
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto/electrocatalysis and photosensitization using metal nanoclusters for green energy and medical applications
Tokuhisa
Kawawaki
 a,
Yuichi
Negishi
*a and
Hideya
Kawasaki
*b
a,
Yuichi
Negishi
*a and
Hideya
Kawasaki
*b
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.kagu.tus.ac.jp
bDepartment of Chemistry and Materials Engineering, Faculty of Chemistry, Materials and Bioengineering, Kansai University, Suita-shi, Osaka 564-8680, Japan. E-mail: hkawa@kansai-u.ac.jp
First published on 18th October 2019
Abstract
Owing to the rapidly increasing demand for sustainable technologies in fields such as energy, environmental science, and medicine, nanomaterial-based photo/electrocatalysis has received increasing attention. Recently, synthetic innovations have allowed the fabrication of atomically precise metal nanoclusters (NCs). These NCs show potential for green energy and medical applications. The present article primarily focuses on evaluation of the recent developments in the photo/electrocatalytic and photosensitizing characteristics of metal and alloy NCs. The review comprises two sections: (i) photo/electrocatalysis for green energy and (ii) photosensitization for biomedical therapy applications. Finally, the challenges associated with the use of metal NCs are presented on the basis of current developments.
 Tokuhisa Kawawaki | Assistant Professor of the Department of Applied Chemistry, Faculty of Science, Tokyo University of Science, received PhD degree (2015) in Applied Chemistry from the University of Tokyo. Since 2016, he worked as Japan Society for the Promotion of Science (JSPS) Postdoctoral fellow (PD) at the University of Melbourne. Since 2017, he worked as JSPS super PD (SPD) at Kyoto University. In 2019, he moved to the current position. His current research topics include synthesis of metal nanoparticles and nanoclusters in solutions and their applications for photoelectrochemistry. |
 Yuichi Negishi | Professor of the Department of Applied Chemistry at Tokyo University of Science. He received his PhD degree in Chemistry in 2001 under the supervision of Prof. Atsushi Nakajima from Keio University. Before joining Tokyo University of Science in 2008, he was employed as an assistant professor at Keio University and at the Institute for Molecular Science. His current research interests include the precise synthesis of stable and functionalised metal nanoclusters and their applications in energy and environmental materials. |
 Hideya Kawasaki | Professor of the Department of Chemistry and Materials Engineering, Kansai University, received Doctor degree (1996) in science from Kyushu University. Since 1999, he worked as an assistant professor at Kyushu University. In 2006, he moved to be an Associate Professor at the Department of Chemistry and Materials Engineering, Kansai University. His current research interests include synthesis of photo-functionalised metal nanoclusters and their applications for catalysis, sensing, and biomedical fields. |
1. Introduction
During the past few decades, photocatalysis and electrocatalysis have received significant attention as a result of the increasing demand for sustainable technologies in the fields of energy, environmental science, and medicine.1–7 Photo/electrocatalytic approaches rely on electronic excitation, and their performance depends on the ability to create electron (e−)–hole (h+) pairs that successively undergo chemical reactions with other compounds via oxidative (e.g., 2H2O(l) → O2(g) + 4H+(aq) + 4e−) and reductive reactions (e.g., 2H+(aq) + 2e− → H2(g)). Many advanced nanomaterial-based photo/electrocatalysts have been synthesized and reported, and their advantages include large surface-to-volume effects, numerous catalytic active sites, quantum size effects and high stability.1–7 These catalysts are considered to be promising for energy and environmental applications, such as photo/electro water splitting to generate hydrogen (H2) and conversion of carbon dioxide (CO2). Furthermore, they are used in the fuel industry and in water treatment and disinfection, air purification, and self-cleaning surfaces.1–6 Usually, the term “photo/electrocatalysis” refers to photo/electrochemical reactions, which involve an electron transfer. Conversely, when energy transfer occurs in a photochemical reaction, the process is often called “photosensitization”. Triplet photosensitizers are used not only for triplet energy transfer, but are also utilized in photodynamic therapy (PDT) for medical applications.7One drawback of these nanomaterial-based photo/electrocatalysts is that they are generally poly-dispersed in size, morphology, and chemical composition. The heterogeneity of photo/electrocatalysts with multiple active sites often results in different catalytic activities and varying catalytic selectivity. Therefore, it is important to fabricate nanomaterial-based photo/electrocatalysts that can be controlled well at the atomic level.
Recent synthetic innovations have allowed the fabrication of atomically precise metal nanoclusters (NCs).8–18 These metal NCs contain a few to hundreds of metal atoms that are protected by organic ligands (e.g., thiolate, phosphine, small molecules, synthetic polymers, and biomolecules). Particularly, thiolate-protected metal NCs and their alloy NCs can be prepared with atomic precision as Mn(SR)m (M = gold (Au), silver (Ag), cupper (Cu), platinum (Pt), palladium (Pd), or other element; SR = thiolate ligand) in a wide range of sizes by varying the n and m values. As mentioned previously, such NCs have potential applications in the fields of energy, environmental science, medicine, and other.9–13 The Mn(SR)m NCs have discrete energy levels due to the quantum size effect, making them significantly different from the larger (>3 nm) plasmonic metal (Au or Ag) nanoparticles (NPs) with continuous energy levels. By adjusting n and m, one can control the discrete electronic/geometrical structure of Mn(SR)m NCs and thereby their photosensitizing and photo/electrocatalytic capability. Recently, several excellent reviews have discussed the syntheses of metal NCs,13 photoluminescence,14 and their biomedical15–17 and catalytic applications.18 However, to the best of our knowledge, there are no publications focusing on the photo/electrocatalytic and photosensitizing characteristics of metal NCs for applications in the green energy and medical fields. Therefore, a survey of recent progress, with a view to improving the photo/electrocatalytic and photosensitizing capability of metal NCs is needed.
In this review, we discuss recent achievements and evaluate the photo/electrocatalytic and photosensitizing characteristics of metal NCs and their alloy NCs. The review comprises two sections: (i) photo/electrocatalysis and photosensitization for green energy applications (specifically solar cell), and (ii) photosensitization for medical therapeutic applications (Fig. 1). We hope to provide a clear picture on how the photo/electrocatalytic and photosensitizing capability of metal NCs can improve their performance and promote the development of new green energy and medical applications.
 | ||
| Fig. 1 Schematic of photo/electrocatalysis and photosensitization using metal NCs for green energy and medical applications. Reprinted with permission from ref. 51 and 75. Copyright American Chemical Society; ref. 127, Copyright 2015 Royal Society of Chemistry. | ||
2. Photo/electrocatalysis of metal NCs for green energy applications
In this section, we discuss recent progress of the photo/electrocatalytic (specifically reaction of water splitting and fuel cells) and photosensitizing applications (specifically solar cell) of metal NCs (Pd, Ag, Pt, or Au) and their alloy NCs. We summarized references used in this section in Table 1.| Section | Reaction | Clusters | References |
|---|---|---|---|
| 2.1.1 | H2 evolution reaction | Pt | 24 and 25 |
| 2.1.1 | H2 evolution reaction | Pd | 26 and 34 |
| 2.1.1 | H2 evolution reaction | Au | 27–34 |
| 2.1.2 | O2 evolution reaction | Pd | 26 |
| 2.1.2 | O2 evolution reaction | Au | 40 and 41 |
| 2.2 | Electrocatalytic reaction for fuel cells | Pt | 45–48 and 55 |
| 2.2 | Electrocatalytic reaction for fuel cells | Au | 49–54 and 56 |
| 2.3 | Photocatalytic water splitting reaction | Pt | 58 and 59 |
| 2.3 | Photocatalytic water splitting reaction | Au | 60–64, 67 and 68 |
| 2.3 | Photocatalytic water splitting reaction | Ag | 65 and 66 |
| 2.4 | Redox reaction for solar cell | Au | 69, 71 and 73–78 |
| 2.4 | Redox reaction for solar cell | Ag | 70 and 72–74 |
| 2.4 | Redox reaction for solar cell | Pt | 71 |
| 2.4 | Redox reaction for solar cell | Pd | 71 |
2.1. Electrocatalytic applications for water splitting
Water electrolysis is one of the cleanest energy-producing processes, generating H2 using only water and electricity, which is regarded as the next-generation energy source or raw material for chemicals. Water electrolysis comprises two half-reactions, which are the H2 evolution reaction (HER) and the O2 evolution reaction (OER). When supplying electricity to metal electrodes, a reduction reaction occurs on the cathode and an oxidation reaction on the anode, and thereby water molecules are split into H2 and O2. However, the reaction does not proceed if the voltage supplied to the electrodes is higher than the redox potential of each reaction (HER: 0 V vs. SHE, OER: 1.23 V vs. SHE) due to the high activation energy. Therefore, investigations have been conducted to enhance the reactions using metal catalysts, such as precious metal NPs. An increased specific surface area of the active sites in the metal catalysts significantly increases the reaction rate per unit area. Hence, research on the improvement of the synthetic techniques to obtain fine metal particles has progressed. According to the Sabatier principle of chemical reactions occurring on the surface of a catalyst, the maximum activity is attained when the free energy of the adsorption and desorption of the reagents on the metal catalyst is the lowest. Consequently, the activity is closely related to the energy level (Fermi level) of the d-band center of each metal and the geometrical structure of the surface, which depends on the crystal plane and interatomic distances. These characteristics result in the tendency to form peaks in the activity volcano plots. The optimum metal types and their alloy NPs have been prepared based on the activity volcano plots obtained using various metal elements. Further reduction of the metal particle sizes induces the appearance of novel electronic and/or geometrical structures, which are different from the bulk metal. Therefore, recently, a number of studies on the electrocatalytic application of metal NCs composed of approximately several tens of atoms or less have been reported.Under acidic conditions, the reaction proceeds as follows:
Volmer reaction:
| H+ + e− ↔ Hads |
Heyrovsky reaction:
| Hads + H+ + e− ↔ H2 |
Tafel reaction:
| 2Hads ↔ H2 |
On the other hand, under alkaline conditions, the following reaction sequence occurs:
Volmer reaction:
| H2O + e− ↔ Hads + OH− |
Heyrovsky reaction:
| Hads + H2O + e− ↔ H2 + OH− |
Tafel reaction:
| 2Hads ↔ H |
In contrast, bulk Au, which belongs to group 11, shows almost no HER activity. Reduction of the size to the cluster region, however, results in changes in the electronic structure, and thereby Au NCs exhibit HER activity. Based on the recent studies, Au NCs can now be synthesized using a very simple and easy approach. Moreover, the sizes can be controlled at atomic precision and their functions can be modulated via ligand exchange and heteroatom doping. In recent years, a number of studies have been conducted on the HER activity of Au NCs. For example, Teranishi and Sakamoto et al. studied the effect of the ligand structure on the HER activity using porphyrin-coordinated Au NCs (Fig. 3).27 In their study, the distance between the porphyrin-ring and the surface sulfur (S) directly bonded to Au NCs was controlled by varying the number of methylene groups between those moieties (Fig. 3A and C). They found that the HER activity increases with decreasing distance between the porphyrin-ring and the Au NC surface (Fig. 3G and H). In addition, it was demonstrated that tetrakis-5α,10α,15α,20α-(2-acetylthiomethylphenyl) porphyrin (SC1P)-protected Au NCs, with a 3.4 Å distance between the porphyrin-ring and the Au NC surface, show a high current density of 460% at 0.4 V vs. RHE, in comparison with Au NCs protected by ordinary phenylethanethiolate (PET). The study also demonstrates that the ligand structure strongly affects the HER activity and that the smaller Aun(SR)m NCs exhibit the higher HER activity.
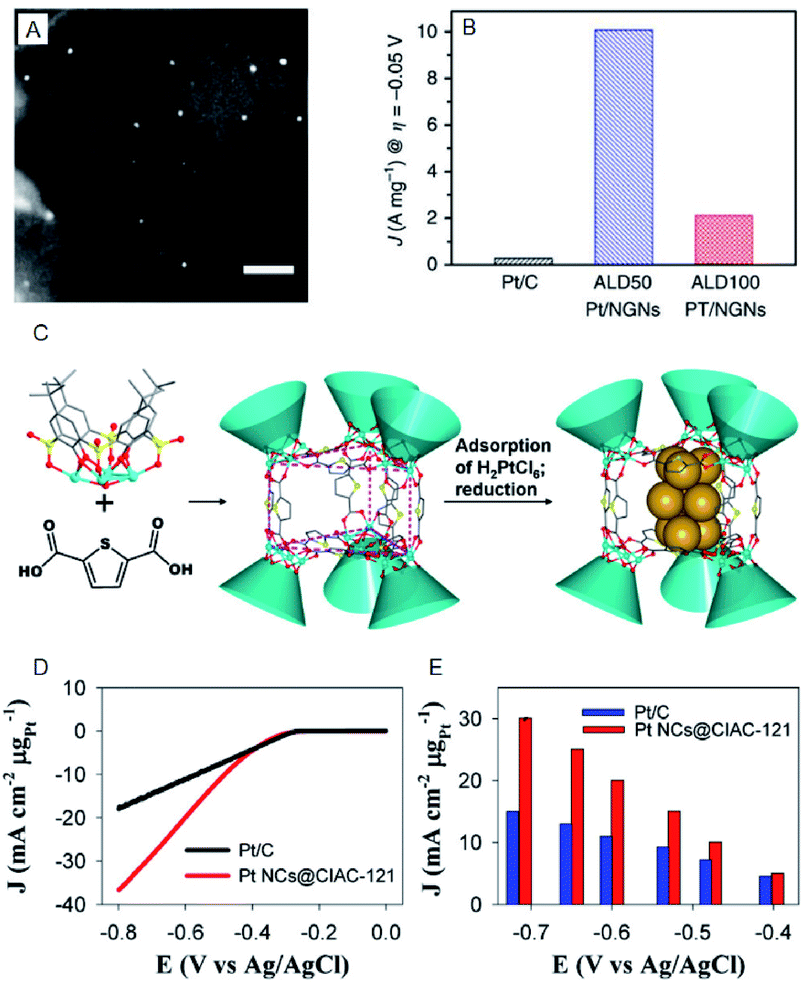 | ||
| Fig. 2 (A) The annular dark-field scanning transmission electron microscopy (SEM) image of ALD50Pt/nitrogen-doped graphene nanosheets (NGNs). Scale bar shows the length of 20 nm. (B) HER current density of Pt/C, ALD50Pt/NGNs and ALD100Pt/NGNs. (C) Illustration of the assembly of trigonal prismatic {Ni24} coordination cage (CIAC-121) and the fabrication of ultrafine Pt NCs. (D) The HER polarization curves. (E) Mass HER activity of Pt NCs@CIAC trigonal prismatic coordination cage ({Ni24(TC4A-SO2)6(TDC)12(H2O)6}, H4TC4A-SO2 = p-tert-butylsulfonylcalix[4]arene; H2TDC = 2,5-thiophenedicarboxylic acid). Panels (a) and (b) are reproduced with permission from ref. 24. Copyright 2016 Springer Nature. Panels (c), (d) and (e) are reproduced with permission from ref. 25. Copyright 2016 American Chemical Society. | ||
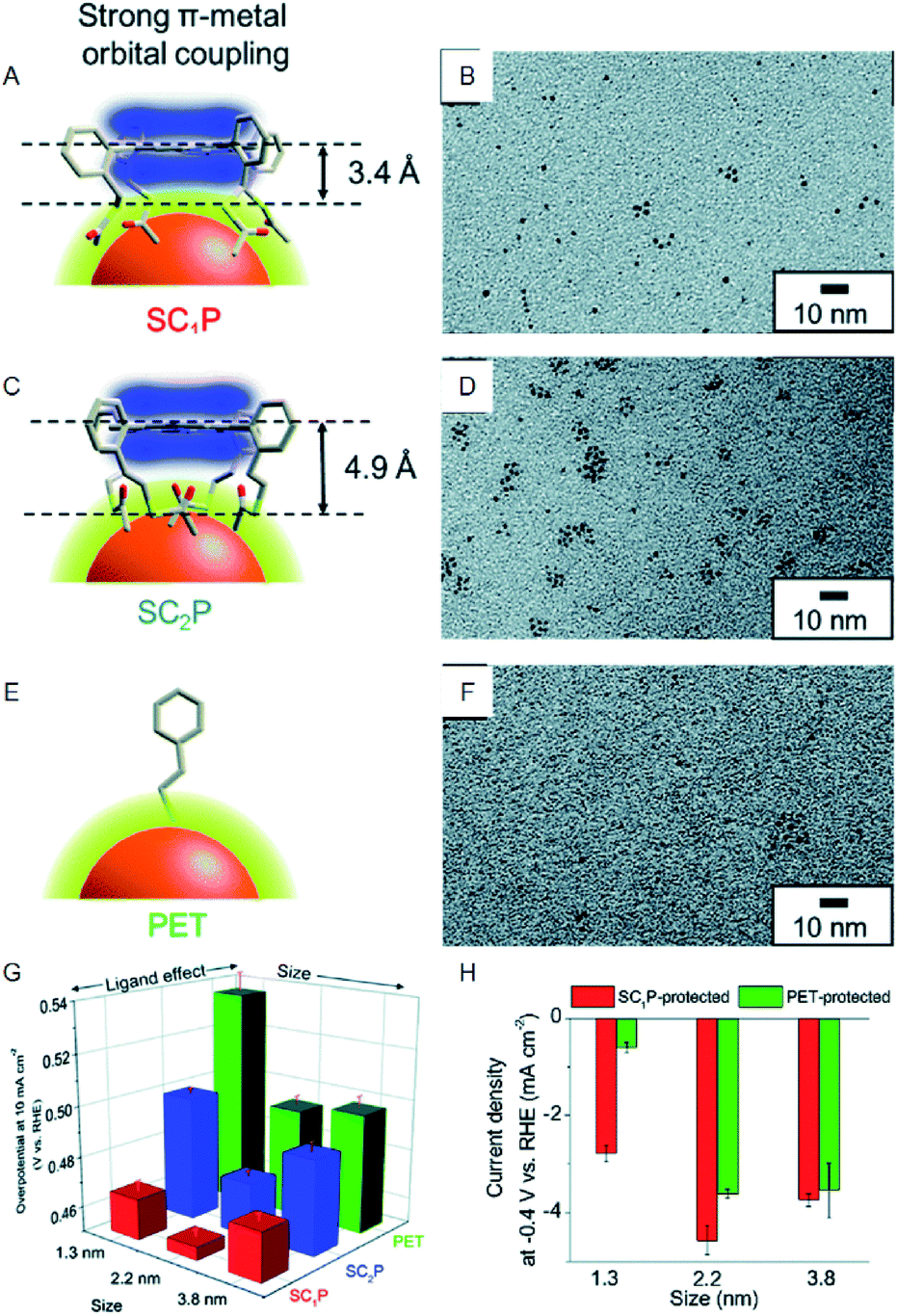 | ||
| Fig. 3 (A), (C), and (E) Schematic of coordination fashions of SC1P. (A) Tetrakis-5α,10α,15α,20α-(2-acetylthioethylphenyl) porphyrin (SC2P; A) and PET (E) at the surface of Au NCs. (B), (D), and (F) TEM images of 1.3, 2.2, and 3.8 nm Au NCs protected with SC1P, respectively. (G) Comparison of overpotential at −10 mA cm−2. (H) Current density at −0.4 V of each size of Au NCs protected with each ligand. Panels (a)−(h) are reproduced with permission from ref. 27. Copyright 2018 Royal Society of Chemistry. | ||
On the other hand, Lee and Jiang et al. investigated the effect of heteroatom doping of Aun(SR)m NCs on the HER activity.28–31 They examined the HER activity of hexanethiolate (C6)-protected Au25 and PtAu24 NCs in a tetrahydrofuran (THF) electrolyte containing 1.0 M trifluoroacetic acid (TFA) (Fig. 4A–C). The obtained results demonstrated that PtAu24 NC has better catalytic activity with a higher starting potential (−0.89 V) than Au25 NC (−1.10 V). The same research group showed that the improved catalyst current and turnover frequency can be obtained even with Pd as the central atom, instead of Pt (PtAu24 > PdAu24 > Au25). The same tendency can be observed for the other-sized C6-protected Au38 NCs (Pt2Au36 > Pd2Au36 > Au38).32 These results are in good agreement with those obtained from DFT calculations, which predicted that the HER activity significantly varies depending on the doping elements. Hence, the control of the electronic structure by alloying at the atomic level greatly affects the HER activity in the cluster size range.
