
Robustness in glycosylation systems: effect of modified monosaccharides, acceptor decoys and azido sugars on cellular nucleotide-sugar levels and pattern of N-linked glycosylation†
Virginia
del Solar
ab,
Rohitesh
Gupta
a,
Yusen
Zhou
a,
Gabrielle
Pawlowski
a,
Khushi L.
Matta
a and
Sriram
Neelamegham
 *a
*a
aDepartment of Chemical & Biological Engineering, Biomedical Engineering and Medicine, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: neel@buffalo.edu; Fax: +1 716-645-4822; Tel: +1 716-645-1200
bDepartment of Chemistry, Brooklyn College, City University of New York, New York, NY 11210, USA
First published on 22nd April 2020
Abstract
Small molecule monosaccharide analogs (e.g. 4F-GlcNAc, 4F-GalNAc) and acceptor decoys (e.g. ONAP, SNAP) are commonly used as metabolic glycoengineering tools to perturb molecular and cellular recognition processes. Azido-derivatized sugars (e.g. ManNAz, GlcNAz, GalNAz) are also used as bioorthogonal probes to assay the glycosylation status of cells and tissue. With the goal of obtaining a systems-level understanding of how these compounds work, we cultured cells with these molecules and systematically evaluated their impact on: (i) cellular nucleotide-sugar levels, and (ii) N-linked glycosylation. To this end, we developed a streamlined, simple workflow to quantify nucleotide-sugar levels using amide-based hydrophilic interaction liquid chromatography (HILIC) separation followed by negative-mode electrospray ionization mass spectrometry (ESI-MS/MS) using an Orbitrap detector. N-Glycans released from cells were also procainamide functionalized and quantified using positive-mode ESI-MS/MS. Results show that all tested compounds changed the baseline nucleotide-sugar levels, with the effect being most pronounced for the fluoro-HexNAc compounds. These molecules depressed UDP-HexNAc levels in cells by up to 80%, while concomitantly elevating UDP-4F-GalNAc and UDP-4F-GlcNAc. While the measured changes in nucleotide-sugar concentration were substantial in many cases, their impact on N-linked glycosylation was relatively small. This may be due to the high nucleotide-sugar concentrations in the Golgi, which far exceed the KM values of the glycosylating enzymes. Thus, the glycosylation system output exhibits ‘robustness’ even in the face of significant changes in cellular nucleotide-sugar concentrations.
1. Introduction
Glycosyltransferases (GTs) catalyze the transfer of saccharides (i.e. glycosyl donors) from activated nucleotide-sugars to nucleophilic acceptors.1 The rate of this transferase reaction controls the final distribution of carbohydrate structures on the cell surface. While a number of factors, including the spatial location of the glycosylating enzymes, regulate the nature of the final product, two important parameters that control the rate of individual reactions include: (1) the concentration of the nucleotide-sugar donor, and (2) the activity of the GTs.2 In this regard, traditionally, the vast majority of studies that focus on determining key rate-limiting steps controlling the repertoire of the final glycan products on cells, focus on measuring the GT activity/expression rather than nucleotide-sugar levels.3–6 This is at least in part due to the ready availability of streamlined Sanger/next-generation sequencing methods to quantify cellular GT mRNA expression,7 and well established radioactivity,8,9 absorbance/fluorescence10,11 and mass spectrometry (MS) methods12 to measure GT activity. Relative to this, methods to measure nucleotide-sugar donor concentrations are fewer.Several sample preparation and analytical approaches have been employed to quantify cellular nucleotide-sugar composition, but a gold-standard technology is yet to be established (reviewed by ref. 13). These methods include strategies that employ high resolution chromatography and UV detection to resolve intra-cellular extracts.14–16 While this line of investigation has demonstrated the ability of both high-performance reverse phase and anion-exchange (HPAEC) liquid chromatography to separate isomeric compounds (e.g. UDP-Gal from UDP-Glc, and UDP-GalNAc from UDP-GlcNAc), the range of quantitation possible using UV is somewhat limited; the method requires authentic standards that are scarce; it sometimes necessitates the use of ion-pairing reagents and salt concentrations that are incompatible with MS; and data interpretation can be confounded by other co-eluting UV-absorbing products. MS-based techniques focussed on negative-ion mode ESI (electrospray ionization) have emerged as an important alternative, especially using Porous Graphitic Carbon (PGC) stationary phase17 and Hydrophilic Interaction Liquid Chromatography (HILIC).18 In this regard, PGC can suffer from retention time instability due to stationary phase modifications, and only a relatively small number of HILIC media have been tested.13,19
In this study, we systematically examined the impact of fluorinated monosaccharide analogs, azido-sugars, and small molecule decoys on both cellular nucleotide-sugar levels and N-glycan biosynthesis. This interest stems from our previous work where the peracetylated monosaccharide analog 4-Fluoro-GalNAc (‘4F-GalNAc’) was applied to truncate O-glycan biosynthesis,20 and our more recent studies where a peracetylated GlcNAc-based thioglycoside (‘SNAP’) acted as an efficient decoy of galactosyltransferase activity.21 Both these compounds reduced selectin-dependent cell adhesion, albeit via different mechanisms. In this regard, others have demonstrated that fluoro- and thio-modified monosaccharide analogs may alter cellular glycan biosynthesis by perturbing nucleotide-sugar levels, and this can impact cell function.22–24 This has been observed for fluoro analogs of fucose, sialic acid22 and GalNAc;23 as well as for peracetylated 5-thio-L-fucose, 5T-Fuc.24 Azido compounds are also commonly used for bioorthogonal chemistry, but their impact on nucleotide-sugar pools and ensuing glycan structures is not well studied.25
In order to obtain a systematic understanding of the relation between nucleotide-sugar concentrations and N-glycan structures, we cultured HL60 promyeloid leukocytes for 48 h with 50 μM of a panel of selected peracetylated fluoro-sugars (4F-GalNAc, 4F-GlcNAc), peracetylated azido-sugars (ManNAz, GalNAz, GlcNAz), and peracetylated GlcNAc-based O- and S-glycoside decoys (ONAP and SNAP). The impact of these compounds on cellular nucleotide-sugar levels was determined using a new HILIC based LC-MS/MS method, where single step acetonitrile (ACN)–water extraction was applied to enable simultaneous cell lysis and protein precipitation, followed by amide base nucleotide-sugar chromatography and MS analysis. The relatively simple method using ACN–water both preserved the stability of the extracted nucleotide-sugars26 and was compatible with MS. In parallel with the nucleotide-sugar analysis, we procainamide labelled N-glycans that were released from cell lysates using PNGaseF. These were analyzed using nano-LC C18 chromatography and MS, in positive ion mode. Such procainamide labelling of glycans reduces the amount of starting material required for structural analysis as it is ionized at 10–30 fold higher intensities compared to 2AB (2-aminobenzoic acid). In this study, we observed significant changes in nucleotide-sugar levels upon addition of most of the modified sugars, including monosaccharide analogs, azido sugars, and carbohydrate decoys. This was, however, accompanied by relatively small changes in N-linked glycosylation. Based on this highly non-linear relationship, we conclude that nature has developed robust mechanisms to deliver similar system output (i.e. glycan profiles) even upon significant perturbation in nucleotide-sugar metabolism. This may be, in part, due to the relatively high nucleotide-sugar concentrations in the Golgi, which far exceed the enzyme KM (Michaelis constant) values of most glycosylating enzymes.
