 in both acid and alkaline media, respectively. The authors attributed the excellent performance of an electrocatalyst to the abundant defects generated from a myriad of Mo+ species introduced in the MoxC IOL structure.
DFT calculations provided a fundamental understanding of the remarkable electrocatalytic activity of MoxC-IOL at the atomic level. Metal–hydrogen (M–H) bond strength was calculated on the surface of Mo2+, Mo3+ and Mo2+/Mo3+ (Fig. 10f). In the acidic solution, the binding energy for the MoxC-IOL was only 0.07 eV smaller than Pt (Fig. 10g). Such hydrogen binding energy was beneficial for proton adsorption, reduction, and H2 release in the acidic solution.137 On the other hand in the alkaline medium, H2O molecules are the reactant to release H2. H2O is first absorbed on the surface of catalysts, followed by subsequent reactions to produce H2. The H2O binding energies of Pt and MoxC-IOL are 0.12 and 1.12 eV, respectively. Higher binding energy of H2O favors capture of more H2O molecules. Hence, the MoxC-IOL electrocatalysts showed significantly enhanced HER performance in alkaline solution.
3.5 Nano-size effect
Electrocatalysts with a nano-structure are one of the most common and popular means to boost electrocatalytic activity.138–140 In view of the significance of exposure of more surface active sites for an electrochemical reaction, a series of electrocatalysts with nano-size were fabricated and exhibited enhanced electrocatalytic activity via experiments and DFT calculations in recent studies.33,104,141–143 Lee and his co-workers75 prepared B and N co-doped Mo2C nanoparticles imbedded in a BCN network via a novel Mo-imidazole complex route. B, N-doping decreased the crystal size of Mo2C NPs from 29.1 nm (pure Mo2C@C) to 17.6 nm (N:Mo2C@NC) and 8.6 nm for B,N:Mo2C@BCN. In alkaline solution, the corresponding overpotentials at 10 mA cm−2 were 270 mV, 150 mV and 100 mV, respectively. Excellent electrocatalytic properties of Mo2C were attributed to both the nano-size effect and co-doped with B and N. In another work, Qu et al.33 prepared 1 nm Mo2C nanoparticles supported on N-doped carbon (Mo2C@NC) nanomesh which exhibited remarkable HER activity with an overpotential of 36 mV at 10 mA cm−2 in 0.5 M H2SO4 solution. In general, the nano-size effect is important to enhance the electrocatalytic performance of the catalyst. However, in practical applications, the size effect often combines with other methods to achieve the optimal electrocatalytic performance, such as doping, morphology, interface engineering, etc.3.6 Surface phase modulation
Tuning a specific phase of a homogeneous catalyst is an effective method to improve the catalytic activity for electrocatalysis. The specific facet, the corner site exposed in a crystal and the surface electronic configuration all depend on the crystalline phase. These parameters identify the adsorption energy of the reaction intermediate on the catalyst surface, in turn controlling the reaction rate. Different phases of Mo-C, including α-MoC1−x, β-Mo2C, η-MoC, and γ-MoC, were successfully prepared via a unique amine–metal oxide composite material by the Lenard research group.144 All these four phases exhibited similar hexagonal crystal structures and different stacking sequences (Fig. 11a). High temperature treatment with appropriate atomic ratios of precursor at different dwelling times is an effective method to obtain carbide ceramic materials with different phases.145,146 The morphology and size of four phases of molybdenum carbide were quite different due to the high temperature processing conditions. The miniature size of carbides contributed to the enhanced surface area. β-Mo2C has the smallest size among four phases of molybdenum carbide and the BET was about 90 m2 g−1, while the BET of γ-MoC was about 25 m2 g−1. The large surface area of β-Mo2C was beneficial for impressive HER activity. β-Mo2C exhibited the best HER activity among the four phases of molybdenum carbide (Fig. 11b). γ-MoC also had similar catalytic activity for the HER owing to the similar valence band shape of β-Mo2C and γ-MoC electrocatalysts to that of Pt. The excellent HER performance of β-Mo2C and γ-MoC may be due to their Pt-like Fermi level energy. However, there is no detailed research to confirm the trends in activity. Additionally, density around the Fermi level of β-Mo2C is higher than γ-MoC. This is the main reason that β-Mo2C exhibits less stability compared to γ-MoC. The Tafel slope of β-Mo2C was 120 mV dec−1 (Fig. 11c) which was marginally lower than that of γ-MoC (121.6 mV dec−1). The exchange current density of β-Mo2C was 17.29 μA cm−2, which was substantially higher than that of γ-MoC (3.2 μA cm−2). Although the HER performance of β-Mo2C is much better than γ-MoC, substantial improvement is needed in comparison with existing carbide catalysts. The hydrogen evolution performance of representative carbides in the past five years is summarized in Table 2.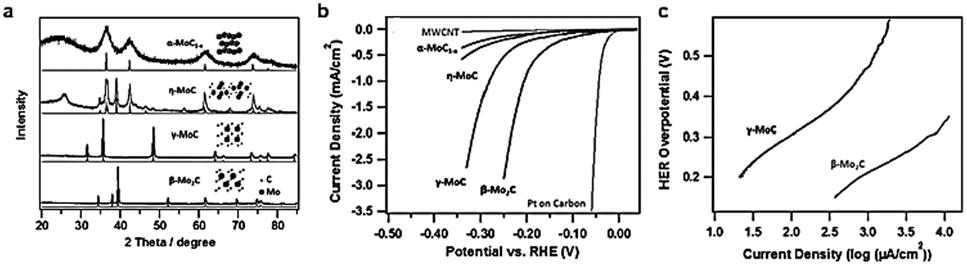 | ||
| Fig. 11 (a) X-ray diffraction (XRD) patterns of different phase MoC. The insets show the corresponding crystal structures. (b) Polarization curves for four phases of molybdenum carbide, Pt on a carbon support, and multiwall carbon nanotubes (MWCNTs) in 0.1 M HClO4. (c) The corresponding Tafel plots for β-MoC and γ-Mo2C. The catalyst loading for all samples is 0.28 mg cm−2 and the scan rate is 2 mV s−1. Ref. 144. Copyright 2014, John Wiley and Sons, Inc. | ||
| Electrocatalyst | Catalyst loading (mg cm−2) | Electrolyte | η 10 (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|
| β-Mo2C/N-doped carbon | 0.35 | 0.5 M H2SO4 | 119 | 72 | J. Mater. Chem. A, 2018, 6, 6054 |
| 2D WC@graphene | 0.00222 | 0.5 M H2SO4 | 120 | 38 | Nano Energy, 2017, 33, 356 |
| Nanoporous Mo2C NMs | 0.21 | 0.5 M H2SO4 | 120 | 53 | Energy Environ. Sci., 2014, 7, 387 |
| V8C7 networks | NA | 0.5 M H2SO4 | 38 | 34.5 | Adv. Energy Mater., 2018, 8, 1800575 |
| 1 M KOH | 47 | 44 | |||
| Mo2C@NC nanomesh | 0.5 | 0.5 M H2SO4 | 36 | 33.7 | Adv. Funct. Mater., 2018, 28, 1705967 |
| Ni3Mo3C@NPC NWs | 0.285 | 0.5 M H2SO4 | 161@η100 | 74.8 | ChemSusChem, 2018, 11, 2717 |
| B,N:Mo2C@BCN | 1 | 1 M KOH | 360@η100 | 62 | ACS Catal., 2018, 8, 8296 |
| Co4Mo2@NC | 0.35 | 1 M KOH | 218 | 73.5 | J. Mater. Chem. A, 2017, 5, 16929 |
| Co-Mo2C nanowires | 0.14 | 0.5 M H2SO4 | 140 | 39 | Adv. Funct. Mater., 2016, 26, 5590 |
| 1 M KOH | 118 | 44 | |||
| Mo2C@NPC/N,P-doped RGO | 0.14 | 0.5 M H2SO4 | 34 | 33.4 | Nat. Commun. 2016, 7, 11204 |
| Mo2C@NC | 0.28 | 0.5 M H2SO4 | 124 | 60 | Angew. Chem., Int. Ed., 2015, 54, 10752 |
| α-Mo2C | 0.102 | 0.5 M H2SO4 | 198 | 56 | J. Mater. Chem. A, 2015, 3, 8361 |
| 1 M KOH | 176 | 58 | |||
| NiMo2C@C | 0.531 | 0.5 M H2SO4 | 72 | 65.8 | Nanoscale, 2018, 10, 6080 |
| Nanoporous Mo carbide | NA | 0.5 M H2SO4 | 229 | 100.7 | Adv. Sci., 2018, 5, 1700601 |
| Fe–Ni3C-2% | 0.15 | 0.5 M H2SO4 | 178 | 36.5 | Angew. Chem., Int. Ed., 2017, 56, 12566 |
| 1 MKOH | 292 | 41.3 | |||
| N@Mo2C-3/CFP | 2 | 0.5 M H2SO4 | 56 | 49 | Adv. Energy Mater., 2018, 8, 1800789 |
| 1 M KOH | 66 | 51 | |||
| Mo2CNWAs/CFP | 20 | 0.5 M H2SO4 | 56 | 68 | Adv. Funct. Mater., 2018, 28, 1804600 |
| 1 MKOH | 52 | 72 | |||
| Mo2C | 1.5 | 0.5 M H2SO4 | 140 | 124 | J. Mater. Chem. A, 2015, 3, 16320 |
| Co/β-Mo2C@N-CNTs | 0.014 | 1 M KOH | 170 | 92 | Angew. Chem., Int. Ed. 2019, 58, 1 |
| β-Mo2C nanotubes | 0.75 | 0.5 M H2SO4 | 172 | 62 | Angew. Chem., Int. Ed., 2015, 54, 15395 |
| 0.1 M KOH | 112 | 55 | |||
| Ni/WC hybrid | 0.7 | 0.5 M H2SO4 | 53 | 43.5 | Energy Environ. Sci., 2018, 11, 2114 |
| Mo2C | 1 | 0.5 M H2SO4 | 35 | 25 | Adv. Energy Mater., 2018, 8, 1801461 |
| Mo2C-N, P-rGO | 0.24 | 0.5 M H2SO4 | 95 | 60 | ChemCatChem, 2018, 10, 2300 |
| 1 M KOH | 71 | 49 | |||
| Mo2N–Mo2C | 0.337 | 0.5 M H2SO4 | 157 | 55 | Adv. Mater., 2018, 30, 1704156 |
| 1 M KOH | 154 | 68 | |||