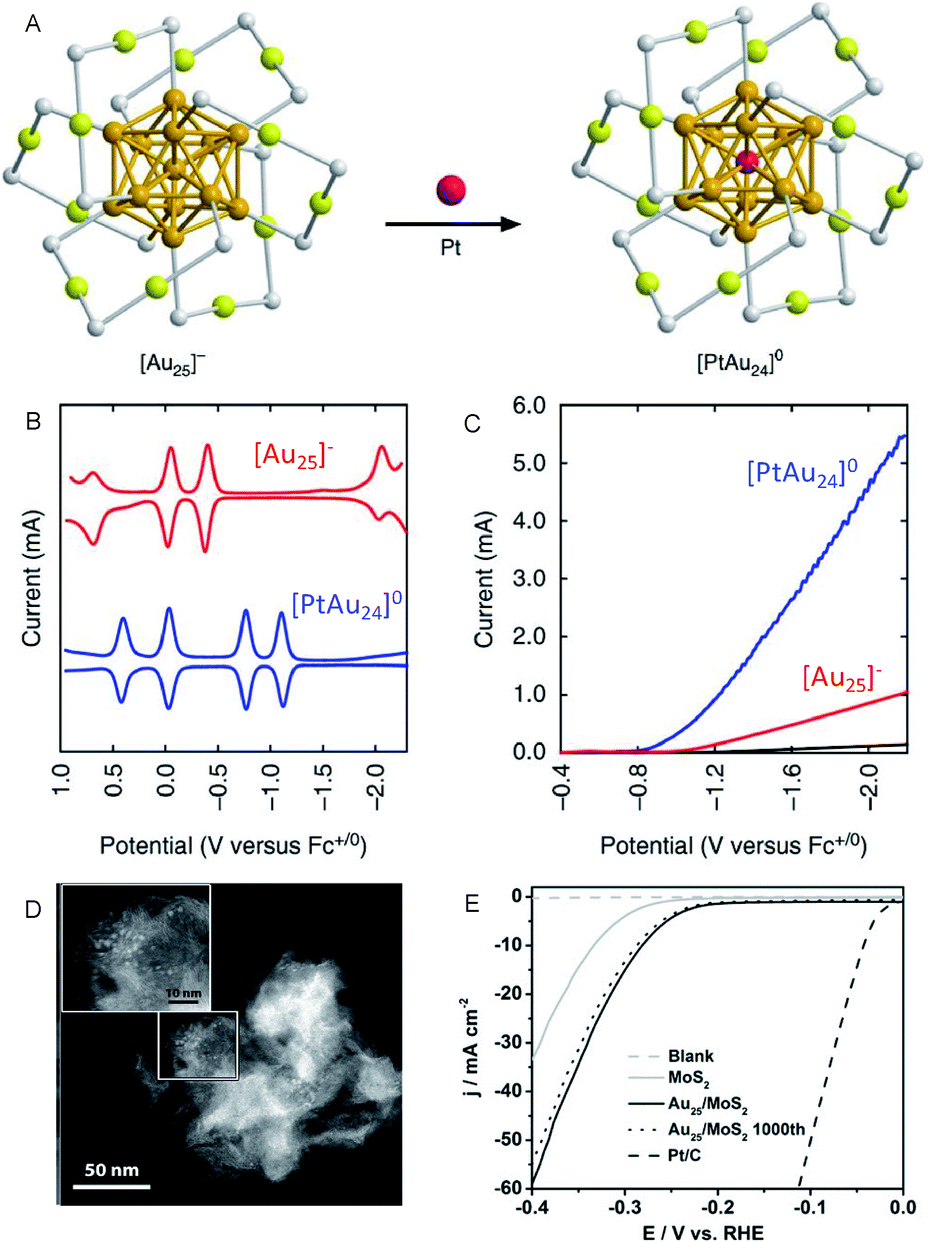 | ||
| Fig. 4 (A) Geometrical structures of C6-protected Au25 and PtAu24 NCs (golden, gold atoms of the core; olive, gold atoms of the shell; grey, sulfur). (B) The square-wave voltammogram. (C) HER polarization curves of C6-protected Au25 and PtAu24 NCs. (D) High angle annular dark-field SEM (HAADF-STEM) images. (E) HER polarization curves of the C6-protected Au25/MoS2 composite. Panels (a)–(c) are reproduced with permission from ref. 29. Copyright 2017 Springer Nature. Panels (d) and (e) are reproduced with permission from ref. 33. Copyright 2017 Wiley-VCH. | ||
In addition to these studies, Jin et al. investigated the HER activity of the composite comprising PET-protected Au25 NCs and molybdenum disulfide (MoS2) nanosheet (MoS2 nanosheet carrying PET-protected Au25; Au25/MoS2; Fig. 4D and E).33 The Au25/MoS2 showed a current density 1.79 times higher than that of MoS2 at −0.4 V, indicating that the improvement of HER activity of MoS2 nanosheet was caused by loading Au25 NCs. X-ray photoelectron spectroscopy analysis showed that a negative shift of ∼0.4 eV in the Mo 3d orbital energy was induced by loading Au25 NCs. This implies that the partial charge transfer occurs from Au25 NCs to MoS2, leading to the increased HER activity. The HER activity of the composite is, therefore, considerably improved by the electronic interaction between the materials. Regarding the HER activity of the composite comprising MoS2, Zhu et al. also evaluated Au2Pd6/MoS2.34
Under acidic conditions, the reaction proceeds as follows:
| H2O → OHads + H+ + e− |
| OHads → Oads + H+ + e− |
| Oads + H2O → OOHads + H+ + e− |
| OOHads → 2Oads + H+ + e− |
| 2Oads → O2 |
On the other hand, under alkaline conditions, the following reaction sequence takes place:
| OH− → OHads + e− |
| OHads + OH− → Oads + H2O + e− |
| Oads + OH− → OOHads + e− |
| OOHads + OH− → 2Oads + H2O + e− |
| 2Oads → O2 |
According to this reaction pathway, the OER activity of the catalyst generally depends on the binding energy of the OER intermediates on the surface (O, OH, OOH, etc.). An appropriate binding energy of the oxygen species (neither too high nor too low) enables the catalyst suitable for OER. For this reason, the metal oxides, such as iridium oxide (IrOx), ruthenium oxide (RuOx), exhibit high activity. Therefore, extensive efforts have been made on their miniaturization, the theoretical prediction of the activity and the mechanism.35–38
Moreover, research regarding cluster-sized particles is progressing. Chen and Gao reported that dodecanethiolate (C12)-protected Pd6/AC exhibits a current density 37 times higher than that of commercially available Pt/C at an applied voltage of 1.85 V (Fig. 5A and B).26 Although ligands were considered to be disadvantageous for electrode reactions, this study revealed that the higher electron density of C12-protected Pd6/AC enhances the desorption process of oxygen atoms or molecules and is favorable for OER. In addition, there are several reports describing the suitability of Mn(SR)m NCs for OER. For example, Hussain and Joya et al. reported that the OER activity of Ni4(PET)8 NC is equal to that of a RuO2 electrocatalyst.39
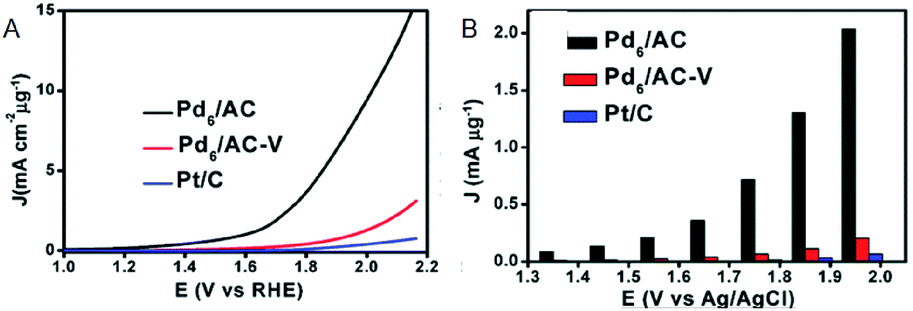 | ||
| Fig. 5 (A) OER polarization curves. (B) Mass OER activity of Pd6/AC, Pd6AC-V and Pt/C at each overpotential. Panels (a) and (b) are reproduced with permission from ref. 26. Copyright 2017 Royal Society of Chemistry. | ||
As with HER activity, OER activity has also been studied for the composites including Au NCs. Jin et al. investigated the OER activity of the composite comprising PET-protected Au25 NCs and cobalt diselenide (CoSe2) nanosheet (CoSe2 nanosheet carrying PET-protected Au25; Au25/CoSe2) and revealed that Au25/CoSe2 shows a current density of 10 mA cm−2 with a small overvoltage of approximately 0.43 V (∼0.52 V with CoSe2 alone) (Fig. 6).40 The group also investigated the effect of Au NC size on OER activity using a series of Aun(SR)m NCs (Au10, Au25, Au144, and Au333). It was revealed that OER activity increases with increasing the Au NC size. Furthermore, Peng and Zeng demonstrated that Au NC/CoSe2 is superior for increasing the OER activity compared to commercially available Ir/C.41
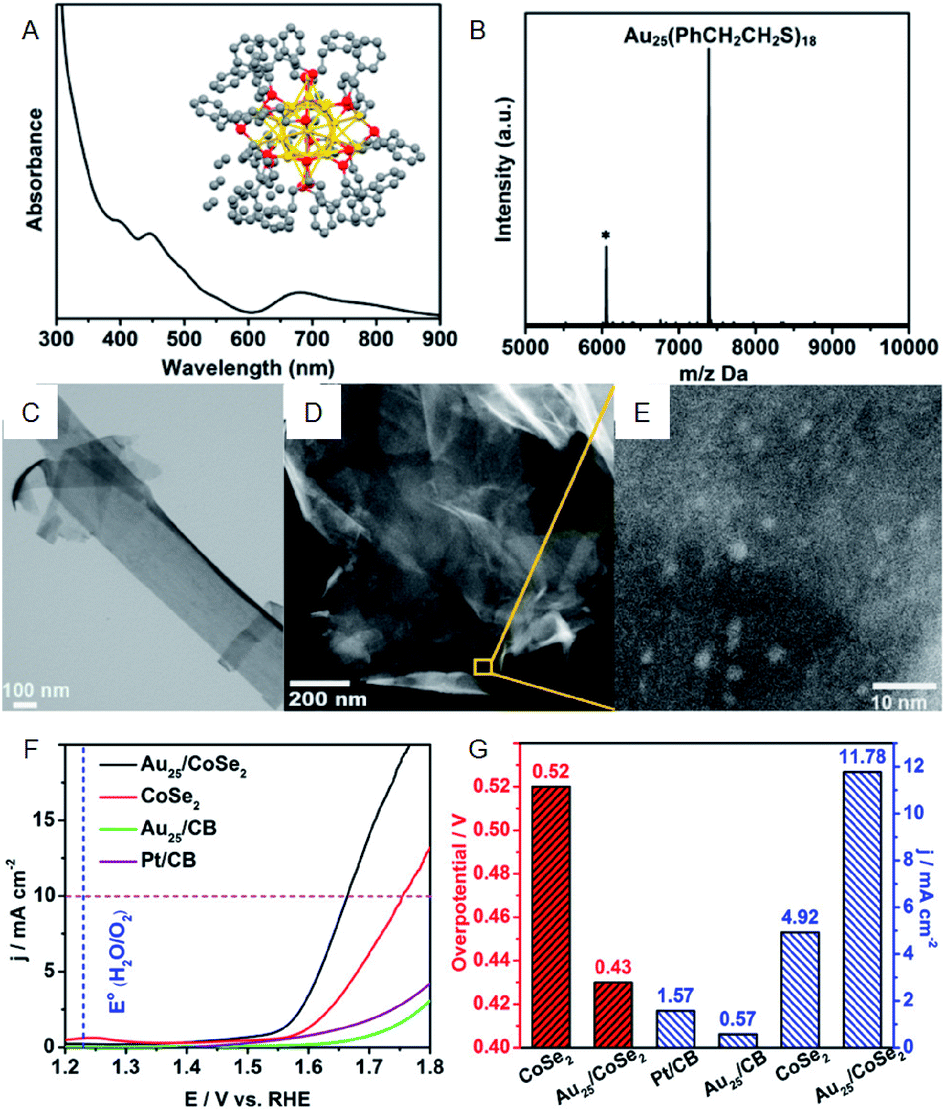 | ||
| Fig. 6 (A) UV-vis and (B) MALDI-mass characterization of Au25(PET)18 NCs. Inset of (A): crystal structure of Au25(PET)18 NCs. (C) TEM image of CoSe2 nanosheets. (D) and (E) HAADF-STEM images of Au25/CoSe2 composite with different magnifications. (F) OER polarization curves. (G) Mass OER activity of Au25/CoSe2 composite, CoSe2, Au25 supported on carbon black (CB) and commercial Pt/CB. Panels (a)–(g) are reproduced with permission from ref. 40. Copyright 2017 American Chemical Society. | ||
2.2. Electrocatalytic application for fuel cells
The ultimate energy-conversion systems used for power generation are fuel cells utilizing materials such as H2 and methanol obtained from natural energy sources. Such methods involve circulating energy systems that do not use any fossil fuels and only release water as the waste product.Fuel cells are roughly classified into those using alcohol and those using H2. The latter involves the H2 oxidation reaction (HOR) and O2 reduction reaction (ORR), which are the reverse reactions of HER and OER, respectively. HOR is a one-electron reaction system where the catalysts displaying HER-activity can be useful. In contrast, ORR is a four-electron reaction system, and since it proceeds through a complex reaction pathway, its reactivity is different from that of OER.