2. Materials and methods
Reagents
Peracetylated 2-acetamido-1,3,6-tri-O-acetyl-4-deoxy-4-fluoro-D-glucopyranose (4F-GlcNAc) and 2-acetamido-1,3,6-tri-O-acetyl-4-deoxy-4-fluoro-D-galactopyranose (4F-GalNAc) were from previous studies.20,27 Peracetylated 1-(naphthalen-2-ylmethanol) 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-1-thio-β-D-glucopyranoside, GlcNAc-β-S-NAP (abbreviated SNAP), and corresponding peracetylated O-glycoside GlcNAc-β-O-NAP (abbreviated ONAP) were recently described.21 Azido-derivatized carbohydrates ManNAz (N-azido acetyl-mannosamine tetraacylated), GalNAz (N-azido acetyl-galactosamine tetraacylated), and GlcNAz (N-azido acetyl-glucosamine tetraacylated) were purchased from ThermoFisher. Nucleotide-sugars and nucleotides were obtained from either Carbosynth (Compton, UK) or Sigma-Aldrich (St. Louis, MO). All other chemicals were from either ThermoFisher or Sigma unless otherwise mentioned.Cell culture and treatment
Human promyelocytic leukemia cells (HL60s) were cultured according to ATCC (Manassas, VA) in Iscove's Modified Dulbecco's Medium (4.5 g L−1 glucose) containing 10% fetal bovine serum (FBS). Peracetylated HexNAc or azido compounds described above were dissolved in anhydrous dimethyl sulfoxide (DMSO) at 40 mM stock and stored at −20 °C until use. During experimentation, these chemicals were added to 1–1.2 × 106 HL60 cells per mL in normal culture medium at a final concentration of 50 μM for 48 h. All runs included vehicle control (VC, 0.2% DMSO). After culture, the cells were collected and washed trice with 1 mL PBS. A portion of the material was used to measure protein concentration using the Bradford assay. The remainder was frozen at −80 °C as cell pellet until use for nucleotide-sugar and N-glycan analysis as explained below.Sugar nucleotide analysis
Cell pellets (formed from ∼107 cells) were resuspended at a final protein concentration of 37.0 mg mL−1 using 75% acetonitrile (ACN, CH3CN) containing 10 μM CMP-Neu5Gc ([M − H]− = 629.13491) internal standard. This suspension was spun down at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g at 4 °C for 10 min to pellet cell mass per proteins. 5 μL of the collected supernatant, obtained from an equivalent of 185 μg of cell protein or 480000 cells, was separated on an online HILIC column (Waters XBridge Amide 3.5 μm, 2.1 × 150 mm, item ID 186004861) prior to injection into a Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo) for MS/MS analysis. The mobile phases were, (A) water containing 5 mM NH4OH and 5 mM NH4OAc, and (B) 90% ACN containing 5 mM NH4OAc. Data were acquired over 60 min at a flow rate of 0.1 mL min−1 at 40 °C using the following gradient: (i) from 85% to 80% B (0–50 min), (iii) 80% to 50% B (50–55 min), and (iv) 50% to 90% B (55–60 min). MS1 data were acquired using the Orbitrap detector (60000 resolution) and MS/MS in HCD mode (28% collision energy). Independent experiments were performed in triplicate.
000 × g at 4 °C for 10 min to pellet cell mass per proteins. 5 μL of the collected supernatant, obtained from an equivalent of 185 μg of cell protein or 480000 cells, was separated on an online HILIC column (Waters XBridge Amide 3.5 μm, 2.1 × 150 mm, item ID 186004861) prior to injection into a Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo) for MS/MS analysis. The mobile phases were, (A) water containing 5 mM NH4OH and 5 mM NH4OAc, and (B) 90% ACN containing 5 mM NH4OAc. Data were acquired over 60 min at a flow rate of 0.1 mL min−1 at 40 °C using the following gradient: (i) from 85% to 80% B (0–50 min), (iii) 80% to 50% B (50–55 min), and (iv) 50% to 90% B (55–60 min). MS1 data were acquired using the Orbitrap detector (60000 resolution) and MS/MS in HCD mode (28% collision energy). Independent experiments were performed in triplicate.
Cellular N-glycomics analysis
Frozen pellets formed from 20 × 106 HL60 cells (described above) were resuspended into 100 μL of 50 mM NH4HCO3 solution. Cells were then heated at 95 °C for 10 min in the presence of 5 mM TCEP (tris (2-caboxyethyl) phosphine), followed by incubation with 20 mM IAM (2-iodoacetamide) at room temperature for 30 min in dark. 15 mM TCEP was then added to neutralize unreacted IAM for 15 min at room temperature. This was followed by PNGaseF (5 μL of 500000 units per mL, New England Biolabs) addition to the solution for 40 h at 37 °C. Proteins were then precipitated by addition of 1 mL MeOH and centrifugation at 12000g for 5 min. The supernatant containing N-glycans was collected and labelled with procainamide under reductive conditions as follows.28 Briefly, the released glycans from the previous step were vacuum dried, and then resuspended with 20 μL of procainamide labelling mix (5 mg procainamide in 100 μL of DMSO:glacial acetic acid (65:35), and 3.3 mg sodium cyanoborohydride) for 3 h at 70 °C. After labelling, samples were purified using Sep-Pak C18 cartridges (Waters, Milford, MA). Here, the cartridge was pre-conditioned with 4 mL MeOH and then with 8 mL nanopure water containing 0.1% formic acid (FA). Procainamide-labelled samples were mixed with 180 μL water containing 0.1% FA, added to the cartridge, washed with 10 mL of nanopure water 0.1% FA. The bound N-glycans were then eluted using 4.5 mL of 15% v/v ACN 0.1% FA. Samples were vacuum dried and resuspended with 50% MeOH 0.1% FA.
1 μL of sample, obtained from an equivalent of 500 μg of cell protein or 1.1 × 106 cells, was analyzed using ESI (electrospray ionization) LC-MS/MS (Orbitrap-XL MS, Thermo) equipped with a nano-LC column (PepMap C18 2 μm; 75 μm × 150 mm, Thermo). The mobile phases for product separation were: (A) water and (B) acetonitrile (CH3CN), both containing 0.1% (v/v) formic acid. Data were acquired over 80 min at a flow rate of 300 nL min−1 using the following linear gradient: (i) increase from 0% to 10% B (0–5 min), (ii) 10% to 40% B (5–45 min), (iii) 40% to 70% B (45–65 min), (iv) 70% to 100% B (65–75 min), and (v) isocratic elution at 100% B (75–80 min). MS1 data were acquired using the Orbitrap detector (60000 resolution), and MS/MS in CID mode (ion trap with 30% collision energy, z = 2–4). Experiments were performed in triplicate.