| Mo2C/N-doped graphene | 0.25 | 1 M KOH | 142 | 101.8 | Electrochim. Acta, 2019, 299, 627 |
| Mo/Mo2C heteronanosheets | 0.285 | 0.5 M H2SO4 | 89 | 70.7 | ACS Energy Lett., 2018, 3, 341 |
| Fe3C–Mo2C/NC | 0.14 | 0.5 M H2SO4 | 116 | 43 | ChemSusChem, 2017, 10, 2597 |
| WxC@WS2 | 0.3 | 0.5 M H2SO4 | 146 | 61 | Adv. Funct. Mater., 2017, 27, 1605802 |
| Inverse opal-like Mo2C | 2.22 | 0.5 M H2SO4 | 117 | 60 | ACS Nano, 2017, 11, 7527 |
| 0.1 M KOH | 82 | 56 | |||
| Co–NC@Mo2C | 0.83 | 0.5 M H2SO4 | 143 | 60 | Nano Energy, 2019, 57, 746 |
| 1 M KOH | 173 | 65 | |||
| Mo2C/GCSs hybrids | 0.36 | 0.5 M H2SO4 | 120 | 62.6 | ACS Catal., 2014, 4, 2658 |
| Mo2C nanoparticles | 0.28 | 0.5 M H2SO4 | 144 | 55 | ACS Nano, 2016, 10, 11337 |
| 1 M KOH | 100 | 65 | |||
| Mo2C | 0.57 | 0.5 M H2SO4 | 70 | 39 | Adv. Sci., 2018, 5, 1700733 |
| 1 M KOH | 66 | 37 | |||
| Mo2C–RGO hybrid | 0.285 | 0.5 M H2SO4 | 70 | 54 | Chem. Commun., 2014, 50, 13135 |
| MoCx-DecoratedCo@N-C | 0.7 | 0.5 M H2SO4 | 187 | 82 | Small, 2018, 14, 1704227 |
| 1 M KOH | 157 | 148 | |||
| MoxC–Ni@NCV | 1.1 | 0.5 M H2SO4 | 75 | 45 | J. Am. Chem. Soc., 2015, 137, 15753 |
| MoP/Mo2C@C | 0.453 | 0.5 M H2SO4 | 89 | 45 | ACS Appl. Mater. Interfaces, 2017, 9, 16270 |
| 1 M KOH | 75 | 58 | |||
| MoCx@C | 0.354 | 0.5 M H2SO4 | 79 | 56 | J. Mater. Chem. A, 2016, 4, 3947 |
| Mo2C nanobelts | 0.5 | 0.5 M H2SO4 | 140 | 51.3 | Appl. Catal., B, 2018, 224, 533 |
| 1 M KOH | 110 | 49.7 | |||
| Ni/Mo2C–NCNFs | 1.4 | 1 M KOH | 143 | 57.8 | Adv. Energy Mater., 2019, 1803185. |
| Porous N@MoPC hybrid | 0.14 | 0.5 M H2SO4 | 139 | 69.4 | Adv. Energy Mater., 2018, 8, 1701601 |
| Mo2C | 0.255 | 0.5 M H2SO4 | 160 | 66 | Electrochim. Acta, 2018, 274, 23 |
| 1 M KOH | 92 | 63 | |||
| N,P-Doped Mo2C@C | 0.9 | 0.5 M H2SO4 | 56 | 56 | ACS Nano, 2016, 10, 8851 |
| 1 M KOH | 47 | 71 | |||
| MoCx | 0.8 | 0.5 M H2SO4 | 142 | 53 | Nat. Commun., 2015, 6, 6512 |
| 1 M KOH | 151 | 59 | |||
| Mo2C/C | 0.3 | 0.5 M H2SO4 | 145 | 60.6 | ChemCatChem, 2017, 9, 1588 |
| Ni–Mo2C/C composite | 1 | 1 M KOH | 99 | 73 | Chem. – Eur. J., 2017, 23, 4644 |
| MoSx@Mo2C | 0.213 | 0.5 M H2SO4 | 178 | 44 | ACS Catal., 2015, 5, 6956 |
| Mo2C-G | 0.8 | 0.5 M H2SO4 | 150 | 57 | Chem. Commun., 2015, 51, 8323 |
| Zn–N porous MoC NSs | 0.357 | 0.5 M H2SO4 | 128 | 52.1 | Nanoscale, 2019, 11, 1700 |
| Mo2C NPs | 0.25 | 0.5 M H2SO4 | 78 | 41 | Angew. Chem., Int. Ed., 2015, 127, 14936 |
| Vanadium carbide nanoparticles | 0.28 | 0.5 M H2SO4 | 98 | 56 | Nano Energy, 2016, 26, 603 |
| MoS2/Mo2C NSs | 1.67 | 0.5 M H2SO4 | 63 | 53 | ACS Catal., 2017, 7, 7312 |
| VC@NCNT | 0.9 | 0.5 M H2SO4 | 161 | 95 | Nanoscale, 2018, 10, 14272 |
| 1 M KOH | 159 | 125 |
4. Interface engineering for the HER
As discussed in the previous section, most TMCs are inert for the electrochemical reactions owing to their limited number of active sites, weak adsorption energy, and low surface area.147 Interface engineering, especially integrating different types of materials into a single nanostructure, is a promising strategy to improve the electrocatalytic performances.148 Interface engineering can facilitate the formation of strong chemical bonds and synergistic interaction between the various components, which may change the electronic DOS and the positions of the valence and conduction bands of the as-prepared electrocatalysts.149,150 Until now, carbon-based materials (such as CNTs, graphene, reduced graphene oxide, and mesoporous carbon),32,151,152 metals,153,154 and metal compounds98,155,156 have been introduced into the TMCs to form hybrid materials with unique physical/chemical properties. The incorporation of other materials not only enhances the electrical conductivity, but also introduces additional active sites.4.1 TMC/carbon-based materials
Owing to the large specific surface, high charge mobility, and good durability, CNTs are usually used as support materials in electrochemistry.157,158 By introducing these materials, the electronic structure of TMCs can be tuned. In particular, the CNTs are reactive C sources for forming nanosized TMCs. For instance, Adzic and co-workers40 synthesized Mo2C/CNTs by in situ carburization of ammonium molybdate on CNTs without using any gaseous carbon source. The XANES results demonstrated that, in the Mo2C/CNTs, there is charge transfer from Mo to C, which downshifts the d-band center of Mo and thus weakens its H-binding energy. This results in the improved HER activity. Recently, the confinement preparation of TMCs on the surface of CNTs was investigated by Lee and co-workers.45 They synthesized ultrasmall and phase-pure W2C nanoparticles, which have higher activity and stability for electrochemical HER than that of conventional WC owing to the greater electronic DOS close to the Fermi level. The authors proposed that the gaseous C sources (e.g., CH4) and the non-volatile solid C sources (e.g., Multi-walled carbon nanotubes (MWCNTs)) might have different effects on the chemical composition and nanostructure of the carburized materials. As displayed in Fig. 12a,45 conventional strategies using CH4 as the C precursors were unable to obtain the highly active W2C owing to the lack of control over the ratio between C atoms and W atoms. Furthermore, the diffusion rate of C atoms from CH4 to W lattice took place too fast to allow for phase tuning. When the MWCNTs were selected as the solid C sources, the C diffusion through the solid–solid interface slowed down owing to their chemical inertness, resulting in the formation of coking-free, nanosized, and phase-pure W2C. The high-resolution transmission electron microscopy (HR-TEM) image demonstrated that the size of the as-prepared W2C nanoparticles ranged between 2 and 5 nm and few smaller clusters had size in the sub-nanometer range (Fig. 12b). Moreover, the XPS and EXAFS spectrum (Fig. 12c–e) indicated that the ultrasmall size and defect-rich structure of the as-obtained W2C could decrease the coordination number of W–C and W–W. When evaluating their performance for the HER, they found that, compared to WC/MWNTs, W2C/MWNTs exhibited better activity with a low onset overpotential of ∼ 50 mV and a small Tafel slope of 45 mV dec−1 (Fig. 12f and g).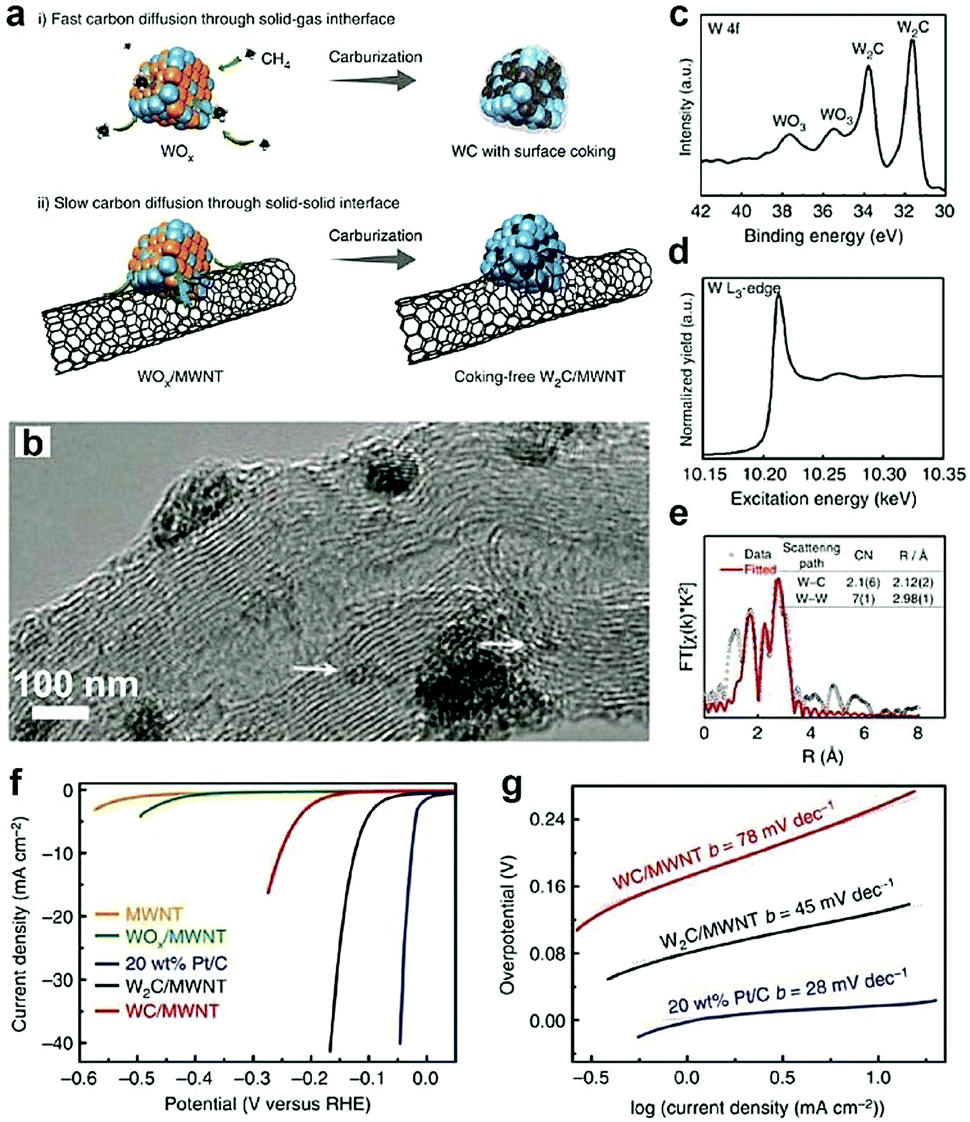 | ||
| Fig. 12 (a) Schematic illustration exhibiting the effect of different carbon precursors. (b) TEM image of W2C/MWNT. (c) W 4f XPS spectrum, (d) W L3-edge XANES spectrum and (e) Fourier transform EXAFS spectrum and associated fitting curve of W2C/MWNT at the W L3-edge. (f) HER polarization curves in 0.5 M H2SO4 and corresponding Tafel plots (g). Reproduced with permission from ref. 45. Copyright 2016, Nature Publishing Group. | ||
Analogously, graphene-based materials, e.g., graphene oxide (GO),159,160 reduced graphene oxide (rGO),41,43 and graphene nanoribbons (GNRs),161 are another class of materials widely applied as the supports to synthesize TMCs. Yang and co-workers42 prepared Mo2C nanoparticles stabilized by a carbon layer on rGO using a general two-step strategy. They found that the outer protection by the carbon layer could increase the stability of the as-prepared materials. Moreover, the synergistic interaction/intimate contact between Mo2C and rGO as well as the carbon layer was the main reason for the improved HER activity. A similar phenomenon has also been reported by Lan and co-workers.43 They fabricated a 2D coupled hybrid consisting of Mo2C nanoparticles encapsulated by N, P-codoped carbon layer and rGO (marked as Mo2C@NPC/NPRGO) with efficient HER activity. They proposed that the improved HER performance is mainly attributed to the small size of Mo2C nanoparticles, the enhanced interaction with H+, the robust conjugation between Mo2C and NPC/NPRGO, and the enhanced electron penetration from Mo2C to rGO. Other hybrid materials such as Fe3C/rGO,41 Fe3C/GRNs,161 and Mo2C/graphene foam162 have been fabricated using a similar principle.