Under acidic conditions, ORR proceeds as follows:
| O2 + 4H+ + 4e− → H2O |
| O2 + 2H+ + 2e− → H2O2 |
| H2O2 + 2H+ + 2e− → 2H2O |
On the other hand, under alkaline conditions, the following reaction sequence takes place:
| O2 + H2O + 4e− → 4OH− |
| O2 + H2O + 2e− → OOH− + OH− |
| OOH− + H2O + 2e− → 3OH− |
The first step in both of these reaction pathways is the scission of the O–O bond. Regarding fuel cell applications, the theoretical redox potential of the direct four-electron pathway is 1.23 V vs. SHE, which is greater than that of the indirect two-electron pathway (0.68 V vs. SHE). Therefore, the direct four-electron pathway is the preferred reaction system, owing to the higher energy-conversion efficiency. Since the ORR on the reduction side is slower than on the oxidation side, the former is the rate-limiting step in the fuel cell. Hence, understanding the factors controlling ORR and thereby the creation of high-performance ORR catalysts are essential for the development of efficient fuel cells.
Since Pt exhibits high ORR activity as well as high OER activity, the development of the miniaturization techniques, prediction of the activity by theoretical calculations, and the mechanism for Pt NCs have been studied extensively.42–44 For example, Yamamoto et al. examined the ORR activity of fine Pt NCs (Ptn NC, n = 12–24) using dendrimer-encapsulated atomically-controlled Ptn NCs (Fig. 7).45 The results demonstrated that Pt19 NC exhibits the highest ORR activity (Fig. 7E). The edge site of Pt19 has an ideal oxygen binding energy, which indicates that in addition to the electronic structure, the geometrical structure affects the ORR activity.46–48
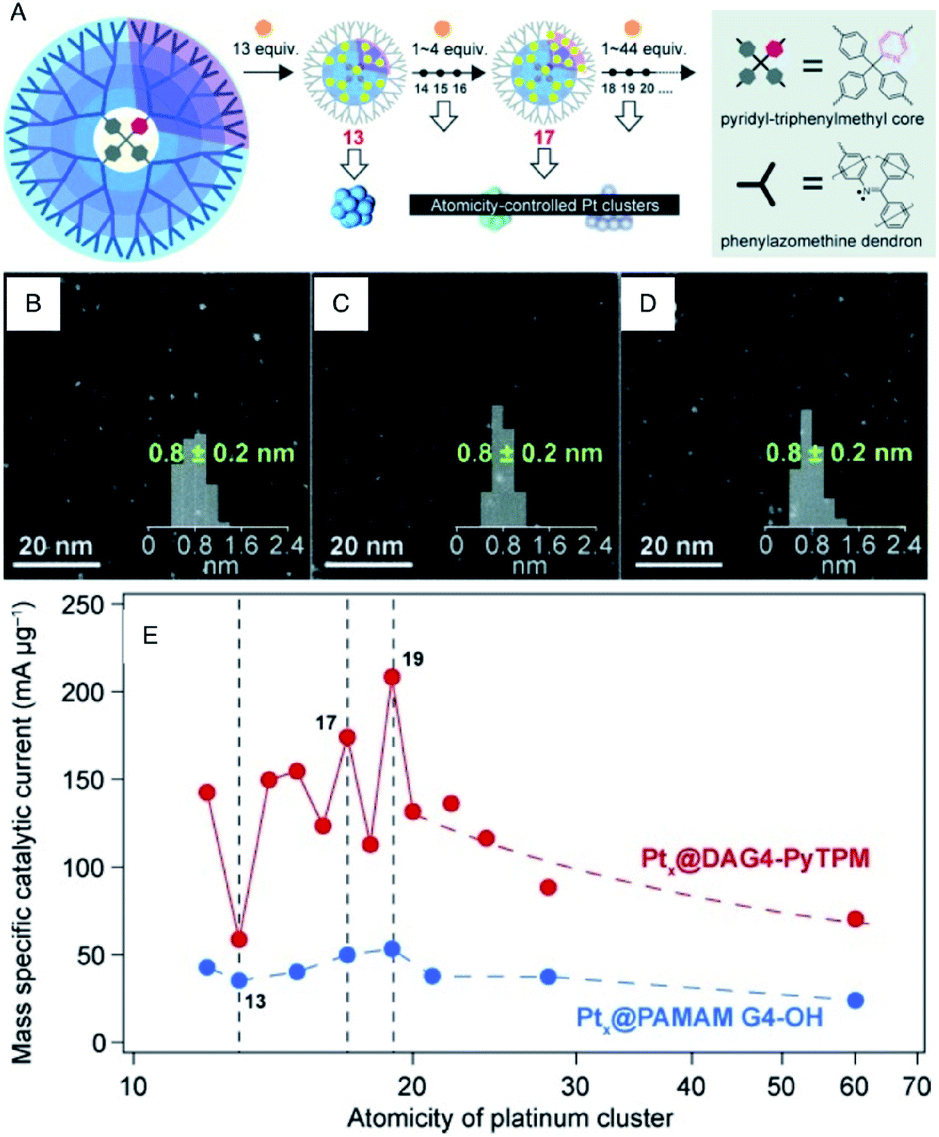 | ||
| Fig. 7 (A) Illustration of the precise synthesis method of atomicity precise Pt NCs using a dendrimer reactor. HAADF-STEM images of (B) Pt13, (C) Pt17 and (D) Pt19 NCs. (E) Variations of the mass ORR activity vs. the atomicity of the Pt NCs. Panels (a)–(e) are reproduced with permission from ref. 45. Copyright 2015 Wiley-VCH. | ||
Moreover, the use of Aun(SR)m NCs as catalysts for ORR has also attracted significant attention as an alternative to expensive Pt catalysts and to evaluate the catalytic reaction mechanism. Chen et al. reported that the ORR activity increases with a decrease of Au NC size (Au25(PET)18 > Au38(PET)24 > Au144(PET)60; Fig. 8A and B).49,50 This increase is suggested to be due to the enhancement of the oxygen adsorption caused by increasing the proportion of surface atoms and shifting the d-band center associated with a decrease of Au NC size. Chakraborty and Dass et al. synthesized a series of 4-tert-butylbenzenethiolate (TBBT)-protected Au NCs and analyzed the ORR activity of Au NCs supported on single-walled carbon nanotubes.51 The study on the reaction rate revealed that the ORR activity increases in the order of Au36(TBBT)24 > Au133(TBBT)52 > Au279(TBBT)84 > Au28(TBBT)20 (Fig. 8C and D). The same research group also used tert-butylthiolate (S-tBu) as a ligand and showed that the ORR activity increases in the order of Au46(S-tBu)29 ≈ Au65(S-tBu)29 > Au30(S-tBu)18 > Au23(S-tBu)16 in the case of S-tBu-protected Au NCs.52
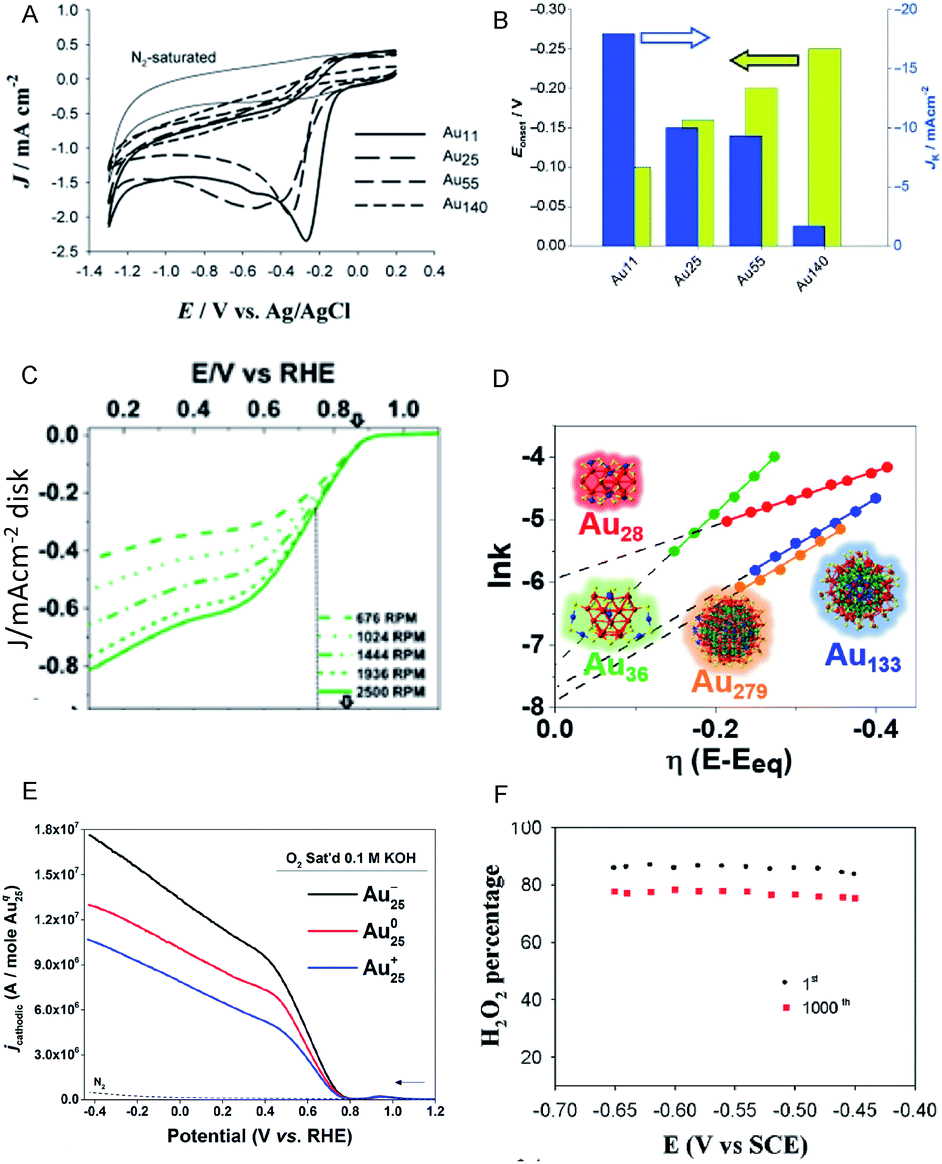 | ||
| Fig. 8 (A) Cyclic voltammograms of the Aux/glassy carbon (GC) electrodes (x = 11, 25, 55 and 140) saturated with O2 and of the Au11/GC electrode saturated with N2 (thin solid curve). (B) Current density and overpotential of ORR activity with each size of Au NCs calculated from (A). (C) Rotating-disk voltammograms recorded for ORR activity by Au36(TBBT)24 NCs/GC electrode at different rotation rates. (D) Reaction rate constant ln(k) vs. overpotential E plots with each size of Au NCs calculated from (C). (E) Rotating-ring-disk voltammograms with 1600 rpm rotation rate. (F) Percentage (or selectivity) of H2O2 using with Au25−, Au250, and Au25+ NCs. Panels (a) and (b) are reproduced with permission from ref. 49. Copyright 2009 Wiley-VCH. Panels (c) and (d) are reproduced with permission from ref. 51. Copyright 2018 American Chemical Society. Panel (e) is reproduced with permission from ref. 54. Copyright 2014 Royal Society of Chemistry. Panel (f) is reproduced with permission from ref. 53. Copyright 2014 Royal Society of Chemistry. | ||
In addition to the NC size, the control of charge state of NCs is also a crucial factor for obtaining high activity. For example, the negatively charged Au25(PET)18 NC shows higher activity in the production of H2O2, which is a two-electron reduction reaction, than the neutral Au25(PET)18 NC (Fig. 8E and F).53,54 Since H2O2 is a useful raw material for a variety of chemical products, the development of a highly selective H2O2 production method is of importance. Likewise, the interaction with the substrate also requires consideration. Pt or Au NCs supported on various substrates, such as indium tin oxide (ITO)55 and reduced graphene oxide (rGO) sheets56 show outstanding electrocatalytic performance in ORR, which was found to be originated in the interaction between metal NC and the substrate.
2.3. Photocatalytic application for water splitting
Artificial photosynthesis, which has attracted significant attention in recent years, is an ultimate energy production technique involving conversion of light into chemical energy. In 1972, Honda and Fujishima et al. discovered that water can be split into H2 and O2 by irradiating the titanium dioxide (TiO2) photocatalyst with ultraviolet light.57 Utilizing this method allows efficient generation of H2 as an energy source using abundant water, without the production of carbon dioxide. In the photocatalytic water splitting reaction, the catalyst comprises a photocatalyst, which absorbs light, and a cocatalyst whose role is to reduce the overpotential of HER and OER. Moreover, the photocatalyst uses light energy instead of electric energy as the driving force for HER or OER. Therefore, the position of the conduction and valence bands and the width of the bandgap are essential factors when considering the photocatalyst.Highly active metal NCs in electrochemical HER and OER are effective cocatalysts in photocatalytic systems. The photocatalytic reaction involves several key steps: (1) generation of the e−–h+ pair in the photocatalyst, (2) transfer of the e− and h+ to the active sites, and (3) generation of H2 and O2 at the active sites. Here the cocatalyst greatly affects processes (2) and (3). The photodeposition and impregnation methods are typical approaches for loading cocatalysts on photocatalysts. They offer several advantages, such as the formation of fine metal particles directly on the photocatalyst. However, the disadvantages of these methods include large size dispersibility.
It is possible to load the controlled NC on photocatalyst and thereby to deepen the understanding on the effect of cocatalyst size on the photocatalytic activity when using an ultra-high vacuum apparatus. Heiz, Feldmann, and Jäckel et al. loaded a series of atomically precise Pt NCs (Pt8, Pt22, Pt34, Pt46, Pt68) on cadmium sulfide (CdS) nanorod utilizing an ultra-high vacuum apparatus (Fig. 9A and B).58,59 They investigated the water-splitting activity of the resulting photocatalysts (Pt NC/CdS) and consequently revealed that Pt46/CdS exhibits the highest activity (Fig. 9C). In the NC region, the bandgap increases with a decrease of the NC size due to the quantum size effect. It was also reported that the control of the lowest unoccupied molecular orbital (LUMO) position of the cocatalyst by controlling the NC size is of great importance for the effective electron transfer from the photocatalyst to the cocatalysts. Furthermore, the group demonstrated that the LUMO position must be lower than the conduction band of the semiconductor photocatalysts and higher than the reduction potential of HER (Fig. 9D).
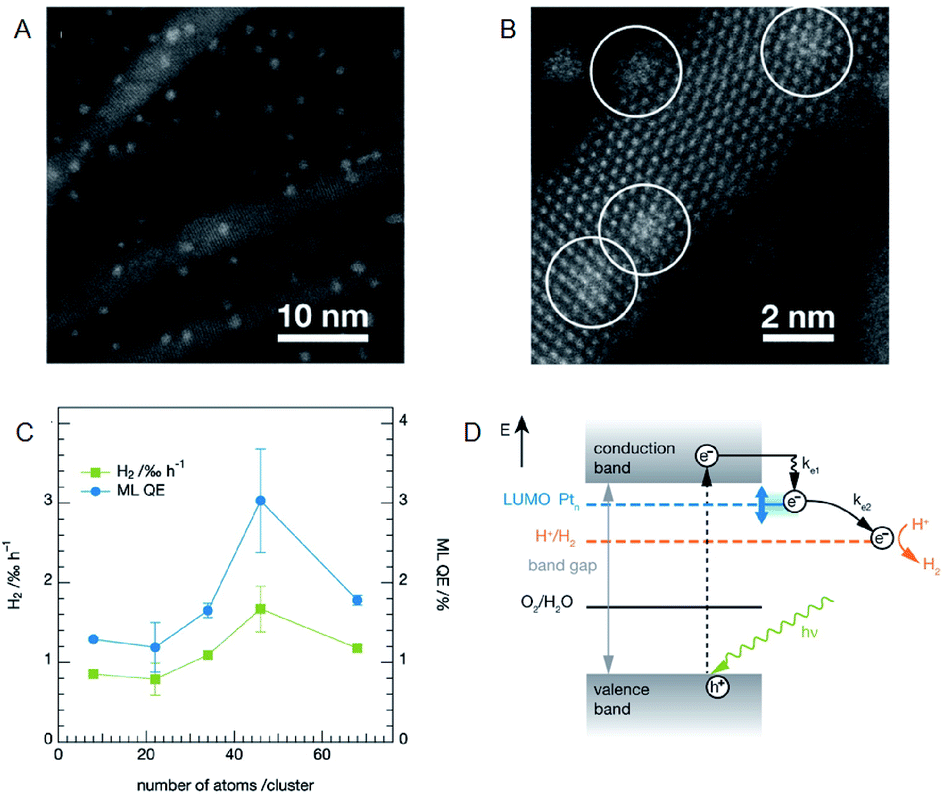 | ||
| Fig. 9 (A) and (B) HAADF-STEM images of CdS nanorods decorated with Pt NCs at different magnification. (C) Photocatalytic activity of the CdS nanorods decorated with size-selected Pt NCs as a function of cluster size. (D) Reaction pathways for the photocatalytic H2 evolution in CdS with Pt NCs. Panels (a)–(d) are reproduced with permission from ref. 58. Copyright 2013 American Chemical Society. | ||
As well as investigation into electrochemical properties of HER and OER, research on photocatalytic water splitting is rapidly progressing also for Aun(SR)m NCs. Negishi and Kudo et al. developed a photocatalytic system comprising atomically precise Au25 NC and the BaLa4Ti4O15 photocatalyst (Fig. 10).60 In this study, the atomically precise Au25 NC was loaded on the photocatalyst by adsorbing glutathionate (SG)-protected Au25 NC on BaLa4Ti4O15 and subsequently eliminating the SG ligands by calcination. TEM analysis of the products confirmed that NC-size change barely occurred during the calcination. The resulting Au25/BaLa4Ti4O15 composite generates both H2 and O2 in a stoichiometric ratio upon light irradiation and the catalytic activity is 2.6 times higher than that of Au NP loaded-BaLa4Ti4O15 (Fig. 10A). The same group also revealed that the smaller the particle size, the higher the activity (Fig. 10B). In their more recent study, it was described that the ORR, which is a reverse reaction, can be suppressed by coating the Au25 NC with Cr2O3, and thereby the activity was improved by 19-fold compared to the non-coated Au25/BaLa4Ti4O15 (Fig. 10C–E).61 The cocatalysts with high HER and OER activities typically display good activity in HOR and ORR.62,63 Overall, as shown in this study, the control of the reverse reaction is an important factor for improving the activity.
