
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeometry-induced protein reorientation on the spikes of plasmonic gold nanostars†
Rosália
Lopes Rodrigues
 *,
Fang
Xie
,
Alexandra E.
Porter
and
Mary P.
Ryan
*
*,
Fang
Xie
,
Alexandra E.
Porter
and
Mary P.
Ryan
*
London Centre for Nanotechnology, Department of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: m.p.ryan@imperial.ac.uk
First published on 21st January 2020
Abstract
Functionalized gold nanostars (AuStrs) are remarkable candidates for drug delivery, photothermal therapy and imaging due to their large surface area to volume ratio and plasmonic properties. In this study, we address the challenge of achieving therapeutically controlled dosing using these high aspect ratio nanoparticle vectors by tailoring the nanostar loading area and protein conformation. We synthesized a library of different Au nanostars with varied geometries for potential biomedical applications. The Au nanostars were subsequently coated with different amounts of transferrin (Tf) and a novel depletion method was devised to measure the amount of Tf bound to the surface of the nanostructures. This methodology allowed us to show that coating thickness could be controllably varied and moulded onto the nanoparticle's high index features, whilst simultaneously preserving the key properties of the particle. The orientation of the Tf was measured on nanostars and spheres using transmission electron microscopy by negatively staining the Tf. The Tf was conformal on the nanostars, and protein packing efficiency increased on the AuStrs by 14-fold due to a geometry-induced protein reorientation at the nanoparticle surface. Interestingly, the reorientation of the transferrin observed at the AuStrs spikes did not occur at the AuStrs tips thus highlighting surface energy effects associated with surface curvature.
1 Introduction
Spikey gold nanomaterials are opening up new approaches for treating and diagnosing a range of diseases by providing tools to specifically target and interact with the diseased cells.1,2 These high aspect ratio nanostructures are plasmonic and, as a consequence, are being utilized in photothermal therapy and fluorescent-based imaging applications. Their shape can be tuned to modulate their plasmonic properties for improved performance in deep tissue imaging and photothermal therapy.3,4 Modification of their geometry will also change their surface area to volume (SA/V) ratio to enable significant loading of surface molecules (i.e. drugs or targeting ligands). Functionalizing gold nanostructures with proteins or antibodies is a widely used technique to tailor the ability of the nanoparticles to bind selectively to specific cell types. Surface ligands are selected that recognize specific binding sites (over)expressed on the target tissue.5–8 Surface loading and orientation (or conformation) of surface ligands will alter significantly the interaction these nanostructures with cells by changing the activity of these ligands, but is rarely considered for rationale design of these particles.9–11 Currently, it is unclear whether protein–gold nanoparticle (AuNP) binding (capacity or conformation) at the nanoparticle surface are dependent on their radius of curvature. It has been suggested that, for spherical, citrate capped AuNPs, surface curvature alters the structure and binding capacity of adsorbed proteins onto their surface, although this issue remains controversial.12–17 A recent report measured binding of GB3 and BCA proteins onto spherical gold nanoparticle surfaces with varied radius of curvature and showed, using NMR, that the adsorbed protein structure is independent of curvature for these proteins.18 However, to our knowledge it is unknown how protein binding and structure compare on the surface of star-shaped (AuStr) and spherical gold nanostructures. In particular, the differences in surface energy, related to the dramatic differences in local curvature are likely to influence protein adsorption along the stem and tips of the AuStr's spikes.We investigate whether varying the geometry of gold nanostructures (star-shaped vs. spherical) can be used to change multilayer protein binding and induce protein unfolding. Tf is used as a model protein as it is overexpressed on several cell types, notably brain endothelial cells at the blood brain barrier, and so is of clinical interest as a cell targeting molecule to target nanoparticles across the blood brain barrier and shuttle them inside the brain.19 To shed light on this process, we first synthesize a panel of AuStrs with a highly controllable spectrum of properties including size, surface spikiness and spike morphology and hence optical properties. A challenge with current synthesis of AuStrs is to control precisely the spike geometry thermodynamically. This can be achieved by adding Ag ions into the synthesis process, however the role by which the Ag+ induces this branched growth is poorly understood. To attain more precise control over the surface spikiness of the gold nanostars, we also establish the role of tuning the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag ratio by adding chloride ions and gold seeds into the reaction to understand how these ions and pre-formed nano-seeds drive the mode of spike-growth. In this study, we demonstrate the correlation between ionic concentration, spike morphology and resulting optical properties. A novel depletion assay is then devised to quantify the amount of transferrin bound to the surface of the gold nanostars and spheres by measuring the unbound fraction of the Tf in solution by UV-visible spectroscopy against a set of known standards. The protein loading efficiency was controllably tuned to achieve maximum surface area (SA) coverage without significantly altering the shape or volume of the nanostructures. Using electron microscopy of negatively stained Tf–AuStrs, we show that the transferrin conformation changes on the sides of the spikes but remains unaltered at the tip of the spikes when the protein concentration is raised. The procedure is first developed and calibrated using isotropic gold nanospheres (AuNP) produced by a standard citrate reduction method and finally translated to the anisotropic AuStrs.20,21
:Ag ratio by adding chloride ions and gold seeds into the reaction to understand how these ions and pre-formed nano-seeds drive the mode of spike-growth. In this study, we demonstrate the correlation between ionic concentration, spike morphology and resulting optical properties. A novel depletion assay is then devised to quantify the amount of transferrin bound to the surface of the gold nanostars and spheres by measuring the unbound fraction of the Tf in solution by UV-visible spectroscopy against a set of known standards. The protein loading efficiency was controllably tuned to achieve maximum surface area (SA) coverage without significantly altering the shape or volume of the nanostructures. Using electron microscopy of negatively stained Tf–AuStrs, we show that the transferrin conformation changes on the sides of the spikes but remains unaltered at the tip of the spikes when the protein concentration is raised. The procedure is first developed and calibrated using isotropic gold nanospheres (AuNP) produced by a standard citrate reduction method and finally translated to the anisotropic AuStrs.20,21
2 Experimental
A more detailed account of the materials and methods is provided on the ESI file.†2.1 Materials
All the chemicals used were purchased from Sigma-Aldrich unless otherwise stated. Milli-Q water was used in all experiments.2.2 Gold nanostructure synthesis
Standard procedures for seed-mediated synthesis were used to prepare the nanoparticles.21,22 The nanostructure designs were obtained through specific modifications to the method (Fig. 1, graphs I to V). Gold nanostructures were conjugated with transferrin via a thiol group and salt titration was used to verify the coating.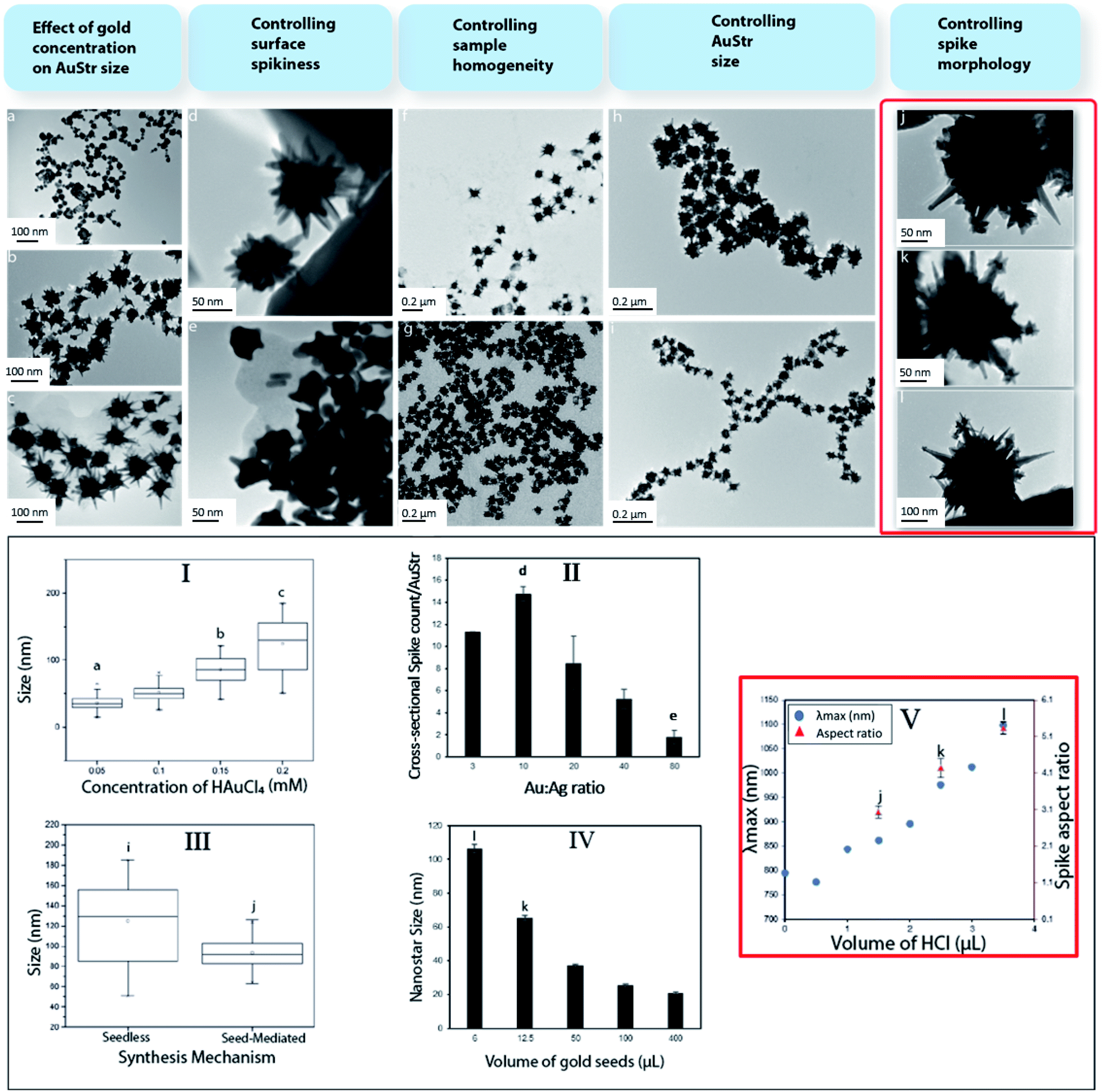 | ||
| Fig. 1 Systematically tailored synthesis of AuStrs to vary their size, spikeness, homogeneity and spike morphology (I–V). AuStr synthesis was performed in the presence or absence of gold seeds. In absence of gold seeds, nanoparticle size control (I) was achieved by varying the total amount of metal precursor (a–c). Cross-sectional spike count per AuStr (II) was controlled by increasing the Au:Ag ratio (from d to e). Introducing seeds in the reaction dramatically improved the AuStr size homogeneity (III) as evidenced by images taken before (f) and after (g) seed addition. Further varying the seed concentrations (IV) proved effective in fine tuning the AuStr final size (h and i). Spike morphology (III) was tailored by increasing HCl in the reaction system (j–l). By changing spike morphology the nanoparticles λmax was tuned in the NIR (V). | ||
2.3 AuStr and AuNP characterization
The size and morphology of the nanostructures made using different synthesis conditions were assessed by using a JEOL JEM-2100F and a FEI Titan TEM, at the acceleration voltage of 200 kV and 300 kV respectively. Negative staining of the nanoparticles was performed using 2% (w/v) solution of uranyl acetate to reveal the presence of a transferrin corona on the surface of the nanoparticle drop-cast on 300 mesh copper grids (Agar Scientific), coated with holey carbon film. The nanoparticles' hydrodynamic diameter was measured in suspension by Dynamic Light Scattering (DLS, Malvern Zetasizer Nano) at 25 °C.2.4 Elemental quantification
The elemental concentration of gold in the samples was measured by ICP-OES (Thermo Scientific iCAP 6000 series). The nanostructures were dissolved in Aqua-Regia, prepared by mixing hydrochloric acid and nitric acid in a 3:1 ratio. The volume of the solution was brought to a final volume of 10 mL with Milli-Q water.
3 Results and discussion
3.1 Controlled synthesis of AuStrs
The influence of different reactant concentrations on AuStr growth in the presence, or absence, of pre-formed seeds was explored first in order to be able to controllably tune the particles size and morphology. The production of nanostructures whose size and shape can be controlled is commonly achieved through the reduction of solution phase metal salts in the presence of stabilizers (surfactants or polymers). During solution-phase chemical synthesis, thermodynamic and kinetic parameters dictate the nanocrystal's final shape through the control of the nucleation and growth stages of metal nanocrystals. Hence, by controlling these factors, crystallographic control may be achieved. Initially, the AuStrs were synthesized in a seedless one-pot reaction based on an approach adapted from Ramsey et al.22 To ensure future bio-compatibility, our method does not employ potentially cytotoxic surfactants (e.g. CTAB). Briefly, HAuCl4 is reduced by ascorbic acid in the presence of silver nitrate to produce AuStrs (Fig. 1a).The effect of the total amount of metal precursor on the nanostructures size was tested while maintaining constant molar ratios between the reactants in a constant reaction volume. Transmission electron microscopy (TEM) of the resulting AuStrs (Fig. 1a to c) revealed that the average AuStr size decreased from 125 ± 6 to 36 ± 2 nm when the concentration of HAuCl4 in the mixture decreased from 0.2 to 0.05 mM (Fig. 1, graph I). At HAuCl4 concentrations as low as 0.005 mM, the synthesis yielded mainly spherical nanoparticles with rough surfaces. When spikes were present (here defined as protrusions longer than 7 nm), these did not exceed one spike per particle. For an HAuCl4 concentration of 0.1 mM, the resulting particles had several spikes with mainly round tips. Increasing the concentration of gold from this critical point generated AuStrs with a more pronounced morphology and spikes of higher aspect ratio (Fig. 1b and c).
The formation of an AuStr includes formation of the core and formation of the spikes. These events could occur either simultaneously or consecutively. Our data show that the gold core is formed first, followed by the spikes. At low gold concentrations of 0.005 mM, spherical gold particles are formed almost exclusively (Fig. 1a). This demonstrates that the spikes do not form simultaneously with, or before, the core. If the amount of metal post-core formation is sufficient, spikes will subsequently form on to the core (Fig. 1b). Ongoing availability of gold will contribute further to spike growth, increasing the probability that they become “sharp” (Fig. 1c).
Additionally, the ratio between Au and Ag was varied while preserving the total amount of metal. Increasing the proportion of silver in the reaction mixture increased the spike count per particle (Fig. 1, graph II). Stars synthesized at Au:Ag of 10 (Fig. 1d) were particularly spiky with an average of 14 cross-sectional spikes per AuStr. Conversely, when the amount of gold overwhelmed that of the silver, the number, size and sharpness of the spikes on the AuStr surface decreased dramatically. At the maximum Au:Ag ratio of 80 (Fig. 1e), AuStrs with a less irregular shape were observed, containing less than 2 cross-sectional spikes per AuStr.
Previous publications support the role of silver ions in asymmetrical growth of particles.23–25 These effects were also observed in this study. Even though a few theories have been used to explain the branching mechanism for spike formation, it remains unclear which factor ultimately drives this mode of growth. One of the most accepted arguments is the underpotential deposition (UDP) of silver on the different facets of gold. A silver monolayer is expected to deposit onto particular gold facets as silver has a lower work function than gold. This deposition may occur preferentially on the gold (110) crystalline facets as these have a higher number of neighbouring gold atoms causing the silver adatom to experience a stronger attractive potential.26,27 This process acts as a symmetry-breaking mechanism, as the growth rate of gold is considerably slowed down on the (110) facets whilst faster growth occurs on facets that are only partially covered with silver.23,28 This growth mechanism is consistent with our study where varying the silver concentration dramatically changed the number of spikes.
Subsequently, pre-formed 12 nm gold citrate seeds were introduced to restrict nucleation events and encourage subsequent particle growth onto the surface of the gold seeds.21,29 TEM of AuStr samples before (Fig. 1f) and after (Fig. 1g) seed addition revealed that the seeds clearly contributed towards the size homogenization of the nanostars (Fig. 1, graph III). Nanostructure size was effectively tuned by varying the ratio of seeds to metal precursor (Fig. 1e and h). It is commonly assumed that in the presence of more seeds, the size of the resulting nanoparticles decreases due to the even distribution of the metal precursor per seed.31 In fact, by decreasing the Au:seed ratio through the addition of more seeds (Fig. 1, graph IV), the AuStr size could be tuned between 103 and 28 nm, which also decreased the spike size and number of spikes per particle.
Finally, hydrochloric acid (HCl) was introduced in the reaction to contribute to the controlled growth of the spikes nanocrystals. The presence of HCl significantly changed the spike morphology (Fig. 1j–l) and noticeably the maximum absorbance wavelength (λmax) shifted towards the near infrared region (Fig. 1, graph V). In the absence of HCl (Fig. 1d), the nanostars contained monocrystal branches that protruded from the core and became gradually thinner, forming spikes. In the presence of HCl (Fig. 1k), the spike morphology is altered and some monocrystals had secondary spikes. The spike aspect ratio also increases in the presence of HCl (Fig. 1j–l).
The altered spike morphology may be attributed to the chloride ions capping particular nanoparticle facets which alters the redox potential of metal ions and even change the degree of UPD of silver on the surfaces of nanocrystals.30 Other halides such as iodide (I−) and bromide (Br−) have previously been reported to influence the shape development of nanostructures.31
The elemental composition of the spikes was analyzed by using spatially resolved Energy Dispersive X-ray Spectroscopy (EDS) in the TEM. The core, primary and secondary spikes were all primarily composed of gold (90.64 atomic%) and minor amounts of silver (9.36 atomic%) (Fig. 2).
 | ||
| Fig. 2 STEM-EDS elemental mapping of AuStrs (top row) showing that both Au (blue) and Ag (red) are constitutive elements of the nanostructure's core and branches. Representative SEM image (bottom row) of AuStrs self-assembled monolayer used for ICP quantification. | ||
3.2 Experimental characterization of nanostar Au content
Given the irregular shape of the nanostars, there is no simple method to convert the particles dimensions into their equivalent mass. Hence, we developed an experimental method to determine the amount of gold per star to thoroughly characterize the nanovehicle. For the reported complex shape nanoparticles, we produced a self-assembled monolayer of nanostars in MPTMS (mercaptopropyl trimethoxysilane) silanized glass substrates (Fig. 2). Finally, the nanoparticles were dissolved in aqua-regia and the amount of gold on the substrates was assessed by inductively coupled plasma optical emission spectrometry (ICP-OES). The gold mass per AuStr was obtained by dividing the total amount of gold by the number of particles in the surface of the substrate from imaging. Using this method, it was determined that the average gold mass per nanostar was 2.4 ± 0.1 × 10−9 μg.3.3 Formation of tunable protein coronas on the AuNPs
The conjugation reaction of transferrin to the nanospheres was conducted in aqueous solution at room temperature under constant stirring, to allow the protein to adsorb homogeneously to all of the nanoparticles. Transferrin itself is soluble in aqueous solutions, which aids in the prevention of flocculation events. By hypothesizing that coating nanoparticles with protein can minimize aggregation events, we prepared transferrin–gold bioconjugates, via thiol–gold chemistry, at different protein to gold ratios. We evaluated their stability in a high ionic strength NaCl (0.1 M) solution tested to aggregate control AuNPs. In the presence of salt, the electrostatic repulsion between nanoparticles is affected as the salt ions bind to the particle surface. The screening of the surface charges halts particle repulsion, predisposing them to aggregation. Conversely, an appropriate protein coating is expected to minimize these events.The stability of the Tf–AuNP complexes was evaluated based on how well the complexes were able to preserve their characteristic localized surface plasmon resonance (LSPR) in aggregating salt conditions. Gold citrate nanoparticles with a size of 12 nm possess a very distinct LSPR peak at 520 nm, allowing changes associated with aggregation events to be easily identified in their spectra.21 Thus, the λmax was used as a measure of stability. The greater the shift in λmax, the more unstable the Tf–AuNP complex. As expected, uncoated AuNPs were the most unstable species, showing a Δλmax of more than 100 nm. The stability of Tf–AuNP complexes increased with increasing volumes of stock protein (Fig. 3b). At optimal protein concentrations, only a small 6–8 nm shift was observed; this is indicative not of aggregation, but of the formation of nanoparticle–protein complexes resulting in the modification of the refractive index of the nanoparticle.
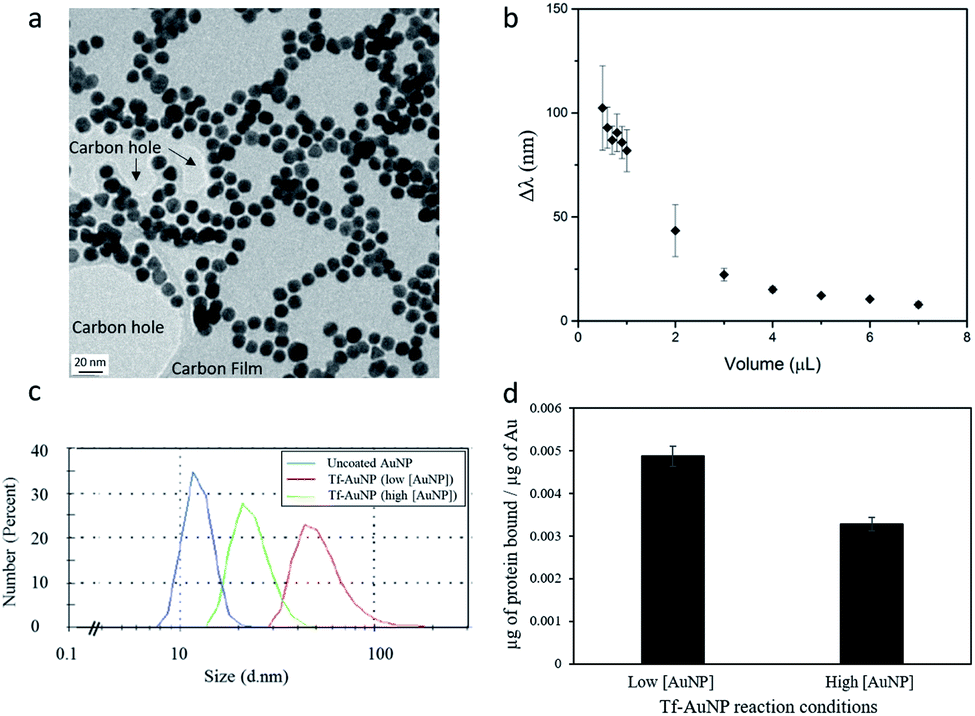 | ||
| Fig. 3 Characterization of AuNPs before and after conjugation with transferrin. TEM of uncoated nanospheres (a) and evaluation of bioconstruct stability as a function of Δλ of TF–AuNP complexes synthesized with different volumes of stock transferrin solution (b). Size distribution of Tf–AuNP constructs obtained by DLS (c) and quantification of transferrin loading (d). The standard synthesis (Tf–AuNP (low[AuNP]) compared with a solution with double the number of particles (Tf–AuNP (high[AuNP]). The data show that even by starting with the same particle size it is possible to control the final size and loading of the bioconstruct by changing the amount of particles in the reaction and fixing the reaction time. | ||
The amount of protein successfully attached to the nanoparticles was obtained by subtracting the amount of unreacted protein, recovered in the sample from that in a control reacted in identical conditions to the sample but in the absence of particles. The unreacted protein in the supernatant is spectrophotometrically measured against a protein calibration curve.
On average, we estimated the binding of approximately 0.226 μg of Tf to 46.36 μg of gold nanoparticles (i.e. Tf:Au ratio of 0.005). This corresponds to a binding of approximately 654 proteins per each of the 2.64 × 1012 gold nanoparticles reacted. Additionally, when double the amount of gold nanoparticles (5.31 × 1012) reacted in the same volume with the same amount of protein, a binding of about 440 proteins per particle was observed. This change in Tf binding can be explained based on the kinetic factors to which the reaction of gold nanoparticles with protein was subjected to. Here, a higher concentration of the reactants results in a higher probability of particle–protein collisions, thus increasing the pressure for the reaction to form bioconstructs to achieve an equilibrium.
In the condition of excess protein, a surface protein coating will occur, the density of which is influenced by the total time of contact between the proteins and the nanoparticle. With a larger number of nanoparticles available, in a fixed reaction time, the total time of contact of a nanoparticle with freely available proteins decreases. Therefore, the density of the coverage also decreases. This explains why doubling the number of gold particles whilst restricting the reaction time decreased the average number of proteins per particle. This may represent an effective way to tailor the amount of protein complexed to the gold nanoparticles.
To establish whether protein binding correlated with a change in particle size, dynamic light scattering (DLS) was used to characterize Tf–AuNP complexes (low[AuNP]) (Fig. 3c) reacted in the presence of excess protein. Tf–AuNP constructs synthesized in identical conditions, but in the presence of double the amount of gold particles (high[AuNP]), had half the hydrodynamic size of the Tf–AuNPs synthesized with the standard amount of spherical AuNPs. Thus, by restricting the reaction time, it was possible to control the hydrodynamic bioconstruct size by changing the protein to nanoparticle ratio.
3.4 Effect of curvature on protein conformation
To study the protein nanoparticle interface at high resolution we imaged negatively stained Tf–AuNP and Tf–AuStr samples in a TEM set at 300 kV. Negative staining was performed because the amorphous protein layer is relatively ineffective in scattering electrons and hence provides insufficient contrast to detect its presence at the particle surface. To generate contrast, the sample was stained using 2% aqueous solution of the heavy metal uranyl acetate. Fig. 4 shows Bright field TEM images of the negatively stained Tf–AuNP (hydrodynamic size of 20 nm) and Tf–AuStr conjugates. | ||
| Fig. 4 Formation of a Tf corona on gold nanostructures. Panels (a–c) show the control unconjugated nanostructures. Images (d and e) show the presence of a protein monolayer corona surrounding the AuNP. On the high aspect ratio topology of the AuStr a conformal monolayer protein coating is observed (f and g). The thickness of the Tf coating and Tf loading (j) increase when the standard amount of reacting protein [1Tf] (f and g) is raised by a factor of ten [10Tf] (h and i). At the spikes tips the protein thickness remained unaltered (k). Where, red arrows indicate the presence of a Tf corona and black arrows represent the Tf binding orientation along its short axis (↑) or long axis (↔). Statistical analysis revealed that the protein corona thickness at the sides of the AuStrs[1Tf] significantly differed from that of the AuStrs[10Tf] (tested by one-tailed t-test, p < 0.0001, n = 47). Conversely, the difference between the protein corona thickness at the tips of the AuStrs[1Tf] and AuStrs[10Tf] was not statistically significant (tested by one-tailed t-test, p = 0.2064, n = 38). | ||
The AuStrs could be successfully bio-conjugated when reacted at the same Tf:Au mass ratio as the AuNPs. The thickness of the protein coating on the nanoparticle surface (Fig. 4f and g) was about 2 nm, similar to previously determined ellipsoidal axial measurements of transferrin of 2.76 nm and 5.52 nm.32 This indicates that a protein monolayer has successfully formed on the nanoparticles' surface. These nanostars bound about 5.23 μg of protein when 229 μg of gold reacted (i.e. Tf:Au ratio of 0.023) which shows that, per microgram of gold reacted, the nanostars bound about 4.6 times more protein than AuNPs. As demonstrated by TEM, the Tf conformally accesses all of the nanostar surface and the above binding ratio is consistent with surface area effects.
In order to assess the effects of excess protein, we dramatically increased the amount of reacting protein by a factor of 10 to test whether the conformal monolayer arrangement could be perturbed. At this extreme condition (Fig. 4h and i), the thickness of the protein coating along the side of the nanostars spikes more than doubled, increasing from 2.1 nm to 5.0 nm. Interestingly, the thickness of the protein layer at the tip of the nanostar's spikes remained undisturbed, irrespective of protein concentration. This behavior is likely due to the protein binding being influenced by changes in nanoparticle surface energy which relates to the local radius of curvature.33 The overall conformal monolayer behaviour of the coating remained, although a multilayer protein packing bridging particularly small inter-spike distances was occasionally observed.
A difference in nanoparticle surface corona thickness between AuStrs reacted at standard (Fig. 4g) and high (Fig. 4i) transferrin concentrations is consistent with an increased surface binding of the protein. The amount of protein that binds to the AuStrs surface in the presence of the high protein concentration was quantified as 78.25 ± 5.08 μg of protein when the same 229 μg of gold were reacted. It is important to note that although the surface binding of protein increased in saturated conditions, the thickness of the corresponding protein coating did not exceed the dimensions of the protein's long axis (5.52 nm), indicating that conformal monolayer behavior is maintained – but the orientation has changed (Fig. 5).
 | ||
| Fig. 5 Manipulation of the orientation of transferrin on the surface of AuStrs. Tf changes binding orientation from horizontal (a) at standard Tf concentrations [1Tf] to a double layer (b) or vertical orientation (c) at high Tf concentrations [10Tf]. | ||
Transferrin can bind to the surface of the gold nanoparticle along its long or short axis. At high Tf concentrations, the thickness of the protein corona is 5.0 nm. The preferential binding of Tf along its longest axis would allow the binding of about two layers of the protein on the surface of the nanoparticle, as the height per layer is 2.76 nm (Tf short axis) (Fig. 5b). The 14 times higher surface binding is however not consistent with a simple bilayer but suggestive of a change in protein orientation, to bind along its shortest axis under the saturation pressures of the environment (Fig. 5c). This would enable more protein packaging on the surface of the nanomaterial. At the AuStrs branch tips, it is likely that the protein remains bound along its long axis as independent of the Tf concentration the thickness of the corona in this region did not change. Alternatively, alterations to the protein's native conformation following nanoparticle binding could also alter the complexity of the binding events influencing the thickness of the protein corona.18
Finally, we evaluated the targeting activity of the standard Tf–AuStrs (100 nm AuStrs [1Tf]) by measured their uptake efficiency in hCMEC/D3 brain endothelial cells know to overexpress Tf receptors.34 To determine uptake, monolayers of hCMEC/D3 were cultured on the surface of multirowell, exposed to the nanostructures and the cell lysates analysed by ICP-OES to measure the intracellular gold content (Fig. 6). We demonstrated that cell uptake efficiency significantly increased by 29% (tested by a one-tailed paired t-test, p = 0.04) for the Tf–AuStrs comparatively to the PEG–AuStrs, demonstrating preservation of protein activity post-conjugation with complex anisotropic nanostructures.
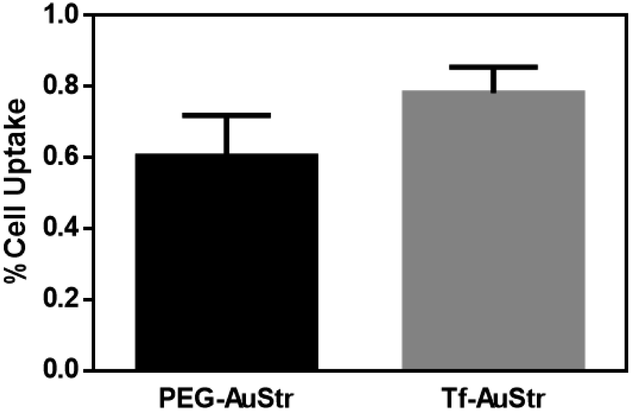 | ||
| Fig. 6 Uptake of PEGylated and Tf conjugated AuStrs into hCMEC/D3 monolayers cultured on multiwell plates. Graph showing the effect of surface conjugation of 100 nm AuStrs on uptake (one-tailed paired t-test, p = 0.04, n = 5). Cells were treated with 50 μg mL−1 nanostructure solution overnight. The amount of gold in the apical media and cellular compartments were quantified by ICP-OES. The values are expressed as percentage of the administered dose of gold. | ||
4 Conclusions
In summary, the flexibility of wet chemistry approaches provides exciting possibilities for the generation of tunable particles. In this study, we demonstrated how gold nanostructures can be finely tailored in a systematic manner to achieve unique morphologies. In particular, we show that silver ions and gold seed concentration alter the star growth morphology. High control over the AuStr geometry allows for the careful tuning of the nanostructure optical properties but also over the surface area for protein loading. Transferrin was successfully and conformally conjugated onto AuStrs with a loading efficiency 4.6 higher than that of AuNPs, as devised by a new depletion method to measure protein binding. The radius of curvature on the spiky nanostructures influenced the binding capacity and protein conformation. The resulting corona coated the nanostructures high index features even at high protein concentrations. The particle Tf loading was tuned by controlling the particle to protein ratio for a fixed reaction time. AuStr uptake studies in hCMEC/D3 cells showed preserved Tf activity of Tf–AuStr bioconstructs. The experimental procedure here studied can be translated and tuned across different nano-topologies and is compatible with applications in which preservation of particle morphology is desired, for example in retention of optical properties or why “spikiness” is related to uptake. The platform here developed constitutes an important addition to the rational design of targeted drug delivery nanovehicles.Conflicts of interest
The authors declare that there is no conflict of interests regarding this work.Acknowledgements
M. P. R. currently holds the Shell/RAEng Research Chair for Interfacial Nanoscience. A. E. P. acknowledges funding from an Elsie Widdowson Fellowship.Notes and references
- T. M. Allen, Nat. Rev. Cancer, 2002, 2, 750–763 CrossRef CAS PubMed.
- M. Kuhn, I. Letunic, L. J. Jensen and P. Bork, Nucleic Acids Res., 2016, 44, D1075–D1079 CrossRef CAS PubMed.
- A. Espinosa, A. K. A. Silva, A. Sánchez-Iglesias, M. Grzelczak, C. Péchoux, K. Desboeufs, L. M. Liz-Marzán and C. Wilhelm, Adv. Healthcare Mater., 2016, 5, 1040–1048 CrossRef CAS PubMed.
- M. Schütz, D. Steinigeweg, M. Salehi, K. Kömpe and S. Schlücker, Chem. Commun., 2011, 47, 4216–4218 RSC.
- M. A. Wronska, I. B. O'Connor, M. A. Tilbury, A. Srivastava and J. G. Wall, Adv. Mater., 2016, 28, 5485–5508 CrossRef CAS PubMed.
- A. Kumar, H. Ma, X. Zhang, K. Huang, S. Jin, J. Liu, T. Wei, W. Cao, G. Zou and X.-J. Liang, Biomaterials, 2012, 33, 1180–1189 CrossRef CAS PubMed.
- M. Zhang, X. Mao, Y. Yu, C.-X. Wang, Y.-L. Yang and C. Wang, Adv. Mater., 2013, 25, 3780–3801 CrossRef CAS PubMed.
- J. J. Richardson, M. Y. Choy, J. Guo, K. Liang, K. Alt, Y. Ping, J. Cui, L. S. Law, C. E. Hagemeyer and F. Caruso, Adv. Mater., 2016, 28, 7703–7707 CrossRef CAS PubMed.
- J. Li, R. Cai, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2015, 3, 5806–5814 RSC.
- S. Sasidharan, D. Bahadur and R. Srivastava, RSC Adv., 2016, 6, 84025–84034 RSC.
- D. H. M. Dam, K. S. B. Culver and T. W. Odom, Mol. Pharm., 2014, 11, 580–587 CrossRef CAS PubMed.
- T. K. Sau, A. L. Rogach, F. Jäckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef CAS PubMed.
- P. Alexandridis, Chem. Eng. Technol., 2011, 34, 15–28 CrossRef CAS.
- S. G. Antimisiaris, S. Mourtas, E. Markoutsa, A. Skouras and K. Papadia, Advanced Healthcare Materials, Wiley-Blackwell, 2014, pp. 87–179 Search PubMed.
- A. J. Mieszawska, W. J. M. Mulder, Z. A. Fayad and D. P. Cormode, Mol. Pharm., 2013, 10, 831–847 CrossRef CAS PubMed.
- Z. Chu, K. Miu, P. Lung, S. Zhang, S. Zhao, H.-C. Chang, G. Lin and Q. Li, Sci. Rep., 2015, 5, 11661 CrossRef PubMed.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
- K. E. Woods, Y. R. Perera, M. B. Davidson, C. A. Wilks, D. K. Yadav and N. C. Fitzkee, J. Phys. Chem. C, 2016, 120, 27944–27953 CrossRef CAS PubMed.
- D. T. Wiley, P. Webster, A. Gale and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8662–8667 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Frens, Nature, 1973, 241, 20–22 CAS.
- J. D. Ramsey, L. Zhou, C. K. Almlie, J. D. Lange and S. M. Burrows, New J. Chem., 2015, 39, 9098–9108 RSC.
- W. Ahmed, E. S. Kooij, A. v. Silfhout and B. Poelsema, Nanotechnology, 2010, 21, 125605 CrossRef PubMed.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef PubMed.
- H. Yuan, W. Ma, C. Chen, J. Zhao, J. Liu, H. Zhu and X. Gao, Chem. Mater., 2007, 19, 1592–1600 CrossRef CAS.
- Y. Yu, Q. Zhang, Q. Yao, J. Xie and J. Y. Lee, Chem. Mater., 2013, 25, 4746–4756 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- S. E. Lohse, N. D. Burrows, L. Scarabelli, L. M. Liz-Marzán and C. J. Murphy, Chem. Mater., 2014, 26, 34–43 CrossRef CAS.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- M. Y. Rosseneu-motreff, F. Soetewey, R. Lamote and H. Peeters, Biopolymers, 1971, 10, 1039–1048 CrossRef CAS PubMed.
- K. K. Nanda, A. Maisels, F. E. Kruis, H. Fissan and S. Stappert, Phys. Rev. Lett., 2003, 91, 106102 CrossRef CAS PubMed.
- B. B. Weksler, E. A. Subileau, N. Perrière, P. Charneau, K. Holloway, M. Leveque, H. Tricoire-Leignel, A. Nicotra, S. Bourdoulous, P. Turowski, D. K. Male, F. Roux, J. Greenwood, I. A. Romero and P. O. Couraud, FASEB J., 2005, 19, 13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00584f |
| This journal is © The Royal Society of Chemistry 2020 |