In addition to the aforementioned as-formed carbon-based materials, there are other carbon supports which can be/are in situ formed during the carburization process. In the past, low-cost polymers (including polydopamine (PDA)163 and PANI160) and renewable biomass (including chitosan164 and glucose162) have been widely used to rationally design efficient TMCs/carbon hybrids. Li and coworkers165 reported a simple two-step method to fabricate Mo2C nanoparticles (∼3nm) uniformly dispersed on N-doped carbon microflowers (denoted as Mo2C/NCF) using the self-polymerization of dopamine. The as-prepared Mo2C/NCF exhibited excellent HER performance with small onset overpotential, low Tafel slopes, and superior durability both in acidic and basic solution. Most recently, ultra-small 1 nm Mo2C nanoparticles confined in mesh-like N-doped C (denoted as Mo2C@NC) were fabricated using dicyandiamide as a C precursor (Fig. 13a).34 As displayed in the HR-TEM image (Fig. 13b), the Mo2C@NC had a fine pore size between 2 and 15 nm, and the corresponding lattice fringes (∼0.32 nm) on the carbon nanolayers around the Mo2C corresponds to the (004) graphene crystal plane. Furthermore, the as-prepared Mo2C@NC offered exceptional HER activity with a low overpotential of 36 mV at the current density of −10 mA cm−2 and a high TOF of 0.046 H2 s−1 at the overpotential of 40 mV (Fig. 13c and d). DFT calculations further demonstrated that the N dopants in the carbon layers close to an Mo atom could withdraw an electron and introduce a fast electron-transfer process, which contributes to the enhanced performance of the HER (Fig. 13e). Mu and co-workers166 developed a novel colloid-confinement method for the preparation of ultrafine Mo2C nanoparticles with sub-2 nm size assembled in the in situ formed amorphous carbon foams (denoted as uf-Mo2C/CF). Uniformly sized SiO2 nanospheres and glucose were chosen as the template and C precursors, respectively. The as-obtained uf-Mo2C/CF exhibited excellent and stable activity for the HER in the whole pH range due to the 3D hierarchical architecture, synergistic effects of ultrasmall size, and low valence states of the Mo atoms on the surface. This work might provide some new insights into the fabrication of ultrafine nanocrystals assembled in the 3D porous frameworks as a class of highly active electrocatalysts.
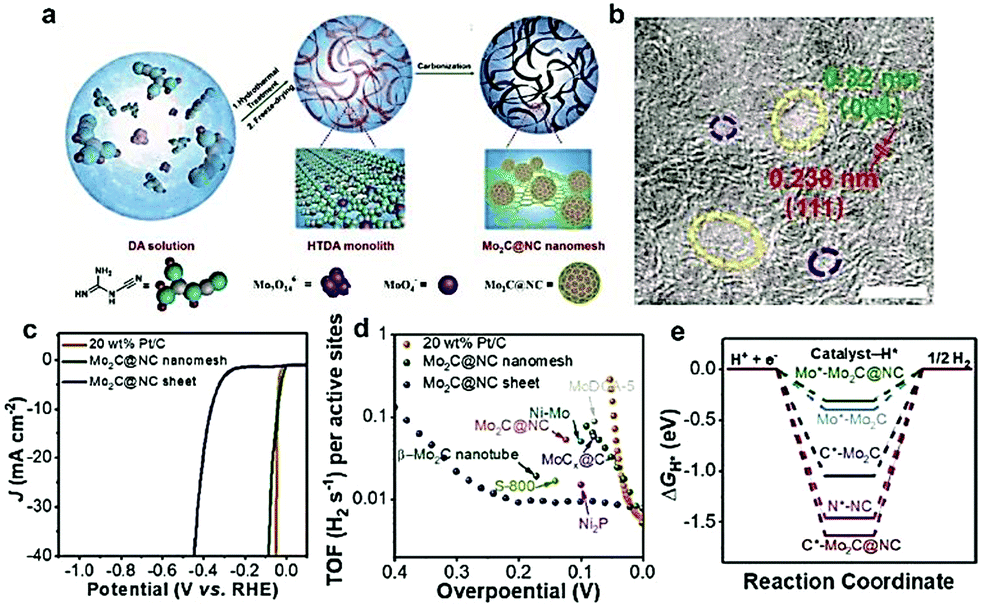 | ||
| Fig. 13 (a) Schematic illustration of the synthesis procedure for the as-prepared Mo2C@NC nanomesh. (b) HRTEM image of the Mo2C@NC nanomesh and corresponding crystal lattice fringes of the Mo2C nanoparticle and N-doped carbon. (c) Polarization curves of the Mo2C@NC nanomesh in comparison with Mo2C@NC sheet and Pt/C. (d) TOF plots of Mo2C@NC nanomesh in comparison with Mo2C@NC sheet, Pt/C, and other reported noble-metal free catalyst. (e) Theoretical calculations of the free adsorption energy of the sites of the catalyst (the * represents the H atoms adsorption site). Reproduced with permission from ref. 34. Copyright 2018, John Wiley and Sons, Inc. | ||
4.2 TMCs/other metal compounds
Apart from the hybridization with carbon-based materials, other metal or metal compounds have also been regarded as a promising alternative to tune the interface of the as-prepared nanocomposites.34,167 Moreover, regulating the chemical composition is a promising method to tune the adsorption/desorption energy of intermediates for the design of highly efficient electrocatalysts.96 In addition, the incorporation of multiple components is also expected to induce strong synergistic effects, such as electronic structure modulation, atomic arrangement, and interfacial stabilization, which are beneficial for the improvement in the electrocatalytic performance.79 For instance, Dai and co-workers156 prepared Mo2C/MoO2 heteronanorods via manipulating the pyrolysis temperature, which displayed high HER activity in the whole pH range. The excellent electrocatalytic performances were mostly contributed by the synergistic effects of the heterointerfaces between MoO2 and Mo2C. Wang and co-workers155 reported the sulfur (S) decoration of Mo2C, which exhibited a great enhancement in the final HER activity due to the synergistic effect between MoSx and Mo2C. Similarly, Xin Wang and co-workers96 prepared a novel Mo2C/WC catalyst with a well-defined and highly robust 1D structure. They demonstrated that the Mo atoms were dissolved into the WC lattice during the carburization process improving the structural stability. Bao and co-workers168 synthesized free-standing vertically aligned MoS2/Mo2C nanosheets by integrating a hydrothermal method with gas-phase carburization (Fig. 14a and b). This MoS2/Mo2C electrocatalyst exhibited excellent activity for the HER, achieving a low overpotential of 63 mV at the current density of 10 mA cm−2 and a small Tafel slope of 48 mV dec−1 (Fig. 14c). DFT calculations demonstrated that the excellent HER activity of the MoS2/Mo2C electrocatalyst was a consequence of enhanced number and activity of Mo-S-C sites formed during the carburization process. Similarly, Tang and co-workers169 developed MoC–Mo2C heteronanowires (denoted as MoC–Mo2C-x, where x refers to the MoC weight percentage) as high performance HER electrocatalysts using MoOx-amine as the precursor and investigated the role of MoC nanocrystals. The interfaces between MoC and Mo2C were visible in the TEM image with clear lattice fringes (Fig. 14d). It is worth noting that the HER activities of the MoCx-based electrocatalysts (such as MoC, MoC–Mo2C-31.4, MoC–Mo2C-68.1, and Mo2C) were highly dependent on the Mo3+/Mo2+ ratio (n3+/2+), evident from the current density at j0 (η = 0 mV) and j150 (η = 150 mV). As displayed in Fig. 14e and f, the MoC–Mo2C-31.4 (n3+/2+ = 3) exhibited higher HER activities compared to Mo2C (n3+/2+ = 0.4), MoC–Mo2C-68.1 MoC (n3+/2+ = 7.2), and MoC (n3+/2+ = 10.9), owing to the optimized H-binding of the MoC/Mo2C interfaces by virtue of the optimized MoC–Mo2C ratio.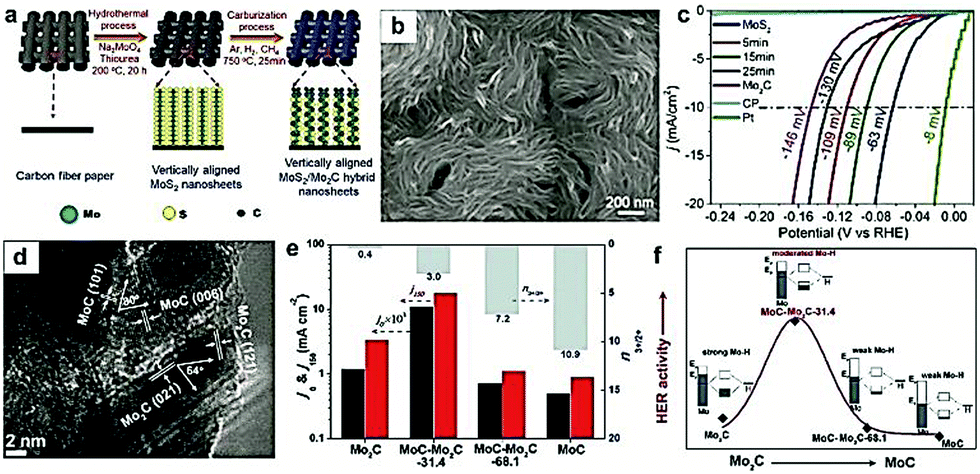 | ||