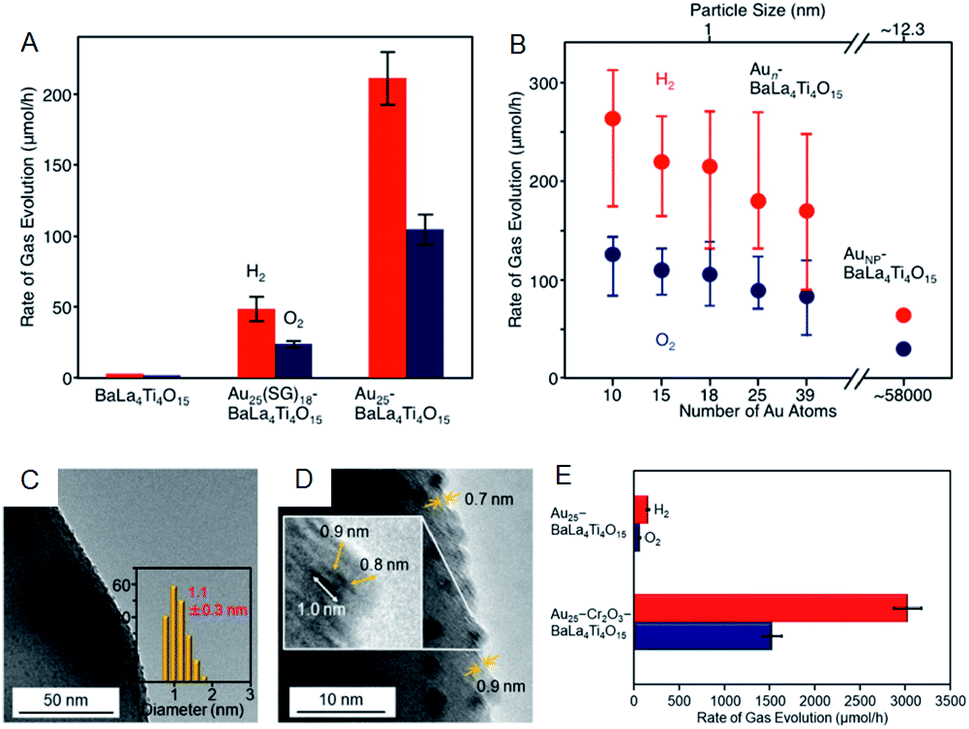 | ||
| Fig. 10 Effect of (A) ligands protection and (B) cluster size on the rates of photocatalytic activity for water splitting studied using Aun/BaLa4Ti4O15 (n = 10–39) and Au NPs/BaLa4Ti4O15. (C) TEM image. (D) High resolution-TEM image. (E) Rates of photocatalytic activity for water splitting of Au25–CrxOy/BaLa4Ti4O15. Panels (a) and (b) are reproduced with permission from ref. 60. Copyright 2015 American Chemical Society. Panels (c)–(e) are reproduced with permission from ref. 61. Copyright 2018 American Chemical Society. | ||
The photocatalytic activity could also be modulated by heteroatom doping on the cocatalysts. Negishi and Yamazoe et al. demonstrated that Pd doping on Au25 NC reduces the water-splitting activity, whereas Pt doping on Au25 NC enhances it (Fig. 11A–F).64 Yang et al. also examined the effect of heteroatom doping on the photocatalytic activity of PtAg24-loaded graphitic carbon nitride (PtAg24/g-C3N4).65 They revealed that PtAg24/g-C3N4 shows higher activity for photocatalytic H2 production than Ag25/g-C3N4 (Fig. 11G and H). In addition to the above studies, there are several reports on water-splitting photocatalytic activity using composites including metal NCs, such as graphitic carbon nitride nanosheets carrying mercaptosuccinic acid-protected Ag9 NC,66 TiO2 carrying SG-protected Au NC, etc.67,68
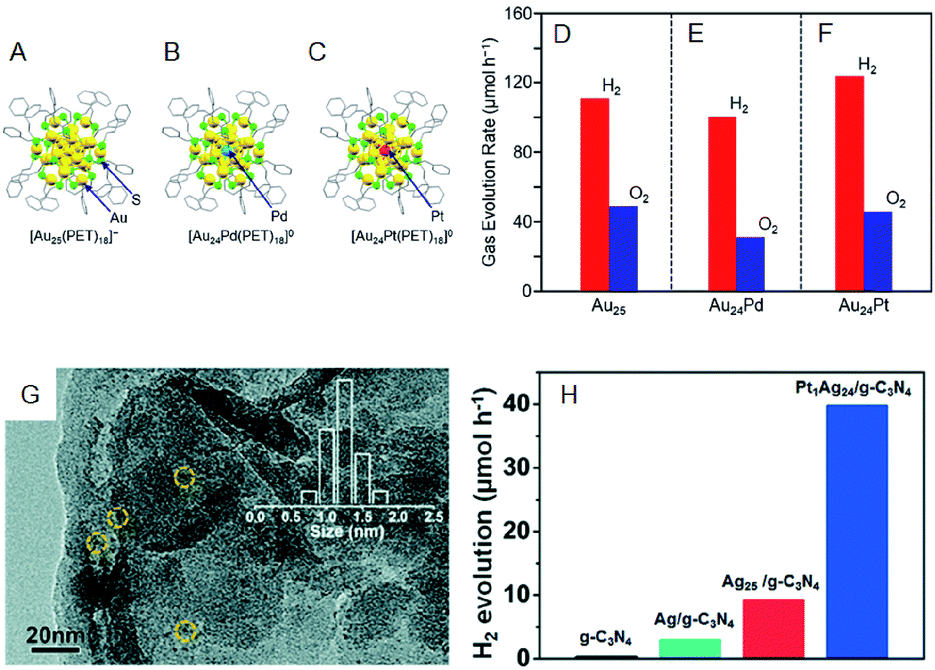 | ||
| Fig. 11 Geometrical structures of (A) Au25(PET)18, (B) Au24Pd(PET)18, and (C) Au24Pt(PET)18. Rates of photocatalytic activity for water splitting with Au24M–BaLa4Ti4O15 for M = (A) Au, (E) Pd, and (F) Pt. (G) TEM image of PtAg24/g-C3N4. (H) The rates of photocatalytic activity of PtAg24/g-C3N4, Ag25/g-C3N4, Ag/g-C3N4, and g-C3N4. Panels (a)–(f) are reproduced with permission from ref. 64. Copyright 2019 American Chemical Society. Panel (g) and (h) are reproduced with permission from ref. 65. Copyright 2017 Royal Society of Chemistry. | ||
2.4. Photosensitization for solar cell applications
Solar power generation systems, which directly convert light energy into electricity, continue to be important renewable energy sources and are the key components of the world's technological advancement. A photoelectrode cell, commonly known as the solar cell, comprises a light absorption layer, which generates e−–h+ pairs, and a working electrode/counter electrode that independently take out e− and h+. The material used for the light absorption layer must effectively absorb light in the visible range, constituting approximately 50% of sunlight. In this respect, bulk gold reflects light due to a large number of free electrons. However, with the decreasing size of the NC, the number of electrons decreases, and discrete energy levels appear. Hence, a metal NC has a HOMO–LUMO gap similar to that of a molecule and exhibits light absorption in the visible to the near-infrared (NIR) region.Tatsuma and Sakai previously demonstrated that metal NCs can be used for the light absorption layer in solar cells.69 They produced a photoelectrode in which a TiO2 electrode was coated with a series of SG-protected Au NC of various sizes (Au NC/TiO2). When hydroquinone was used as an electron donor, a photo-response was observed (Fig. 12A and B) and its photocurrent behavior was consistent with photocurrent action spectrum and absorption spectrum (Fig. 12C and D). These results indicate that an electron is excited and promoted from the HOMO to the LUMO by photoirradiation in Au NC and that the photoexcited electron of LUMO is transferred to the CB of TiO2. In this way, the Au NC functions like a dye in dye-sensitized solar cells (DSSCs). The same research group observed the generation of photovoltaic power even when the other element NCs (Ag, Pt and Pd NCs) were employed.70,71 In addition, various other metal and alloy cluster sensitized solar cells have been reported by a number of research groups.72–74
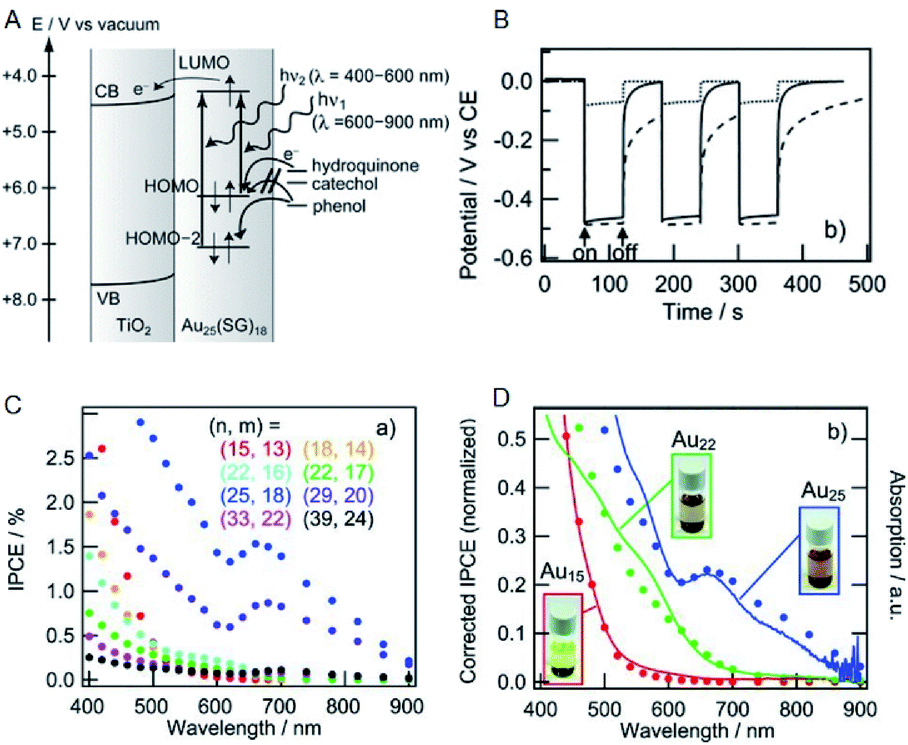 | ||
| Fig. 12 (A) Mechanism of Au NCs sensitized solar cells. (B) Photopotential and (C) incident photons conversion efficiency vs. irradiation light wavelength (incident power conversion efficiency (IPCE) spectra) of Au NCs sensitized solar cells. (D) Normalized IPCE spectra and absorption spectra of Au NCs sensitized solar cells. Panels (a)–(d) are reproduced with permission from ref. 69. Copyright 2010 Wiley-VCH. | ||
Regarding the solar cells, Kamat et al. achieved a high external quantum efficiency (IPCE) of 70% at 400–425 nm by using a Co(bpy) redox pair (Co(bpy)3(PF6)2/Co(bpy)3(PF6)3) in SG-protected Au NC/TiO2 system (Fig. 13).75 This value compares favorably even with the CdS-based quantum dot solar cells. The power conversion efficiency (PCE) was 2.36% at maximum, indicating that metal NCs can act as a high-performance sensitizer in sensitized solar cells. Bang, Kang, and Lee et al. achieved a PCE value as high as 3.8% when investigating SG-protected Au18 NC/TiO2 solar cells using the I−/I3− redox pair (Fig. 14).76 They reported that Au18 NC barely underwent exciton recombination and absorbed light in a wide wavelength range, resulting in high solar cell characteristics (Fig. 14A–C).
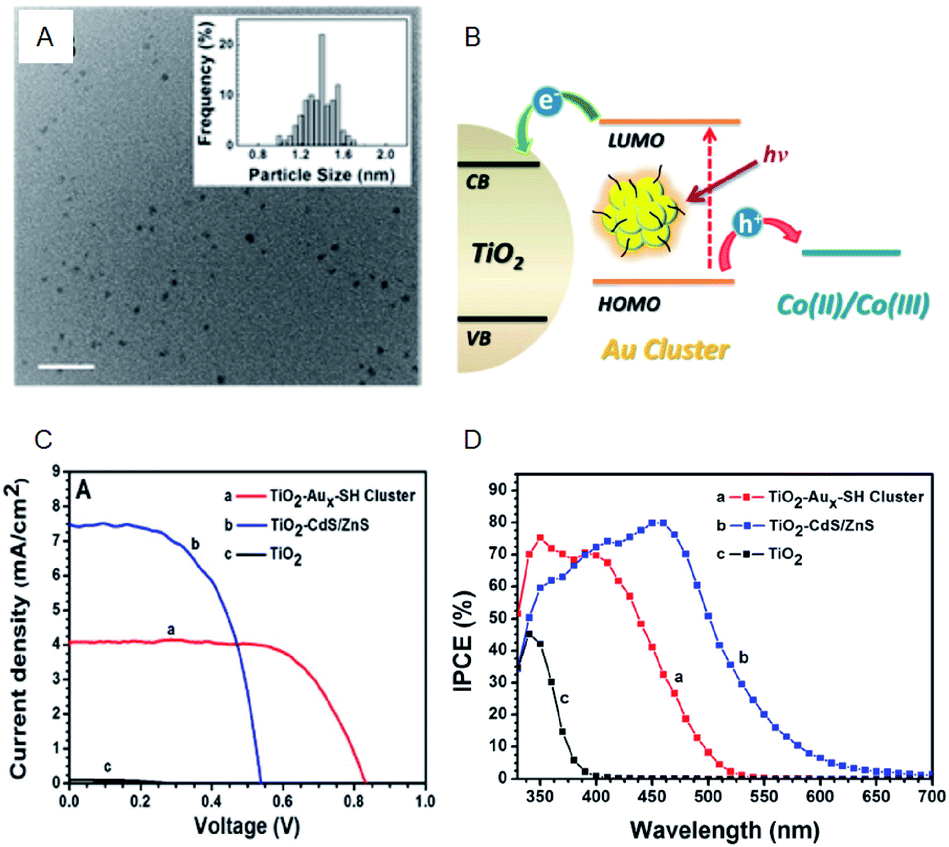 | ||
| Fig. 13 (A) TEM image of Aux-SG cluster. Scale bar shows the length of 10 nm. (B) Schematic of the working principle of metal-NC-sensitized solar cells. (C) J–V characteristics. (D) IPCE spectra of TiO2 modified with Aux-SH cluster, TiO2 modified with CdS/ZnS and TiO2 electrodes. Panels (a)–(d) are reproduced with permission from ref. 75. Copyright 2013 American Chemical Society. | ||
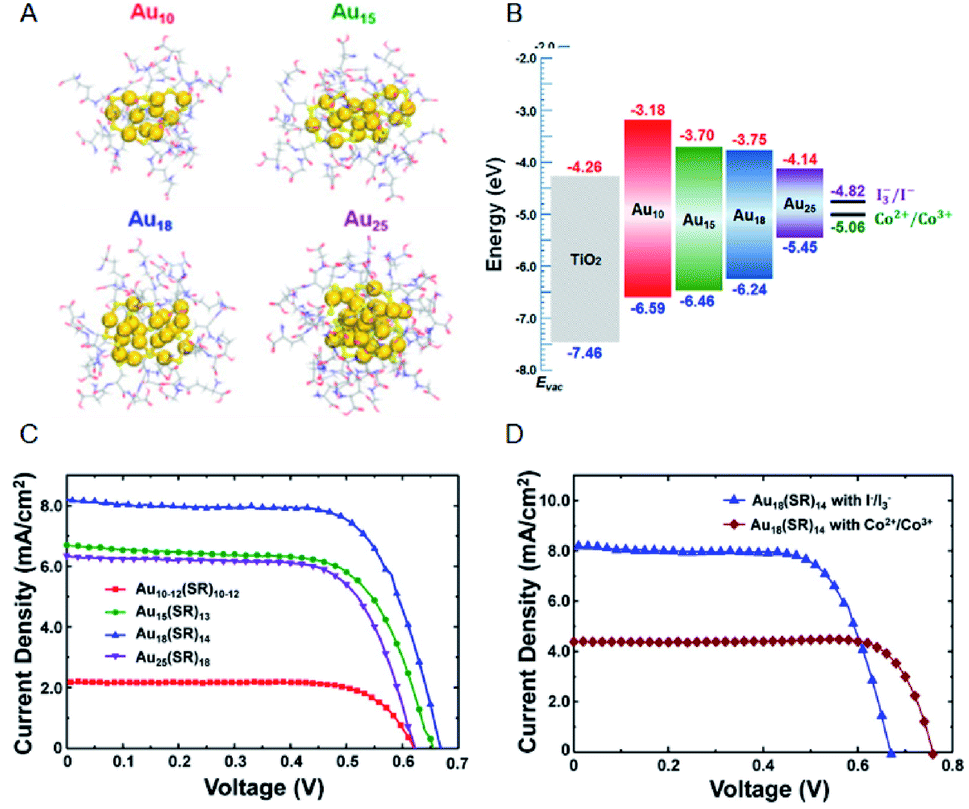 | ||
| Fig. 14 (A) Geometrical structures of Aux(SG)y NCs (x = 10, 15, 18 and 25). (B) Energy diagrams of the Aux(SG)y NCs sensitized solar cells. J–V characteristics of (C) Aux(SG)y NCs sensitized solar cells (x = 10, 15, 18 and 25) with I−/I3− redox pair and (D) Au18(SG)14 NCs sensitized solar cells with I−/I3− or Co2+/Co3+ redox pair. Panels (a)–(d) are reproduced with permission from ref. 76. Copyright 2016 American Chemical Society. | ||
However, to improve the stability of the system, other ligand-protected metal NCs should be used. Pradeep et al. compared the photovoltaic performance of solar cells using various ligand-protected metal NCs, such as bovine serum albumin, TBBT, and 4-mercaptobenzoic acid.73 They reported that the Au30 NC protected with bovine serum albumin has highest PCE (0.35%).
In the described systems, there is a correlation between the expansion of the light absorption wavelength range of the metal NCs and the decrease in opening photovoltage accompanying the bandgap decrease. To further improve the PCE, combination with other light-absorbing materials is required. DSSC using a squalene dye as the co-sensitizer was reported to provide a PCE of 4%,77 and a maximum PCE of 9.15% (ref. 78) by incorporating Au38 clusters in organic thin-film solar cells.
3. Photosensitization of metal NCs for therapeutic applications
3.1. Photo-based therapeutic applications of water-soluble metal NCs
Water-soluble noble metal (such as Au and Ag) NCs are composed of a metal core with several to a few hundred atoms, capped by water-soluble ligands such as amino acids, peptides, proteins, deoxyribonucleic acid (DNA), and hydrophilic thiolate ligands with different functional groups (e.g., –COOH, –OH, and –NH2). These water-soluble metal NCs exhibit unique physico-chemical properties including high photostability, low toxicity, and luminescence. In addition, the good penetration of such ultra-small metal NCs into the cells, the ability to target tumors through proper surface modification, and their superior renal clearance make the water-soluble metal NCs promising materials for utilization in the field of biomedicine, e.g., bioimaging, sensing, drug delivery, and diagnostic and therapeutic applications.Cancer is a class of common life-threatening diseases, understanding of which is incomplete, despite significant efforts and extensive research. Developed therapies against cancer include chemotherapy (chemo-T), radiotherapy (RT), surgery, immunotherapy, NPs-based therapy, photothermal therapy (PTT), and PDT.79 Among them, PDT is considered clinically reliable and non-invasive. In this method reactive oxygen species (ROS) generated by a photoexcited photosensitizer kill the tumor cells.
Traditional PDT utilizes organic photosensitizers with organic dye molecules such as porphyrin and its derivatives, chlorin e6, methylene blue, rose bengal, eosin Y, and indocyanine green. However, organic photosensitizers suffer from common drawbacks including low water-solubility, poor selectivity, toxicity, and photo-instability. Meanwhile, known inorganic photosensitizers include quantum dots, silicon nanocrystals, fullerene (C60), and metal NPs.80–83 More recently, Aun(SR)m NCs have been considered as promising photosensitizers for PDT. Their electronic structures can be optimized for efficient singlet oxygen (1O2) production using atomically precise size control. Several fundamental properties of the Aun(SR)m NCs are also desirable for PDT, such as good biocompatibility in dark conditions, photosensitization under near-infrared light irradiation, good resistance to photobleaching, and target specificity via surface modification. The following section focuses on the photosensitizing ability of Aun(SR)m NCs toward PDT applications. A number of recent reviews on other biomedical applications of metal NCs have also been published.15–17
3.2. Basic principle of photosensitization for PDT
Upon absorbing the appropriate light, the photosensitizer transitions from the ground singlet state (S0) to the excited singlet state (S1). Through non-radiative transition, it subsequently changes the multiplicity from S1 to a triplet state (T1) via intersystem crossing (ISC). The photosensitizer in the T1 state can then decay via two distinctive processes: Type I and Type II (Fig. 15). In Type I, the excited photosensitizer undergoes single-electron transfer with the substrate to produce a radical or a radical ion, such as hydroxyl radical (˙OH) and superoxide (O2˙−). These highly reactive radical species can further react with biological substrates to modify their structure and/or function. In the Type II process, however, 1O2 produced via the energy transfer from the photosensitizer to oxygen, readily reacts with biological molecules (e.g., lipids, proteins, and DNA), which are the main components of cells and nuclear membranes. The majority of the effective photosensitizers are characterized by high quantum yields of their T1 state, since a relatively longer living T1 state allows more chance for energy and/or electron transfer. Thus, ROS such as O2˙−, ˙OH, and 1O2 are key species in the PDT process.84,85 | ||
| Fig. 15 Schematic Jablonski's diagram of Type I and Type II reactions in PDT. PS: photosensitizer. | ||
The lifetime of 1O2 in most biological environments is in the range of a few microseconds. Thus, 1O2 produced from Type II photosensitization can affect biological substrates within a moderate distance from the photosensitizer itself (0.02–0.15 μm). In contrast, ˙OH produced in Type I photosensitization is highly reactive and interacts with biological molecules within a range of less than 5 nm, restricting its effects to the location where it is produced.84,85 Thus, 1O2 formed from the Type II reaction is thought to be primarily responsible for the biological effect in PDT.
The direct confirmation of 1O2 generation in photosensitization reactions comes from detecting phosphorescence at around 1270 nm resulting from the spontaneous decay of 1O2 to its ground state. Other methods of 1O2 detection are based on analyzing the spectral change of a probe molecule upon its interaction with 1O2, using electron spin resonance (ESR), UV-vis absorption, or fluorescence. The various 1O2 detection techniques and their probes are summarized in Table 2.
| Method | Probe | Detection |
|---|---|---|
| Phosphorescence | 1O2 | Phosphorescence of 1O2 at 1270 nm |
| UV-vis spectroscopy | 9,10-Diphenyl-anthracene (DPA) | Decrease peak at 355 nm |
| UV-vis spectroscopy | 9,10-Anthracenediyl-bis(methylene) dimalonic acid (ABDA) | Decrease peak at 382 nm |
| UV-vis spectroscopy | 9,10-Anthracenedipropionic acid (ADPA) | Decrease peak at 400 nm |
| UV-vis spectroscopy | Anthracene-9,10-bisethanesulfonic acid (AES) | Decrease peak at 360, 378 and 400 nm |
| UV-vis spectroscopy | 1,3-Diphenyl-isobenzofuran (DPBF) | Decrease peak at 410 nm |
| UV-vis spectroscopy | p-Nitrosodimethylaniline imidazole (RNO method) | Decrease peak at 440 nm |
| Fluorescence | Singlet oxygen sensor Green (SOSG) | Increased fluorescence at 540 nm |
| Fluorescence | Methotrexate (MTX) | Increased fluorescence at 450 nm |
| ESR | 2,2,6,6-Tetramethyl-piperidine (TEMP) | Change in enhanced permeability and ESR spectrum |
1O2 scavengers can also be used to confirm the 1O2 generation. Azide ion and histidine are often utilized as quenchers for 1O2. They rapidly and preferentially interact with 1O2 and convert it to the ground state O2 before reacting with any other molecules in the system. Further confirmation of 1O2 can be obtained by replacing water with deuterated water, which extends the lifetime of 1O2 by as much as 15-fold.
3.3. Au NC-based photosensitizer for PDT
In previous studies, protein-stabilized Au NCs and Au NCs embedded in polymer matrix were reported to produce 1O2 upon photoexcitation at 330 and 532 nm (UV and visible light, respectively).86,87 Following these reports, Kawasaki and Jin et al. described the formation of 1O2 through direct sensitization of Au25(SR)18 (H–SR = phenylethanethiol or captopril) under visible/NIR light (532, 650, and 808 nm) irradiation.88 The generation of 1O2 by photoexcited Au25(SR)18 was confirmed by observation of the 1O2 emission at approximately 1270 nm, 1O2-selective probes, and the use of a 1O2 quencher.88 The larger HOMO–LUMO gap of Au25(SR)18 NCs (1.3 eV) than the energy of 1O2 (0.97 eV), the long lifetime of the electronic excitations, and the well-defined O2 adsorption sites are suggested to be the key factors promoting the energy transfer from the Au25(SR)18 NCs to molecular oxygen resulting in the formation of 1O2. Photodynamic activity of water-soluble Au25(Capt)18 NCs toward cancer cells under NIR light (808 nm) was also demonstrated (Fig. 16A). Moreover, Miyata and Miyaji et al. developed Au25(Capt)18 NCs exhibiting photodynamic activity toward oral bacteria (Fig. 16B).89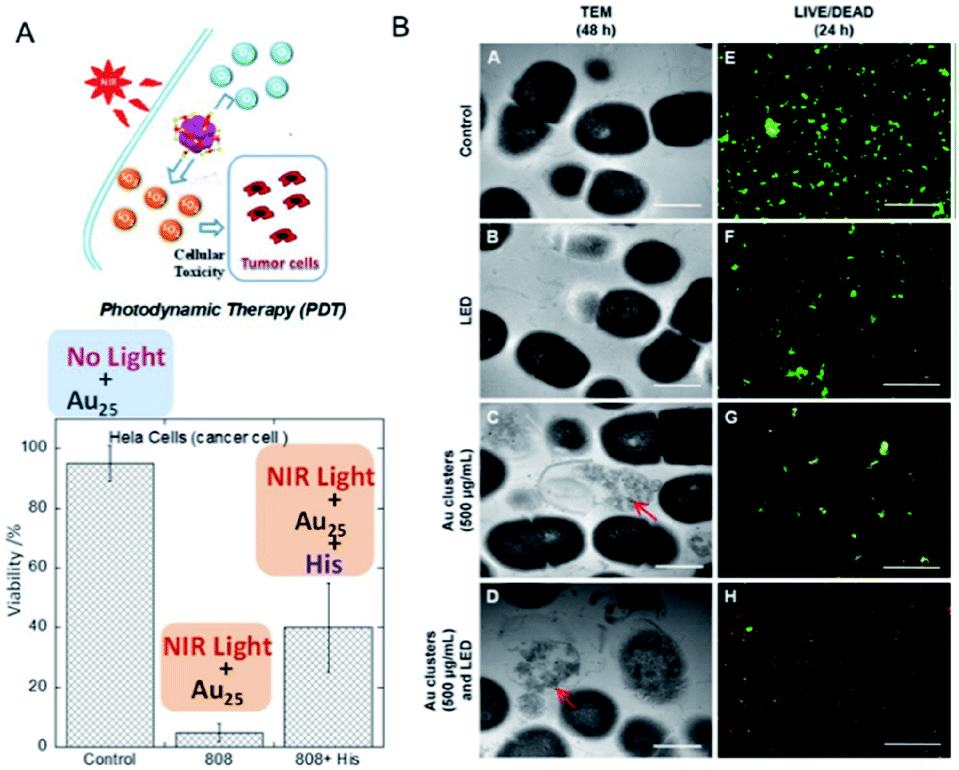 | ||
| Fig. 16 Photodynamic action of Au25(Capt)18 NCs. (A) HeLa cancer cells under NIR light in the presence/absence of histidine, a 1O2 scavenger. (B) TEM images of Streptococcus mutans after 48 h incubation, and LIVE (green)/DEAD (red) results of S. mutans after 24 h incubation. Panel (a) is reproduced with permission from ref. 88. Copyright 2014 American Chemical Society. Panel (b) is reproduced with permission from ref. 89. Copyright 2017 Dove Medical Press Ltd. | ||
The following section includes discussion of several strategies to improve the 1O2 generation efficiency and PDT activity of Au NCs by using: (i) atomically precise size control, (ii) NIR activation for deep-tissue treatments, (iii) aggregation-induced emission (AIE), (iv) resonance energy transfer (RET), and (v) forming nanocomposites with other nanomaterials for multimodal synergistic therapy (Fig. 17).