Glycomics data were analyzed using a variant of the glycoinformatics toolbox, GlycoPAT,29,30 that was customized to handle glycan workflows. Whereas the original program was solely designed for glycoproteomics, the search library and fragmentation rules were now modified to accommodate: (i) arbitrary anomeric modifications/groups, (ii) adducts including Na+ and H+, (iii) glycan-specific fragmentation rules, and (iv) related scoring algorithms (manuscript in preparation). The specific search library that we used for this project included 261 N-linked glycans with unique masses, including 5 high-mannose glycans, various hybrid structures, and bi-, tri-, tetra-antennary, and bisecting glycans. Core fucose was allowed, and both Lewis-X and sialyl Lewis-X (sLeX) structures were permitted on each of the lactosamine chains. Thus, the search library included 62 fucosylated, 37 sialylated, and 133 fucosylated plus sialylated structures, with the largest glycan being a tetra-antennary, bisecting, core-fucosylated N-glycan with sLeX on each antenna. Polylactosamine and LacdiNAc structures were not included since these are not commonly observed in HL-60s.31
Following automated data analysis to match MS1 mass and MS/MS scoring based on a previously described metric called Ensemble Score (ES,29,30), all spectrum-matches with ES > 0.4 were manually inspected and annotated. This involved grouping sets of MS/MS spectra corresponding to a single glycan based on: (i) common monoisotopic MS1 mass, (ii) MS/MS fragmentation patterns, and (iii) LC retention time. Glycan rearrangement was considered as a part of this manual inspection, and whenever possible Na+ adduct spectra were annotated along with protonated MS/MS spectra. Glycan abundance was then quantified by manual integration of area under the curve (AUC) in the corresponding MS1 ion current profile. This was performed using Xcalibur Qual Browser 3.0.63 (Thermo). Here, glycan AUC equalled the sum of the corresponding areas for the same species at different charge states (z = 2–4), including all adducts (Na+, H+). In-source fragmentation was typically <10%, and this was considered during AUC estimation. In this regard, in-source fragments were usually observed to be due to galactose loss, since the whole glycan and agalactosylated ions often appeared at the same retention times, with similar MS/MS fragmentation patterns. Structural isomers were distinguished based on: (i) LC retention time and (ii) MS/MS spectra analysis, though the interpretation is limited as authentic standards were not available. Glycans showing different retention times but similar MS/MS fragmentation were considered to be isomers. When glycans could not be unambiguously distinguished or inferred based on biological knowledge, bracket notations were used as described in the Symbol Nomenclature For Glycans (SNFG,32). All carbohydrate drawings were rendered using DrawGlycan-SNFG software.33 Supplemental figure provide examples of completely annotated structures. Legends to Fig. 4 explain the method used to quantify/group glycans into high mannose, hybrid, complex, branched structures, fucose containing moieties, and chains terminated by sialic acid, galactose, or GlcNAc/GalNAc.
Search was also performed for azido-derivatized sialic acid (modified Neu5Ac, called ‘Az’) for samples treated with peracetylated ManNAz, GalNAz, and GlcNAz. Here, the Neu5Ac present in the original search library was changed to a modified form of sialic acid (Az) in a combinatorial manner. This resulted in a search library with 500 candidates. In such analysis, Az contains an amine at the C-5 position due to the reducing conditions utilized for procainamide labelling. During data analysis, the AUC of each glycan was enumerated along with the number of chains that were terminated by either Neu5Ac or Az% Azido incorporation was then quantified by weighting the fraction of chains on each glycan terminated by Neu5Ac/Az (NeuAc/Az-fraction) by the AUC according to: %Az = 100 × [∑i(Az-fraction × AUC)/∑i(Az-fraction × AUC + Neu5Ac-fraction × AUC)].
Statistics
All error bars represent standard deviations. Analysis of variance followed by the Bonferroni post-test was used for multiple comparisons.3. Results and discussion
An optimized LC-MS/MS strategy to quantify cellular nucleotide-sugar levels
We performed amide-based HILIC separation using a mixture of standards (Fig. 1A). Satisfactory separation could be achieved to distinguish between different nucleotide-sugars, monosaccharides, and individual nucleotides, over a 60 min gradient. The gradient was designed to maximize separation of UDP-galactose/glucose and UDP-GalNAc/GlcNAc pairs, while taking into account their structures (hexoses are stereoisomers), as well as their pKa values. High content of organic phase (80–85%), along with NH4OAc as additive in both mobile phases, helped the separation of these stereoisomer pairs; for one side, ammonium might mimic the behaviour of an ionic liquid and, on the other side, the additive and pH from the mobile phase could modulate the pKa of analytes. In addition to the gradient and additives, the separation was carried out at 40 °C to increase interaction between mobile and stationary phase. The chromatography was reproducible. MS ion count measured based on the elution MS1 profile of chemical standards yielded a linear relationship with injection dose, with a limit of detection of 1–10 pmol (Fig. 1B and Table S1, ESI†).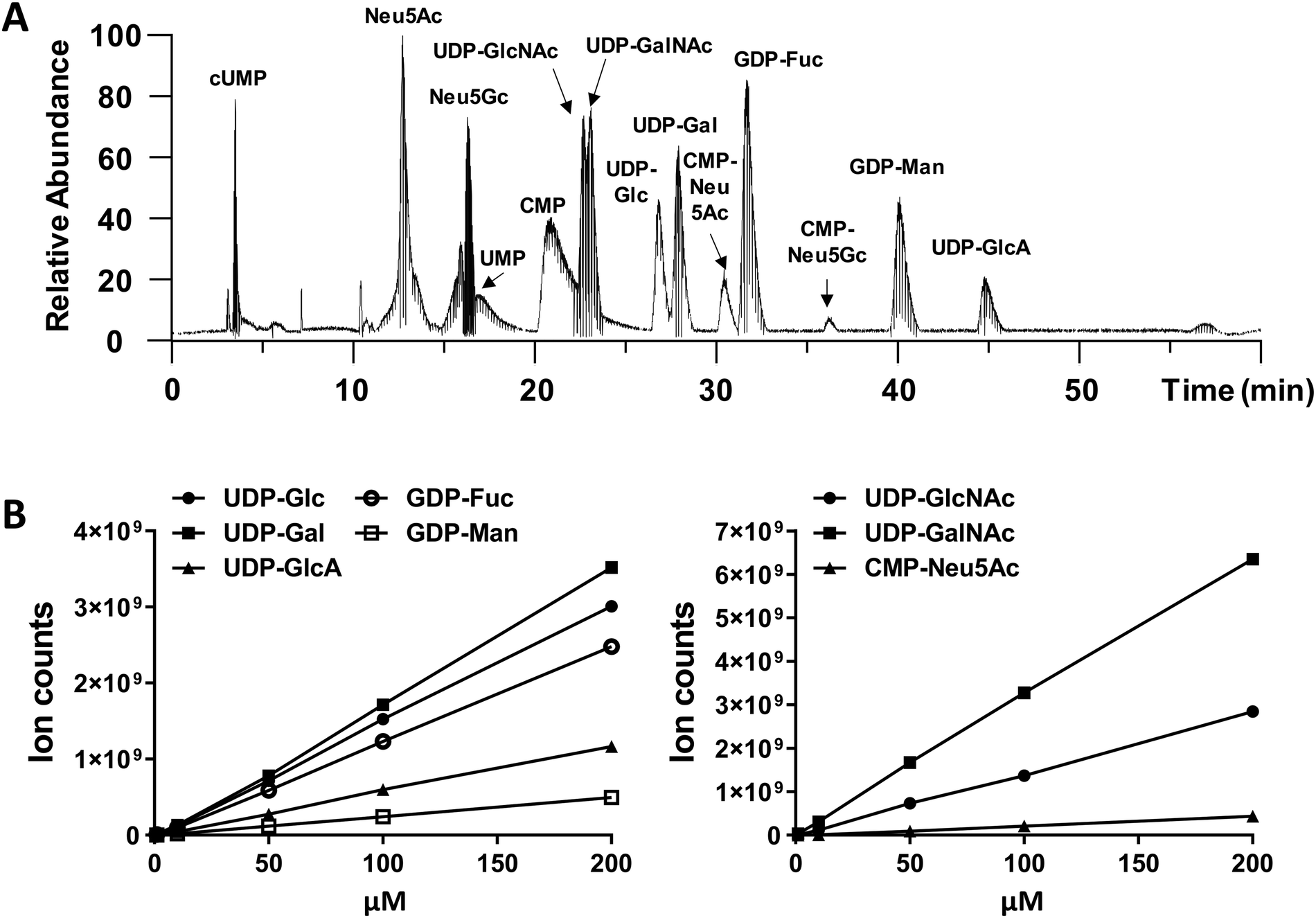 | ||
| Fig. 1 LC-MS chromatogram of nucleotide-sugar standard mixture and their corresponding calibration curves. (A) Total ion chromatogram in negative ion mode based on HILIC-ESI-MS/MS analysis of a nucleotide-sugar mixture. All structures were verified using MS1 mass match and MS/MS fragmentation. Nucleotide-sugars used include 5 μM UDP-GalNAc/GlcNAc, 5 μM UDP-Gal/Glc, 15 μM GDP-Fuc, 10 μM UDP-GlcA, 20 μM CMP-Neu5Ac, and 50 μM GDP-Man. (B) Calibration curves of standard nucleotide-sugars. Samples were injected into LC-MS in triplicates and results are shown as average ± SD. Error bars are too small to be visible in many cases. | ||
While we did not perform specific runs to compare nucleotide-sugar quantitation in standard runs vs. cell lysates, we note that sample complexity was low in the timeframe (22–48 min) where the nucleotide-sugars eluted with few co-eluting compounds with m/z > 500 even in complex HL60 cell lysates (Fig. S1, ESI†). For example, in the case of UDP-Hex (Fig. S1, ESI†), only two other compounds (m/z = 445, 547) co-eluted with UDP-Hex, and the intensity of these other molecules was low compared to the nucleotide-sugar. Thus, matrix effects may not be significant. Additionally, note that the studies below examine the effect of a panel of small molecules on individual nucleotide-sugars rather than between different species.
Changes in nucleotide-sugar concentration upon addition of monosaccharide analogs, azido sugars and acceptor decoys
Cells were cultured for 2 days with 50 μM of various small molecules that were variants of either HexNAc or Mannose. At the study end point, cell extracts were prepared using one-step ACN–water based lysis and protein precipitation. These were injected into LC-MS, along with CMP-Neu5Gc internal chemical standard. CMP-Neu5Gc was used, as this nucleotide-sugar is not produced by human cells due to absence of cytidine monophospho-N-acetylneuraminic acid hydroxylase (CMAH) activity. Particular attention was placed to ensure that the retention time of individual compounds followed the profile in Fig. 1A. In this plot, the elution pattern is primarily controlled by the nucleotide polarity, independent of the sugar moiety, except for the case of sugar-acids. Thus, less polar nucleotide-sugars containing UDP eluted first, before GDP-sugars. Neither the nucleotide nor phosphate group appear to control sugar-acid retention (GlcA, Neu5Ac, and Neu5Gc), which also eluted with increased polarity of bonds and ionic character. Due to this, CMP-Neu5Ac and CMP-Neu5Gc presented higher retention times compared to the UDP-nucleotide sugars, with UDP-GlcA exhibiting maximum retention. Among the sialic acids, CMP-Neu5Gc containing an extra hydroxyl group had higher retention due greater polarity with respect to CMP-Neu5Ac.Additional structural confirmation was obtained using the MS/MS fragmentation profile of compounds in cell extract, which were nearly identical to chemical standards (Fig. S2, ESI†). These spectra display strong peaks due to the nucleotide, as the phosphate–sugar bond is frangible. Following this, are the peaks due to the phosphorylated pentose and phosphate groups. All spectra also presented characteristic water-loss peaks from the phosphate moiety as well as nucleobase fragments.
The nucleotide-sugar concentrations measured for vehicle control samples was in the range of 2.5–10 μM in the injected sample, which translates to ∼2500–10000 μM concentration within the Golgi [e.g.: 3 μM = 15 pmol since MS injection volume was 5 μL. This injection volume was made of 480000 cells each with an estimated Golgi volume of 10 μm3 or 0.01 pL. Thus, total Golgi conc. is ∼15 pmol/(0.01 pL × 480000 × 106) = 3125 μM]. These values are in agreement with previous estimates of Golgi nucleotide-sugar concentrations.34 These concentrations are also consistent with experimental measured KM (∼2–8 μM) values of various mammalian nucleotide-sugar antiporters belonging to the SLC35 family,35,36 and the notion that donor concentrations within the Golgi may exceed cytoplasmic levels by a factor of 20–100.37
In studies that measured the effect of monosaccharide analogs on cellular nucleotide-sugar levels, we observed that both the 4F-HexNAc analogs, 4F-GalNAc and 4F-GlcNAc, acted via similar mechanisms. Both compounds decreased the intra-cellular concentration of UDP-GalNAc and UDP-GlcNAc compared to vehicle (Fig. 2A). This decrease was more dramatic upon culture with 4F-GalNAc (75–80%) compared to 4F-GlcNAc (55–70%). These fluoro-molecules also boosted UDP-4F-GalNAc and UDP-4F-GlcNAc levels in cells, such that the relative abundance of the UDP-4F-HexNAc analogs was comparable to normal UDP-HexNAc in vehicle control (Fig. 3A). Based on the observation that Glc-based nucleotide-sugars typically appear at an earlier retention time compared to corresponding Gal-based entities upon amide-based separation (Fig. S3, ESI†), we estimate that 4F-GalNAc may augment UDP-4F-GlcNAc levels to a greater extent, with 4F-GlcNAc more prominently increasing cellular UDP-4F-GalNAc concentrations. Both fluorinated nucleotide-sugars were present in both the 4F-HexNAc runs, suggesting that UDP-4F-GlcNAc and UDP-4F-GalNAc may be epimerized by the UDP-glucose 4-epimerase (GALE). A small decrease in cellular CMP-Neu5Ac and GDP-Man (∼25–30%) was also noted upon addition of the 4F-HexNAc compounds.
 | ||
| Fig. 2 Nucleotide-sugar levels in HL-60s after monosaccharide analog/decoy treatment. 1.2 × 106 HL60s were cultured for 48 h with VC (vehicle control) or 50 μM of (A) 4F-HexNAcs, (B) HexNAz/ManNAz azido compounds, or (C) GlcNAc based decoys SNAP/ONAP. Nucleotide-sugars were then extracted from cell lysates, and their concentrations determined using LC-MS/MS. Concentrations were calculated based on the area under the curve in MS1 and the corresponding calibration curve of each species. As UDP-Xyl standard data were not available, its concentration was determined based on GDP-Fuc calibration data. All nucleotide-sugars were identified by MS/MS. Data are represented as average ± SD (n = 3–6). *Represents P < 0.05, **Represents P < 0.01, ***Represents P < 0.001. | ||
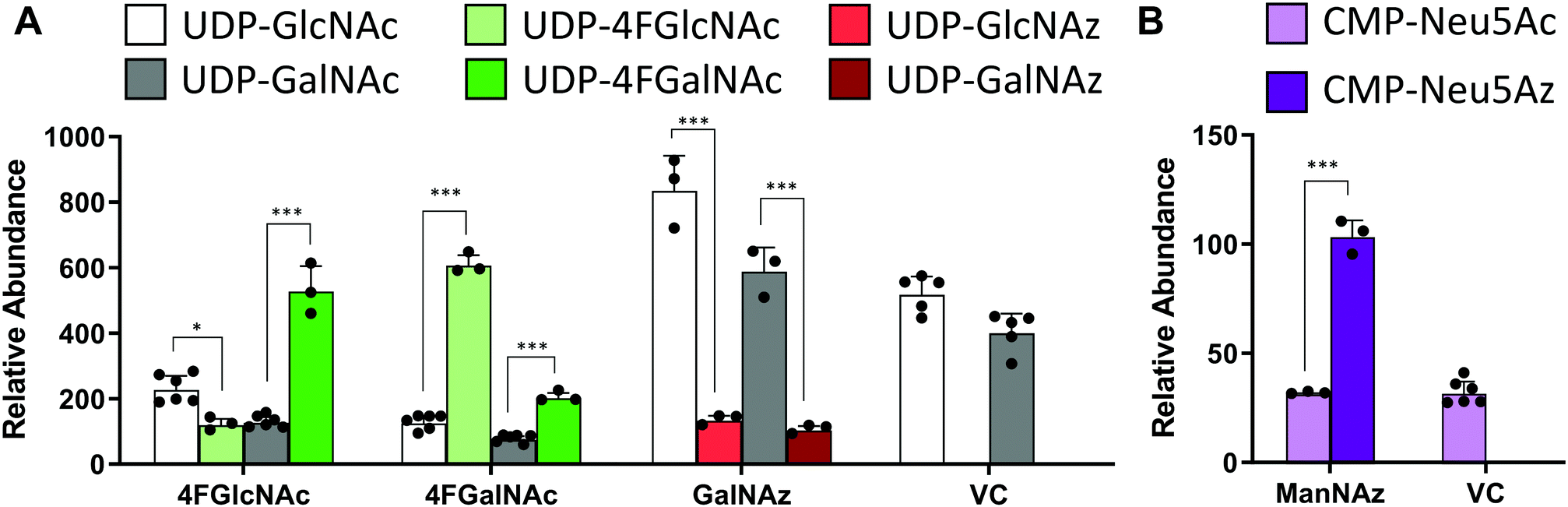 | ||
| Fig. 3 Nucleotide-sugar levels in HL-60s after HexNAc analog and azido compound treatment. HL60s were cultured with small molecules for 48 h. Nucleotide-sugar concentrations in cell extracts were determined using LC-MS/MS. Graph shows relative abundance of area under MS1 curve for individual species, normalized with respect to internal standard (CMP-Neu5Gc). All nucleotide-sugars were identified by MS/MS. Data are presented as ‘relative abundance’ rather than absolute concentration due to absence of authentic chemical standards for fluoro- and azido-compounds. (A) Two UDP-4F-HexNAc peaks with identical MS1 mass and MS/MS profiles were detected. Since UDP-Glc appears before UDP-Gal, and UDP-GlcNAc before UDP-GalNAc, UDP-4F-GlcNAc was also presumed to encompass the first peak with UDP-4F-GalNAc constituting the second. (A and B) Azido derivatized nucleotide-sugars were observed upon GalNAz and ManNAz treatment, but not upon cell culture with GlcNAz. Data are represented as average ± SD (n = 3–6). *Represents P < 0.05, ***Represents P < 0.001. | ||
Compared to the 4F-HexNAc compounds, the azido entities had a relatively minor effect on cellular nucleotide-sugar levels (Fig. 2B). Here, ManNAz, GalNAz and GlcNAz increased UDP-GlcNAc levels by ∼70%, and they augmented UDP-GalNAc by ∼50%, with minor changes also being observed for UDP-Glc. Changes in CMP-Neu5Ac concentration was not detected. The azido compounds could be converted into corresponding nucleotide-sugars. This was observed for GalNAz, which appeared both as UDP-GalNAz and UDP-GlcNAz, two isomeric compounds with different retention times, though the extent of formation was low (Fig. 3A). Similarly, cell culture with ManNAz resulted in substantial formation of CMP-Neu5Az, which exceeded the relative abundance of CMP-Neu5Ac by 3-fold (Fig. 3B). UDP-HexNAz was not detected upon culture with GlcNAz. CMP-Neu5Az was also not observed upon addition of GalNAz.
In the last runs, two GlcNAc based glycoside-decoys with naphthalenemethanol (NAP) anomeric groups were added to HL60s in culture (Fig. 2C). In ONAP, the GlcNAc is attached to NAP via an O-glycosidic linkage whereas this is an S-glycosidic bond in SNAP.21 Here, only minimal changes in nucleotide-sugar levels were observed upon addition of SNAP. In contrast, ONAP resulted in a 2.3-fold increase in both UDP-GlcNAc and UDP-GalNAc concentrations. A 25% increase in CMP-NeuAc was also noted. This may be due to the instability of ONAP as it can be readily hydrolysed within cells to produce free GlcNAc. We speculate that this free GlcNAc may be converted into the corresponding UDP-sugar via the salvage pathway, using NAGK (N-acetyl-D-glucosamine kinase) to convert GlcNAc into GlcNAc-6-phosphate (GlcNAc-1-P), PGM3 (phosphoacetylglucosamine mutase) to convert GlcNAc-6-P to GlcNAc-1-P, and finally UAP1 (UDP-N-acetylhexosamine pyrophosphorylase) aiding transforming into UDP-GlcNAc. UDP-GalNAc may then form using GALE activity. Excess UDP-GlcNAc may also be converted into CMP-Neu5Ac using UDP-GlcNAc 2-epimerase (GNE), sialic acid synthase (NANS) and N-acylneuraminate cytidylyltransferase (CMAS). Overall, all chemical treatments caused alterations in nucleotide-sugar levels with the effects being most prominent for 4F-HexNAc analogs and ONAP.
Effect of small molecules on cellular N-linked glycosylation
We determined the impact of changes in cellular nucleotide-sugar concentrations on N-linked glycosylation (the system output). This was performed using nano-LC-MS/MS analysis of procainamide-functionalized N-glycans that were released from cell lysates (Fig. 4 and Fig. S4, ESI†). Here, the N-glycans eluted mainly between 10–40 min of the LC run, with the glycan elution pattern depending on the carbohydrate structure. In this regard, the glycans eluted in the sequence of increasing polarity: (i) high mannose, (ii) non-fucosylated glycans, (iii) fucosylated glycans, (iii) monosialylated glycans, (iv) di-sialylated glycans, and finally (v) tri and tetrasialylated glycans. Within these groups, sialylated glycans eluting earlier were devoid of fucose, while later compounds contained both sialic acid and fucose (Fig. S5, ESI†).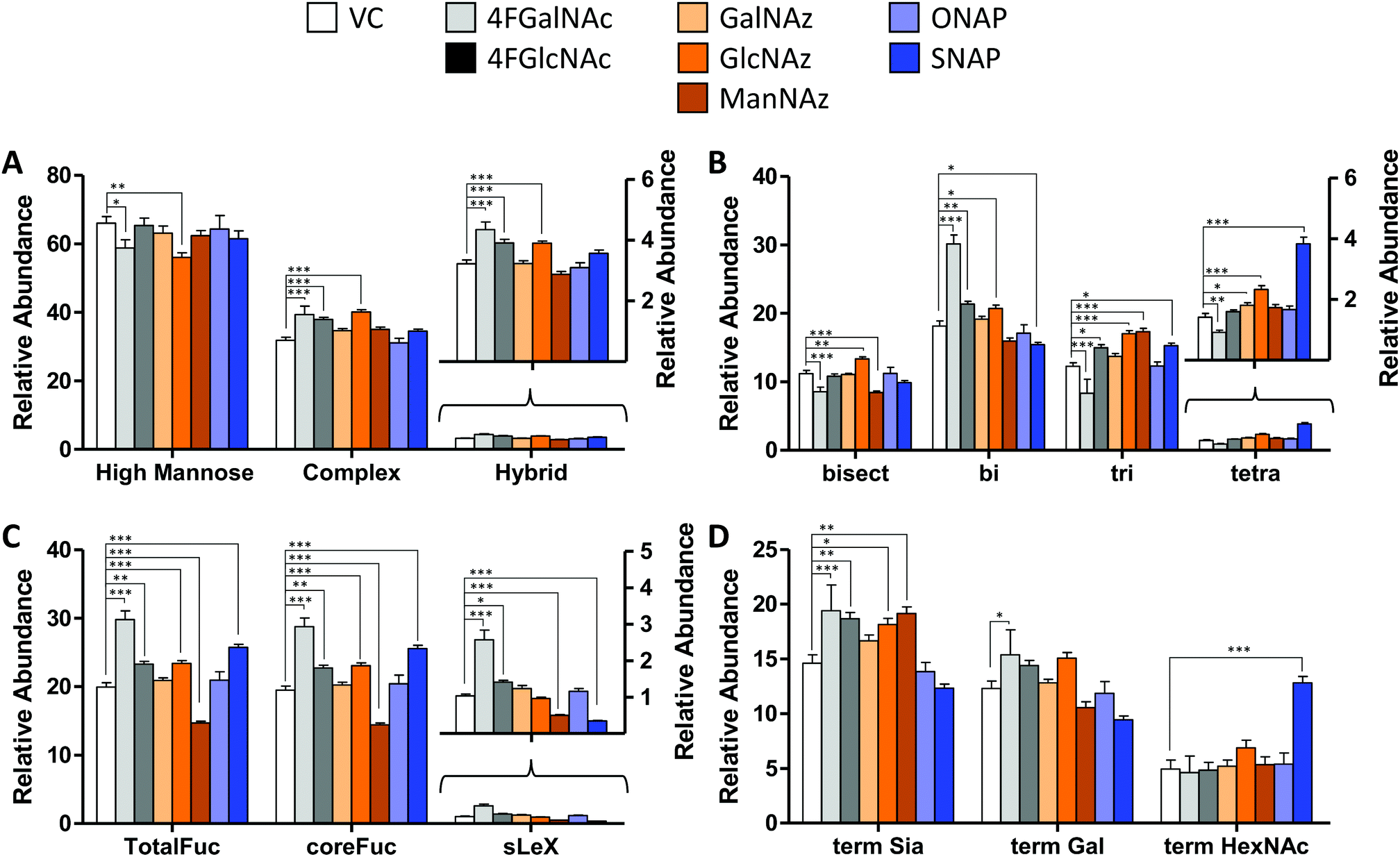 | ||
| Fig. 4 N-Glycan distribution following HexNAc analog and azido compound treatment. N-Glycans released from HL60s cultured with various small molecules for 48 h, were assayed using LC-MS/MS. Detailed analysis was performed to verify individual structures and calculate corresponding MS1 area under the curve (AUC) to estimate ‘relative abundance (% of vehicle control)’ (Fig. S6, ESI†). These data were grouped as follows: (A) sum of relative abundance of glycans appearing as high mannose, hybrid and complex structures. These data were normalized so that the sum of the relative abundance for these 3 groups for each individual small molecule treatment equals 100. Glycans in each of these groups are similar for all treatments (±20%). (B) The relative abundance of complex structures in panel A was broken into either bi-, tri-, and tetra- antennary glycans (i.e. sum of relative abundance of bi-, tri-, plus tetra = relative abundance of complex glycans). The portion of complex that contained bisecting structures is indicated in the first bar. The most remarkable change was the reduction in glycan branching upon addition of 4F-GalNAc, and slightly higher level of tetraantennary structures upon SNAP addition. (C) A portion of complex structures contained fucose (at either terminal and/or core), a vast majority of which included core fucose, and a much smaller fraction contained sialyl Lewis-X (sLeX). (D) Complex glycans can have terminal structures that end with Sia, Gal, or GlcNAc/GalNAc. To calculate this, we weighted each of the complex glycan AUC values by the fraction of chains terminated by either Sia, Gal, or HexNAc. Thus, sum of term Sia, term Gal plus term HexNAc equals relative abundance of complex in panel (A) SNAP increased appearance of terminal HexNAc chains by usurping galactosyltransferase activity. All data are represented as average ± SD. *Represents P < 0.05, **P < 0.01, ***P < 0.001. | ||
In addition to following this retention pattern, all glycan identifications were confirmed manually based on MS1 monoisotopic peak matching and detailed MS/MS spectra annotation. This included accounting for different species charges and adducts (e.g. in Fig. S4A and B, ESI†). Additionally, while not all structural features could be unambiguously elucidated due to absence of chemical standards, high mannose glycans could be easily identified base on their characteristic MS/MS spectra: (i) the sequential loss of mannoses at charge +2, and (ii) the loss of core GlcNAc (GlcNAc attached to procainamide) followed by the terminal carbohydrate losses as charge +1 (Fig. S4C, ESI†). Additionally, we could distinguish core fucose from terminal fucose modifications based on the appearance of peaks at m/z = 587 for core fucose compounds (Fig. S4D, ESI†), m/z = 512 for Lewis-type glycans (Fig. S4E, ESI†), and m/z at 803 for sLex structures (Fig. S4F, ESI†). The remaining structures were based on biochemical knowledge and our previous work where permethylated HL60 N-glycans were identified using MALDI-TOF.31,38 Detailed annotations with ambiguous identifications indicated using SNFG brackets are presented in Fig. S6 (ESI†).
In general, the N-glycans formed in all experimental conditions were not very different when these structures were classified into high mannose, hybrid, and complex groupings (Fig. 4A). Upon examining the breakdown of complex glycans into bi-, tri-, and tetra-antennary structures, however, 4F-GalNAc exhibited a 50% increase in biantennary glycans compared to vehicle control, and smaller amounts of branched structures (Fig. 4B). This observation is consistent with the 75–80% decrease in cellular UDP-HexNAc levels, though the changes in glycan structures were smaller than would be predicted based on the reduced nucleotide-sugar concentrations. 4F-GlcNAc was previously reported to exhibit similar reduction in N-glycan branching,27 and it is possible that increasing the dose of 4F-GlcNAc (50 μM currently) to 80–100 μM would result in similar observations as the current 55–70% decrease in UDP-HexNAc levels is insufficient to affect N-glycan changes. In this analysis, all branches with singleton HexNAc were assumed to be bisecting, as our workflow is unable to precisely distinguish single HexNAc attached to the β(1,4) Mannose arm with similar attachment on other branches. Upon quantifying fucosylation, we observed that a majority of complex glycans contained core fucose (Fig. 4C). N-Glycan sLeX antigens were reduced upon SNAP addition, which is consistent with previous work.21 Azido-derivatized monosaccharides did not have a large effect on glycan distribution, though ManNAz appeared to reduce the extent of core-fucosylation by ∼25%. Upon searching for azido-derivatized N-glycans, we noted that ∼1/20th of the chains terminated by sialic acid were capped by Neu5Az while the rest were Neu5Ac terminated. These azido groups were found in MS/MS spectra as amine derivatives due to the reductive conditions used during procainamide labelling (Fig. S4H, ESI†). Thus, despite the substantially higher concentrations of CMP-Neu5Az in cells compared to CMP-Neu5Ac (Fig. 3), their integration into N-glycan biosynthesis was low. ManNAz exhibited higher incorporation into N-glycans compared to HexNAz, which appeared on both fucosylated and non-fucosylated glycans (Fig. S4J, ESI†). Additional investigations are necessary to study transferase activity in the presence of CMP-Neu5Az. Finally, consistent with prior work,21 SNAP addition resulted in a greater number of glycan chains terminated by HexNAc (Fig. 4D and Fig. S6, ESI†).
4. Conclusions
In this manuscript, carbohydrate-based small molecules were observed to alter cellular nucleotide-sugar levels. Among the families tested, 4F-HexNAc members depressed both UDP-GalNAc and UDP-GlcNAc. Addition of the decoy ONAP, which contains a hydrolysable O-glycosidic linkage but not SNAP with a S-glycosidic linkage, increased UDP-HexNAc levels by 2.3 fold. Many of the azido sugars tested also formed corresponding azido-modified sugar donors, and these had a modest effect on endogenous nucleotide-sugar concentrations.Whereas the effect of these compounds on cellular nucleotide-sugars was sometimes large, their impact on N-linked glycosylation was relatively small with only some aspects altered. For example, a 75–80% decrease in UDP-GlcNAc and UDP-GalNAc, and a concomitant increase in UDP-4F-HexNAc levels upon culture with peracetylated 4F-GalNAc, only resulted in a ∼50% change in N-glycan branching and some increase in fucosylation, with less impact on lactosamine chain extension. Cell treatment with 4F-GlcNAc had an even smaller effect, despite the 55–70% reduction in overall UDP-HexNAc levels. Increasing UDP-HexNAc concentrations by 2.3 fold using ONAP also did not change the output N-glycosylation profile. Together, these observations highlight a highly non-linear relationship between nucleotide-sugar levels and N-linked glycosylation. They suggest that activated sugar-donor concentrations may not be a major determinant of glycosylation rates, unless substantially depleted. Consistent with this, whereas we estimate that the Golgi compartment of cells contain ∼2500–10000 μM of individual nucleotide-sugars, the Michaelis constant (KM) of enzymes regulating N-glycan biosynthesis is typically smaller, <500 μM (Table 1). Additionally, it is possible that alterations in nucleotide-sugars may result in system response and changes in glycosyltransferase expression which would represent another control mechanism.54 Due to these effects, biological systems appear to be robust to changes in nucleotide-sugar levels, at least for HexNAc derivatives.
| Gene name | Enzyme name/function | K M (μM) | Species | Ref. |
|---|---|---|---|---|
| Man1b1 | Mannosyl-oligosaccharide 1,2-α-mannosidase IA | 100–300 | R. norvegicus; S. cerevisiae | 39 and 40 |
| MAN2A1 | α-Mannosidase 2 | 200 | Homo sapiens | 41 |
| MGAT1 | α1,3-Mannosyl-glycoprotein 2-β-N-GlcNAcT | 483 | Homo sapiens | 42 |
| MGAT2 | α1,6-Mannosyl-glycoprotein 2- β-N-GlcNAcT | 130–550 | Homo sapiens | 43 |
| MGAT3 | β-1,4-Mannosyl-glycoprotein 4-β-N-GlcNAcT | 4700–6800 | Homo sapiens | 44 |
| MGAT4A | α-1,3-Mannosyl-glycoprotein 4-β-N-GlcNAcT-A | 118–242 | Homo sapiens | 45 |
| MGAT5 | α-1,6-Mannosylglycoprotein 6-β- GlcNAcT | 150 | Homo sapiens | 46 |
| B4GALT1 | β-1,4-Galactosyltransferase 1 | 170 | Homo sapiens | 47 |
| B3galt1 | β-1,3-Galactosyltransferase | 34 | Rattus norvegicus | 48 |
| FUT8 | α-(1,6)-Fucosyltransferase | 12.90 | Homo sapiens | 49 |
| FUT1 | Galactoside 2-alpha-L-fucosyltransferase 1 | 110 | Homo sapiens | 50 |
| FUT4 | α-(1,3)-Fucosyltransferase 4 | 380–650 | Homo sapiens | 51 |
| ST3GAL3 | CMP-N-acetylneuraminate-β-galactosamide-α-2,3-sialylT | 140–830 | Homo sapiens | 52 |
| ST6GAL1 | β-Galactoside alpha-2,6-sialylT 1 | 200–1500 | Homo sapiens | 53 |
This study could have broad implications for our understanding of glycan biosynthesis regulation. It may explain why low concentrations of orally ingested fucose is sufficient to correct defects in the SLC35C1 transporter of leukocyte adhesion deficiency-II (LAD-II) patients,55 as this amount is sufficient to restore enough GDP-Fuc in blood cells. This may also represent a control mechanism to limit alterations in N-glycan branching, as high branching of such structures can promote growth factor activation and oncogenic transformation.56
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Supported by NIH grants HL103411, GM133195 and GM126537 (to SN). VdS was supported by the University at Buffalo GEM program. We gratefully thank Dr Alan Friedman for assistance with mass spectrometry. All MS data are available from: https://glycopost.glycosmos.org/entry/GPST000066.References
- T. Naoyuki, K. Honke, M. Fukuda, H. Narimatsu, Y. Yamaguchi and T. Angata, Handbook of glycosyltransferases and related genes, Springer Reference, Tokyo, 2nd edn, 2014 Search PubMed.
- S. Neelamegham and G. Liu, Glycobiology, 2011, 21, 1541–1553 CrossRef CAS PubMed.
- S. Akiyoshi, M. Iwata, F. Berenger and Y. Yamanishi, Mol. Inf., 2020, 39, e1900112 CrossRef PubMed.
- R. R. Drake, C. McDowell, C. West, F. David, T. W. Powers, T. Nowling, E. Bruner, A. S. Mehta, P. M. Angel, L. A. Marlow, H. W. Tun and J. A. Copland, J. Mass Spectrom., 2019, e4490, DOI:10.1002/jms.4490.
- I. G. Ferreira, M. Carrascal, A. G. Mineiro, A. Bugalho, P. Borralho, Z. Silva, F. Dall’olio and P. A. Videira, Int. J. Oncol., 2019, 55(5), 1033–1048, DOI:10.3892/ijo.2019.4886.
- D. D. Marathe, E. V. Chandrasekaran, J. T. Lau, K. L. Matta and S. Neelamegham, FASEB J., 2008, 22, 4154–4167 CrossRef CAS PubMed.
- A. V. Nairn, W. S. York, K. Harris, E. M. Hall, J. M. Pierce and K. W. Moremen, J. Biol. Chem., 2008, 283, 17298–17313 CrossRef CAS PubMed.
- E. V. Chandrasekaran, J. Xue, J. Xia, R. D. Locke, S. A. Patil, S. Neelamegham and K. L. Matta, J. Proteome Res., 2012, 11, 2609–2618 CrossRef CAS PubMed.
- S. A. Patil, E. V. Chandrasekaran, K. L. Matta, A. Parikh, E. S. Tzanakakis and S. Neelamegham, Anal. Biochem., 2012, 425, 135–144 CrossRef CAS PubMed.
- H. S. Lee and J. S. Thorson, Anal. Biochem., 2011, 418, 85–88 CrossRef CAS PubMed.
- Z. L. Wu, C. M. Ethen, B. Prather, M. Machacek and W. Jiang, Glycobiology, 2011, 21, 727–733 CrossRef CAS PubMed.
- A. Sanchez-Ruiz, S. Serna, N. Ruiz, M. Martin-Lomas and N. C. Reichardt, Angew. Chem., Int. Ed., 2011, 50, 1801–1804 CrossRef CAS PubMed.
- M. Rejzek, L. Hill, E. S. Hems, S. Kuhaudomlarp, B. A. Wagstaff and R. A. Field, Methods Enzymol., 2017, 597, 209–238 CAS.
- K. Nakajima, S. Kitazume, T. Angata, R. Fujinawa, K. Ohtsubo, E. Miyoshi and N. Taniguchi, Glycobiology, 2010, 20, 865–871 CrossRef CAS PubMed.
- J. Rabina, M. Maki, E. M. Savilahti, N. Jarvinen, L. Penttila and R. Renkonen, Glycoconjugate J., 2001, 18, 799–805 CrossRef CAS PubMed.
- N. Tomiya, E. Ailor, S. M. Lawrence, M. J. Betenbaugh and Y. C. Lee, Anal. Biochem., 2001, 293, 129–137 CrossRef CAS PubMed.
- M. Pabst, J. Grass, R. Fischl, R. Leonard, C. Jin, G. Hinterkorner, N. Borth and F. Altmann, Anal. Chem., 2010, 82, 9782–9788 CrossRef CAS PubMed.
- S. U. Bajad, W. Lu, E. H. Kimball, J. Yuan, C. Peterson and J. D. Rabinowitz, J. Chromatogr. A, 2006, 1125, 76–88 CrossRef CAS PubMed.
- X. Qin and X. Wang, J. Pharm. Biomed. Anal., 2018, 158, 280–287 CrossRef CAS PubMed.
- D. D. Marathe, A. Buffone, Jr., E. V. Chandrasekaran, J. Xue, R. D. Locke, M. Nasirikenari, J. T. Lau, K. L. Matta and S. Neelamegham, Blood, 2010, 115, 1303–1312 CrossRef CAS PubMed.
- S. S. Wang, X. Gao, V. D. Solar, X. Yu, A. Antonopoulos, A. E. Friedman, E. K. Matich, G. E. Atilla-Gokcumen, M. Nasirikenari, J. T. Lau, A. Dell, S. M. Haslam, R. A. Laine, K. L. Matta and S. Neelamegham, Cell Chem. Biol., 2018, 25, 1519–1532 CrossRef CAS PubMed.
- C. D. Rillahan, A. Antonopoulos, C. T. Lefort, R. Sonon, P. Azadi, K. Ley, A. Dell, S. M. Haslam and J. C. Paulson, Nat. Chem. Biol., 2012, 8, 661–668 CrossRef CAS PubMed.
- X. M. van Wijk, R. Lawrence, V. L. Thijssen, S. A. van den Broek, R. Troost, M. van Scherpenzeel, N. Naidu, A. Oosterhof, A. W. Griffioen, D. J. Lefeber, F. L. van Delft and T. H. van Kuppevelt, FASEB J., 2015, 29, 2993–3002 CrossRef CAS PubMed.
- W. F. Zandberg, J. Kumarasamy, B. M. Pinto and D. J. Vocadlo, J. Biol. Chem., 2012, 287, 40021–40030 CrossRef CAS PubMed.
- C. T. Campbell, S. G. Sampathkumar and K. J. Yarema, Mol. BioSyst., 2007, 3, 187–194 RSC.
- I. J. del Val, S. Kyriakopoulos, K. M. Polizzi and C. Kontoravdi, Anal. Biochem., 2013, 443, 172–180 CrossRef CAS PubMed.
- S. R. Barthel, A. Antonopoulos, F. Cedeno-Laurent, L. Schaffer, G. Hernandez, S. A. Patil, S. J. North, A. Dell, K. L. Matta, S. Neelamegham, S. M. Haslam and C. J. Dimitroff, J. Biol. Chem., 2011, 286, 21717–21731 CrossRef CAS PubMed.
- D. J. Harvey, Rapid Commun. Mass Spectrom., 2000, 14, 862–871 CrossRef CAS PubMed.
- K. Cheng, G. Pawlowski, X. Yu, Y. Zhou and S. Neelamegham, Bioinformatics, 2020, 36(6), 1942–1943, DOI:10.1093/bioinformatics/btz819.
- G. Liu, K. Cheng, C. Y. Lo, J. Li, J. Qu and S. Neelamegham, Mol. Cell. Proteomics, 2017, 16, 2032–2047 CrossRef CAS PubMed.
- N. Mondal, A. Buffone, Jr., G. Stolfa, A. Antonopoulos, J. T. Lau, S. M. Haslam, A. Dell and S. Neelamegham, Blood, 2015, 125, 687–696 CrossRef CAS PubMed.
- S. Neelamegham, K. Aoki-Kinoshita, E. Bolton, M. Frank, F. Lisacek, T. Lutteke, N. O'Boyle, N. H. Packer, P. Stanley, P. Toukach, A. Varki, R. J. Woods and S. D. Group, Glycobiology, 2019, 29, 620–624 CrossRef.
- K. Cheng, Y. Zhou and S. Neelamegham, Glycobiology, 2017, 27, 200–205 CrossRef CAS PubMed.
- F. J. Krambeck, S. V. Bennun, S. Narang, S. Choi, K. J. Yarema and M. J. Betenbaugh, Glycobiology, 2009, 19, 1163–1175 CrossRef CAS PubMed.
- P. Berninsone, H. Y. Hwang, I. Zemtseva, H. R. Horvitz and C. B. Hirschberg, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3738–3743 CrossRef CAS PubMed.
- T. Suda, S. Kamiyama, M. Suzuki, N. Kikuchi, K. Nakayama, H. Narimatsu, Y. Jigami, T. Aoki and S. Nishihara, J. Biol. Chem., 2004, 279, 26469–26474 CrossRef CAS PubMed.
- M. Perez and C. B. Hirschberg, J. Biol. Chem., 1985, 260, 4671–4678 CAS.
- N. Mondal, G. Stolfa, A. Antonopoulos, Y. Zhu, S. S. Wang, A. Buffone, Jr., G. E. Atilla-Gokcumen, S. M. Haslam, A. Dell and S. Neelamegham, Arterioscler., Thromb., Vasc. Biol., 2016, 36, 718–727 CrossRef CAS PubMed.
- I. Tabas and S. Kornfeld, J. Biol. Chem., 1979, 254, 11655–11663 CAS.
- F. Lipari and A. Herscovics, Glycobiology, 1994, 4, 697–702 CrossRef CAS PubMed.
- E. V. Chandrasekaran, M. Davila, D. Nixon and J. Mendicino, Cancer Res., 1984, 44, 4059–4068 CAS.
- K. Fujiyama, Y. Ido, R. Misaki, D. G. Moran, I. Yanagihara, T. Honda, S.-I. Nishimura, T. Yoshida and T. Seki, J. Biosci. Bioeng., 2001, 92, 569–574 CrossRef CAS PubMed.
- H. Schachter, F. Reck and H. Paulsen, in Methods in Enzymology, Academic Press, 2003, vol. 363, pp. 459–475 Search PubMed.
- E. Y. Song, S. K. Kang, Y. C. Lee, Y. G. Park, T. H. Chung, D. H. Kwon, S. M. Byun and C. H. Kim, Cancer Invest., 2001, 19, 799–807 CrossRef CAS PubMed.
- S. Oguri, A. Yoshida, M. T. Minowa and M. Takeuchi, Glycoconjugate J., 2006, 23, 473–480 CrossRef CAS PubMed.
- K. Sasai, Y. Ikeda, T. Fujii, T. Tsuda and N. Taniguchi, Glycobiology, 2002, 12, 119–127 CrossRef CAS PubMed.
- K. Nakazawa, K. Furukawa, H. Narimatsu and A. Kobata, J. Biochem., 1993, 113, 747–753 CrossRef CAS PubMed.
- D. K. Chou and F. B. Jungalwala, J. Neurochem., 1994, 62, 307–314 CrossRef CAS PubMed.
- H. Ihara, Y. Ikeda and N. Taniguchi, Glycobiology, 2005, 16, 333–342 CrossRef.
- H. Masutani and H. Kimura, J. Biochem., 1995, 118, 541–545 CrossRef CAS PubMed.
- M. Jezequel-Cuer, H. N'Guyen-Cong, D. Biou and G. Durand, Biochim. Biophys. Acta, 1993, 1157, 252–258 CrossRef CAS.
- R. Gupta, K. L. Matta and S. Neelamegham, Biochem. Biophys. Res. Commun., 2016, 469, 606–612 CrossRef CAS PubMed.
- M. Nakamura, A. Tsunoda, K. Yanagisawa, Y. Furukawa, J. Kikuchi, S. Iwase, T. Sakai, G. Larson and M. Saito, J. Lipid Res., 1997, 38, 1795–1806 CAS.
- K. Bork, W. Weidemann, B. Berneck, M. Kuchta, D. Bennmann, A. Thate, O. Huber, V. S. Gnanapragassam and R. Horstkorte, Gene Expression Patterns, 2017, 23-24, 52–58 CrossRef CAS.
- T. Marquardt, K. Luhn, G. Srikrishna, H. H. Freeze, E. Harms and D. Vestweber, Blood, 1999, 94, 3976–3985 CrossRef CAS.
- K. S. Lau, E. A. Partridge, A. Grigorian, C. I. Silvescu, V. N. Reinhold, M. Demetriou and J. W. Dennis, Cell, 2007, 129, 123–134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mo00023j |
| This journal is © The Royal Society of Chemistry 2020 |