| Fig. 14 (a) Schematic illustration of the steps for preparation of vertically aligned MoS2/Mo2C nanosheets. (b) SEM image of the MoS2/Mo2C hybrid nanosheets. (c) LSV curves of vertically aligned MoS2/Mo2C nanosheets and the 5, 15, 25, and 60 min (Mo2C) samples. Reproduced with permission from ref. 182. Copyright 2017, American Chemical Society. (d) HRTEM image of the as-prepared MoC–Mo2C-31.4 heterojunction nanowires. (e) j0 (current density at η = 0 mV) and j150 (current density at η = 150 mV) for Mo2C, MoC–Mo2C-31.4, MoC–Mo2C-68.1, and MoC as a function of Mo3+/Mo2+ ratio in 0.5 M H2SO4. (f) Schematic illustration of the HER activity depending on the electron density of Mo in Mo2C, MoC–Mo2C-31.4, MoC–Mo2C-68.1, and MoC electrocatalysts. Reproduced with permission from ref. 168. Copyright 2017, American Chemical Society. | ||
Most recently, MOFs have been widely used as both the precursor and template to construct highly active multiple interfaces due to their well-defined nanostructure, high surface area, and uniformly dispersed metal ions.170–172 In this regard, Luo and co-workers173 fabricated a CoP-Mo2C heterojunction using the Co-based ZIF-67 and an Mo-based compound as the precursor. The as-prepared CoP/Mo2C–NC hybrid catalyst exhibited a significantly improved HER activity over the whole pH range with a small overpotential of ∼55.7 mV at the current density of 10 mA cm−2. The XPS analysis confirmed that the electron could transfer from Co to Mo through the Co–P–Mo bond, leading to the generation of a higher Co valence and lower Mo valences, which could effectively optimize the H binding energy during the electrochemical HER process. In addition, Arne Thomas and co-workers44 developed a metal–organic coordination-precursor assisted method to prepare mesoporous Mo-based carbon catalysts (denoted as mC-Mo) consisting of uniformly distributed Mo2C–Mo2N heterojunctions. As shown in Fig. 15a, distinct from other investigations on Mo-based precursors via MOF precursors, this novel strategy was a fast and controlled aqueous self-assembly process based on dopamine (DA) hydrochloride and sodium molybdate. The introduced Si nanoparticles not only acted as a template but also played a crucial role in stabilizing the formed microspheres, thus resulting in the homogeneous assembly of isolated organic–inorganic microspheres. The TEM image demonstrated that the as-prepared mC-Mo exhibited a uniform spherical morphology (Fig. 15b). As shown in Fig. 15c, a large amount of ultrasmall overlapping crystals of Mo2C or Mo2N were formed in the boundary regions between Mo2C and Mo2N, thus indicating the in situ generation of heterojunctions in the electrocatalysts. Benefiting from the Mo2C/Mo2N heterojunctions and the mesoporous structure, the as-prepared mC-Mo electrocatalysts exhibited an impressive HER activity with a low overpotential of ∼173 mV at the current density of 10 mA cm−2 and excellent stability (Fig. 15d and e). Other efforts have been devoted to constructing an efficient interface between the metal and metal carbides. For example, Yangguang Li and co-workers79 reported multi-interfacial Ni/WC hybrid nanoparticles anchored on N-doped carbon sheets (denoted as Ni/WC@NC) using a two-step strategy as shown in Fig. 16a. Polyoxometalate (POM) molecular cages [Ni(en)2(H2O)2]6{Ni6(Tris)(en)3(BTC)1.5(B-αPW9O34)}8·12en·54H2O (denoted as Ni54W72) were chosen as the metal source as the Ni and W elements in this POM cluster were naturally intimately linked with each other, which might facilitate the in situ formation of a Ni/WC interwoven architecture with multiple interfaces. Moreover, the introduced rGO not only favored the good dispersion of POM clusters but also promoted the deoxygenation and collapse of Ni54W72. The TEM image confirmed that the Ni/WC hybrid nanoparticles had a fine size between 4 and 14 nm and intimately anchored in the N-doped carbon sheets (Fig. 16b and c). The HRTEM image of one Ni/WC nanoparticle confirmed the formation of multiple interfaces between Ni and WC (Fig. 16d), which might benefit the mass and charge transfer of the hybrid. Furthermore, the loose N-doped carbon sheets around Ni/WC nanoparticles not only offered rich defect sites but also stabilized the Ni/WC during the electrochemical process. The as-prepared Ni/WC@NC exhibited an excellent HER activity in the acidic electrolyte (0.5 M H2SO4) with a low overpotential of 53 and 125 mV to reach 10 and 50 mA cm−2, respectively, which is very close to that of commercial Pt/C (Fig. 16e). The DFT calculations demonstrated that the Ni/WC@NC had a weaker ΔGH* than either of the parent surfaces (Fig. 16f), confirming optimized electronic interaction between Ni and WC. Importantly, the in situ X-ray adsorption spectroscopy (XAS) further verified a conceivable electron transfer process (from WC to Ni) and a synergistic mass transport process occurring in Ni/WC@NC during the electrocatalytic HER process. Analogously, a similar improvement mechanism whereby multiple interfaces could facilitate the electrochemical activity was also verified in the Mo/Mo2C heteronanosheets.174 Xiaofeng Lu and co-workers183 synthesized N-doped carbon nanofibers integrated with Ni and Mo2C nanocrystals (denoted as Ni/Mo2C–NCNFs) and investigated the relationship between the interfaces and the improved electrocatalytic activity. They proposed that the close interaction of Ni (weak H binding energy) with Mo2C (strong H binding energy), could form a moderate metal–H binding energy, which would benefit the adsorption and desorption of Hads. Most importantly, Ni/Mo2C–NCNFs possessed a higher Ni valence and lower Mo valence caused by the electron transfer from Ni to Mo2C in their interfaces, which could further facilitate the electrocatalytic activity. Their study further confirmed that constructing hetero-interfaces is an effective method to increase the active sites of the as-prepared hybrids and thus enhance their electrocatalytic performances.