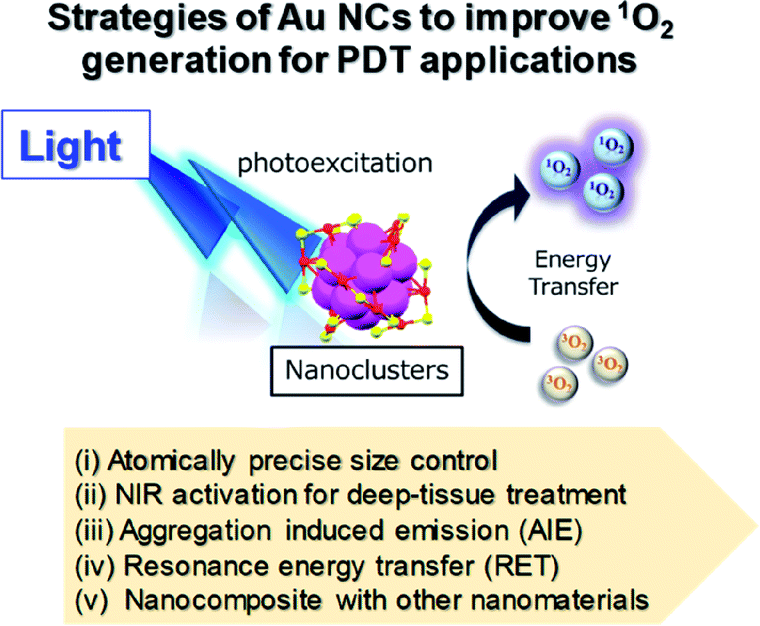 | ||
| Fig. 17 Schematic illustration of different approaches to improve 1O2 generation from Au NCs for PDT. | ||
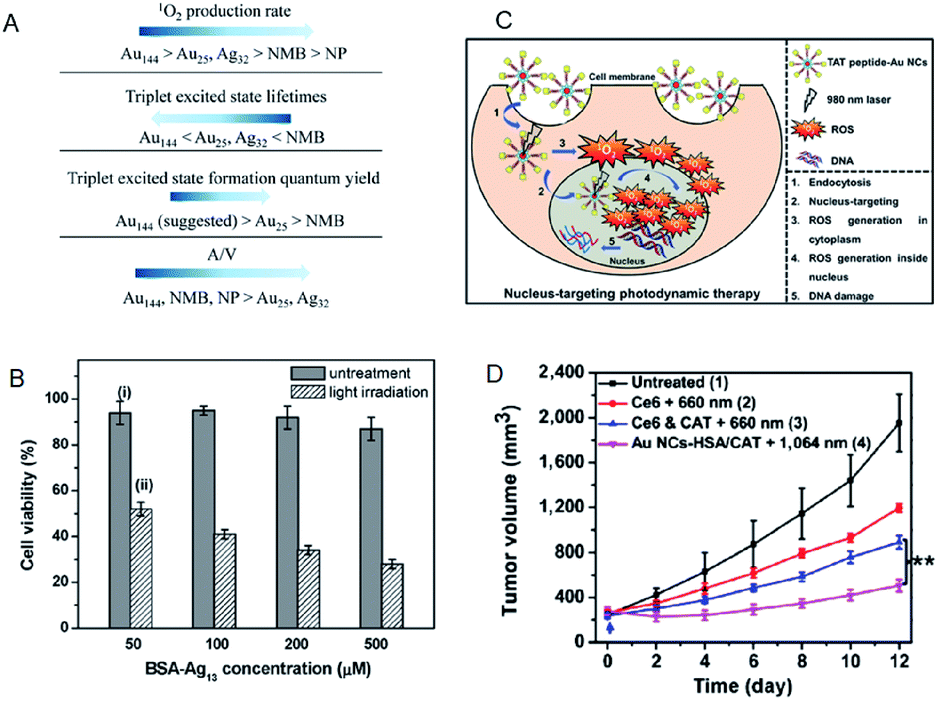 | ||
| Fig. 18 (A) Summary of the photosensitizing properties of Au NCs, Ag NCs, Au NPs, and new methylene blue for producing 1O2. (B) MTT assay histogram showing the viability of MCF-7 cancer cells treated with BSA-Ag13 NCs at concentrations ranging from 50 to 500 × 10−6 M (Ag basis) with and without light irradiation. (C) Schematic of the PDT mechanism mediated by nucleus-targeting TAT peptide–Au NCs (peptide sequence: TAT = N-GRKKRRQRRR-C). (D) NIR-II triggered PDT on tumors. Growth curves of large tumors in mice after different treatments: (1) untreated control; (2) Ce6 + 660 nm laser; (3) Ce6 + CAT + 660 nm laser; (4) AuNC@HSA/CAT + 1064 nm laser. Panel (a) is reproduced with permission from ref. 91. Copyright 2017 American Chemical Society. Panel (b) is reproduced with permission from ref. 93. Copyright 2016 Wiley-VCH. Panel (c) is reproduced with permission from ref. 97. Copyright 2015 Wiley-VCH. Panel (d) is reproduced with permission from ref. 98. Copyright 2018 Springer Nature. | ||
Compared to the Au NC-based photosensitizers, reports on Ag NC-based photosensitizers are limited due to their low photostability.92 Yu et al. examined bovine serum albumin (BSA)-protected Ag13 NC as a photosensitizer. Due to a large population in the triplet state, the BSA-Ag13 NCs showed high 1O2 generation quantum efficiency (∼1.26 using rose bengal as the standard).93 Owing to the good cellular uptake and high 1O2 generation efficiency, the BSA-Ag13 can effectively kill the MCF-7 breast cancer cells following uptake and white light treatment (Fig. 19B). Tominaga and Kawasaki et al. demonstrated that Ag7(MBISA)6 (MBISA = 2-mercapto-5-benzimidazolesulfonic acid sodium salt) NCs also generate 1O2 with high efficiency under white light irradiation. The following order was suggested for the 1O2 generation efficiency on different Ag NCs: Ag7(MBISA)6 > BSA-Ag13 > Ag75(SG)40 > Ag35(SG)18 > BSA-Ag8 (not detected).94
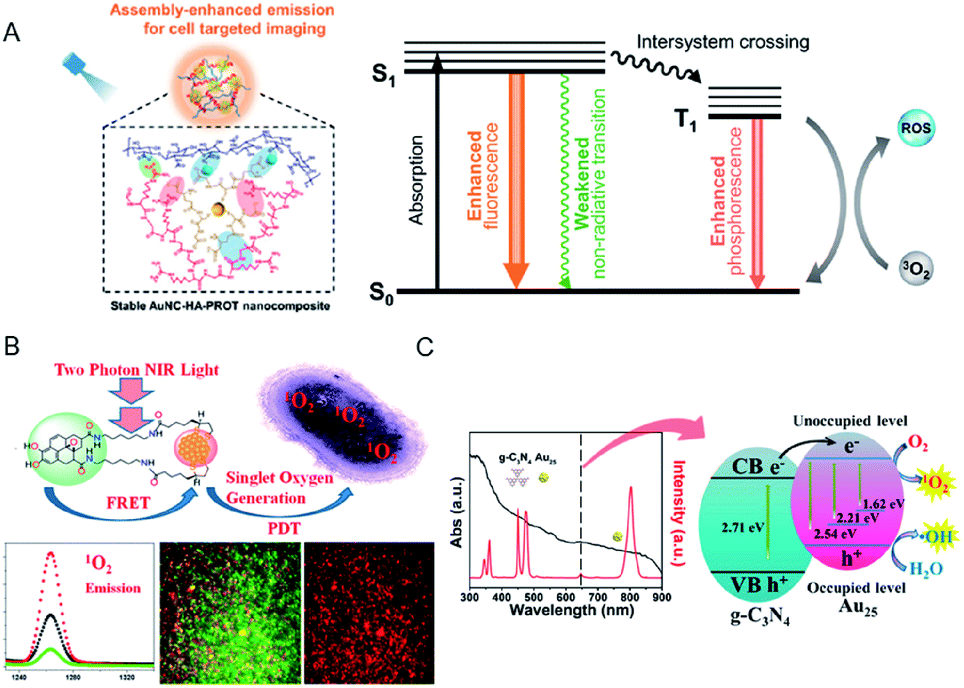 | ||
| Fig. 19 (A) Schematic of the application of AuNC–hyaluronic acid (HA)–protein protamine (PROT) nanocomposites with enhanced emission and ROS generation through a self-assembly approach. (B) FRET-based two-photon excited theranostic nanoplatform using Au NCs attached to GQD for two-photon imaging and two-photon induced PDT killing of MDRB. 1O2 emissions under two-photon excitation at ∼1270 nm from the nanoplatform. (C) UV-vis absorption spectrum (black) and emission spectrum (red) of Tm3+-activated up-conversion NPs (UCNPs)@g-C3N4–Au25–PEG upon 980 nm NIR laser excitation, and schematic of the ROS generation mechanism. Panel (a) is reproduced with permission from ref. 106. Copyright 2019 American Chemical Society. Panel (b) is reproduced with permission from ref. 115. Copyright 2018 American Chemical Society. Panel (c) is reproduced with permission from ref. 118. Copyright 2017 American Chemical Society. | ||
Recently, X-ray was also combined with Au NCs as “radiosensitizer” for cancer radiotherapy. The use of X-ray as the light source is most suitable for activating the Au NCs for deep-tissue cancer treatment and biomedical imaging applications, due to its nearly unlimited penetration depth in living tissues and organisms. An interesting review,99,100 as well as various other recent publications101–103 covering the applications of Au NCs as radiosensitizers are available.
The use of chromophore ligands can control photo-functions of metal nanoparticles and metal NCs.110–113 Two-photon absorption of ligand-protected metal NCs shows a nonlinear optical property that is attractive for applications such as biological imaging and PDT.114 Vangara et al. demonstrated two-photon PDT under 860 nm NIR excitation for multiple drug resistance bacteria (MDRB) using a nanocomposite of α-lipoic acid stabilized Au NCs and graphene quantum dots (GQDs) (Au NCs–GQDs nanocomposite),115 where the GQDs with high NIR absorption act as two-photon donor-chromophores, while the Au NCs act as acceptors. Due to the FRET process between the Au NCs and the GQDs, the 1O2 generation capability of Au NCs–GQD nanocomposite enhanced tremendously, which was higher than that of rose bengal, which is an efficient organic photosensitizer (1O2 quantum yield of rose bengal, ΦRB = 0.75) (Fig. 19B). The Au NCs–GQDs nanocomposite allowed bright two-photon bioimaging and two-photon PDT against MDRB and carbapenem-resistant Escherichia coli.
The singlet fission (SF) is a photophysical process in which interaction between one singlet-excited state (S1) and ground state (S0) species generates two triplet-excited species (T1 + T1).116 The SF enables the maximum yield of triplet excitons to be 200% different from simple intersystem crossing (ISC). Saegusa and Hasobe et al. newly synthesized a series of mixed tetracene (TC)-protected Au NCs prepared from a TC-modified heterodisulfide with two different chain lengths, which achieved a high-yield SF (ΦSF −90%) and individual triplet yields (ΦT −160%).117 High-yield and long-lived triplet excited states through SF-based metal NCs may be also used for PDT application.
Feng et al. fabricated a dual-photosensitizer nanocomposite by coating a mesoporous layer of graphitic-phase carbon nitride (g-C3N4) on Tm3+-activated up-conversion NPs (UCNPs), followed by attachment of Au25(MHA)18 (MHA = 6-mercaptohexanoic acid) and polyethylene glycol molecules (UCNPs@g-C3N4–Au25–PEG) for PDT and bioimaging under NIR light.118 The higher PDT efficiency of this nanocomposite in comparison with any single modality was demonstrated by the increased ROS production via the energy transfer in the UCNPs@g-C3N4–Au25–PEG system when excited by NIR light (980 nm). In addition, the effective separation of photogenerated electron–hole pairs in the nanocomposite could also promote the production of ˙OH (Fig. 19C) and, consequently, cancer cell death. The therapeutic efficacy of the nanocomposite in animal experiments and tissue section analysis demonstrated the ability to inhibit tumor growth without damaging the major organs.
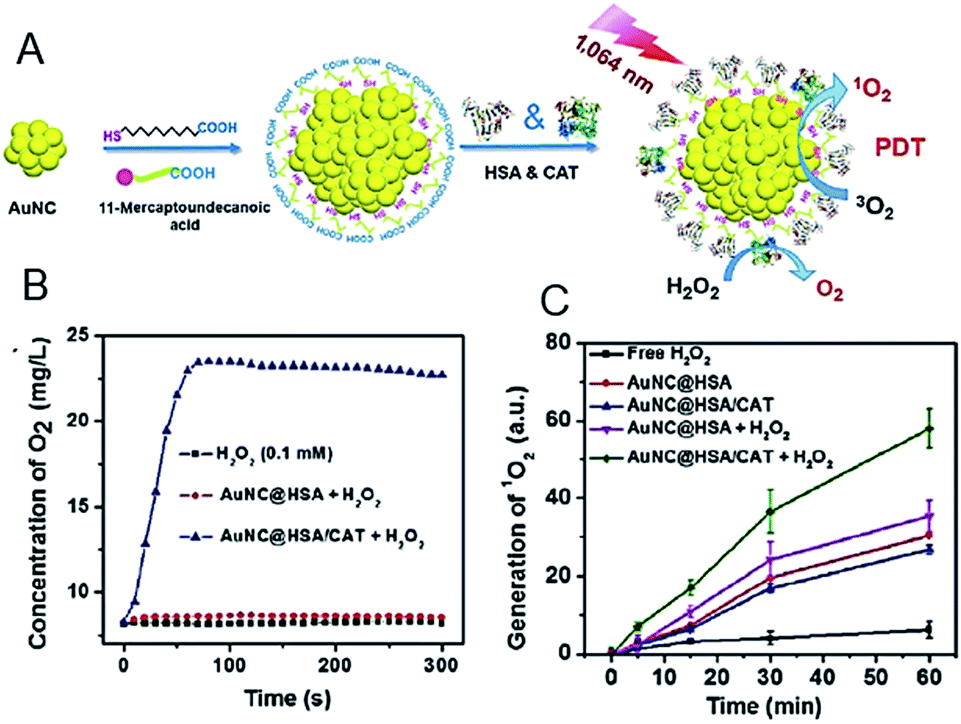 | ||
| Fig. 20 (A) Synthesis and characterization of AuNC@HSA/CAT NPs. (B) Oxygen generation in H2O2 solutions (100 μM) incubated with AuNC@HSA or AuNC@HSA/CAT. (C) The generation of 1O2 determined by the increased SOSG fluorescence from AuNC@HSA or AuNC@HSA/CAT, with or without addition of H2O2. Panels (a)–(c) are reproduced with permission from ref. 98. Copyright 2018 Springer Nature. | ||
To overcome the hypoxia limitation in Type II PDT, an O2 independent Type I PDT has recently been developed, as these photosensitizers produce HO˙.119,120 HO˙ is a stronger oxidant than 1O2 and could react with virtually any biological molecule, including DNA, proteins, lipids, and carbohydrates (Fig. 21).123 Yang et al. fabricated Au25 NCs anchored on black anatase TiO2−x nanotubes (Au25/B-TiO2−x NTs) for Type I PDT.124 Under NIR light, the Au25/B-TiO2−x NTs could achieve separation of the e−–h+ pair, leading to the production of HO˙ and O2˙− radicals. In vivo and in vitro experiments demonstrated an obviously enhanced PDT effect of Au25/B-TiO2−x NTs. Cheng et al. also fabricated titanium dioxide NP-Au NCs–graphene heterogeneous nanocomposites to induce a large production of HO˙ and O2˙− radicals, which could serve as Type I PDT for B16F1 melanoma cells.125
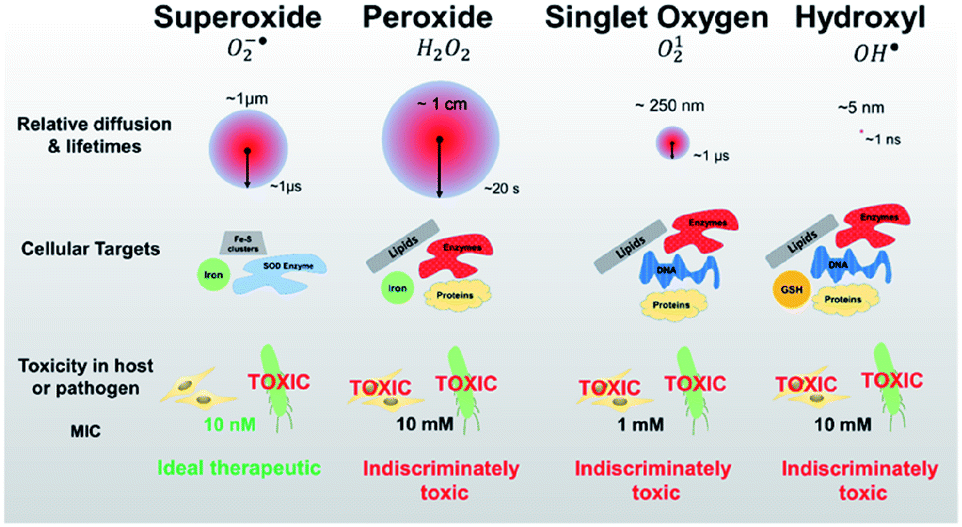 | ||
| Fig. 21 Identifying pathogen vulnerability using redox perturbation with different ROS. Compared to singlet oxygen and hydroxyl radicals, superoxide and peroxide radicals have much longer diffusion lengths and half-lives in the cellular environment (red circles, not to scale). This figure is reproduced with permission from ref. 123. Copyright 2019 Springer Nature. | ||
He et al. assembled Au25(Capt)18 onto mesoporous silica-coated Nd3+-sensitized up-conversion NPs (UCNPs@MS-Au25) to form a multifunctional platform for photoacoustic (PA) imaging and dual phototherapy (PTT/PDT).126 The UCNPs@MS-Au25 exhibited considerable PTT effect, by combining its intrinsic PDT effect to further enhance in vivo tumor inhibition under 808 nm laser.
Yang et al. fabricated metal–organic frameworks (MOFs) of Fe3O4/ZIF-8-Au25 (IZA) nanospheres.127 The IZA nanospheres not only exhibited PTT effects upon NIR light irradiation to effectively kill tumor cells, but also proved useful in targeting and magnetic resonance imaging (MRI). The Au25(Capt)18 in IZA nanospheres produced highly reactive 1O2 to cause PDT as well as PTT effects under NIR light irradiation. As a result, the IZA nanospheres exhibited synergistic therapeutic effect superior to any single therapy both in vitro and in vivo (Fig. 22A). Tumor-bearing mice treated only with PDT (Au25(Capt)18 NCs under NIR) or PTT (IZ under NIR) displayed much less inhibition in tumor growth than those treated by the multimodal synergistic therapy with combined PDT, PTT, and MRI approaches (Fig. 22A).
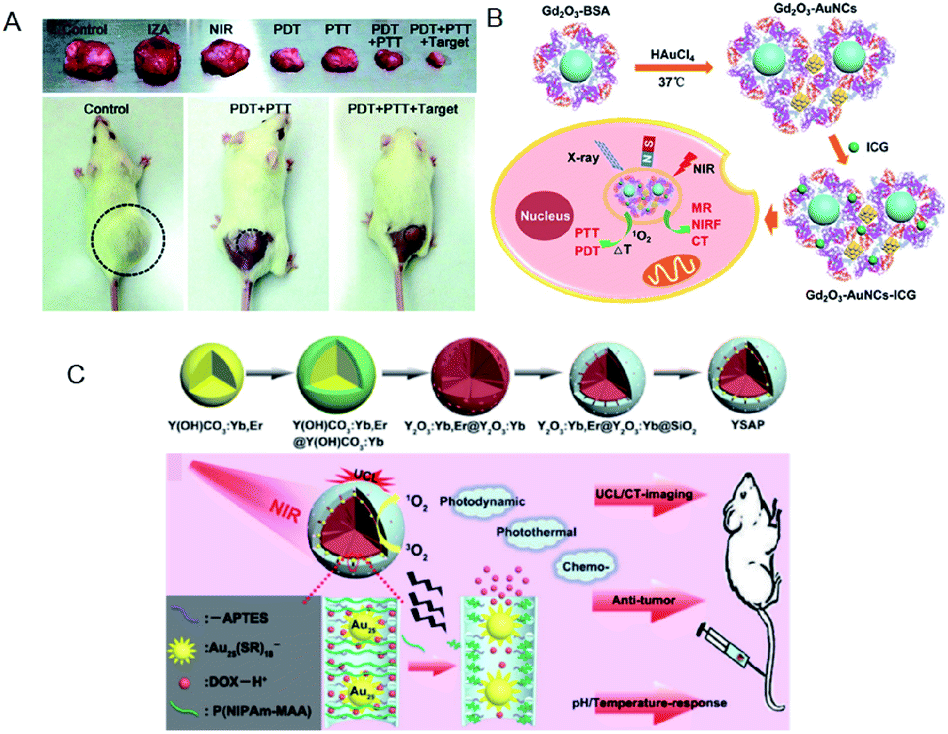 | ||
| Fig. 22 (A) Photographs of tumor tissues excised from mice on the 14th day of treatment using normal saline, pure NIR laser, IZA:MOFs of Fe3O4/ZIF-8-Au25, PDT (pure Au25(Capt)18 NCs under NIR), PTT (IZA under NIR), PDT + PTT, and PDT + PTT + magnetic stimulus. Bottom: photographs of representative tumor-bearing mice on the 14th day. (B) Schematic of Gd2O3–AuNCs–ICG nanohybrid as theragnostic agent for imaging-guided cancer therapy. (C) Schematic for the formation of Y2O3:Yb,Er@Y2O3:Yb@mSiO2–Au25 P(NIPAm-MAA) and the imaging-guided synergistic multimodal anti-cancer therapy. Panel (a) is reproduced with permission from ref. 127. Copyright 2015 Royal Society of Chemistry. Panel (b) is reproduced with permission from ref. 134. Copyright 2017 American Chemical Society. Panel (c) is reproduced with permission from ref. 129. Copyright 2015 Elsevier Ltd. | ||
Han et al. described a BSA-stabilized multifunctional theragnostic nanoplatform, gadolinium oxide–Au NCs–indocyanine green (ICG) hybrid (Gd2O3–BSA-AuNCs–ICG). The Gd2O3–AuNCs–ICG nanocomposites demonstrated excellent in vivo triple imaging capability for NIR fluorescence (NIRF), magnetic resonance, and computed topography, as well as efficacy in combined PDT and PTT (Fig. 22B).134 Furthermore, Cui et al. constructed a nanocomposite of BSA-Au NCs–ICG to allow multimodal synergistic therapy, namely dual-modal NIRF/PA imaging, cancer treatment by synergistic action of PDT/PTT, and real-time therapeutic monitoring based on FRET.135 Multimodal synergistic therapy using this nanocomposite resulted in 95% cancer cell death and complete tumor disappearance. Lv et al. produced a core/shell structured nanocomposite by conjugating Au25(SR)18 NC (a PDT/PTT agent), the pH/temperature-responsive polymer P(NIPAm-MAA), and the anti-cancer drug doxorubicin (DOX) onto the surface of mesoporous silica-coated core–shell up-conversion NPs (UCNPs) (Fig. 22C).129 The controlled DOX release in the cancer cells was triggered by a high temperature from the PTT under NIR irradiation and low pH. The combined PDT, PTT, and pH/temperature-responsive chemo-T could significantly improve the therapeutic efficacy, as confirmed by both in vitro and in vivo assays. Moreover, the nanocomposite showed dual-modal imaging properties (computer tomography and up-conversion luminescence) when irradiated with 980 nm NIR light, demonstrating the potential for imaging-guided therapy.
In addition to their highly efficient 1O2 generation, the physiological properties of Au NCs in biological environments must be further evaluated in terms of the toxicity, renal clearance, and stability for in vivo PDT applications.136–147 As for the renal clearance, NPs larger than 6 nm are eliminated from the blood stream by the reticuloendothelial system (RES; liver, spleen, etc.), while those smaller than 6 nm can be eliminated via the kidneys by passing through the glomerular capillary wall (GCW; i.e., the kidney filtration) (Fig. 23A).136,146 The longer circulation time of Au10,11 NCs in the blood compared to Au25 NCs allowed their accumulation in cancerous tissues through EPR (Fig. 23B).145 The unique renal clearance behavior of the sub-nm Au NCs was explained by the filtration mechanism of GCW like-size separation utilizing size exclusive chromatography. Compared to the neutral NCs, it is more difficult for the negatively charged NCs to cross the GCW, since the GCW is negatively charged. Zwitterionic or non-ionic surface ligands (e.g., SG or PEG) are desirable for the efficient urinary elimination of small NCs from the body.138,142,144,147
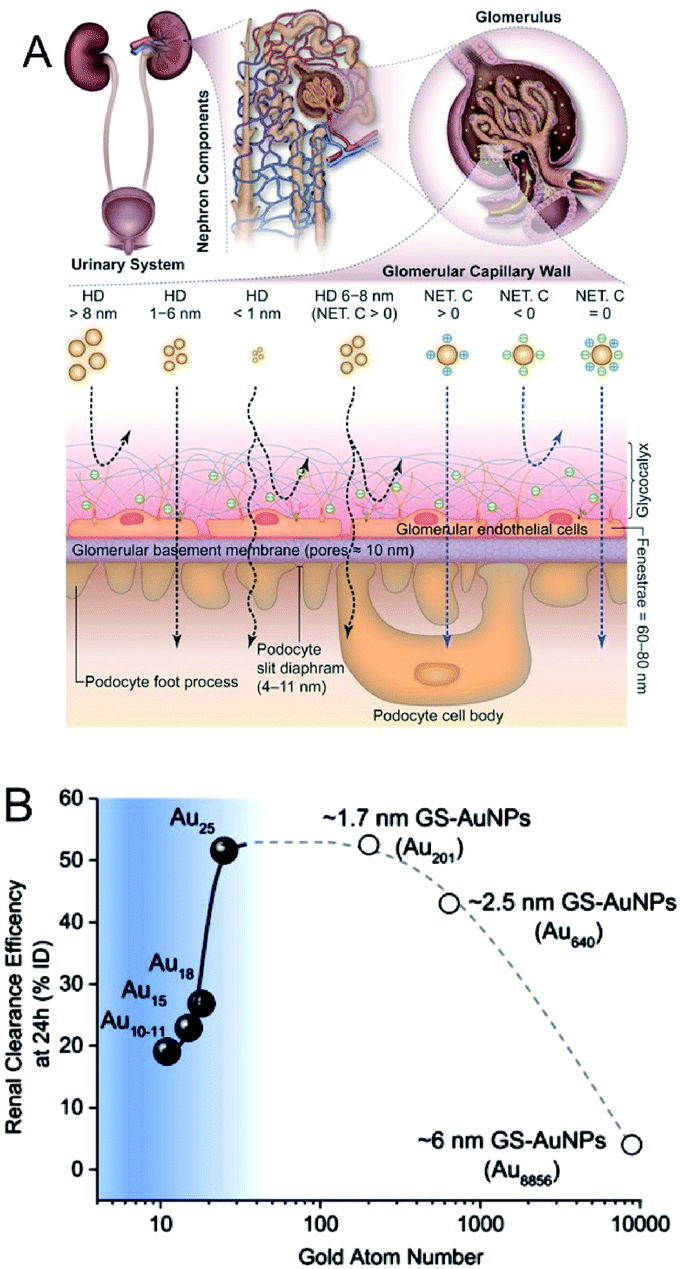 | ||
| Fig. 23 (A) Transport of small NPs of different sizes and charges in the urinary system. A schematic shows three layered components of GCW, and the fate of small nanoparticles when reaching the capillary endothelium. (B) The relationship between Au atom numbers and renal clearance efficiency of SG-protected Au NPs (GS–Au NPs) at 24 h. The renal clearance efficiency dramatically decreased when the size of NPs <1 nm. Panel (a) is reproduced with permission from ref. 146. Copyright 2018 Wiley-VCH. Panel (b) is reproduced with permission from ref. 145. Copyright 2017 Springer Nature. | ||
4. Conclusions and perspectives
In summary, we have reviewed the recent advances in the fields of photo/electrocatalysis and photosensitization using metal NCs, and their related green energy and medical applications. Owing to the unique properties of metal NCs, including their attractive photo-/electro-functional properties, as well as recent developments in their atomically precise synthesis, these functionalized moieties show excellent potential for the abovementioned applications.Considering the use of metal NCs in photo/electrocatalysis and photosensitization in green chemistry, we summarized several strategies to improve conversion efficiency and selectivity of the following materials: (i) electrocatalysts for water splitting and fuel cells, (ii) photocatalysts for water splitting, and (iii) solar cells. As described in this review, atomically precise control can contribute to the improvement of these materials. However, considering the practical applications, further developments are expected for simple and precise synthesis and isolation of metal NCs, as well as their loading. In addition, the following studies are envisaged for the improvement of these materials.
(1) Deeper understanding of the electronic structures of the loaded mono-metal and alloy NCs is essential for all of the applications. The electronic structures of the small NCs strongly depend on the number of the constituent atoms and the types of heteroatoms. Recent studies revealed the relationship between the electronic structures and these factors for the isolated NCs. However, the NCs loaded on the substrate seem to show different electronic structures as a result of the interaction between the NCs and the substrate, especially when the ligands are removed from the NC surface. It is expected that future studies would reveal the electronic structures of the loaded NCs for their application as active site on the substrate. In addition, the method for controlling the heteroatom position in the loaded alloy NCs is also expected to be developed in the future studies.
(2) For the improvement of photocatalysts, an effective method for separating e−–h+ pairs should be established. The Fermi energy of the cocatalysts strongly affects the charge transfer of the excited electron. Therefore, the electronic structure of cocatalysts, which effectively facilitates the charge transfer from photocatalysts to cocatalysts must be designed. In addition, although most studies concentrate on the loading of cocatalysts only for the reduction reaction, the future studies should conduct the co-loading of the cocatalysts for both reduction and oxidation reactions. It is also important to deepen the understanding on the reaction intermediates, the mechanism, and the methods for suppression of the reverse reactions.
(3) Regarding the photosensitization application for solar cells, NCs, which are able to absorb the visible and infrared light to effectively use the sunlight, should be fabricated and utilized. The stability of NCs should also be improved for this application. In addition, improvements in the energy-conversion efficiency must be made by combining several systems.
Considering photosensitization of metal NCs for therapeutic applications, we summarized several strategies to improve the 1O2 generation efficiency and PDT activity of metal NCs: (i) atomically precise size control, (ii) NIR activation for deep-tissue treatments, (iii) AIE, (iv) RET, and (v) forming nanocomposites with other nanomaterials for multimodal synergistic therapy. Despite these efforts, further research into the following aspects is necessary:
(4) By adjusting n and m, the discrete electronic/geometrical structures of metals, and thereby their photosensitizing capability, can be controlled. Extensive studies on the syntheses of organo-soluble metal NCs with atomic precision have been described, while the synthesis of water-soluble metal NCs remains undeveloped. Therefore, further research into the synthesis of water-soluble metal NCs is required for therapeutic applications. Ligand exchange method using organo-soluble metal NCs may be useful for the synthesis of water-soluble metal NCs and alloy metal NCs would be valuable for the exploration of their photosensitizing capability.
(5) A photosensitizer is primarily required to possess the following properties:148 (1) constant composition, (2) simplicity of synthesis, (3) lack of toxicity in the dark, (4) target specificity, (5) having a triplet state energy higher than 0.97 eV (the energy of single state oxygen, 1O2), (6) photostability, (7) high triplet state quantum yield, (8) fast clearance from the body, and (9) minimal self-aggregation. Metal NCs satisfy the abovementioned requirements; however, their 1O2 generation efficiencies are lower than those of organic photosensitizers. The high triplet state quantum yield and inhibition of energy dissipation through non-radiative pathways would be the key factors to improve the 1O2 generation efficiency of the metal NCs. Since water-soluble thiolated metal NCs have multi-functionality such as imaging, drug delivery, and multiple sensitizing capability (photo-sensitizer, radiosensitizer, and sonosensitizer), it would be useful as a multipurpose ROS mediated nanomedicine84,85,149,150
(6) Regarding the in vivo PDT applications, the use of specific ligands for metal NCs increases the selectivity toward the target (e.g., cancerous tumors), an approach called “active targeting”. Targeting ligands on metal NCs are responsible for the selective interaction between the metal NCs and specific receptors on the surface of tumor cells, but not healthy cells, thus increasing the internalization of the NCs. Various types of surface modification of metal NCs, such as aptamer, specific peptides, glucose, DNA, and folic acid have been developed for efficient delivery of the metal NCs to the target (e.g., cancerous tumors) for in vivo applications.151–156 Further studies involving active targeting of metal NCs would be important for in vivo PDT applications.
Conflicts of interest
There are no conflicts to declare.Authorship contributions
Photo/electrocatalysis of metal nanoclusters in green energy applications: Tokuhisa Kawawaki and Yuichi Negishi. Photosensitization of metal nanoclusters for therapeutic applications: Hideya Kawasaki. Introduction and conclusion: equal contribution by all authors.Notes and references
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- C. He, J. Cheng, X. Zhang, M. Douthwaite, S. Pattisson and Z. Hao, Chem. Rev., 2019, 119, 4471–4568 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- O. Stroyuk, A. Raevskaya and N. Gaponik, Chem. Soc. Rev., 2018, 47, 5354–5422 RSC.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- J. Yang, F. Wang, H. Yuan, L. Zhang, Y. Jiang, X. Zhang, C. Liu, L. Chai, H. Li and M. Stenzel, Nanoscale, 2019, 11, 17967–17980 RSC.
- S. Yougbare, T.-K. Chang, S.-H. Tan, J.-C. Kuo, P.-H. Hsu, C.-Y. Su and T.-R. Kuo, Int. J. Mol. Sci., 2019, 20, 2924 CrossRef PubMed.
- H. Yu, B. Wao, W. Jiang, S. Yang and M. Zhu, Coord. Chem. Rev., 2019, 378, 595–617 CrossRef CAS.
- T. Higaki, Q. Li, M. Zhou, S. Zhao, Y. Li, S. Li and R. Jin, Acc. Chem. Res., 2018, 51, 2764–2773 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- X. Jiang, B. Du, Y. Huang and J. Zheng, Nano Today, 2018, 21, 106–125 CrossRef CAS.
- X. Dou, X. Chen, H. Zhu, Y. Liu, D. Chen, X. Yuan, Q. Yao and J. Xie, Dalton Trans., 2019, 48, 10385–10392 RSC.
- Y. Su, T. Xue, Y. Liu, J. Qi, R. Jin and Z. Lin, Nano Res., 2019, 12, 1251–1265 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.8b00726.
- D. Wang, Z.-P. Liu and W.-M. Yang, ACS Catal., 2018, 8, 7270–7278 CrossRef CAS.
- J. Xing, H. B. Jiang, J. F. Chen, Y. H. Li, L. Wu, S. Yang, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 15258–15264 RSC.
- D. Wang, Z.-P. Liu and W.-M. Yang, ACS Catal., 2017, 7, 2744–2752 CrossRef CAS.
- Y.-G. Wang, Y. Yoon, V.-A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS PubMed.
- H. Chen, P. Li, N. Umezawa, H. Abe, J. Ye, K. Shiraishi, A. Ohta and S. Miyazaki, J. Phys. Chem. C, 2016, 120, 5549–5556 CrossRef CAS.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T.-K. Sham, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- S. Wang, X. Gao, X. Hang, X. Zhu, H. Han, W. Liao and W. Chen, J. Am. Chem. Soc., 2016, 138, 16236–16239 CrossRef CAS PubMed.
- X. Gao and W. Chen, Chem. Commun., 2017, 53, 9733–9736 RSC.
- D. Eguchi, M. Sakamoto and T. Teranishi, Chem. Sci., 2018, 9, 261–265 RSC.
- K. Kwak, W. Choi, Q. Tang, D.-e. Jiang and D. Lee, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- G. Hu, Q. Tang, D. Lee, Z. Wu and D.-e. Jiang, Chem. Mater., 2017, 29, 4840–4847 CrossRef CAS.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-e. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef PubMed.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- G. Mattioli, P. Giannozzi, A. A. Bonapasta and L. Guidoni, J. Am. Chem. Soc., 2013, 135, 15353–15363 CrossRef CAS.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS.
- K. S. Joya, L. Sinatra, L. G. AbdulHalim, C. P. Joshi, M. N. Hedhili, O. M. Bakr and I. Hussain, Nanoscale, 2016, 8, 9695–9703 RSC.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- X. Zhao, P. Gao, Y. Yan, X. Li, Y. Xing, H. Li, Z. Peng, J. Yang and J. Zeng, J. Mater. Chem. A, 2017, 5, 20202–20207 RSC.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Phys. Chem. Chem. Phys., 2014, 16, 26539–26545 RSC.
- A. Mahata, P. Bhauriyal, K. S. Rawat and B. Pathak, ACS Energy Lett., 2016, 1, 797–805 CrossRef CAS.
- K. A. Kacprzak, J. Akola and H. Häkkinen, Phys. Chem. Chem. Phys., 2009, 11, 6359–6364 RSC.
- T. Imaoka, H. Kitazawa, W.-J. Chun and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9810–9815 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397 CrossRef CAS.
- M. Nesselberger, M. Roefzaad, R. Fayçal Hamou, P. Ulrich Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 12, 919–924 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed.
- L. Wang, Z. Tang, W. Yan, H. Yang, Q. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 20635–20641 CrossRef CAS PubMed.
- L. Sumner, N. A. Sakthivel, H. Schrock, K. Artyushkova, A. Dass and S. Chakraborty, J. Phys. Chem. C, 2018, 122, 24809–24817 CrossRef CAS.
- T. C. Jones, L. Sumner, G. Ramakrishna, M. b. Hatshan, A. Abuhagr, S. Chakraborty and A. Dass, J. Phys. Chem. C, 2018, 122, 17726–17737 CrossRef CAS.
- Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 RSC.
- D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Chem. Sci., 2014, 5, 3151–3157 RSC.
- A. von Weber, E. T. Baxter, H. S. White and S. L. Anderson, J. Phys. Chem. C, 2015, 119, 11160–11170 CrossRef CAS.
- H. Yin, H. Tang, D. Wang, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- F. F. Schweinberger, M. J. Berr, M. Döblinger, C. Wolff, K. E. Sanwald, A. S. Crampton, C. J. Ridge, F. Jäckel, J. Feldmann, M. Tschurl and U. Heiz, J. Am. Chem. Soc., 2013, 135, 13262–13265 CrossRef CAS PubMed.
- M. J. Berr, F. F. Schweinberger, M. Döblinger, K. E. Sanwald, C. Wolff, J. Breimeier, A. S. Crampton, C. J. Ridge, M. Tschurl, U. Heiz, F. Jäckel and J. Feldmann, Nano Lett., 2012, 12, 5903–5906 CrossRef CAS.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- Y. H. Li, J. Xing, Z. J. Chen, Z. Li, F. Tian, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 2500 CrossRef PubMed.
- Y. H. Li, J. Xing, X. H. Yang and H. G. Yang, Chem.–Eur. J., 2014, 20, 12377–12380 CrossRef CAS PubMed.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- X. L. Du, X. L. Wang, Y. H. Li, Y. L. Wang, J. J. Zhao, L. J. Fang, L. R. Zheng, H. Tong and H. G. Yang, Chem. Commun., 2017, 53, 9402–9405 RSC.
- K. Sridharan, E. Jang, J. H. Park, J.-H. Kim, J.-H. Lee and T. J. Park, Chem.–Eur. J., 2015, 21, 9126–9132 CrossRef CAS.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed.
- B. Weng, K.-Q. Lu, Z. Tang, H. M. Chen and Y.-J. Xu, Nat. Commun., 2018, 9, 1543 CrossRef.
- N. Sakai and T. Tatsuma, Adv. Mater., 2010, 22, 3185–3188 CrossRef CAS.
- N. Sakai, S. Nakamura and T. Tatsuma, Dalton Trans., 2013, 42, 16162–16165 RSC.
- N. Sakai, T. Ikeda, T. Teranishi and T. Tatsuma, ChemPhysChem, 2011, 12, 2415–2418 CrossRef CAS.
- M. S. Kim, M. A. Abbas and J. H. Bang, Bull. Korean Chem. Soc., 2016, 37, 791–792 CrossRef CAS.
- V. Jeseentharani, N. Pugazhenthiran, A. Mathew, I. Chakraborty, A. Baksi, J. Ghosh, M. Jash, G. S. Anjusree, T. G. Deepak, A. S. Nair and T. Pradeep, ChemistrySelect, 2017, 2, 1454–1463 CrossRef CAS.
- Y. Wang, X.-H. Liu, S. A. Kovalenko, Q.-Y. Chen and N. Pinna, Chem.–Eur. J., 2019, 25, 4814–4820 CrossRef CAS.
- Y.-S. Chen, H. Choi and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 8822–8825 CrossRef CAS.
- M. A. Abbas, T.-Y. Kim, S. U. Lee, Y. S. Kang and J. H. Bang, J. Am. Chem. Soc., 2016, 138, 390–401 CrossRef CAS.
- H. Choi, Y.-S. Chen, K. G. Stamplecoskie and P. V. Kamat, J. Phys. Chem. Lett., 2015, 6, 217–223 CrossRef CAS PubMed.
- D. C. Lim, B. Y. Seo, S. Nho, D. H. Kim, E. M. Hong, J. Y. Lee, S.-Y. Park, C.-L. Lee, Y. D. Kim and S. Cho, Adv. Energy Mater., 2015, 5, 1500393 CrossRef.
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- J. Hu, Y. Tang, A. H. Elmenoufy, H. Xu, Z. Cheng and X. Yang, Small, 2015, 11, 5860–5887 CrossRef CAS.
- M. Rizwan, T. Rasheed, A. Raza, M. Bilal, R. Yahya, M. Yar and H. M. N. Iqbal, J. Drug Delivery Sci. Technol., 2019, 51, 70–82 CrossRef CAS.
- J. Krajczewski, K. Rucińska, H. E. Townley and A. Kudelski, Photodiagn. Photodyn. Ther., 2019, 26, 162–178 CrossRef CAS PubMed.
- P. G. Calavia, G. Bruce, L. Pérez-García and D. A. Russell, Photochem. Photobiol. Sci., 2018, 17, 1534–1552 RSC.
- S. Kwon, H. Ko, D. G. You, K. Kataoka and J. H. Park, Acc. Chem. Res., 2019, 52, 1771–1782 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- M. Sakamoto, T. Tachikawa, M. Fujitsuka and T. Majima, Langmuir, 2009, 25, 13888–13893 CrossRef CAS.
- T. Das, P. Ghosh, M. S. Shanavas, A. Maity, S. Mondal and P. Purkayastha, Nanoscale, 2012, 4, 6018–6024 RSC.
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777–2788 CrossRef CAS.
- S. Miyata, H. Miyaji, H. Kawasaki, M. Yamamoto, E. Nishida, H. Takita, T. Akasaka, N. Ushijima, T. Iwanaga and T. Sugaya, Int. J. Nanomed., 2017, 12, 2703–2716 CrossRef CAS PubMed.
- C. Talavera and P. V. Kamat, J. Chem. Sci., 2018, 130, 1–7 CrossRef CAS.
- R. Ho-Wu, S. H. Yau and T. Goodson, J. Phys. Chem. B, 2017, 121, 10073–10080 CrossRef CAS.
- C. Tominaga, H. Hasegawa, K. Yamashita, R. Arakawa and H. Kawasaki, RSC Adv., 2016, 6, 73600–73604 RSC.
- Y. Yu, J. Geng, E. Y. X. Ong, V. Chellappan and Y. N. Tan, Adv. Healthcare Mater., 2016, 5, 2528–2535 CrossRef CAS.
- C. Tominaga, D. Hikosou, I. Osaka and H. Kawasaki, Acta Phys.-Chim. Sin., 2018, 34, 805–811 CAS.
- Z. Liu, J. Wang, K. Qiu, X. Liao, T. W. Rees, L. Ji and H. Chao, Chem. Commun., 2019, 55, 6523–6526 RSC.
- Q. Wang, Y. Dai, J. Xu, J. Cai, X. Niu, L. Zhang, R. Chen, Q. Shen, W. Huang and Q. Fan, Adv. Funct. Mater., 2019, 29, 1901480 CrossRef.
- R. Vankayala, C.-L. Kuo, K. Nuthalapati, C.-S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2015, 25, 5934–5945 CrossRef CAS.
- Q. Chen, J. Chen, Z. Yang, L. Zhang, Z. Dong and Z. Liu, Nano Res., 2018, 11, 5657–5669 CrossRef CAS.
- X. Chen, J. Song, X. Chen and H. Yang, Chem. Soc. Rev., 2019, 48, 3073–3101 RSC.
- N. Goswami, Z. Luo, X. Yuan, D. T. Leonga and J. Xie, Mater. Horiz., 2017, 4, 817–831 RSC.
- X.-D. Zhang, Z. Luo, J. Chen, S. Song, X. Yuan, X. Shen, H. Wang, Y. Sun, K. Gao, L. Zhang, S. Fan, D. T. Leong, M. Guo and J. Xie, Sci. Rep., 2015, 5, 8669 CrossRef CAS.
- T.-T. Jia, G. Yang, S.-J. Mo, Z.-Y. Wang, B.-J. Li, W. Ma, Y.-X. Guo, X. Chen, X. Zhao, J.-Q. Liu and S.-Q. Zang, ACS Nano, 2019, 13, 8320–8328 CrossRef CAS.
- D. Luo, X. Wang, S. Zeng, G. Ramamurthy, C. Burda and J. P. Basilion, Small, 2019, 15, 1900968 CrossRef.
- F. Hu, S. Xu and B. Liu, Adv. Mater., 2018, 30, 1–29 Search PubMed.
- D. Hikosou, S. Saita, S. Miyata, H. Miyaji, T. Furuike, H. Tamura and H. Kawasaki, J. Phys. Chem. C, 2018, 122, 12494–12501 CrossRef CAS.
- J. Xia, X. Wang, S. Zhu, L. Liu and L. Li, ACS Appl. Mater. Interfaces, 2019, 11, 7369–7378 CrossRef CAS.
- H. Cao, Y. Qi, Y. Yang, L. Wang, J. Sun, Y. Li, J. Xia, H. Wang and J. Li, Chem.–Asian J., 2018, 13, 3540–3546 CrossRef CAS.
- X. Zhang, Y. Hu, X. Yang, Y. Tang, S. Han, A. Kang, H. Deng, Y. Chi, D. Zhu and Y. Lu, Biosens. Bioelectron., 2019, 138, 111314 CrossRef CAS PubMed.
- M. Yamamoto, K. Shitomi, S. Miyata, H. Miyaji, H. Aota and H. Kawasaki, J. Colloid Interface Sci., 2018, 510, 221–227 CrossRef CAS.
- K. G. Thomas and P. V. Kamat, J. Am. Chem. Soc., 2000, 122, 2655–2656 CrossRef CAS.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS.
- F. Stellacci, C. A. Bauer, T. Meyer-Friedrichsen, W. Wenseleers, S. R. Marder and J. W. Perry, J. Am. Chem. Soc., 2003, 125, 328–329 CrossRef CAS.
- K. Pyo, N. H. Ly, S. M. Han, M. b. Hatshan, A. Abuhagr, G. Wiederrecht, S.-W. Joo, G. Ramakrishna and D. Lee, J. Phys. Chem. Lett., 2018, 9, 5303–5310 CrossRef CAS.
- V. Bonačić-Koutecký and R. Antoine, Nanoscale, 2019, 11, 12436–12448 RSC.
- A. Vangara, A. Pramanik, Y. Gao, K. Gates, S. Begum and P. C. Ray, ACS Appl. Bio Mater., 2018, 1, 298–309 CrossRef CAS.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS.
- T. Saegusa, H. Sakai, H. Nagashima, Y. Kobori, N. V. Tkachenko and T. Hasobe, J. Am. Chem. Soc., 2019, 141, 14720–14727 CrossRef CAS.
- L. Feng, F. He, Y. Dai, B. Liu, G. Yang, S. Gai, N. Niu, R. Lv, C. Li and P. Yag, ACS Appl. Mater. Interfaces, 2017, 9, 12993–13008 CrossRef CAS.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–11531 CrossRef CAS PubMed.
- A. R. Lippert, G. C. Van De Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS.
- C.-P. Liu, T.-H. Wu, C.-Y. Liu, K.-C. Chen, Y.-X. Chen, G.-S. Chen and S.-Y. Lin, Small, 2017, 13, 1700278 CrossRef.
- M. Levy, P. P. Chowdhury and P. Nagpal, J. Biol. Eng., 2019, 13, 1–12 CrossRef.
- D. Yang, A. Gulzar, G. Yang, S. Gai, F. He, Y. Dai, C. Zhong and P. Yang, Small, 2017, 13, 1703007 CrossRef.
- Y. Cheng, Y. Chang, Y. Feng, N. Liu, X. Sun, Y. Feng, X. Li and H. Zhang, Small, 2017, 13, 1603935 CrossRef.
- F. He, G. Yang, P. Yang, Y. Yu, R. Lv, C. Li, Y. Dai, S. Gai and J. Lin, Adv. Funct. Mater., 2015, 25, 3966–3976 CrossRef CAS.
- D. Yang, G. Yang, S. Gai, F. He, G. An, Y. Dai, R. Lv and P. Yang, Nanoscale, 2015, 7, 19568–19578 RSC.
- S. K. Katla, J. Zhang, E. Castro, R. A. Bernal and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 75–82 CrossRef CAS.
- R. Lv, P. Yang, F. He, S. Gai, G. Yang, Y. Dai, Z. Hou and J. Lin, Biomaterials, 2015, 63, 115–127 CrossRef CAS.
- F. Xia, W. Hou, C. Zhang, X. Zhi, J. Cheng, J. M. de la Fuente, J. Song and D. Cui, Acta Biomater., 2018, 68, 308–319 CrossRef CAS.
- X. Zan, Q. Li, Y. Pan, D. J. Morris, P. Zhang, P. Li, H. Yu and M. Zhu, ACS Appl. Nano Mater., 2018, 1, 6773–6781 CrossRef CAS.
- L. V. Nair, S. S. Nazeer, R. S. Jayasree and A. Ajayaghosh, ACS Nano, 2015, 9, 5825–5832 CrossRef CAS.
- F. Gao, W. Zheng, L. Gao, P. Cai, R. Liu, Y. Wang, Q. Yuan, Y. Zhao and X. Gao, Adv. Healthcare Mater., 2017, 6, 1601453 CrossRef.
- L. Han, J.-M. Xia, X. Hai, Y. Shu, X.-W. Chen and J.-H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 6941–6949 CrossRef CAS.
- H. Cui, D. Hu, J. Zhang, G. Gao, Z. Chen, W. Li, P. Gong, Z. Sheng and L. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 25114–25127 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2009, 6, 12–21 CrossRef.
- C. Zhou, M. Long, Y. Qin, X. Sun and J. Zheng, Angew. Chem., Int. Ed., 2011, 50, 3168–3172 CrossRef CAS.
- X. D. Zhang, D. Wu, X. Shen, P.-X. Liu, F.-Y. Fan and S.-J. Fan, Biomaterials, 2012, 33, 4628–4638 CrossRef CAS.
- O. A. Wong, R. J. Hansen, T. W. Ni, C. L. Heinecke, W. S. Compel, D. L. Gustafson and C. J. Ackerson, Nanoscale, 2013, 5, 10525–10533 RSC.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978–4981 CrossRef CAS.
- J. Liu, M. Yu, X. Ning, C. Zhou, S. Yang and J. Zheng, Angew. Chem., Int. Ed., 2013, 52, 12572–12576 CrossRef CAS.
- X.-D. Zhang, Z. Luo, J. Chen, H. Wang, S.-S. Song, X. Shen, W. Long, Y.-M. Sun, S. Fan, K. Zheng, D. T. Leong and J. Xie, Small, 2014, 11, 1683–1690 CrossRef.
- X. Ning, C. Peng, E. S. Li, J. Xu, R. D. Vinluan, M. Yu and J. Zheng, APL Mater., 2017, 5, 053406 CrossRef.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS.
- J. Wang and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 3008–3010 CrossRef CAS.
- L. Gong, Y. Chen, K. He and J. Liu, ACS Nano, 2019, 13, 1893–1899 CrossRef CAS.
- R. Bakalova, H. Ohba, Z. Zhelev, M. Ishikawa and Y. Baba, Nat. Biotechnol., 2004, 22, 1360–1361 CrossRef CAS.
- Z. Lin, J. Song, X. Chen and H. Yang, Angew. Chem., Int. Ed., 2019 DOI:10.1002/ange.201906823.
- K. Kawamura, D. Hikosou, A. Inui, K. Yamamoto, J. Yagi, S. Saita and H. Kawasaki, J. Phys. Chem. C, 2019 DOI:10.1021/acs.jpcc.9b06849.
- F. Ghahremani, D. Shahbazi-Gahrouei, A. Kefayat, H. Motaghi, M. A. Mehrgardi and S. H. Javanmard, RSC Adv., 2018, 8, 4249–4258 RSC.
- B. N. P. Kumar, N. Puvvada, S. Rajput, S. Sarkar, M. K. Mahto, M. M. Yallapu, A. Pathak, L. Emdad, S. K. Das, R. L. Reis, S. C. Kundu, P. B. Fisher and M. Mandal, Mol. Pharm., 2018, 15, 2698–2713 CrossRef.
- F. Su, Q. Jia, Z. Li, M. Wang, L. He, D. Peng, Y. Song, Z. Zhang and S. Fang, Microporous Mesoporous Mater., 2019, 275, 152–162 CrossRef CAS.
- J. Lan, X. Wu, L. Luo, J. Liu, L. Yang and F. Wang, Talanta, 2019, 197, 86–91 CrossRef CAS.
- A. Kefayat, F. Ghahremani, H. Motaghi and A. Amouheidari, Nanomed. Nanotechnol. Biol. Med., 2019, 16, 173–184 CrossRef CAS PubMed.
- G. S. Heo, Y. Zhao, D. Sultan, X. Zhang, L. Detering, H. P. Luehmann, X. Zhang, R. Li, A. Choksi, S. Sharp, S. Levingston, T. Primeau, D. E. Reichert, G. Sun, B. Razani, S. Li, K. N. Weilbaecher, F. Dehdashti, K. L. Wooley and Y. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 19669–19678 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |