
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergic effects of the decoration of nickel oxide nanoparticles on silicon for enhanced electrochemical performance in LIBs†
Ujjwala V.
Kawade
a,
Sunil R.
Kadam
b,
Milind V.
Kulkarni
*a and
Bharat B.
Kale
 *a
*a
aCentre for Materials for Electronics Technology (C-MET), Ministry of Electronics and Information Technology (MeitY), Panchavati, Pune 411008, India. E-mail: milind@cmet.gov.in
bBen-Gurion University of the Negev, Department of Chemistry, Beer-Sheva, Israel
First published on 6th January 2020
Abstract
Significant efforts continue to be directed toward the construction of anode materials with high specific capacity and long cycling stability for lithium-ion batteries (LIBs). In this context, silicon is preferred due to its high capacity even though it has a problem of excessive volume expansion during electrochemical reactions as well as poor cyclability due to a reduction in conductivity. Hence, the hybridization of silicon with suitable materials could be a promising approach to overcome the abovementioned problems. Herein, we demonstrate the uniform decoration of nickel oxide (NiO) nanoparticles (15–20 nm) on silicon nanosheets using bis(cyclopentadienyl) nickel(II) (C10H10Ni) at low temperatures, taking advantage of the presence of two unpaired electrons in an antibonding orbital in the cyclopentadienyl group. The formation and growth mechanism are discussed in detail. The electrochemical study of the nanocomposite revealed an initial delithiation capacity of 2507 mA h g−1 with a reversible capacity of 2162 mA h g−1, having 86% retention and better cycling stability for up to 500 cycles. At the optimum concentration, NiO nanoparticles facilitate Li+-ion adsorption, which in turn accelerates the transport of Li+-ions to active sites of silicon. The Warburg coefficient and Li+-ion diffusion at the electrodes confirm the enhancement in the charge transfer process at the electrode/electrolyte interface with NiO nanoparticles. Further, the NiO nanoparticles with uniform distribution suppress the agglomeration of Si nanosheets and provide sufficient space to accommodate a volume change in Si during cycling, which also reduces the diffusion path length of the Li-ions. It also helps to strengthen the mechanical stability, which might be helpful in preventing the cracking of silicon due to volume expansion and maintains the Li-ion transport pathway of the active material, resulting in enhanced cycling stability. Due to the synergic effect between NiO nanoparticles and Si sheets, the nanocomposite delivers high reversible capacity.
1. Introduction
The development of high-performance negative electrode materials for LIBs in energy storage applications represents one of the most important goals of current energy research. Nanostructuring is considered among the key strategies for enhancing the performance of active materials in various electrochemical applications such as batteries, supercapacitors and electrocatalysts. Carbon-based nanomaterials have received much attention and have been vigorously studied by many research groups for LIB applications because of their natural abundance, eco-friendliness and electrical conductivity with high stability.1–3 Graphite is one of the important negative electrode materials in commercially available LIBs, but it is inadequate to fulfil the energy demands in next-generation energy storage devices due to its low theoretical capacity.4,5 Additionally, graphite has some safety issues because its operation potential is very close to that of lithium metal and it is very reactive toward certain electrolytes.6,7 In commercial LIBs, silicon has been investigated as a promising anode material and a substitute for graphite anodes due to its high theoretical capacity of 4200 mA h g−1 with low operating potential, abundance and low cost.4,8–10 In this context, the surface engineering of silicon nanocomposites is an important feature of nanostructuring that can lead to innovation in LIBs.11 Despite the high theoretical capacity of silicon anodes, the electrochemical behavior of Si anodes during applications has some problems in LIBs such as lower electrical conductivity and Li+ diffusivity, as well as significant volume expansion (∼400%). These issues can cause Si pulverization, loss of electrical contact, and consumption of active Li associated with the unstable evolution of solid electrolyte interphases (SEIs). As a result, Si-based anodes generally exhibit low coulombic efficiency, poor cycle stability, and rate capability.12–15 Therefore, to overcome these issues, one effective approach is to use Si nanoparticles for the facile accommodation of large volume changes. However, the high specific surface area associated with nanomaterials causes the aggregation of silicon during cycling and provides plenty of active surface sites to form SEIs.6,16 Furthermore, the repeated intercalation of Li-ions in Si nanoparticles causes cracks and unrestrained SEI formation. Therefore, in Si nanoparticle-based anodes in LIBs, due to the continuous consumption of Li+-ions, electrolytes can still lead to increased impedance and this, in turn, results in continuous capacity fading with low coulombic efficiency.17 The Si nanocomposite materials are addressable if the challenges mentioned above can be overcome. The hybridization of silicon with different materials having particular properties for LIBs could be an efficient approach to easing the abovementioned problems.18–24 Researchers are focusing on different structural design approaches for the enhancement of the Li-ion storage capabilities of silicon.Many researchers have systematically studied surface engineering strategies to conquer the volume expansion issues of silicon during Li-ion intercalation.17 These strategies include the surface modification of silicon with the incorporation of different metal oxides and sulfides, alloys with Li-inactive metals such as Cu, as well as nanocomposites with a variety of polymers, and different carbonaceous materials such as CNT, amorphous carbon and graphene to boost the lithium storage capability.17,25–31 These approaches are useful for avoiding structural changes, improving the mechanical stability and simultaneously maintaining excellent electrical conductivity. Therefore, it is crucial to develop a novel approach to obtain nanocomposites of Si anodes with physically stable and conductive materials to provide mechanical stability during pulverization with enhanced specific capacity. Among the transition metal oxides, nickel oxide (NiO) is one of the active materials with the greatest potential for use in catalysts, capacitors and batteries. Nickel oxide exhibits good theoretical capacity i.e. 800–1000 mA h g−1 for LIBs,32 however, NiO electrodes also suffer from volume expansion problems due to its fairly poor electrical conductivity during lithiation and delithiation. Nickel oxide composites with graphene as well as its different nanostructures such as nanosheets/films/membranes have shown high capacity with good cyclability.33–37
Due to their high theoretical capacity, lower cost, abundant resources and eco-friendly nature, Si and NiO-based materials are another smart choice for anodes in LIBs. However, very limited research has been carried out on Si–NiO nanocomposite materials and hence, it is a challenge to develop silicon–NiO based nanocomposite materials with good electrochemical performance for LIBs. In view of this, herein, NiO@Si was prepared by a solvent impregnation technique using bis(cyclopentadienyl) nickel(II) (C10H10Ni) and silicon in chloroform medium. The electrochemical study of NiO@Si nanocomposite electrodes with the synergic effect of NiO nanoparticles on Si nanosheets is reported herein. It is demonstrated that varying the concentration of NiO controls the surface thickness, which has a strong influence on the volume expansion of silicon during the intercalation of Li, which is responsible for stable electrochemical performance. The present approach is important for the design of high-performance nanocomposite active materials for batteries as well as diverse applications.
2. Experimental methods
2.1 Material preparation
All reagents were of analytical grade and were used without purification. A stoichiometric amount of bis(cyclopentadienyl) nickel(II) (C10H10Ni) was dissolved in chloroform. After dissolving completely, silicon nanopowder was added to this solution. The solution was impregnated on the silicon particles and the mixture was dried at room temperature. The thermal conversion of the precursors was performed at 400 °C for 3 h. The heating rate was kept constant at 5 °C min−1 during synthesis. The weight ratios of NiO to Si in the nanocomposite were 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1 and 1.5:1 in order to achieve comparable nickel oxide contents on silicon and were denoted as SNO1, SNO2 and SNO3, respectively.
:1, 1:1 and 1.5:1 in order to achieve comparable nickel oxide contents on silicon and were denoted as SNO1, SNO2 and SNO3, respectively.
2.2 Material characterization
Structural investigations were carried out using an X-ray diffractometer (Model-D8, Advance and Bruker AXS) with Cu Kα radiation. The morphology was observed by field emission scanning electron microscopy (FESEM, Hitachi S-4800). The morphology and crystal size were also confirmed by high-resolution transmission electron microscopy (HRTEM, JEOL) at an accelerating voltage of 200 kV. Samples for HRTEM analysis were prepared by drop-casting the solution onto carbon-coated copper grids followed by vacuum drying. The elemental composition and chemical bond formation in the nanocomposite was examined by X-ray photoelectron spectroscopy (XPS). Room temperature Raman scattering was performed using an HR 800-Raman Spectrometer (Horiba Jobin Yvon, France) with excitation at 532 nm. The surface area and porosity of the powders were characterized using Brunauer–Emmett–Teller analysis (BET: Quantachrome TouchWin v1.11).2.3 Electrochemical measurements
To perform electrochemical measurements the pyrolyzed powders (active material) were ground and mixed with carbon black as the conducting additive and carboxymethyl cellulose (CMC) binder dissolved in deionised water in the weight ratio of 70:20:10. The slurry was homogenized and tape-cast on the rough side of a copper foil current collector using a doctor blade. The tape was dried in a vacuum oven for 12 h at 120 °C, in order to remove the solvent, and was further used as a working electrode. Electrodes of 16 mm diameter were cut and vacuum-dried at 100 °C for 24 h before transferring to an argon-filled glove box for cell assembly. To perform electrochemical measurements, 2032-type coin cells were used with metallic lithium (Li-tape 99.9% purity, 0.75 mm thickness, Aldrich) as reference and counter electrodes, 1 M LiPF6 in ethylene carbonate (EC):dimethyl carbonate (DMC) (1:1 by volume, BASF) was the electrolyte, Whatman quartz fibre filter (Whatman, UK) was the separator and the 16 mm round discs were used as the working electrodes. The electrochemical measurements (galvanostatic charge–discharge tests) were performed using an MTI battery analyzer with the cut off potential window of 0.01 and 3 V (vs. Li/Li+) at room temperature. Cyclic voltammetry studies of the cell were performed on an Autolab potentiostat/galvanostatic instrument (Metrohm Autolab) with a scan rate of 0.1 mV S−1 between 0.01 V and 3 V. Electrochemical impedance spectroscopy was performed using an amplitude of 5 mV with a frequency range from 0.1 Hz to1 MHz.
3. Results and discussion
The NiO nanoparticles were decorated on Si sheets with the weight ratios 0.5:1 (SNO1), 1:1 (SNO2) and 1.5:1 (SNO3) in NiO@Si nanocomposites. The crystal structures of the as-synthesized nanocomposites were examined by X-ray diffraction (XRD) and are shown in Fig. 1. For the composites including both Si and NiO, peaks were observed at 2θ values of 28.28, 47.13, 56.00, 68.98 and 76.22, which can be assigned to the (111), (220), (311), (400) and (331) planes of the tetragonal silicon phase (JCPDS no. 00-005-0565), respectively. In addition to the silicon phase, the peaks at 2θ values of 37.08, 42.93 and 62.51 can be assigned to the (111), (200) and (220) lattice planes of cubic NiO, respectively (JCPDS no. 01-078-0423). This confirms the presence of crystalline Si and NiO in the composite matrix. The broader peaks of NiO exhibit the nanocrystalline nature of the sample. The crystallite sizes of NiO were calculated using the well known Scherrer's equation and were observed to be 15–20 nm. X-ray diffraction patterns demonstrated the existence of silicon and nickel oxide in the nanocomposite. The SNO1, SNO2 and SNO3 samples showed similar XRD patterns.
 | ||
| Fig. 1 XRD patterns of NiO@Si samples: 0.5:1 (SNO1), 1:1 (SNO2) and 1.5:1 (SNO3). | ||
The surface morphology of the as-prepared SNO2 nanocomposite was analyzed with FE-SEM. The FESEM micrographs of the pristine Si, SNO1, SNO2 and SNO3 (NiO@Si with 0.5:1, 1:1 and 1.5:1) are depicted in Fig. S1 (ESI†) and 2a. Pristine silicon showed a sheet-like structure with smooth surfaces (Fig. S1a and b†). The nanocomposites showed silicon nanosheets that were uniformly decorated with NiO nanoparticles. The nanoparticles were less than 10 nm and hence, agglomerated clusters were observed on the surface of the silicon nanosheets, which may shorten the transport path of lithium ions and electrons. The surface morphology and size of NiO in all nanocomposites are similar. For further clarification, we performed HRTEM of the sample. The morphology of the NiO@Si nanocomposites was elucidated under high-resolution TEM (HRTEM) for a typical sample SNO2 (Fig. 2b–j). As seen in Fig. 2, high-resolution images indicate that the silicon sheets are homogeneously coated with NiO nanoparticles having a size less than 10 nm. The HRTEM of the SNO2 sample depicts the inter-planar spacing of 0.24 nm and 0.30 nm, corresponding to the (111) plane of NiO and Si with the spinel and cubic structure, respectively. Furthermore, the SAED pattern (Fig. 2e) exhibited clear diffraction rings demonstrating the polycrystalline nature of the NiO and Si phase in the NiO@Si nanocomposite. These can be indexed to the (111), (200) and (220) planes of the NiO and (220) of silicon, which is consistent with the XRD results.
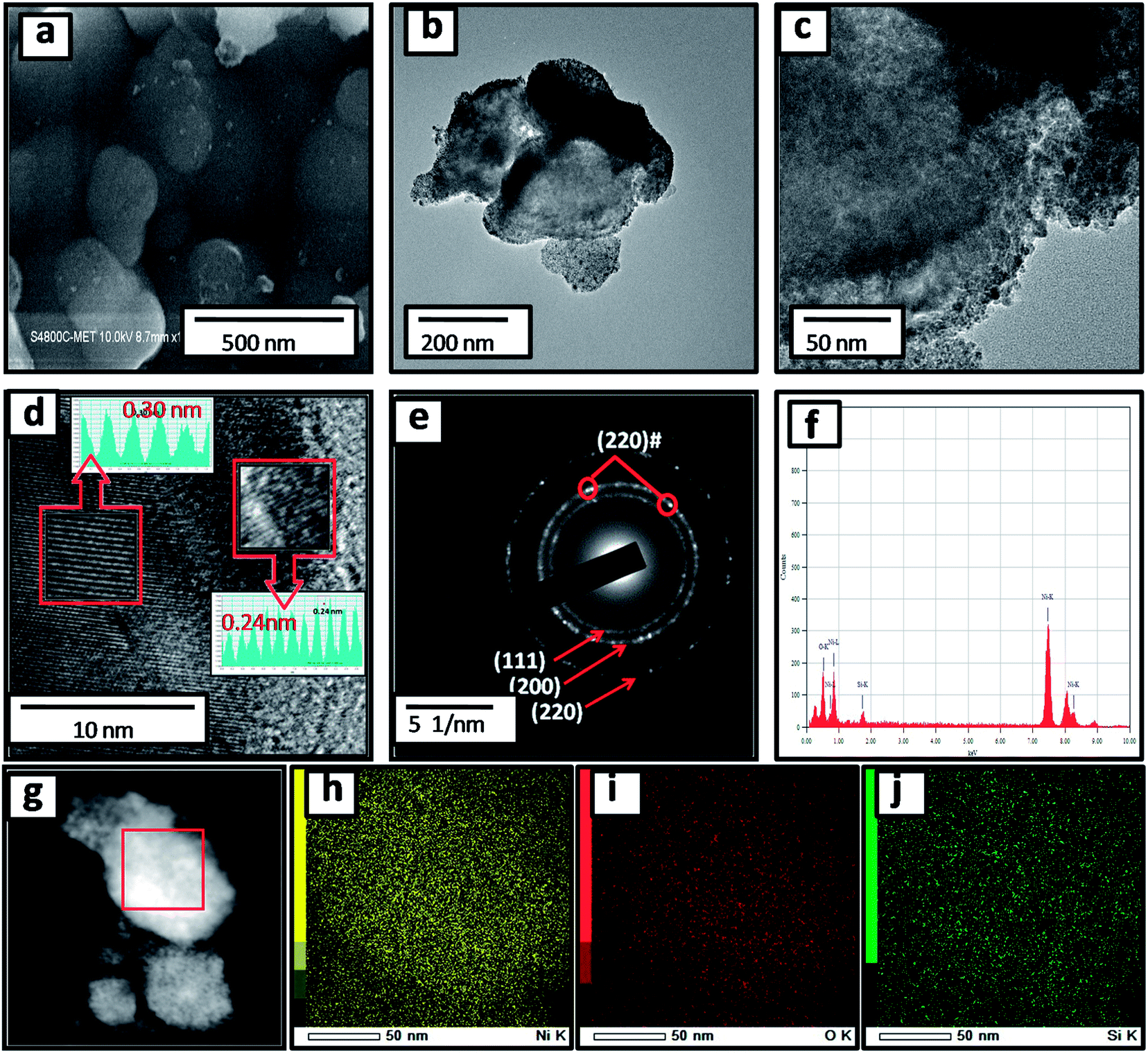 | ||
| Fig. 2 (a) FESEM image of SNO2; (b and c) HRTEM images of SNO2 at different magnifications; (d) HRTEM image and (e) the corresponding SAED pattern; (f) EDS spectrum of SNO2; (g) elemental mapping image and colour map of (h) Si (i) Ni and (j) O. | ||
SNO2 was further analyzed using energy dispersive X-ray spectrometry (EDS) and is shown in Fig. 2f. Ni, O and Si peaks present in the sample confirmed the composition. Further, the elemental mapping (Fig. 2g–j) indicated the distribution of active materials in the nanocomposite. The compositional mapping demonstrated that Ni, O and Si were distributed homogeneously. The brighter mapping image of NiO clearly indicates NiO coating over the Si nanosheets. The background of silicon nanosheets can be clearly observed in the silicon image.
Further, the NiO@Si nanocomposite (SNO2) was examined using X-ray photoelectron spectroscopy (XPS) for elemental composition (Fig. 3a–c), which clearly shows the existence of Si, Ni and O elements. The Ni 2p peaks in the spectrum were deconvoluted by Gaussian curves, consisting of two peaks. These peaks correspond to the spin–orbit levels of Ni (2p3/2) and Ni (2p1/2) located at 854.96 and 872.75 eV, respectively (Fig. 3a). At higher binding energies than Ni (2p3/2) and Ni (2p1/2) peaks, i.e. ∼6.33 eV and ∼5.70 eV, the two satellite peaks were observed. The major singlet peak at 854.96 eV of Ni (2p3/2) associated with their satellite peak, 861.29 eV, corresponds to Ni cations with +2 oxidation state.37,38 The observed energy separation, i.e. spin–orbit splitting between Ni (2p3/2) and Ni (2p1/2) peaks was 17.79 eV. These results confirm the presence of the pure NiO phase with Ni2+ in the composite.39Fig. 3b shows that the deconvoluted XPS Gaussian curves of O(1s) have three peaks, i.e. 528.6 eV, 530.85 and 533.0 eV. Of these, the peaks at 528.6 eV and 533.0 eV are associated with the O 1s binding energy. Obviously, each O(1s) XPS spectrum has two peaks assigned to the O(1s) core levels of the O2− anions. These peaks, attributed to O–Ni and O–C, may have resulted from the surface contamination and defect sites of the NiO surface.38Fig. 3c shows deconvoluted peaks of silicon in which the Si 2p peak located at 99.75 depicts Si. However, peaks at 101.9 (Fig. 3c) and 530.85 eV (Fig. 3b) illustrate the Si–O bond due to the surface oxidation of silicon. These results confirm the existence of silicon and nickel oxide components within the SNO2 sample.
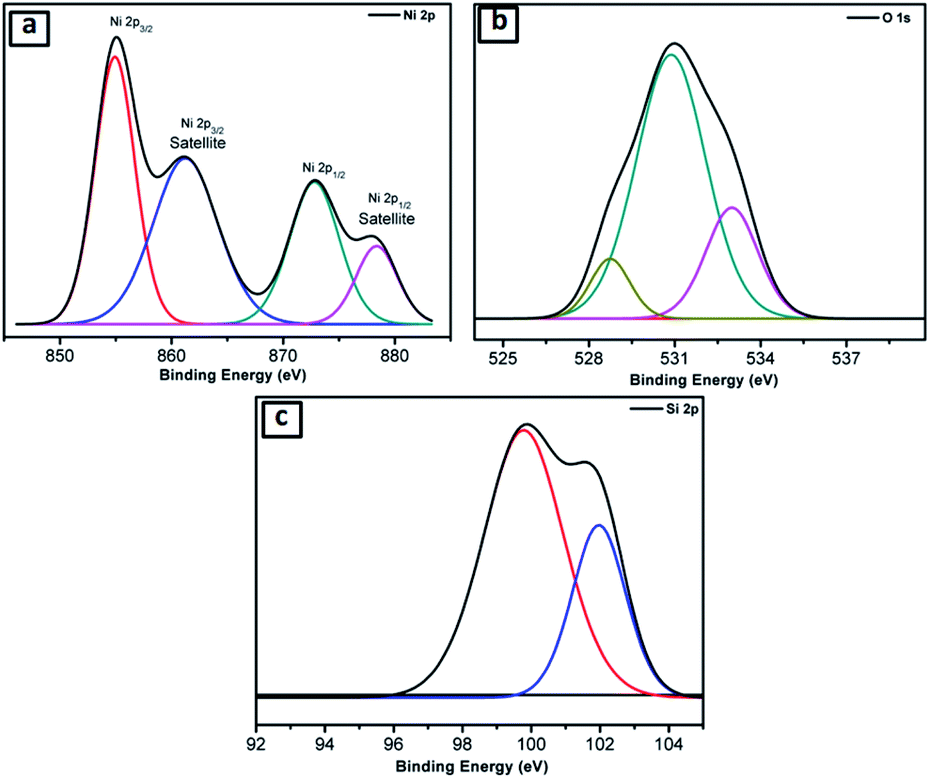 | ||
| Fig. 3 (a–c) XPS spectra of SNO2 for (a) Ni, (b) O, and (c) Si. | ||
Fig. 4a and b presents the Raman spectra of SNO1, SNO2 and SNO3. In the NiO@Si nanocomposite, the Raman peaks were observed at 226, 292, 421, 508 and 933 cm−1. The Raman peaks at 226, 292, 421 and 508 cm−1 were assigned to the first-order LO phonon scattering,40 while the broader peak at 933 resulting from TO + LO phonon (2P) scattering occurring in NiO might be due to nickel vacancy defects.41 The overlapping of the peaks at 508 and 933 cm−1, which are assigned to the Si–Si stretching mode, and a small peak at 933 cm−1, is due to the stretching mode of the amorphous Si–Si of silicon.42 This is in good agreement with the XRD results and also confirms the existence of NiO and Si in NiO@Si nanocomposites. The absence of the D (disordered) band and the G (graphite) band of carbon, indicates the formation of NiO without carbon from the bis(cyclopentadienyl) nickel complex.
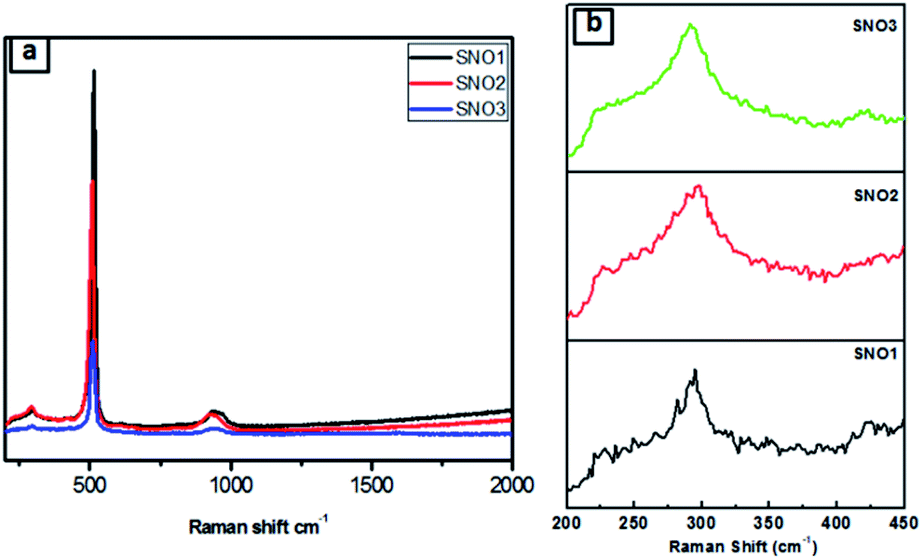 | ||
| Fig. 4 (a) Raman spectra of all samples. (b) Magnified Raman signature. | ||
The specific surface areas and porosity measurements of pristine Si, SNO1, SNO2 and SNO3 were performed. Adsorption–desorption isotherms of N2 from BET analysis and Barrett–Joyner–Halenda (BJH) pore-size distribution for the adsorbent are depicted in Fig. 5 and S2 (ESI†), respectively. BET analysis displays the type (IV) adsorption/desorption isotherm curve and type H3 hysteresis loop at high relative pressure, which demonstrate that there is a layered porous structure caused by the multi-layer adsorption of mesoporous materials.43–45 The observed specific surface areas, pore radii and pore volumes of Si, SNO1, SNO2 and SNO3 are presented in Table 1.
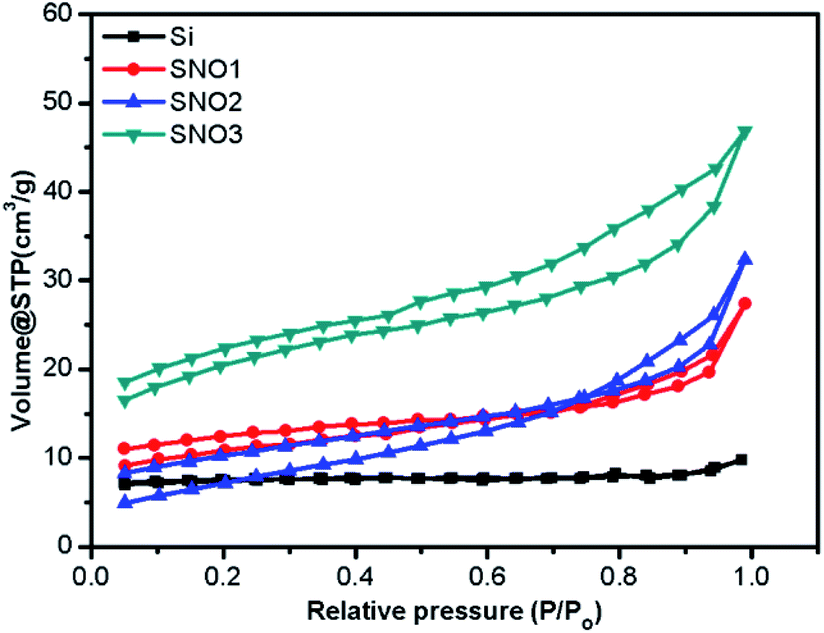 | ||
| Fig. 5 BET adsorption–desorption isotherms of pristine Si, SNO1, SNO2 and SNO3. | ||
| Sample code | Surface area (m2 g−1) | Pore radius (nm) | Pore volume (cc g−1) |
|---|---|---|---|
| Si | 23.08 | 1.3137 | 1.5165 × 10−2 |
| SNO1 | 35.1082 | 2.8570 | 4.2447 × 10−2 |
| SNO2 | 39.9312 | 2.1260 | 5.0151 × 10−2 |
| SNO3 | 68.971 | 2.1058 | 7.2618 × 10−2 |
The pristine Si has a lower surface area as compared to the samples and the overall surface area increased with NiO in the nanocomposites, which is quite obvious. The graphical plot of the BJH pore size distribution indicates that the composite material comprises smaller mesopores <10 nm and a mixture of mesopores and macropores in the range of 10–100 nm. Larger macropores might be attributed to large voids formed by the cross-linking of the silicon and NiO frameworks.
In LIBs, polarization is caused by slow lithium diffusion in the active material and increases in the resistance of the electrolyte when the charging–discharging rate is increased. Faradaic reactions occur at the surface of an electrode, i.e., mass and charge transfer through the electrode; therefore, the surface area and the transport distance play important roles in determining the performance of the battery. The composite nanomaterials (SNO1, SNO2 and SNO3) exhibited greater surface areas and more porous microstructures. Synthesized nanocomposites provide a high surface area, which ultimately provides short diffusion paths (short distance for mass and charge) for ionic transport and electronic conduction. Also, pore structures can facilitate freedom for volume change in materials during lithium-ion intercalation; therefore, composites buffer the Si nanoparticle expansion during lithiation/delithiation processes. In short, high surface area and pore structure may provide improved energy storage capacity and charge–discharge kinetics, as well as better cyclic stabilities.
Based on the above characterization data, Fig. 6 depicts a scheme of the formation mechanism of the NiO@Si composite nanostructure. The NiO@Si was prepared by the solvent impregnation technique using bis(cyclopentadienyl) nickel(II) (C10H10Ni) and silicon in chloroform medium. The NiO–silicon hybrid material was formed upon calcination of the impregnated mass at 400 °C under an air atmosphere in which NiO nanoparticles were uniformly distributed on silicon. Fig. 6 shows the schematic illustration of the preparation of the NiO@Si nanocomposite and its Li-ion battery application.
 | ||
| Fig. 6 Schematic illustration of the preparation of the NiO@Si nanocomposite and its Li-ion battery application. | ||
It is quite well-known that the bis(cyclopentadienyl) nickel(II) complex produces NiO after decomposition in air as per the following equation.46
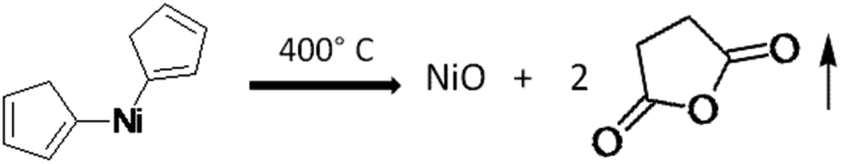
Metallocenes usually adopt structures in which a metal ion is sandwiched between two parallel cyclopentadienyl rings (cp). In the solid-state, the molecule has D5h symmetry, wherein the two rings are eclipsed. The Ni centre has a formal +2 charge, and the Cp rings are usually assigned as cyclopentadienyl anions (Cp−), related to cyclopentadiene by deprotonation. In bis(cyclopentadienyl) nickel(II) (C10H10Ni), there are ten valence electrons from Ni, as well as from the two C2H5 groups, which bond with two protons. Since it does not follow the 18 electron rule, the compound exhibits poor stability. Three pairs of d electrons on nickel are allocated to the three d orbitals involved in Ni–Cp bonding, i.e. dxy, dx2−y2, and dz2. Two remaining d-electrons reside in each of the dyz and dxz orbitals. The low-temperature decomposition is ascribed to the presence of two unpaired electrons in an antibonding orbital in the cyclopentadienyl group. Further, the oxidation of nickelocene leads to a monocation, which in turn causes its decomposition at lower temperatures. The orbital of Ni, being smaller in size, causes insufficient overlap of the π orbitals of electrons in the cyclopentadienyl ring, which accelerates the facile decomposition in an oxidizing medium. In the present case, the metal oxide-catalysed oxidation of cp produced the volatile maleic anhydride, which resulted in the formation of pure NiO (without carbon). Hence, in the present investigation, we have used bis(cyclopentadienyl) nickel(II) for the synthesis of NiO@Si at lower temperatures. Further, this nanocomposite was used as an anode material for the Li-ion battery.
4. Electrochemical study
The lithium-ion half cells have been fabricated using the above nanocomposites to investigate the electrochemical behavior. The electrochemical performances of individual as-synthesized nanocomposite samples were identically investigated using a coin-type (2032) half cell. Fig. 7a displays the first three representative continual cyclic voltammetry cycles of SNO2 in the voltage range of 0.01–3.0 V with a scan rate of 0.1 mV s−1.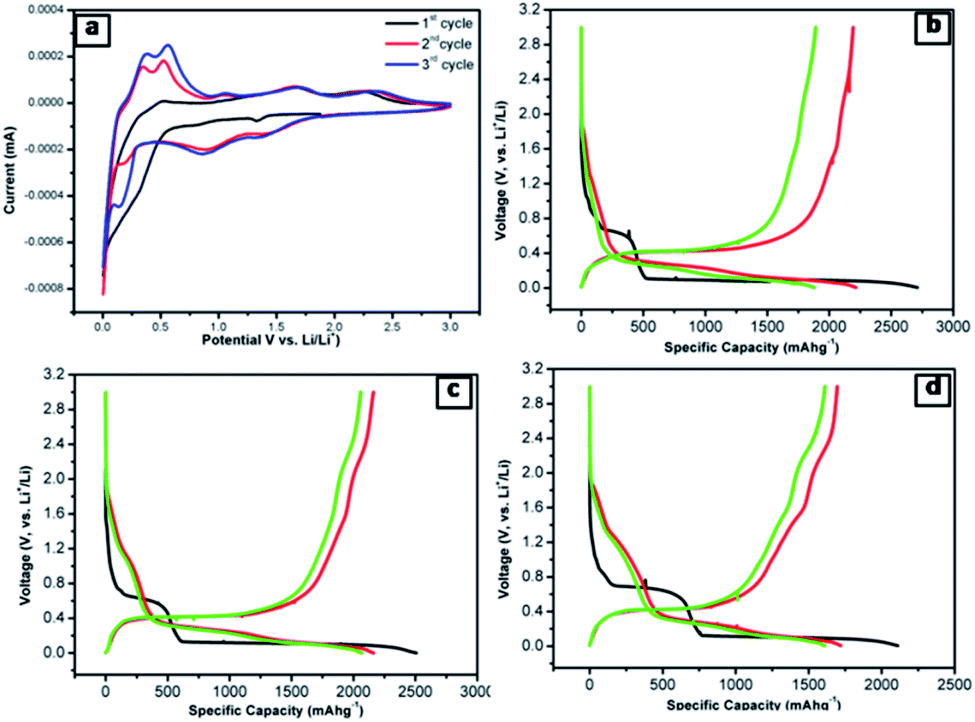 | ||
| Fig. 7 (a) CV curves of the SNO2 electrode at a scan rate of 0.1 mV s−1. (b–d) The initial discharge–charge profiles at the current density of 20 mA g−1 (b) SNO1 (c) SNO2 (d) SNO3 between 0.01 and 3 V. | ||
During the cathodic scan, there was a small reduction peak at ∼0.30 V, which could be related to a multi-step electrochemical reduction process and the formation of a partial reversible solid electrolyte interface (SEI) layer. In the first cathodic scan, peaks at 1.39 V and 0.88 V correspond to the initial reduction of NiO to Ni within the amorphous Li2O matrix (eqn (1)) and the formation of a partially reversible solid electrolyte interface (SEI) layer.47–51
| NiO + 2Li+ + 2e− → Ni + 2Li2O | (1) |
| Ni + Li2O → NiO + 2Li+ + 2e− | (2) |
NiO + 2Li+ + 2e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Ni + Li2O Ni + Li2O | (3) |
In addition, the cathodic part of the second cycle showed peaks at 0.13 V, which can be attributed to the formation of the Li–Si alloy phase and its transition from the Li–Si phase to the subsequent transition to the Li15Si4 phase (eqn (4) and (5)).52
| xLi+ + xe− + Si(crystalline) LixSi(amorphous) | (4) |
| LixSi(amorphous) + (3.75 − x)Li+ + (3.75 − x)e− → Li15Si4(crystalline) | (5) |
| Li15Si4(crystalline) ⇌ 4Si(amorphous) + 15Li+ + 15e− | (6) |
During the anodic scan, the peaks at 0.37 and 0.56 V were due to the extraction of lithium ions, i.e. the de-alloying of Li–Si alloys (LixSi) (eqn (6)). The results are consistent with the previous reports of Si-based electrodes.53,54 The two oxidation peaks located at about 1.69 and 2.38 V in the first anodic scan curve correspond to the decomposition of Li2O and the oxidation of nickel to NiO, respectively (eqn (2)). The abovementioned processes are well-known mechanisms for the reversible reaction (eqn (3)) and the partial composition/decomposition of the SEI layer.47,48 The well-stable and overlapping CV curves imply good electrochemical reversibility and stability during electrochemical reactions.
To further clarify the electrochemical behaviour, the galvanostatic discharge/charge performances of the NiO@Si nanocomposites as an anode material for LIBs was investigated. The first three galvanostatic discharge/charge voltage profiles of the prepared composites are shown in Fig. 7b–d within the voltage range from 0.01 to 3 V against Li0/Li+. In this study, the specific capacity was calculated based on the total mass of the NiO@Si composite as an active material. The first discharge capacities of samples SNO1, SNO2 and SNO3 were 2711, 2507 and 2110 mA h g−1 with reversible charging capacities of 2192, 2162 and 1696 mA h g−1, respectively. The respective retention efficiencies were observed to be 80, 86 and 80%. The lower retention for the SNO1 and SNO3 samples in the first cycle is attributed to the formation of more solid electrolyte interface (SEI) film as compared to SNO2. The SEI layer formation takes place due to the decomposition of the electrolyte as well as the formation of semiconductive metallic Si and Ni during the irreversible electrochemical reactions of Si and NiO, respectively.
There are a number of problems with the existing Si-based materials, such as agglomeration, poor conductivity, cracking of the matrix and the unstable interface between Si and the electrolyte upon long-term cycling. The nanocomposite plays a significant role in nanostructuring during the surface engineering of silicon, which can lead to innovation in LIBs for high and stable capacity. In this work, we introduce nanostructured NiO for synergetic protection to overcome the drawbacks of the silicon anode. With the increase in the NiO content, the surface area increases within the nanocomposites and this NiO provides short diffusion paths for ionic transport and electronic conduction. NiO enhances the electronic pathways and results in better lithium diffusion kinetics i.e. shortening the transport path of lithium ions and electrons. Also, the larger surface area with the porous structure accommodates the volume change, which prevents the pulverization of electrodes, resulting in enhanced cycling stability.
The FESEM and HRTEM images indicated that NiO nanoparticles were dispersed uniformly and anchored on silicon nanosheets. Silicon was completely encapsulated with NiO nanoparticles, which prevented the agglomeration, large volume expansion and contraction in the process of lithation/delithiation. Additionally, the intimate contact of NiO nanoparticles with silicon enhanced the mechanical stability of the electrode, which might prevent cracking due to volume expansion upon the repeated intercalation of Li+ ions. With lower NiO content in the nanocomposite, sharp capacity fading is attributed to the agglomeration of Si particles, the poorly preserved electrical contact between the active materials and the current collector, as well as the pulverization of active substances owing to the large volume variation during charging/discharging. However, the SNO2 exhibited stable charging/discharging performance. This could be attributed to there being less agglomeration of Si nanoparticles, and the porous structure, which are favourable for faster lithium-ion transfer.
Fig. 8a shows the AC impedance spectra of the SNO1, SNO2 and SNO3 electrodes, which were measured under identical conditions in the frequency range of 0.01 to 100 kHz. The charge transfer resistances (Rct) determined in the medium-frequency region from the semicircles of SNO1, SNO2 and SNO3 were 36, 32 and 43 Ω, respectively. The effect of surface modification with NiO on silicon, in terms of Li+-ion diffusion, was calculated from the relationship between Zre and ω−1/2 in the low-frequency region using the following equations:55
 | (7) |
| Z′ ∝ σω−1/2 | (8) |
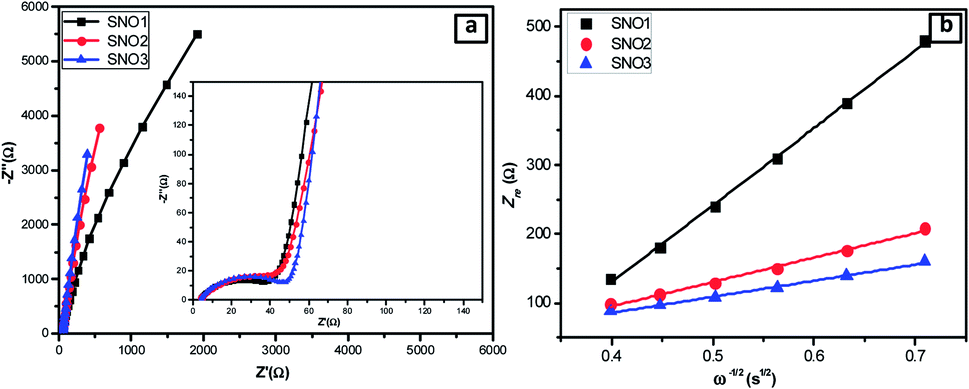 | ||
| Fig. 8 (a) Nyquist plots of the all NiO@Si nanocomposites. (b) The relationship of Z′ and ω−1/2 in the low-frequency region of SNO1, SNO2 and SNO3. | ||
The linear fitting between Z′ and ω−1/2 is shown in Fig. 8b where the low-frequency slope is ascribed to the Warburg impedance associated with the diffusion of Li ions in the bulk of the electrode material. The Li-ion diffusion coefficient D can be calculated from the slope using the above equation. The estimated Warburg factor for SNO1, SNO2 and SNO3 samples are 1119.92, 353.11 and 233.08, respectively. Accordingly, the respective Li-ion diffusion coefficients (D) calculated from the above equation were 4.93 × 10−18, 4.96 × 10−17 and 1.14 × 10−16 cm2 s−1. The results reveal that the NiO nanoparticles play an important role in Li-ion diffusion. It clearly shows that with increasing the concentration of NiO, the Warburg coefficient decreases but the Li-ion diffusion coefficient increases. The NiO@Si with 1:1.5 shows the highest Li-ion diffusion coefficient, most likely due to the availability of channels for Li-ions. The ionic conductivity was found to be increased due to the decoration of NiO, which reduced the electrochemical polarization within silicon. This clearly shows a linear relationship between diffusion coefficients with the concentration of NiO in the nanocomposite. However, even though the amount of NiO was 1.5 times that of silicon in the SNO3 sample, it still showed lower electronic conductivity as compared to SNO1 and SNO2. Generally, nanomaterials exhibit higher electron transfer rates than the bulk but there are some disadvantages. Nanoparticles have low density and higher interparticle resistance, which decreases the electrical conductivity of the material.57 The low tap density leads to the formation of a thick electrode at mass loading as compared to the bulk. This makes it difficult to maintain electrical contact between the electrode material and the current collector, which increases the charge transfer resistance in SNO3. In the case of SNO3, the ionic conductivity was enhanced but the electronic conductivity decreased dramatically because the transport of the electrons to the interface was hindered, which is responsible for lower capacity. Additionally, in SNO3, the amount of silicon is less than NiO, which is also responsible for the lower capacity than SNO2.
To investigate the stability of the electrode, the galvanostatic discharge/charge behaviors of the pristine silicon, obtained SNO1, SNO2 and SNO3 electrode cells were determined as shown in Fig. 9 and S3 (ESI†). During this study, the coin cell was activated at a current density of 20 mA g−1 and the electrode was cycled at 400 mA g−1. The tenth discharge capacities of pristine Si, SNO1, SNO2 and SNO3 were 89, 225, 1479 and 921 mA h g−1, respectively. Fig. S3† demonstrates the continuous capacity fading for Si, SNO1 and SNO3 cells. Fig. 9b shows the stable behavior of the SNO2 electrode; it was, therefore, further cycled up to 500 cycles at a current density of 400 mA g−1. During this study, the coin cell was activated at the current density of 20 mA g−1 and the electrode was further cycled at 400 mA g−1 for 500 cycles. The specific capacity was observed to be 1260 mA h g−1 with 80% retention after 500 cycles (Fig. 8b) along with 98% efficiency. In pristine Si and with lower NiO loading in the composite (SNO1), drastic capacity fading was observed up to the 10th cycle. In the case of sample SNO2, NiO nanoparticles offer a high surface area (Table 1) fast Li+ ion diffusion with superficial electron pathway and good strain accommodation sustainability. Hence, it confers excellent reversibility with highly stable capacity. At the same time, due to the high percentage of NiO nanoparticles, as compared to silicon, in sample SNO3, more NiO clusters were formed over the silicon and resulted in a decrease in the capacity, which was also more stable than SNO1. This might be due to overall lower content of pristine Si. Hence, moderate NiO nanoparticle content is essential for enhancing the capacity.
 | ||
| Fig. 9 Electrochemical properties of SNO2: (a) the rate performance at different current densities and (b) cycling performance between 0.01 and 3 V. | ||
Considering the higher retention of sample SNO2, the detailed galvanostatic discharge/charge behaviors were investigated at different current densities. This NiO@Si electrode was first tested at 20 mA g−1 and further at 50, 100, 200, 400, 800, 1600 and 3200 mA g−1, then at 50 mA g−1 for the last five cycles. At 3200 mA g−1, the specific capacity was above 500 mA h g−1, which indicates excellent rate capability (Fig. 9a). In a nutshell, the NiO coating is a stable active material surface/interface that acts as a protective layer on the surface of the Si, which prevents parasitic side reactions with the electrolyte. The silicon nanocomposite electrode successfully overcame the pulverization problem of the electrode due to volume expansion and retained the reversibility. Due to the synergic effect between NiO and Si, the nanocomposite provides an effectual electrical contact, shortens lithium-ion diffusion distances, enhances Li-ion transport and facile strain relaxation during delithiation. The results suggest better stability characteristics with the SNO2 (NiO@Si with 1:1) as compared to the SNO1 and SNO3. This was supported by the FESEM images of the electrodes after cycling, which revealed differences in pulverization caused by volume changes during Li-ion intercalation/deintercalation for samples Si, SNO1, SNO2 and SNO3.
Fig. 10 shows the FESEM images and corresponding EDS spectra of the SNO2 electrode after ten (a–c) and five hundred (d–f) cycles. Careful investigation showed that the SNO2 electrode had more cracks after five hundred cycles as compared to ten cycles. The high magnification images (Fig. 10b and e) clearly show volume expansion after cycling. Fig. S4 in the ESI† shows the FESEM images and EDS spectra of pristine Si, SNO1 and SNO3 electrodes after ten cycles. Among these, Fig. S4a and b† shows more cracks as compared to SNO1 and SNO3. This reveals that the NiO nanoparticles provide mechanical strength and prevent cracking during the volume expansion of Si. The EDS spectra clearly indicate the existence of silicon and nickel content in composite samples, indicating better stability characteristics of silicon with NiO as compared to the pristine silicon.
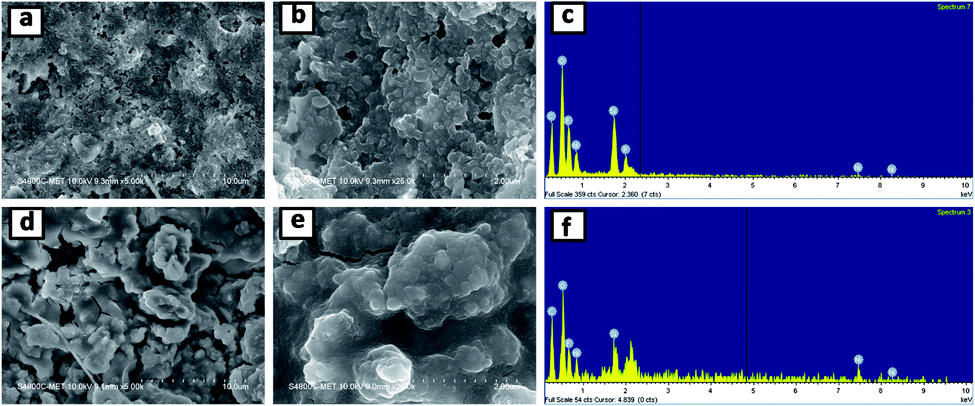 | ||
| Fig. 10 FESEM images and EDS spectra of the SNO2 electrode after ten (a–c) and five hundred (d–f) cycles. | ||
Overall, this study demonstrates that the optimum concentration of NiO with silicon facilitates Li-ion adsorption, which in turn accelerates the transport of Li+ to active sites. Long-term cycling stability suggests that NiO successfully suppressed the agglomeration of silicon during the electrochemical reaction. Simultaneously, NiO in the composite induced a decrease in the polarization and an increase in the electroactive surface area, which led to the enhancement of the Li-ion cell performance. In a nutshell, the enhancement in the performance of the silicon anode with NiO concentration has been presented and compared. The capacities obtained for the existing nanocomposite were compared with earlier reports (see Table S1 in the ESI†).
5. Conclusions
In summary, silicon embedded in NiO was synthesized using a facile solvent impregnation method and used as an anode material for LIBs. Various NiO@Si nanocomposites were fabricated in different weight ratios of NiO and Si. Among them, the SNO2 (1:1) nanocomposite of silicon and nickel oxide exhibited a high reversible capacity (2162 mA h g−1 at 20 mA g−1) and a superior rate capability (500 mA h g−1 at 3200 mA g−1). It is notable that the material was capable of maintaining a high lithium storage capacity 1260 mA h g−1 even after 500 cycles at 400 mA g−1. This study reveals that the optimum concentration of NiO with silicon facilitated Li-ion adsorption, which benefits the transport of Li+ to the active sites. Long-term cycling stability showed that NiO successfully suppressed the agglomeration of silicon during the electrochemical reaction. At the same time, NiO nanoparticles enhanced the mechanical stability of the electrode, which probably helped to prevent the cracking of silicon due to volume expansion upon repeated lithiation/delithiation. This solvent impregnation technique can be applied in the synthesis of various nano-sized active materials using different metallocene precursors for batteries and other applications.
Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.Acknowledgements
Ms Ujjwala would like to thank Department of Science and Technology for financial support towards the implementation of the project (No. SR/WOS-A/CS-73/2017) under woman scientist fellowship scheme. Also, author would like to thank C-MET Pune for providing research facilities and nanocrystalline materials group for kind support.Notes and references
- Z. Zhu, H. Zuo, S. Li, J. Tu, W. Guan, W.-L. Song, J. Zhao, D. Tian and S. Jiao, J. Mater. Chem. A, 2019, 7, 7533–7540 RSC.
- T. Deng and X. Zhou, Mater. Lett., 2016, 176, 151–154 CrossRef CAS.
- B. Xing, C. Zhang, Y. Cao, G. Huang, Q. Liu, C. Zhang, Z. Chen, G. Yi, L. Chen and J. Yu, Fuel Process. Technol., 2018, 172, 162–171 CrossRef CAS.
- K. Feng, M. Li, W. Liu, A. G. Kashkooli, X. Xiao, M. Cai and Z. Chen, Small, 2018, 14, 1702737 CrossRef PubMed.
- Y.-J. Li, C.-Y. Fan, H.-H. Li, K.-C. Huang, J.-P. Zhang and X.-L. Wu, Chem.–Eur. J., 2018, 24, 9606–9611 CrossRef CAS PubMed.
- L. Liu, J. Lyu, T. Li and T. Zhao, Nanoscale, 2016, 8, 701–722 RSC.
- D.-S. Liu, D.-H. Liu, B.-H. Hou, Y.-Y. Wang, J.-Z. Guo, Q.-L. Ning and X.-L. Wu, Electrochim. Acta, 2018, 264, 292–300 CrossRef CAS.
- G. Zheng, Y. Xiang, L. Xu, H. Luo, B. Wang, Y. Liu, X. Han, W. Zhao, S. Chen, H. Chen, Q. Zhang, T. Zhu and Y. Yang, Adv. Energy Mater., 2018, 8, 1801718 CrossRef.
- S. Chen, Z. Chen, X. Xu, C. Cao, M. Xia and Y. Luo, Small, 2018, 14, 1703361 CrossRef PubMed.
- X. Huang, X. Sui, H. Yang, R. Ren, Y. Wu, X. Guo and J. Chen, J. Mater. Chem. A, 2018, 6, 2593–2599 RSC.
- Y. Cen, R. Sisson Jr, Q. Qin and J. Liang, Journal of Carbon Research, 2018, 4, 18 CrossRef.
- A. Tokranov, R. Kumar, C. Li, S. Minne, X. Xiao and B. W. Sheldon, Adv. Energy Mater., 2016, 6, 1502302 CrossRef.
- S. Wu, N. Du, H. Wu, C. Xiao, W. Zhao and D. Yang, RSC Adv., 2016, 6, 109649–109656 RSC.
- X. Cheng, R. Na, X. Wang, N. Xia, Z. Shan and J. Tian, Inorg. Chem. Front., 2019, 6, 1996–2003 RSC.
- D.-H. Liu, H.-Y. Lü, X.-L. Wu, J. Wang, X. Yan, J.-P. Zhang, H. Geng, Y. Zhang and Q. Yan, Nanoscale Horiz., 2016, 1, 496–501 RSC.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 RSC.
- M. A. Rahman, G. Song, A. I. Bhatt, Y. C. Wong and C. Wen, Adv. Funct. Mater., 2016, 26, 647–678 CrossRef CAS.
- W. Luo, X. Chen, Y. Xia, M. Chen, L. Wang, Q. Wang, W. Li and J. Yang, Adv. Energy Mater., 2017, 7, 1701083 CrossRef.
- X. Ma, Y. Gao, M. Chen and L. Wu, ChemElectroChem, 2017, 4, 1463–1469 CrossRef CAS.
- P.-K. Lee, M. H. Tahmasebi, S. Ran, S. T. Boles and D. Y. W. Yu, Small, 2018, 14, 1802051 CrossRef PubMed.
- B. D. Son, J. K. Lee and W. Y. Yoon, Nanoscale Res. Lett., 2018, 13, 58 CrossRef PubMed.
- L. Zhang, R. Rajagopalan, H. Guo, X. Hu, S. Dou and H. Liu, Adv. Funct. Mater., 2016, 26, 440–446 CrossRef CAS.
- L. Zhang, H. Guo, R. Rajagopalan, X. Hu, Y. Huang, S. X. Dou and H. K. Liu, J. Mater. Chem. A, 2016, 4, 4056–4061 RSC.
- D. Jia, X. Li and J. Huang, ChemistrySelect, 2017, 2, 5667–5676 CrossRef CAS.
- C. Martin, O. Crosnier, R. Retoux, D. Bélanger, D. M. Schleich and T. Brousse, Adv. Funct. Mater., 2011, 21, 3524–3530 CrossRef CAS.
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H.-J. Kleebe and R. Riedel, Adv. Funct. Mater., 2014, 24, 4097–4104 CrossRef CAS.
- J.-I. Lee, Y. Ko, M. Shin, H.-K. Song, N.-S. Choi, M. G. Kim and S. Park, Energy Environ. Sci., 2015, 8, 2075–2084 RSC.
- A. R. Park, D.-Y. Son, J. S. Kim, J. Y. Lee, N.-G. Park, J. Park, J. K. Lee and P. J. Yoo, ACS Appl. Mater. Interfaces, 2015, 7, 18483–18490 CrossRef CAS PubMed.
- J. K. Lee, C. Oh, N. Kim, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 5366–5384 RSC.
- Y. Li, Z. Long, P. Xu, Y. Sun, K. Song, X. Zhang and S. Ma, Inorg. Chem. Front., 2017, 4, 1996–2004 RSC.
- J. Wang, D.-H. Liu, Y.-Y. Wang, B.-H. Hou, J.-P. Zhang, R.-S. Wang and X.-L. Wu, J. Power Sources, 2016, 307, 738–745 CrossRef CAS.
- Y. Zou and Y. Wang, Nanoscale, 2011, 3, 2615–2620 RSC.
- Y. Mai, S. J. Shi, D. Zhang, Y. Lu, C. Gu and J. Tu, J. Power Sources, 2012, 204, 155–161 CrossRef CAS.
- Y. Liu, C. Gao, Q. Li and H. Pang, Chem.–Eur. J., 2018, 25(9), 2141–2160 CrossRef.
- Y. Wang, W. Zhou, L. Zhang, G. Song and S. Cheng, RSC Adv., 2015, 5, 63012–63016 RSC.
- A. Khalil, B. S. Lalia and R. Hashaikeh, J. Mater. Sci., 2016, 51, 6624–6638 CrossRef CAS.
- H.-q. Wang, X.-p. Fan, X.-h. Zhang, Y.-g. Huang, Q. Wu, Q.-c. Pan and Q.-y. Li, RSC Adv., 2017, 7, 23328–23333 RSC.
- R. A. Patil, C.-P. Chang, R. S. Devan, Y. Liou and Y.-R. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 9872–9880 CrossRef CAS PubMed.
- G. George and S. Anandhan, RSC Adv., 2014, 4, 62009–62020 RSC.
- G. Vijayaprasath, P. Sakthivel, R. Murugan, T. Mahalingam and G. Ravi, AIP Conf. Proc., 2016, 1731, 080033 CrossRef.
- N. Mironova-Ulmane, A. Kuzmin, I. Steins, J. Grabis, I. Sildos and M. Pärs, J. Phys.: Conf. Ser., 2007, 93, 012039 CrossRef.
- M.-H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- L. Xing, Y. Bai, M. Wang, G. Wang, Y. Ma, Y. Huang and J. Zheng, Nanomaterials, 2019, 9(4), 650 CrossRef PubMed.
- S. Fang, Z. Tong, P. Nie, G. Liu and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 18766–18773 CrossRef CAS PubMed.
- Z.-F. Li, H. Zhang, Q. Liu, Y. Liu, L. Stanciu and J. Xie, ACS Appl. Mater. Interfaces, 2014, 6, 5996–6002 CrossRef CAS PubMed.
- W. Yeh and M. Matsumura, Jpn. J. Appl. Phys., Part 1, 36, 6884–6887 CrossRef CAS.
- X. Sun, C. Yan, Y. Chen, W. Si, J. Deng, S. Oswald, L. Liu and O. G. Schmidt, Adv. Energy Mater., 2014, 4, 1300912 CrossRef.
- J. H. Pan, Q. Huang, Z. Y. Koh, D. Neo, X. Z. Wang and Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 6292–6299 CrossRef CAS PubMed.
- X. Wang, Z. Yang, X. Sun, X. Li, D. Wang, P. Wang and D. He, J. Mater. Chem., 2011, 21, 9988–9990 RSC.
- T. Li, S. Ni, X. Lv, X. Yang and S. Duan, J. Alloys Compd., 2013, 553, 167–171 CrossRef CAS.
- W. Liu, C. Lu, X. Wang, K. Liang and B. K. Tay, J. Mater. Chem. A, 2015, 3, 624–633 RSC.
- M. K. Datta and P. N. Kumta, J. Power Sources, 2009, 194, 1043–1052 CrossRef CAS.
- Y. Chen, S. Zeng, J. Qian, Y. Wang, Y. Cao, H. Yang and X. Ai, ACS Appl. Mater. Interfaces, 2014, 6, 3508–3512 CrossRef CAS PubMed.
- T. Xu, N. Lin, W. Cai, Z. Yi, J. Zhou, Y. Han, Y. Zhu and Y. Qian, Inorg. Chem. Front., 2018, 5, 1463–1469 RSC.
- C. Tang, Y. Liu, C. Xu, J. Zhu, X. Wei, L. Zhou, L. He, W. Yang and L. Mai, Adv. Funct. Mater., 2018, 28, 1704561 CrossRef.
- U. V. Kawade, M. S. Jayswal, A. A. Ambalkar, S. R. Kadam, R. P. Panmand, J. D. Ambekar, M. V. Kulkarni and B. B. Kale, RSC Adv., 2018, 8, 38391–38399 RSC.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00727j |
| This journal is © The Royal Society of Chemistry 2020 |