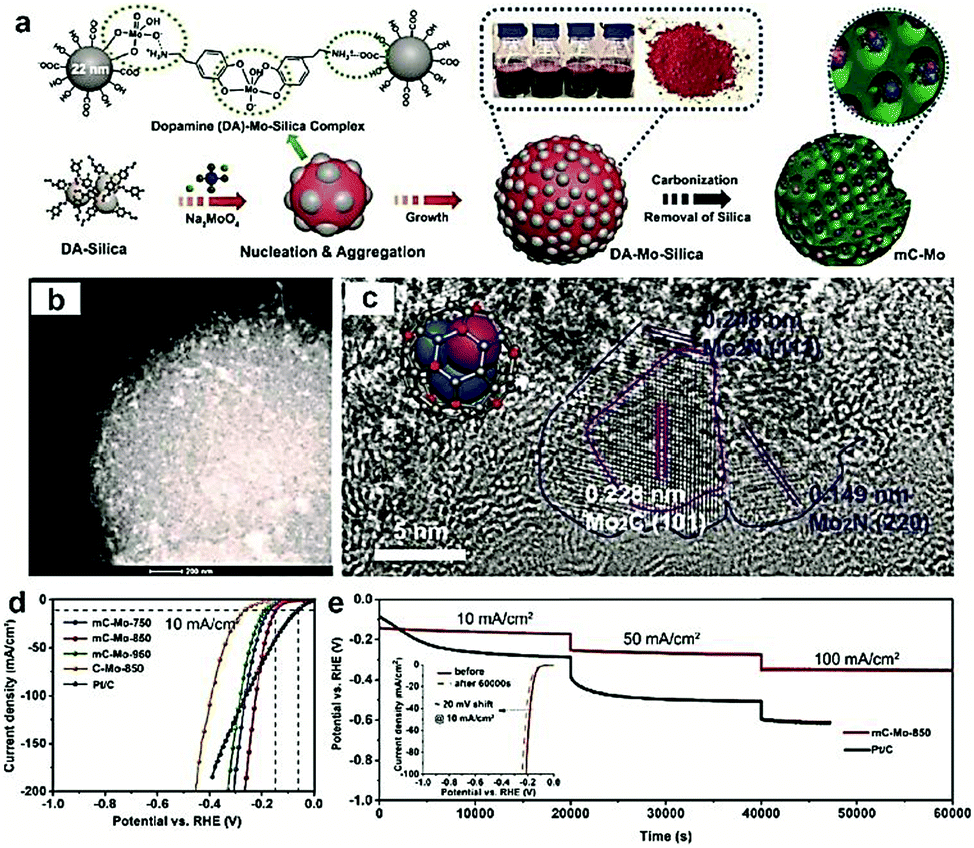 | ||
| Fig. 15 (a) Schematic illustration of the synthesis procedure of Mo2C/Mo2N-doped mesoporous carbon spheres. (b) TEM and (c) HRTEM images of the as-prepared hybrid. (d) Polarization curves of mC-Mo-750, mC-Mo-850, mC-Mo-950, and Pt/C in 1 M KOH. (e) Long-term stability of the mC-Mo-850 and Pt/C electrocatalysts under the various current densities of 10, 50, and 100 mA cm−2, and the inset image exhibits the LSV curves of mC-Mo-850 electrocatalyst before and after stability tests. Reproduced with permission from ref. 44. Copyright 2019, John Wiley and Sons, Inc. | ||
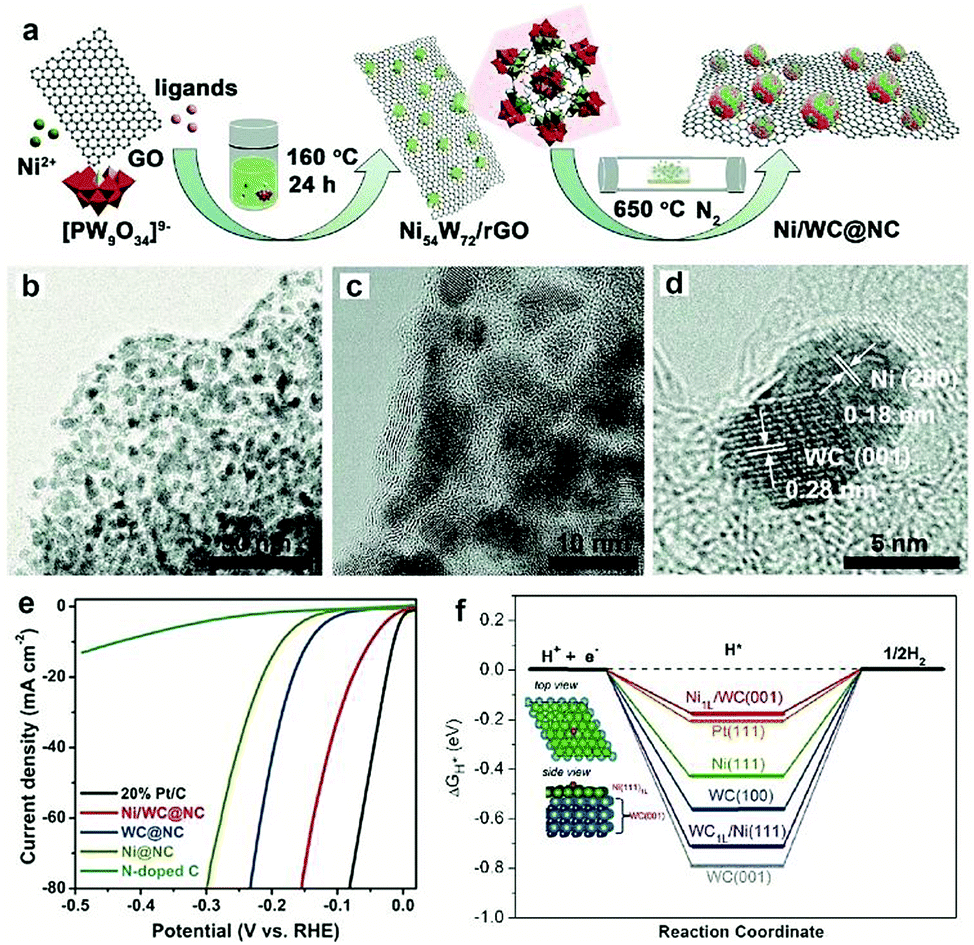 | ||
| Fig. 16 (a) Schematic illustration of the preparation of a Ni/WC@NC catalyst. (b) TEM image and (c and d) HRTEM images of Ni/WC@NC. (e) Polarization curve of Ni/WC@NC Ni@NC, WC@NC, N-doped C and Pt/C in a 0.5 M H2SO4 electrolyte. (f) Gibbs free energy diagram of the HER on different surfaces. Reproduced with permission from ref. 79. Copyright 2018, Royal Society of Chemistry. | ||
5. Surface and interface structure engineering for the NRR
It is well known that theoretical studies and computational modelling are regarded as an effective strategy to predict promising electrocatalysts, including catalysts for the NRR. Furthermore, theoretical investigations might provide a fundamental insight into the experimental rational design of active TMC-based electrocatalysts toward the NRR. For example, Sun and his co-workers175 investigated N2 capture capability of d2 (M = Ti, Zr, and Hf), d3 (M = V, Nb, and Ta), and d4 (M = Cr and Mo) MXenes, considering that TMC materials with predominant capability of N2 capture are in favor of activation for electrochemical conversion into ammonia. Calculation results confirmed that the bonding energy between d2 MXenes and CO2 was much higher than that of N2 and H2O. However, the chemical adsorption capacity of d3 and d4 MXenes to N2 was higher than that of CO2 and H2O. Videlicet, d3 and d4 MXenes displayed favorable selectivity to N2-philicity, which encouraged their applications for electrocatalytic NRR under ambient conditions. Similarly, Garzon and co-workers176 demonstrated that higher index surfaces and defects could render the MoC with the capability of driving the NRR in an aqueous or humid environment using DFT calculations. The comparison between the adsorption energies of N-adatoms and H-adatoms exhibited that MoC(111) is the only surface at which the HER process might be suppressed at low potentials. According to the free energy diagrams for electrocatalytic N2 to ammonia on the surface of MoC(111), a low cell potential of −0.3 V relative to the standard hydrogen electrode (SHE) was required to catalyse the NRR process. Furthermore, the introduction of C vacancies, namely increasing the ratio of Mo to C atoms, could mitigate the HER and thus enhance the affinity of the active surface for both N2 and N-adatoms. Recently, Sun and his colleagues177 proposed that H-terminated molybdenum carbides were potential catalysts for electrocatalytic nitrogen reduction. They demonstrated that doping highly electronegative atoms at pre-defined locations is a unique approach to design non-metal terminated carbides with tailored N2 adsorption energy suitable for NH3 release. DFT calculations confirmed that the interaction between Mo sites and N2 molecules weakened in the H terminated Mo2C(100) surface, which further promoted NH3 release during an NRR process. As mentioned above, the same electrocatalyst would often have active sites for NRR and HER. Therefore, selectivity should be considered. When considering the electrocatalyst design, an effective NRR catalyst should have desired surface characteristics, textural structure, and crystal structure which enhance the strong binding with N-adatoms rather than H-adatoms.178,179From such a perspective, Wang et al.102 prepared Mo2C nanodots embedded on ultrathin carbon nanosheets (Mo2C/C) and explored their nitrogen reduction performance (Fig. 17a). The Mo2C nanodots with a size of 3.5 nm anchored on the ultra-thin carbon (Fig. 17b–d). The inlaid structure of Mo2C dots could availably reduce the coverage of H spillover on the electrocatalyst surface, thus offering more chance for N2 diffusion and adsorption on the surface of the electrocatalyst. As a result, the as-synthesized Mo2C/C exhibited superior electrocatalytic performances with a maximum NH3 yield rate of 11.3 μg h−1 mg−1 at an applied voltage of −0.3 V vs. RHE (Fig. 17e) and the highest faradaic efficiency of 1.6% occurred at −0.2 V vs. RHE (Fig. 17f). Meanwhile, Mo2C/C also displayed the good NRR electrocatalytic stability owing to its excellent structural stability (Fig. 17g). Beyond the regulation of dimension tuning, research indicated that carbides with a porous structure are potential nitrogen reduction catalysts. For instance, Sun and his co-workers reported that porous Mo2C nanorods displayed excellent NRR performance.103 It provided important experimental evidence of the applications of TMC materials for electrocatalytic NRR. The yield of NH3 was 95.1 μg h−1 mgcat−1 at −0.3 V in 0.1 M HCl and the faradaic efficiency was up to 8.13%. In addition, porous Mo2C nanorods exhibited remarkable long-term stability and good selectivity of NH3. Thus, experimental and theoretical calculation results indicated that porous carbide structures are promising electrocatalysts for nitrogen fixation under mild conditions. Up to now, only a few TMC-based electrocatalysts can catalyse N2 reduction, although, there is still much space for improvement of underexplored reactions.
 | ||
| Fig. 17 (a) Illustration of the molten salt synthesis route for Mo2C/C nanosheets. (b) AFM images of the Mo2C/C nanosheets. (c) TEM images of the Mo2C/C nanosheets. (d) HRTEM image of the Mo2C/C nanosheets, the inset in (d) is the corresponding fast-Fourier-transform (FFT) pattern. (e) NH3 synthesis yield rate. (f) Corresponding faradaic efficiency. (g) NH3 synthesis yield rate and faradaic efficiency of each cycle at −0.3 V versus RHE. Ref. 102. Copyright 2018, John Wiley and Sons, Inc. | ||
6. Conclusions and outlook
Electrochemical energy-related technologies, such as water splitting, the nitrogen cycle, and the carbon cycle, play an important role in next-generation clean energy devices. TMC materials due to their appealing properties of high conductivity, high chemical stability and thermal stability have been highly explored in electrocatalytic hydrogen evolution reaction and nitrogen reduction reaction.180 In this review, we summarize the effects of surface engineering and interface engineering on the electrocatalytic performance of the HER and NRR of carbides from theoretical calculations and experiments. Surface and interface engineering strategies are beneficial for adjusting the unique physical/chemical properties of TMCs, such as phase modulation, constructing porous structures with different morphology, preparing nano-sized catalysts, tailoring high index crystal facets, doping with heteroatoms, fabricating surface defects, and forming interfaces between carbides and carbon materials, metals, and metal compounds. The surface and interface engineering strategies mentioned in this review provide an important guiding principle for the design and preparation of HER and NRR electrocatalysts in the future, to a certain degree. However, challenges prevail for the design of highly active catalysts due to lack of in-depth understanding of the reaction process and mechanism of the HER and NRR. In view of the electrocatalytic HER, carbides exhibit splendid catalytic activity and stability under both acid and basic conditions. Their overpotential at 10 mA cm−2 (η10) was only tens to hundreds of millivolts. Some of them were comparable to the performance of precious metal electrocatalysts. The electrocatalytic HER performance of TMC electrocatalysts is summarized in Table 2. It is worth mentioning here that η10 cannot serve as a sole criterion for judging the HER performance of the catalysts, as η10 is greatly affected by the testing setup, electrocatalysts loading, and the choice of counter electrode.181,182Judging the hydrogen evolution performance based on Tafel slope and exchange current density along with η10 is particularly important. Furthermore, Pt contamination is worth paying more attention. The use of an ion exchange membrane to separate the working electrode and Pt counter electrode or using carbon rod to replace Pt is recommended. Highly dispersed and nano-sized carbide materials facilitate their use in electrochemically related reactions due to the easy availability of bulk materials for carbides at increased temperatures.
Low activity and poor selectivity are the primary challenges for nitrogen reduction reactions. In the last few years, metals,183 metal oxides,184 sulphides,185 carbides,102 nitrides,186 non-metallic materials,187etc. have been used for the electrocatalytic nitrogen reduction reaction, and the corresponding Faraday efficiency for producing ammonia is not more than 15%. The electrochemical performance properties of some typical nitrogen reduction electrocatalysts in an aqueous electrolyte are shown in Table 3. The ammonia yield of the existing nitrogen reduction electrocatalysts is extremely low, and ammonia is ubiquitous in a laboratory environment. Thus, ammonia contamination should be taken very seriously. 15N isotope labeling experiments should be carefully performed to confirm that the ammonia is only derived from nitrogen.188 Besides, more accurate product detection methods such as ion chromatography and nuclear magnetic resonance spectroscopy should be greatly recommended in electrocatalytic nitrogen reduction reactions.
| Electrocatalyst | Electrolyte | FE (%) | Yield rate | Ref. | |
|---|---|---|---|---|---|
| Carbides | Mo2C/C | 0.5 M Li2SO4 | 7.80 | 11.30 μg h−1 mgMo2C−1 | Adv. Mater., 2018, 30, 1803694 |
| Mo2C nanorod | 0.1 M HCl | 8.13 | 95.10 μg h−1 mgcat−1 | ACS Cent. Sci., 2019, 5, 116 | |
| MXene (Ti3C2Tx) nanosheets | 0.01 M HCl | 5.78 | 4.72 μg h−1 mg−1 | Joule, 2019, 3, 279 | |
| Metal | THH Au NRs | 0.1 M KOH | 4 | 1.648 μg h−1 cm−2 | Adv. Mater., 2017, 29, 1604799 |
| Bi nanocrystals | 0.5 M K2SO4 | 66 | 200 mmol g−1 h−1 | Nat. Catal., 2019, 2, 448 | |
| 0.052 mmol cm−2 h−1 | |||||
| Mo catalysts | 0.1 M KOH | 14.60 ± 1.60 | 34.0 ± 3.6 μg h−1 mgcat−1 | Angew. Chem., Int. Ed., 2019, 58, 2321 | |
| Ru SAC@ZrO2/NC | 0.1 M HCl | 21 | 3.665 mg h−1 mgRu−1 | Chem, 2019, 5, 204 | |
| Fe/NC | 0.1 M PBS | 18.60 ± 0.80 | 62.9 ± 2.7 μg h−1 mgcat−1 | Nano Energy, 2019, 61, 420 | |
| Ag triangular nanoplates | 0.5 M K2SO4 | 25 | 58.5 mg gAg−1 h−1 | Chem. Commun., 2019 | |
| Sulfide | MoS2 | 0.1 M Na2SO4 | 1.17 | 8.08 × 10−11 mol s−1 cm−1 | Adv. Mater., 2018, 30, 1800191 |
| Sn/SnS2 | 0.1 M PBS | 6.50 | 23.8 μg h−1 mg−1 | Small, 2019, 1902535 | |
| Oxide | MoO3 nanosheets | 0.1 M HCl | 1.90 | 29.43 mg h−1 mgcat−1 | J. Mater. Chem. A, 2018, 6, 12974 |
| MoO2 | 0.1 M HCl | 8.20 | 12.20 μg h−1 mg−1 | Nano Energy, 2019, 59, 10. | |
| Nb2O5 nanofiber | 0.1 M HCl | 9.26 | 43.6 μg h−1 mgcat−1 | Nano Energy, 2018, 52, 264 | |
| Nitride | Mo2N nanorod | 0.1 M HCl | 4.50 | 78.4 μg h−1 mgcat−1 | Chem. Commun., 2018, 54, 8474 |
| VN nanowire | 0.1 M HCl | 3.58 | 2.48 × 10−10 mol−1 s−1 cm−2 | Chem. Commun., 2018, 54, 5323 | |
| Non-metallic material | Polymeric carbon nitride | pH = 1 HCl | 11.59 | 8.09 μg h−1 mgcat−1 | Angew. Chem., Int. Ed., 2018, 57, 10246 |
| Nitrogen-doped porous carbon | 0.1 M HCl | 1.45 | 15.7 μg h−1 mgcat−1 | J. Mater. Chem. A, 2018, 6, 7762 | |
| N,P co-doped carbon | 0.1 M HCl | 4.20 | 0.97 μg h−1 mgcat−1 | Chem. Commun., 2019, 55, 687 | |
| Other | Bi4V2O11/CeO2 hybrid | pH = 1 HCl | 10.16 | 23.21 μg h−1 mgcat−1 | Angew. Chem., Int. Ed., 2018, 57, 6073 |
| Zr4+-doped anatase TiO2 | 0.1 M KOH | 17.30 | 8.90 μg h−1 cm−2 | Nat. Commun., 2019, 10, 2877 | |
| SeO/Te nanrod | 0.1 M HCl | 24.56 | 21.54 μg h−1 mg−1 | Adv. Sci., 2019, 1901627 | |
At present, the investigation of the HER and NRR is still in the laboratory stage. Information on the reaction rate descriptor, reactive active sites and reaction mechanisms is urgently needed to guide design of the electrocatalyst. Hence, we propose that the following potential research opportunities need to be addressed in the field of carbide applications of the HER and NRR. First of all, the importance of the catalyst–electrolyte interface in addition to the electrocatalysts, which can be explored in-depth by many in situ or in operando analytical methods and optimized by surface modification of electrocatalysts, regulation of electrolyte composition, etc. Combination of experimental and theoretical calculations to probe into the electrocatalytic hydrogen evolution and nitrogen reduction mechanism and confirm the reactive sites is of great importance for the design of electrocatalysts. Next, the above-mentioned surface and interface engineering have not been widely used in carbide electrocatalysts. Thus, designing an ideal structure that can overcome the drawbacks of carbides to provide desired electro-chemical performance is very attractive and important for next-generation clean energy devices.
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors gratefully acknowledge the financial support from Singapore MOE AcRF Tier 1 under grant No. RG119/16 and 2017-T1-002-009, Singapore MOE AcRF Tier 2 under Grant No. 2017-T2-2-069 and 2018-T2-01-010, and Guoxuan-NTU joint lab.References
- J. Liu, D. Zhu, Y. Zheng, A. Vasileff and S. Z. Qiao, ACS Catal., 2018, 8, 6707 CrossRef CAS.
- M. Kuang and G. Zheng, Small, 2016, 12, 5656 CrossRef CAS.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439 CrossRef CAS.
- Y. Liu, Y. Li, H. Kang, T. Jin and L. Jiao, Mater. Horiz., 2016, 3, 402 RSC.
- K.-H. Liu, H.-X. Zhong, S.-J. Li, Y.-X. Duan, M.-M. Shi, X.-B. Zhang, J.-M. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64 CrossRef CAS.
- L. Tao, M. Qiao, R. Jin, Y. Li, Z. Xiao, Y. Wang, N. Zhang, C. Xie, Q. He, D. Jiang, G. Yu and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 1019 CrossRef CAS.
- N. Cao and G. Zheng, Nano Res., 2018, 11, 2992 CrossRef CAS.
- Z.-L. Wang, K. Sun, J. Henzie, X. Hao, Y. Ide, T. Takei, Y. Bando and Y. Yamauchi, Mater. Horiz., 2018, 5, 1194 RSC.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012 RSC.
- C. Guo, J. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45 RSC.
- M. Kuang, P. Han, L. Huang, N. Cao, L. Qian and G. Zheng, Adv. Funct. Mater., 2018, 28, 1804886 CrossRef.
- C.-S. Li, Y. Sun, W.-H. Lai, J.-Z. Wang and S.-L. Chou, ACS Appl. Mater. Interfaces, 2016, 8, 27710 CrossRef CAS.
- S. Laha, Y. Lee, F. Podjaski, D. Weber, V. Duppel, L. M. Schoop, F. Pielnhofer, C. Scheurer, K. Müller and U. Starke, Adv. Energy Mater., 2019, 1803795 CrossRef.
- M. Wu, Q. Wei, G. Zhang, J. Qiao, M. Wu, J. Zhang, Q. Gong and S. Sun, Adv. Energy Mater., 2018, 8, 1801836 CrossRef.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215 CrossRef CAS.
- M. Zeng, Y. Chen, J. Li, H. Xue, R. G. Mendes, J. Liu, T. Zhang, M. H. Ruemmeli and L. Fu, Nano Energy, 2017, 33, 356 CrossRef CAS.
- T. Ouyang, Y. Q. Ye, C. Y. Wu, K. Xiao and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 4923 CrossRef CAS.
- Y. Luo, G.-F. Chen, L. Ding, X. Chen, L.-X. Ding and H. Wang, Joule, 2019, 3, 279 CrossRef CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675 CrossRef CAS.
- P. Yu, F. Wang, T. A. Shifa, X. Zhan, X. Lou, F. Xia and J. He, Nano Energy, 2019, 58, 244 CrossRef CAS.
- J. Pang, J. Sun, M. Zheng, H. Li, Y. Wang and T. Zhang, Appl. Catal., B, 2019, 254, 510 CrossRef CAS.
- Y. Liu, T. G. Kelly, J. G. Chen and W. E. Mustain, ACS Catal., 2013, 3, 1184 CrossRef CAS.
- Y. Zhong, X. Xia, F. Shi, J. Zhan, J. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef.
- L. S. Macedo, R. R. Oliveira Jr, T. van Haasterecht, V. T. da Silva and H. Bitter, Appl. Catal., B, 2019, 241, 81 CrossRef.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407 CrossRef CAS.
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem. – Eur. J., 2017, 23, 10947 CrossRef CAS.
- X. Zang, W. Chen, X. Zou, J. N. Hohman, L. Yang, B. Li, M. Wei, C. Zhu, J. Liang and M. Sanghadasa, Adv. Mater., 2018, 30, 1805188 CrossRef.
- K. Zhang, G. Zhang, J. Qu and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 2451 CrossRef CAS.
- S. Y. Bae, I. Y. Jeon, J. Mahmood and J. B. Baek, Chem. – Eur. J., 2018, 24, 18158 CrossRef CAS.
- J. Jia, T. Xiong, L. Zhao, F. Wang, H. Liu, R. Hu, J. Zhou, W. Zhou and S. Chen, ACS Nano, 2017, 11, 12509 CrossRef CAS.
- A. M. Gómez–Marín and E. A. Ticianelli, Appl. Catal., B, 2017, 209, 600 CrossRef.
- W. Yuan, Q. Huang, X. Yang, Z. Cui, S. Zhu, Z. Li, S. Du, N. Qiu and Y. Liang, ACS Appl. Mater. Interfaces, 2018, 10, 40500 CrossRef CAS.
- J. Guo, Z. Mao, X. Yan, R. Su, P. Guan, B. Xu, X. Zhang, G. Qin and S. J. Pennycook, Nano Energy, 2016, 28, 261 CrossRef CAS.
- Z. Cheng, Q. Fu, Q. Han, Y. Xiao, Y. Liang, Y. Zhao and L. Qu, Adv. Funct. Mater., 2018, 28, 1705967 CrossRef.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590 CrossRef CAS.
- S. Wang, J. Wang, M. Zhu, X. Bao, B. Xiao, D. Su, H. Li and Y. Wang, J. Am. Chem. Soc., 2015, 137, 15753 CrossRef CAS.
- H. Jin, J. Chen, S. Mao and Y. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 22094 CrossRef CAS.
- L. Zhang, H. Yang, D. K. Wanigarathna and B. Liu, Small Methods, 2018, 2, 1700353 CrossRef.
- Y. Hu, B. Yu, W. Li, M. Ramadoss and Y. Chen, Nanoscale, 2019, 11, 4876 RSC.
- W.-F. Chen, C.-H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. Muckerman, Y. Zhu and R. Adzic, Energy Environ. Sci., 2013, 6, 943 RSC.
- S. K. Kim, Y. Qiu, Y.-J. Zhang, R. Hurt and A. Peterson, Appl. Catal., B, 2018, 235, 36 CrossRef CAS.
- L. F. Pan, Y. H. Li, S. Yang, P. F. Liu, M. Q. Yu and H. G. Yang, Chem. Commun., 2014, 50, 13135 RSC.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS.
- S. Li, C. Cheng, A. Sagaltchik, P. Pachfule, C. Zhao and A. Thomas, Adv. Funct. Mater., 2019, 29, 1807419 CrossRef.
- Q. Gong, Y. Wang, Q. Hu, J. Zhou, R. Feng, P. N. Duchesne, P. Zhang, F. Chen, N. Han, Y. Li, C. H. Jin, Y. G. Li and S.-T. Lee, Nat. Commun., 2016, 7, 13216 CrossRef CAS.
- X. Yang, Y. C. Kimmel, J. Fu, B. E. Koel and J. G. Chen, ACS Catal., 2012, 2, 765 CrossRef CAS.
- R. Levy and M. Boudart, Science, 1973, 181, 547 CrossRef CAS.
- Q. Zhang, Z. Jiang, B. M. Tackett, S. R. Denny, B. Tian, X. Chen, B. Wang and J. G. Chen, ACS Catal., 2019, 9, 2415 CrossRef CAS.
- L. Peng, J. Shen, L. Zhang, Y. Wang, R. Xiang, J. Li, L. Li and Z. Wei, J. Mater. Chem. A, 2017, 5, 23028 RSC.
- X. Zhao, W. Sun, D. Geng, W. Fu, J. Dan, Y. Xie, P. R. Kent, W. Zhou, S. J. Pennycook and K. P. Loh, Adv. Mater., 2019, 31, 1808343 CrossRef PubMed.
- Z. Chen, X. Lang and Q. Jiang, J. Mater. Chem. A, 2018, 6, 9623 RSC.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1 Search PubMed.
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942 RSC.
- J. Li and G. Zheng, Adv. Sci., 2017, 4, 1600380 CrossRef.
- M. Kuang, P. Han, Q. Wang, J. Li and G. Zheng, Adv. Funct. Mater., 2016, 26, 8555 CrossRef CAS.
- M. Kuang, Q. Wang, P. Han and G. Zheng, Adv. Energy Mater., 2017, 7, 1700193 CrossRef.
- S. BibliographyC., C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef.
- S. Anantharaj, S. Ede, K. Karthick, S. S. Sankar, K. Sangeetha, P. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744 RSC.
- G. F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L. X. Ding and H. Wang, Small Methods, 2018, 1800337 Search PubMed.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531 RSC.
- B. Hu, M. Hu, L. C. Seefeldt and T. L. Liu, ACS Energy Lett., 2019, 4, 1053 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57 CrossRef CAS.
- E. Skulason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235 RSC.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706 CrossRef CAS.
- G. F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L. X. Ding and H. Wang, Small, Methods, 2019, 3, 1800337 Search PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh and B. A. Rohr, Nature, 2019, 570, 504 CrossRef CAS PubMed.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166 RSC.
- J. Halim, S. Kota, M. R. Lukatskaya, M. Naguib, M.-Q. Zhao, E. J. Moon, J. Pitock, J. Nanda, S. J. May, Y. Gogotsi and M. W. Barsoum, Adv. Funct. Mater., 2016, 26, 3118 CrossRef CAS.
- W.-F. Chen, S. Iyer, S. Iyer, K. Sasaki, C.-H. Wang, Y. Zhu, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2013, 6, 1818 RSC.
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Am. Chem. Soc., 2015, 137, 110 CrossRef CAS.
- H. Fan, H. Yu, Y. Zhang, Y. Zheng, Y. Luo, Z. Dai, B. Li, Y. Zong and Q. Yan, Angew. Chem., Int. Ed., 2017, 56, 12566 CrossRef CAS.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590 CrossRef CAS.
- S. T. Hunt, M. Milina, Z. Wang and Y. Román-Leshkov, Energy Environ. Sci., 2016, 9, 3290 RSC.
- T. G. Kelly, S. T. Hunt, D. V. Esposito and J. G. Chen, Int. J. Hydrogen Energy, 2013, 38, 5638 CrossRef CAS.
- F. Yu, Y. Gao, Z. Lang, Y. Ma, L. Yin, J. Du, H. Tan, Y. Wang and Y. Li, Nanoscale, 2018, 10, 6080 RSC.
- Y.-Y. Ma, Z.-L. Lang, L.-K. Yan, Y.-H. Wang, H.-Q. Tan, K. Feng, Y.-J. Xia, J. Zhong, Y. Liu, Z.-H. Kang and Y.-G. Li, Energy Environ. Sci., 2018, 11, 2114 RSC.
- Y. Ma, G. Guan, C. Shi, A. Zhu, X. Hao, Z. Wang, K. Kusakabe and A. Abudula, Int. J. Hydrogen Energy, 2014, 39, 258 CrossRef CAS.
- M. A. R. Anjum, M. H. Lee and J. S. Lee, ACS Catal., 2018, 8, 8296 CrossRef CAS.
- L. Ji, J. Wang, X. Teng, H. Dong, X. He and Z. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 14632 CrossRef CAS.
- Z. Shi, K. Nie, Z.-J. Shao, B. Gao, H. Lin, H. Zhang, B. Liu, Y. Wang, Y. Zhang, X. Sun, X.-M. Cao, P. Hu, Q. Gao and Y. Tang, Energy Environ. Sci., 2017, 10, 1262 RSC.
- Y.-Y. Chen, Y. Zhang, W.-J. Jiang, X. Zhang, Z. Dai, L.-J. Wan and J.-S. Hu, ACS Nano, 2016, 10, 8851 CrossRef CAS.
- H. Ang, H. T. Tan, Z. M. Luo, Y. Zhang, Y. Y. Guo, G. Guo, H. Zhang and Q. Yan, Small, 2015, 11, 6278 CrossRef CAS.
- R. Michalsky, Y.-J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909 CrossRef CAS.
- K. Xiong, L. Li, L. Zhang, W. Ding, L. Peng, Y. Wang, S. Chen, S. Tan and Z. Wei, J. Mater. Chem. A, 2015, 3, 1863 RSC.
- C. Wan and B. M. Leonard, Chem. Mater., 2015, 27, 4281 CrossRef CAS.
- Q. Cao, L. Zhao, A. Wang, L. Yang, L. Lai, Z.-L. Wang, J. Kim, W. Zhou, Y. Yamauchi and J. Lin, Nanoscale, 2019, 11, 1700 RSC.
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Adv. Energy Mater., 2017, 7, 1700020 CrossRef.
- Y. Shi, Y. Zhou, D.-R. Yang, W.-X. Xu, C. Wang, F.-B. Wang, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479 CrossRef CAS.
- X. Wang, R. Su, H. Aslan, J. Kibsgaard, S. Wendt, L. Meng, M. Dong, Y. Huang and F. Besenbacher, Nano Energy, 2015, 12, 9 CrossRef CAS.
- H. Zhang, X. Huang, O. Noonan, L. Zhou and C. Yu, Adv. Funct. Mater., 2017, 27, 1606023 CrossRef.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387 RSC.
- P. Xiao, X. Ge, H. Wang, Z. Liu, A. Fisher and X. Wang, Adv. Funct. Mater., 2015, 25, 1520 CrossRef CAS.
- S. Jing, L. Zhang, L. Luo, J. Lu, S. Yin, P. K. Shen and P. Tsiakaras, Appl. Catal., B, 2018, 224, 533 CrossRef CAS.
- P. Xiao, Y. Yan, X. Ge, Z. Liu, J.-Y. Wang and X. Wang, Appl. Catal., B, 2014, 154, 232 CrossRef.
- J. Wan, J. Wu, X. Gao, T. Li, Z. Hu, H. Yu and L. Huang, Adv. Funct. Mater., 2017, 27, 1703933 CrossRef.
- G. Humagain, K. MacDougal, J. MacInnis, J. M. Lowe, R. H. Coridan, S. MacQuarrie and M. Dasog, Adv. Energy Mater., 2018, 8, 1801461 CrossRef.
- C. He and J. Tao, J. Catal., 2017, 347, 63 CrossRef CAS.
- H. Cheng, L. X. Ding, G. F. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef.
- X. Ren, J. Zhao, Q. Wei, Y. Ma, H. Guo, Q. Liu, Y. Wang, G. Cui, A. M. Asiri, B. Li, B. Tang and X. P. Sun, ACS Cent. Sci., 2018, 5, 116 CrossRef.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. D. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS.
- S. Li, C. Cheng, A. Sagaltchik, P. Pachfule, C. Zhao and A. Thomas, Adv. Funct. Mater., 2019, 29, 1807419 CrossRef.
- F. Li, X. Zhao, J. Mahmood, M. S. Okyay, S.-M. Jung, I. Ahmad, S.-J. Kim, G.-F. Han, N. Park and J.-B. Baek, ACS Nano, 2017, 11, 7527 CrossRef CAS.
- A. Mahmood, W. Guo, H. Tabassum and R. Zou, Adv. Energy Mater., 2016, 6, 1600423 CrossRef.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- P. Chen, Y. Tong, C. Wu and Y. Xie, Acc. Chem. Res., 2018, 51, 2857 CrossRef CAS.
- Z. L. Wang, J. Phys. Chem. B, 2000, 140, 1153 CrossRef.
- Y.-N. Wen and J.-M. Zhang, Solid State Commun., 2007, 144, 163 CrossRef CAS.
- P. Swetha and S.-P. Feng, Electrochem. Commun., 2018, 94, 64 CrossRef CAS.
- N. Tian, Z.-Y. Zhou, N.-F. Yu, L.-Y. Wang and S.-G. Sun, J. Am. Chem. Soc., 2010, 132, 7580 CrossRef CAS.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS.
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2013, 46, 191 CrossRef CAS.
- N. Zhang, L. Bu, S. Guo, J. Guo and X. Huang, Nano Lett., 2016, 16, 5037 CrossRef CAS.
- S. Luo and P. K. Shen, ACS Nano, 2017, 11, 11946 CrossRef CAS.
- Z. Kou, K. Xi, Z. Pu and S. Mu, Nano Energy, 2017, 36, 374 CrossRef CAS.
- K. Hata, T. Ikoma, K. Hirakawa, T. Okano, A. Kawazu, T. Ueda and M. Akiyama, J. Appl. Phys., 1994, 76, 5601 CrossRef CAS.
- D. A. Ersoy, M. J. McNallan and Y. Gogotsi, Mater. Res. Innovations, 2001, 5, 55 CrossRef CAS.
- L. Chen, G. Behlau, Y. Gogotsi and M. McNallan, Carbide derived carbon (CDC) coatings for tyranno ZMI SiC fibers, John Wiley & Sons, 2003, vol. 4 Search PubMed.
- A. N. Zelikman and L. M. Leonova, Tsvetn. Met., 1978, 19, 54 Search PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T.-K. Sham, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS.
- M. Li, Q. Ma, W. Zi, X. Liu, X. Zhu and S. Liu, Sci. Adv., 2015, 1, e1400268 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- H. Yang, S. Luo, X. Li, S. Li, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 18499 RSC.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807 CrossRef CAS.
- L. Yang, S. Wang, J. Mao, J. Deng, Q. Gao, Y. Tang and O. G. Schmidt, Adv. Mater., 2013, 25, 1180 CrossRef CAS.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2017, 12, 441 CrossRef CAS.
- H. Mistry, A. S. Varela, S. Kuehl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- S. Cao, F. F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747 RSC.
- B. E. Hayden, Acc. Chem. Res., 2013, 46, 1858 CrossRef CAS.
- W. Han, L. Chen, B. Ma, J. Wang, W. Song, X. Fan, Y. Li, F. Zhang and W. Peng, J. Mater. Chem. A, 2019, 7, 4734 RSC.
- S. T. Hunt, T. Nimmanwudipong and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2014, 53, 5131 CAS.
- Y. Hu, B. Yu, W. Li, M. Ramadoss and Y. Chen, Nanoscale, 2019, 11, 4876 RSC.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407 CrossRef CAS.
- K. Hackett, S. Verhoef, R. A. Cutler and D. K. Shetty, J. Am. Ceram. Soc., 2009, 92, 2404 CrossRef CAS.
- L. Limeng, Y. Feng, Z. Yu and Z. Zhiguo, J. Am. Ceram. Soc., 2010, 93, 2945 CrossRef.
- L. Yang, X. Li, Y. Ouyang, Q. Gao, L. Ouyang, R. Hu, J. Liu and M. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 19987 CrossRef CAS.
- M. Kuang, Q. Wang, H. Ge, P. Han, Z. Gu, A. M. Al-Enizi and G. Zheng, ACS Energy Lett., 2017, 2, 2498 CrossRef CAS.
- J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Adv. Funct. Mater., 2019, 19, 1808367 CrossRef.
- K. X. Wang, X. H. Li and J. S. Chen, Adv. Mater., 2015, 27, 527 CrossRef CAS PubMed.
- R. Ma, Y. Zhou, Y. Chen, P. Li, Q. Liu and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14723 CrossRef CAS.
- J. Jia, W. Zhou, Z. Wei, T. Xiong, G. Li, L. Zhao, X. Zhang, H. Liu, J. Zhou and S. Chen, Nano Energy, 2017, 41, 749 CrossRef CAS.
- C. Wu, D. Liu, H. Li and J. Li, Small, 2018, 14, 1704227 CrossRef.
- X. Xu, F. Nosheen and X. Wang, Chem. Mater., 2016, 28, 6313 CrossRef CAS.
- C. Tang, W. Wang, A. Sun, C. Qi, D. Zhang, Z. Wu and D. Wang, ACS Catal., 2015, 5, 6956 CrossRef CAS.
- M. Liu, Y. Yang, X. Luan, X. Dai, X. Zhang, J. Yong, H. Qiao, H. Zhao, W. Song and X. Huang, ACS Sustainable Chem. Eng., 2018, 6, 14356 CrossRef CAS.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603 CrossRef CAS.
- J. W. Wilder, L. C. Venema, A. G. Rinzler, R. E. Smalley and C. Dekker, Nature, 1998, 391, 59 CrossRef.
- H. Wei, Q. Xi, X. A. Chen, D. Guo, F. Ding, Z. Yang, S. Wang, J. Li and S. Huang, Adv. Sci., 2018, 5, 1700733 CrossRef.
- C. Lu, D. Tranca, J. Zhang, F. N. R. Hernández, Y. Su, X. Zhuang, F. Zhang, G. Seifert and X. Feng, ACS Nano, 2017, 11, 3933 CrossRef CAS.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407 CrossRef CAS.
- J. Wang, H. Xia, Z. Peng, C. Lv, L. Jin, Y. Zhao, Z. Huang and C. Zhang, ChemSusChem, 2016, 9, 855 CrossRef CAS.
- F. X. Ma, H. B. Wu, B. Y. Xia, C. Y. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 15395 CrossRef CAS.
- Z. Pu, M. Wang, Z. Kou, I. S. Amiinu and S. Mu, Chem. Commun., 2016, 52, 12753 RSC.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337 CrossRef CAS.
- Z. Kou, T. Wang, Y. Cai, C. Guan, Z. Pu, C. Zhu, Y. Hu, A. M. Elshahawy, J. Wang and S. Mu, Small Methods, 2018, 2, 1700396 CrossRef.
- L. Huo, B. Liu, Z. Gao and J. Zhang, J. Mater. Chem. A, 2017, 5, 18494 RSC.
- Z. Zhao, F. Qin, S. Kasiraju, L. Xie, M. K. Alam, S. Chen, D. Wang, Z. Ren, Z. Wang, L. C. Grabow and J. M. Bao, ACS Catal., 2017, 7, 7312 CrossRef CAS.
- H. Lin, Z. Shi, S. He, X. Yu, S. Wang, Q. Gao and Y. Tang, Chem. Sci., 2016, 7, 3399 RSC.
- Z. Liang, C. Qu, W. Guo, R. Zou and Q. Xu, Adv. Mater., 2018, 30, 1702891 CrossRef.
- J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 10889 CrossRef CAS.
- R. Wang, X. Y. Dong, J. Du, J. Y. Zhao and S. Q. Zang, Adv. Mater., 2018, 30, 1703711 CrossRef.
- X. Luo, Q. Zhou, S. Du, J. Li, L. Zhang, K. Lin, H. Li, B. Chen, T. Wu, D. Chen, M. L. Chang and Y. L. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 42335 CrossRef CAS.
- J. Xiong, J. Li, J. Shi, X. Zhang, N.-T. Suen, Z. Liu, Y. Huang, G. Xu, W. Cai and X. Lei, ACS Energy Lett., 2018, 3, 341 CrossRef CAS.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545 RSC.
- I. Matanovic and F. H. Garzon, Phys. Chem. Chem. Phys., 2018, 20, 14679 RSC.
- Q. Li, S. Qiu, L. He, X. Zhang and C. Sun, Phys. Chem. Chem. Phys., 2018, 20, 23338 RSC.
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, A. N. Simonov, C. McDonnell-Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516 RSC.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706 CrossRef CAS.
- R. Wang, X. Y. Dong, J. Du, J. Y. Zhao and S. Q. Zang, Adv. Mater., 2018, 30, 1703711 CrossRef.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957 CrossRef CAS.
- R. Wei, M. Fang, G. Dong and J. C. Ho, Sci. Bull., 2017, 62, 971 CrossRef CAS.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao and E. Stavitski, Angew. Chem., Int. Ed., 2019, 58, 2321 CrossRef CAS.
- G. Zhang, Q. Ji, K. Zhang, Y. Chen, Z. Li, H. Liu, J. Li and J. Qu, Nano Energy, 2019, 59, 10 CrossRef CAS.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef.
- X. Zhang, R.-M. Kong, H. Du, L. Xia and F. Qu, Chem. Commun., 2018, 54, 5323 RSC.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246 CrossRef CAS.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |