
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnAl2O4 decorated Al-doped ZnO tetrapodal 3D networks: microstructure, Raman and detailed temperature dependent photoluminescence analysis†
Joana
Rodrigues
 *a,
Matthias
Hoppe
b,
Nabiha
Ben Sedrine
a,
Niklas
Wolff
c,
Viola
Duppel
d,
Lorenz
Kienle
c,
Rainer
Adelung
b,
Yogendra K.
Mishra
*e,
Maria R.
Correia
a and
Teresa
Monteiro
a
*a,
Matthias
Hoppe
b,
Nabiha
Ben Sedrine
a,
Niklas
Wolff
c,
Viola
Duppel
d,
Lorenz
Kienle
c,
Rainer
Adelung
b,
Yogendra K.
Mishra
*e,
Maria R.
Correia
a and
Teresa
Monteiro
a
ai3N & Physics Department, Universidade de Aveiro, 3810-193 Aveiro, Portugal. E-mail: joana.catarina@ua.pt
bFunctional Nanomaterials, Institute for Materials Science, Kiel University, Kaiserstr. 2, D-24143, Kiel, Germany
cSynthesis and Real Structure, Institute for Materials Science, Kiel University, Kaiserstr. 2, D-24143, Kiel, Germany
dMax Planck Institute for Solid State Research, Heisenbergstr. 1, D-70569 Stuttgart, Germany
eMads Clausen Institute, NanoSYD, University of Southern Denmark, Alsion 2, 6400 Sønderborg, Denmark. E-mail: mishra@mci.sdu.dk
First published on 19th March 2020
Abstract
3D networks of Al-doped ZnO tetrapods decorated with ZnAl2O4 particles synthesised by the flame transport method were investigated in detail using optical techniques combined with morphological/structural characterisation. Low temperature photoluminescence (PL) measurements revealed spectra dominated by near band edge (NBE) recombination in the UV region, together with broad visible bands whose peak positions shift depending on the ZnO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al mixing ratios. A close inspection of the NBE region evidences the effective doping of the ZnO structures with Al, as corroborated by the broadening and shift of its peak position towards the expected energy associated with the exciton bound to Al. Both temperature and excitation density-dependent PL results pointed to an overlap of multiple optical centres contributing to the broad visible band, with the peak position dependent on the Al content. While in the reference sample the wavelength of the green band remained unchanged with temperature, in the case of the composites, the deep level emission showed a blue shift with increasing temperature, likely due to distinct thermal quenching of the overlapping emitting centres. This assumption was further validated by the time-resolved PL data, which clearly exposed the presence of more than one optical centre in this spectral region. PL excitation analysis demonstrated that the luminescence features of the Al-doped ZnO/ZnAl2O4 composites revealed noticeable changes not only in deep level recombination, but also in the material's bandgap when compared with the ZnO reference sample. At room temperature, the ZnO reference sample exhibited free exciton resonance at ∼3.29 eV, whereas the peak position for the Al-doped ZnO/ZnAl2O4 samples occurred at ∼3.38 eV due to the Burstein–Moss shift, commonly observed in heavily doped semiconductors. Considering the energy shift observed and assuming a parabolic conduction band, a carrier concentration of ∼1.82 ×1019 cm−3 was estimated for the Al-doped ZnO/ZnAl2O4 samples.
:Al mixing ratios. A close inspection of the NBE region evidences the effective doping of the ZnO structures with Al, as corroborated by the broadening and shift of its peak position towards the expected energy associated with the exciton bound to Al. Both temperature and excitation density-dependent PL results pointed to an overlap of multiple optical centres contributing to the broad visible band, with the peak position dependent on the Al content. While in the reference sample the wavelength of the green band remained unchanged with temperature, in the case of the composites, the deep level emission showed a blue shift with increasing temperature, likely due to distinct thermal quenching of the overlapping emitting centres. This assumption was further validated by the time-resolved PL data, which clearly exposed the presence of more than one optical centre in this spectral region. PL excitation analysis demonstrated that the luminescence features of the Al-doped ZnO/ZnAl2O4 composites revealed noticeable changes not only in deep level recombination, but also in the material's bandgap when compared with the ZnO reference sample. At room temperature, the ZnO reference sample exhibited free exciton resonance at ∼3.29 eV, whereas the peak position for the Al-doped ZnO/ZnAl2O4 samples occurred at ∼3.38 eV due to the Burstein–Moss shift, commonly observed in heavily doped semiconductors. Considering the energy shift observed and assuming a parabolic conduction band, a carrier concentration of ∼1.82 ×1019 cm−3 was estimated for the Al-doped ZnO/ZnAl2O4 samples.
1 Introduction
Due to their importance in several technological fields, namely photocatalysis, gas and bio-sensing applications, functionalised zinc oxide (ZnO) micro- and nanostructures constitute an important topic in current worldwide research.1–13 The disrupted lattice periodicity at ZnO micro- and nanosurfaces is known to cause an increase in the surface state density, which constitutes the main driving paths of sensing-based applications.3,14–19 Doping effects, thermal treatments under different atmospheres and coverage of as-grown micro- and nano-ZnO surfaces with continuous dielectric media are known to result in strong modifications in the electronic energy levels inside the bandgap and band structure of the semiconductor oxide, with a notable influence on the optical and electrical material response.15,20–23 In the case of ZnO, which has one of the largest free exciton binding energy values (∼60 meV23,24), a bandgap energy of ∼3.4 eV (at low temperature23,25) and an exciton Bohr radius of ∼1.8 nm,23,26,27 surface-related influence has been reported in several optical studies, from cleaved bulk samples28 to low dimensional structures.20,29–33 Additionally, decorating ZnO micro- and nanostructures with other metal oxides is known to result in numerous heterojunctions with enhanced properties, as in the case of gas sensor applications.3,4,15,21,34,35 Despite the technological relevance of such hybrid materials, the investigation of fundamental optically active defects and their role in the composite's properties is still scarce. In particular, contactless spectroscopic measurements, such as steady state and transient photoluminescence (PL), are powerful tools to investigate the influence of bulk and surface/interface defects in such complex hybrid structures. Low temperature PL measurements are of extreme relevance in characterisation of semiconductor materials, allowing, for instance, assessment of excitonic features, which are known to dissociate near room temperature (RT) in a wide number of semiconductors, as well as investigation of defects related to radiative processes, carrier transport dynamics and localised states in semiconductor materials.5,36 These phenomena usually play an important role in the optical and electrical performance of the final devices; thus an adequate understanding of their behaviour is mandatory. In the case of ZnO, it is well established that high quality bulk materials usually exhibit well-resolved free (FX) and donor-bound (D0X) exciton recombination lines located in the high-energy spectral range, near the band edge (NBE) of the material bandgap.5,23,25,37–39 Additionally, depending on the growth/synthesis method, visible deep level recombination broad bands in the green, yellow, orange and red spectral regions are commonly identified.24,40–47 Moreover, for Al-doped ZnO it is well recognised that a neutral donor-bound exciton is responsible for the observed I6 luminescence transition at ca. 3.36 eV, corresponding to a donor binding energy of ∼52 meV.23 Furthermore, heavily Al-doped ZnO is known to promote widening of the material bandgap which can be well accounted by the Burstein–Moss and bandgap renormalization effects.48–50 Recently, it was also pointed out that Al doping led to an enhancement of the ratio between the ultraviolet and the deep level emission in ZnO.51 On the other hand, by decreasing the semiconductor dimensionality, with a subsequent increase of the surface-to-volume ratio, new and modified spectral features have been reported related to surface electronic states. In particular, native defects at the ZnO surface may act as binding sites for chemical species that are able to trap electron and hole carriers, resulting in variations of the ZnO carriers' concentration.11,29,30,32,52–55 For instance, excitons bound to surface defects (SXs) due to adsorbed surface species have been reported in the ultraviolet spectral region.56 Besides the ultraviolet region, noticeable dissimilarities have also been observed in the broad visible emission bands of ZnO micro- and nanostructures when compared with their bulk counterparts.29,31,57–59 Even though the deep level emission in nanostructured ZnO has been widely scrutinised, there is still no consensus in the literature about the main origin of the defect-related luminescence. Nevertheless, native defects (Zni, VO, VZn, Oi) have been pointed out to assume a preponderant role in explaining the characteristics of the broad luminescence bands.24,29,60In Al-doped ZnO 3D tetrapodal networks decorated with ZnAl2O4, like the ones reported here, besides the aforementioned processes, the optical PL response should also be influenced by the role of the Al concentration in the ZnO host, the spinel aluminate itself, and the micro- and nano-ZnAl2O4/ZnO heterojunctions promoted during the synthesis procedure. Thus, in-depth knowledge of the luminescence outcome of decorated networks is fundamental for reliable and reproducible tuning of materials properties for a myriad of applications. In this work, the optical properties of Al-doped ZnO 3D tetrapodal networks decorated with ZnAl2O4 particles synthesised by the flame transport method were investigated. The recombination processes on the Al-doped ZnAl2O4/ZnO structures are compared with those on a reference sample composed only by ZnO tetrapods. The modifications in the optical properties are discussed based on the changes induced by the composite formation, as well as by the Al incorporation into the ZnO lattice.
2 Experimental details
2.1 Materials' synthesis
Tetrapodal ZnO (ZnO-T) structures have been synthesised by the flame transport synthesis (FTS) process developed at Kiel University.61–63 Briefly, a mixture of Zn microparticles (diameter ∼ 10 μm) and sacrificial polyvinyl butyral (PVB) powder in a 1:2 weight ratio is burned in a muffle oven at 900 °C for 30 minutes under normal environmental conditions. During burning, the metallic Zn particles are converted into atomic vapour and, in the presence of native oxygen, the growth of tetrapodal-shaped ZnO micro- and nanostructures (in form of white powder) takes place via a solid–vapour–solid growth process, as discussed in previous reports.61–63 For doping and composite formation, an aluminium based salt (aluminum acetate supplied by Sigma Aldrich) was wet mixed in ethanol with the ZnO-T particles in ZnO:Al ratios of 2:0; 2:0.5; 2:1 and 2:1.5, by weight. After evaporation of the liquid, the organic residues were eliminated by a heating step at 550 °C in a muffle oven. To improve handling for further investigations, cylindrical tablets with a radius of 5 mm and a height of 6 mm were pressed to a density of 1 g cm−3. These samples were subjected to an additional heating procedure for 5 h at 1100 °C to increase their structural integrity.
2.2 Materials' characterisation
The morphological investigations of the doped ZnO tetrapodal structures were carried out using a Carl Zeiss scanning electron microscope (SEM, 10 keV, 5 μA). The elemental composition within the Al-doped ZnO network was investigated by energy-dispersive X-ray (EDX) analysis with a SEM machine equipped with a Si/Li detector (Noran, Vantage System). Additionally, information about the morphology, crystal structure, crystal orientation, interfaces and chemical composition was obtained by transmission electron microscopy (TEM). Two microscopes were used in this study: (1) a Philips CM 30 ST operating with a LaB6 cathode at 300 kV and a spherical aberration coefficient of CS = 1.15 mm for structure determination by diffraction experiments and high-resolution microscopy and (2) a FEI Tecnai F30 (300 kV, EDAX detector system) for chemical mapping in the scanning (S)TEM mode. Precession electron diffraction (PED) experiments were performed by using a NanoMEGAS precession spinning star interface adapted to the TEM. By using PED, a more kinematic recording of the diffraction pattern with increased reciprocal resolution can be obtained.64 High-resolution (HR)TEM micrographs were recorded on oriented crystals to achieve a direct view of the crystal interface by tilting the specimen with a double tilt holder. EDX in TEM mode and chemical mapping were conducted using the Thermo Fisher, Noran System Seven on the Philips instrument and the Si/Li detector (EDAX) system on the Tecnai F30 instrument.The Al-doped ZnAl2O4/ZnO tetrapodal networks and the reference sample (ZnO-T) were analysed by Raman spectroscopy, steady-state PL and PL excitation (PLE) spectroscopy at RT. Furthermore, excitation density-dependent and temperature-dependent (from 14 K to RT) PL studies were performed. In the first case, the samples were kept at RT and excited with the 325 nm (∼3.81 eV) line of a cw He–Cd laser (power density I0 < 0.6 W cm−2), controlling the excitation density by using neutral density filters. In the second case, the samples were placed in a cold finger He cryostat and excited with the same He–Cd laser line. The luminescence radiation was dispersed by a Spex 1704 monochromator (1 m, 1200 grooves per mm) and detected with a cooled Hamamatsu R928 photomultiplier. RT PLE and energy-dependent PL experiments were conducted in a Fluorolog-3 Horiba Scientific set-up with a double additive grating Gemini 180 monochromator (1200 grooves per mm and 2 × 180 mm) for excitation and a triple grating iHR550 spectrometer for emission (1200 grooves per mm and 550 mm). A 450 W Xe lamp was used as the excitation source. The PLE was measured by setting the monochromator in the maxima of the emission bands and, afterwards, the excitation was scanned to higher energies. RT time resolved spectroscopy (TRPL) spectra were acquired with the same Fluorolog-3 system using a pulsed Xe lamp (operating at up to 25 Hz) coupled to the same monochromator and with excitation fixed at 325 nm. The TRPL signal was measured by setting a sample window of 10 ms, with 20 ms of time per flash and a flash count of 100. Time delays after flash were varied between 0.05 and 10 ms.
The RT Raman spectra were obtained on a Horiba Jobin Yvon HR800 spectrometer equipped with a 600 grooves per mm grating, under the incidence of a 442 nm line from a cw He–Cd laser (Kimmon IK Series). The experiments were conducted by focusing the laser beam with an objective of ×50 magnification.
3 Results and discussion
3.1 Morphological and structural analysis
Fig. 1 shows the typical SEM/EDX images of all Al-doped (increasing concentration) ZnO 3D tetrapodal networks prepared and investigated in the present work. As can be seen in the pictures, the samples have different morphologies, with predominance of tetrapodal micro- and nanostructures, even though a large number of plate-like structures can also be observed. In the case of the samples obtained from the mixture of the ZnO-T with the Al-based salt (Fig. 1(b)–(d)), the elemental mapping investigations show that Al is quite randomly distributed through the whole surface of the samples, presenting some Al-related agglomerates too, which are more noticeable in the samples with a high ZnO:Al mixing ratio. Indeed, Fig. 1(e)–(g) evidence that these agglomerates correspond to very small nanoparticles decorating the surface of the ZnO-T arms. These small nanoparticles correspond to a ZnAl2O4 crystalline phase, as supported by the TEM measurements (Fig. 2). Their density at the surface of the ZnO structures seems to increase with the ZnO:Al ratio, as expected. Moreover, in this case, the tetrapods' branches exhibit the formation of sharp wrinkled rings, which is mainly due to a high temperature-induced surface stabilization mechanism in the flame transport synthesis process. These results are in line with the ones previously reported by M. Hoppe et al.34 for similar samples. As in that case, the samples reported here exhibit a fairly homogeneous distribution of ZnAl2O4 small crystalline particles at the surface of the ZnO-T, which is particularly evident in the case of the sample prepared with a mixing ratio of 2:1.5 (Fig. 1(g)). These particles display dimensions in the range of 50–100 nm.
 | ||
| Fig. 1 Representative SEM images (left) and the corresponding EDX elemental maps (right) of the synthesised ZnAl2O4/ZnO networks at various ZnO:Al-salt fractions. (a) 2:0, (b) 2:0.5, (c) 2:1, and (d) 2:1.5. (e–g) SEM images of the ZnAl2O4/ZnO composite with an initial mixing ratio of 2:1.5 (ZnO:Al-salt) at increasing magnifications, where the formation of sharp wrinkled rings decorated with very small nanoparticles at the surface of tetrapods' arms is clearly seen. | ||
 | ||
Fig. 2 (a and b) TEM images corresponding to the ZnO:Al-salt sample with a ratio of 2:1.5, showing the ZnO surface decorated with nanocrystals of ZnAl2O4. (c) HAADF image of the nanocrystals on the ZnO surface and the corresponding EDX elemental maps of the components Zn, Al and O. (d) HRTEM micrograph showing the edge-on view of the boundary between the ZnO surface and the ZnAl2O4 crystal. (e) Corresponding PED pattern and (f) kinematic simulation of diffracted intensities. The crystal orientations of ZnO are indexed to [2![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] and those of ZnAl2O4 to [110] having shared (011) and (13) planes at the interface. Intensity in the PED pattern arising from double diffraction is missing in the simulation. 0] and those of ZnAl2O4 to [110] having shared (011) and (13) planes at the interface. Intensity in the PED pattern arising from double diffraction is missing in the simulation. | ||
Nanostructure investigation of the nanocrystals decorating the ZnO tetrapod arms was also performed by TEM in combination with nanoprobe chemical analysis and structural information from electron diffraction experiments. Individual nanocrystals sitting on one tetrapod arm are depicted in Fig. 2(a) and (b) with different magnifications, showing clear interfaces. As was determined by TEM, the size of these nanocrystals is typically in the range of 40–100 nm. Chemical analysis was carried out by collecting the element specific X-ray photons excited by the electron–matter interaction. To map the elemental distribution of nanocrystal clusters on the ZnO surface, EDX was performed in scanning TEM mode, demonstrating that the nanocrystals are composed of Zn, Al and O in considerable amounts (Fig. 2(c)). At the positions of pure ZnO and pure nanocrystal clusters, EDX spectra were gathered and quantified to the ratios of 1:1 (Zn and O) and 1:2:4 (Zn, Al and O) (see Fig. S1†). Further, electron diffraction analysis proved that the crystalline structure of these nanocrystals can be described by the cubic spinel phase of ZnAl2O4 (space group: Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m).65 The orientation relationships between the ZnO hexagonal lattice and the ZnAl2O4 cubic lattice were observed by the combination of HRTEM and PED on individual ZnAl2O4 nanocrystals. When tilting the ZnO crystal to its [20] zone axis, two distinct cases could be identified regarding the attachment of the nanocrystal with its {111} planes being either parallel or perpendicular to the (0001) facet of ZnO. The noise-filtered HRTEM micrograph given in Fig. 2(d) shows the interface of the ZnAl2O4 nanocrystal with its (1) facet interfacing with the ZnO (010) facet. The PED pattern (Fig. 2(e)) and the superposition of the simulated kinematic diffracted intensity pattern (Fig. 2(f)) reflect this [20]‖[110] orientation relationship. The diamond marked diffraction maxima show the superposition of diffracted intensities from both lattices corresponding to lattice planes (011) and (13) with similar lattice constants, which tend to describe the interfacial planes. The second orientation relationship when the ZnAl2O4 {111} facets are grown on the (0001) facet of ZnO is described as [100]‖[211],34 and is evidenced to match with orientation relationships observed between GaN and MgAl2O4.66
m).65 The orientation relationships between the ZnO hexagonal lattice and the ZnAl2O4 cubic lattice were observed by the combination of HRTEM and PED on individual ZnAl2O4 nanocrystals. When tilting the ZnO crystal to its [20] zone axis, two distinct cases could be identified regarding the attachment of the nanocrystal with its {111} planes being either parallel or perpendicular to the (0001) facet of ZnO. The noise-filtered HRTEM micrograph given in Fig. 2(d) shows the interface of the ZnAl2O4 nanocrystal with its (1) facet interfacing with the ZnO (010) facet. The PED pattern (Fig. 2(e)) and the superposition of the simulated kinematic diffracted intensity pattern (Fig. 2(f)) reflect this [20]‖[110] orientation relationship. The diamond marked diffraction maxima show the superposition of diffracted intensities from both lattices corresponding to lattice planes (011) and (13) with similar lattice constants, which tend to describe the interfacial planes. The second orientation relationship when the ZnAl2O4 {111} facets are grown on the (0001) facet of ZnO is described as [100]‖[211],34 and is evidenced to match with orientation relationships observed between GaN and MgAl2O4.66
Fig. 3 depicts the Raman spectra of all studied samples, evidencing the typical vibrational modes of the ZnO hexagonal wurtzite structure that are active in Raman. According to the group theory, the optical phonon modes at the first Brillouin zone centre (Γ point) can be described by the irreducible representation Γopt = A1 + E1 + 2E2 + 2B1, where the B1 modes are silent and the remaining ones are Raman active (A1 and E1 polar modes are also infrared active). These modes were identified in the present spectra, as well as their overtones and combined ones.67 Moreover, acoustic overtones and optical and acoustic phonon combinations were also detected. The differences observed in the relative intensity of the vibrational modes between the samples may arise from polarization effects caused by the different orientations of the ZnO structures regarding the incident wave vector of the laser beam. It is worth mentioning that the samples were explored in different spots to evaluate their structural uniformity. Probing the samples with a 442 nm laser line reveals the presence of additional modes corresponding to the cubic spinel ZnAl2O4 crystalline phase.
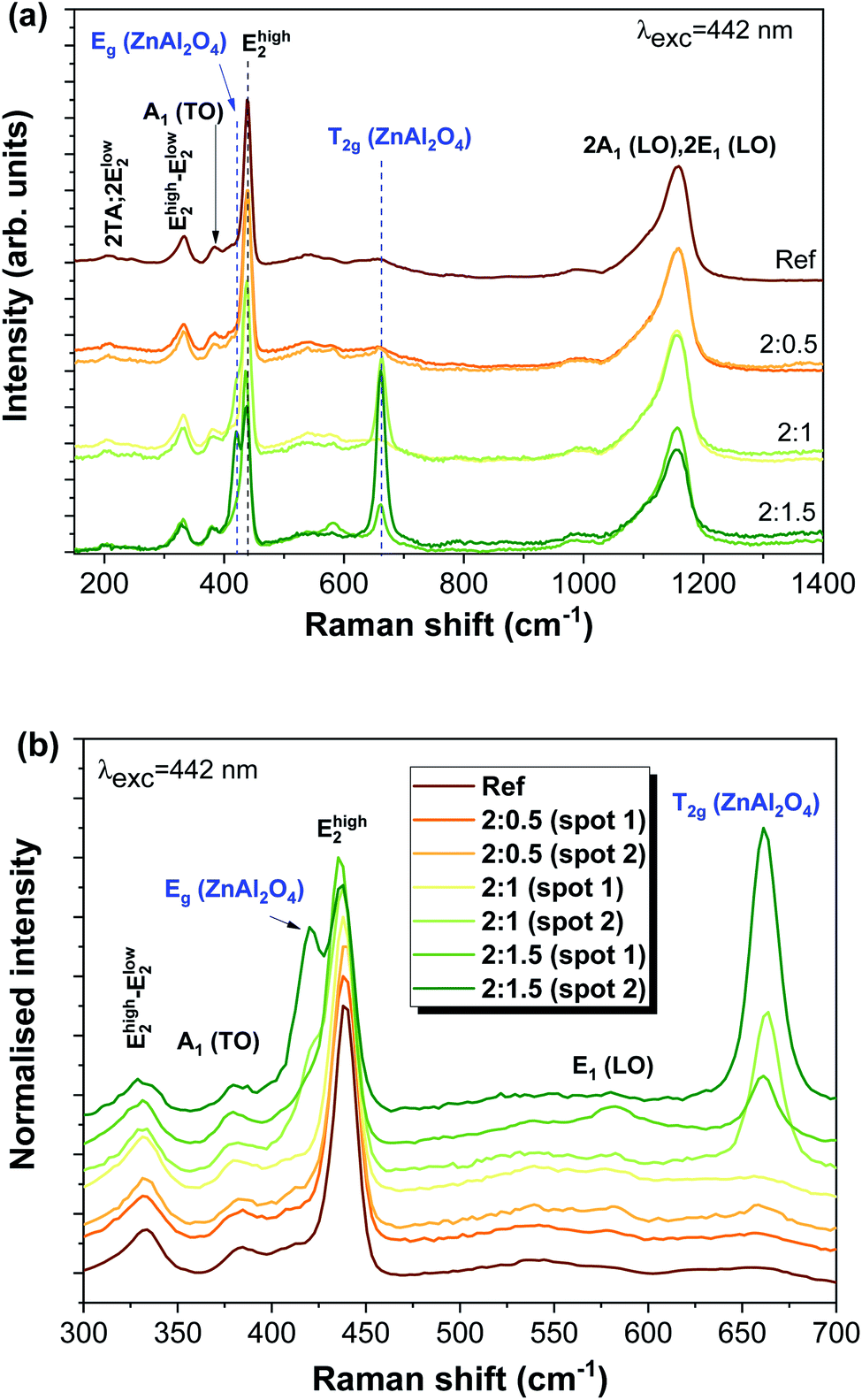 | ||
| Fig. 3 (a) RT Raman spectra of the Al-doped ZnAl2O4/ZnO tetrapodal networks with increasing ZnO:Al ratios, obtained with 442 nm laser line excitation. The spectra were shifted vertically for clarity. (b) Enlarged view of the E2 (high) mode spectral region. | ||
For the cubic spinel ZnAl2O4 phase, the predicted phonon modes at the Γ point are Γ = A1g + Eg + T1g + 3T2g + 2A2u + 2Eu + 4T1u + 2T2u, of which five are Raman active, A1g, Eg, and 3T2g.34,68 In the samples reported here, we were able to clearly identify the modes corresponding to Eg, at ∼420 cm−1 (see the enlarged view in Fig. 3(b)), and one of the T2g modes, peaked at ∼663 cm−1, in accordance with the reported values 418 cm−1 for the Eg mode and 659 cm−1 for T2g reported in the literature.68,69 In previous work by M. Hoppe et al.,34 under the same excitation conditions, the authors could also observe three low intensity shoulders in the Raman spectra at 200, 518 and 653 cm−1, attributed to the T2g vibrational modes. The Raman data agree with the measured X-ray diffraction patterns found in the literature,34 where the diffraction maxima were indexed to both the zinc aluminate spinel and zinc oxide wurtzite structures.
3.2 Optical characterisation
Fig. 4 depicts the low temperature (14 K) PL spectra for all the analysed samples obtained under UV excitation (325 nm laser line, ∼3.81 eV). It is worth noting that this energy corresponds to above bandgap excitation for ZnO and resonant excitation for ZnAl2O4 (Eg ∼ 3.8 eV).3,70 In all cases, the spectra are dominated by visible broad bands, whose peak position is seen to vary with the ZnO:Al ratio (Fig. 4(a)). Moreover, a well-defined NBE recombination is clearly visible at such temperatures for all samples, even for the ones with a higher Al content. Fig. 4(b) displays the high-resolution spectra in this region. In the case of the ZnO reference sample, transitions related to the FX and (D0X were identified at ∼368.3 nm (3.367 eV) and ∼368.8 nm (3.362 eV), respectively, as well as the two electron satellite (TES) recombination at ∼373 nm (∼3.324 eV). As mentioned in the Introduction, D0X-related transitions (I lines) are commonly observed in ZnO structures with different dimensionalities and have been previously assigned to different impurities such as H (I4), Al (I6), Ga (I8) and In (I9).23,71 In addition, Grabowska et al.56 also reported the presence of a SX recombination in this spectral region, near 3.366 eV.
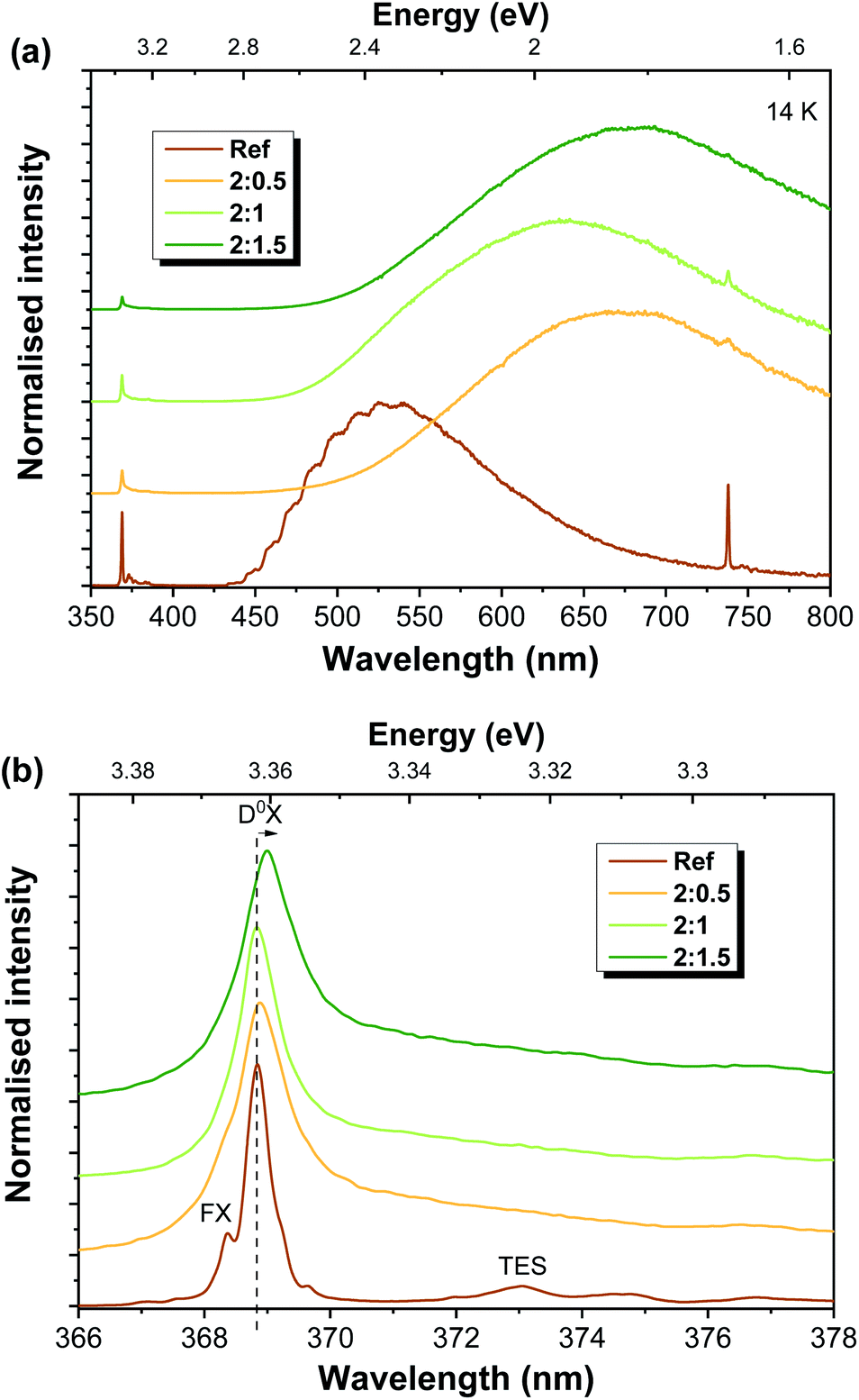 | ||
| Fig. 4 Low temperature PL spectra obtained under 325 nm laser excitation for Al-doped ZnAl2O4/ZnO structures (a) in the UV-Vis range and (b) with high resolution in the NBE region. | ||
Contrary to recently published results,51 in the present case, a decrease in the relative intensity of the UV emission (when compared to the visible one) is observed with increasing ZnO:Al mixing ratios. Moreover, there is a broadening of the emission line associated with the D0X transitions, accompanied by a slight shift of its peak position towards lower energies (longer wavelengths), specially noted in the case of the 2:1.5 sample. Such behaviour indicates the effective incorporation of the Al ions into the ZnO lattice. The observed broadening is likely due to an increase in the contribution of the neutral exciton bound to Al (I6 line) to the overlapped D0X transitions. Indeed, for the 2:1.5 sample, the peak position of the D0X transition is peaked at ∼3.36 eV, which corresponds to the expected position for the transition associated with the recombination of the I6 line.23,71
Regarding the visible spectral region, green luminescence (GL) was detected for the reference sample (peaking at ∼533 nm/∼2.33 eV), exhibiting a structured shape due to two vibrionic progression assisted by a ∼72 meV LO phonon60,72 and with a full width at half maximum (FWHM) close to ∼0.5 eV. In the case of ZnO, the nature of the green emission (far more common and studied than the yellow or orange-red ones) has been widely discussed and associated with several types of defects. Host external impurities, as in the case of Cu,43,44 are frequently associated with the structured green emission (seen at low temperature, as in the case of the present reference sample), while native defects such as oxygen vacancies, ZnO antisites, transitions among interstitials and vacancy-related defects involving Zn species, as well surface related defects, are typically correlated with structureless green emission.24,31,54 The proposed nature of these defects is strongly dependent on the production/synthesis methods and even bands peaking at the same energy position and with similar spectral shapes may be ascribed to different origins.24,60 Moreover, proper assignment to a specific defect is further complicated by the presence of multiple contributions overlapped in the same spectral range, making it rather difficult to assess the origin of each emission.31,73 Nevertheless, compelling evidence that linked structured GL to Cu-related impurities has been presented by Dingle and other authors.24,43,44,60,74,75 According to Garces et al.,44 Cu impurities can be responsible for two distinct mechanisms giving rise to GL, depending on the Cu charge state. In their work, two different GL bands were observed: one unstructured band peaked near 2.48 eV (∼500 nm), arising in the as-grown samples (with Cu+ present), and another peaking at ∼2.43 eV (∼510 nm) with the characteristic vibronic structure reported by Dingle43 and present in thermally annealed samples (with Cu2+). The charge state of the copper ions was assessed by electron paramagnetic resonance (EPR) measurements. Thus, the structured GL accounts for Cu2+-related emission. It is important to stress that even very low impurity/defect concentrations (trace impurities) can be accounted for emissions observed in PL measurements due to the highly sensitive character of the technique. In fact, Dahan et al.76 observed this structured GL band in high-quality unintentionally doped ZnO crystals where copper contents as low as 10–250 ppm were found. Therefore, such trace impurities may also be present in the ZnO-T reported here, leading to the green luminescence features observed on the reference sample. Nevertheless, the possible presence of other centres contributing to this GL should not be neglected. For instance, if the cavity periodicity is at the same scale as the dimensions of the structures, this can also lead to the modulation of the PL emission band, due to light internally reflected, which, under resonance conditions, lead to whispering gallery mode (WGM) resonances.77,78 In fact, Reimer et al.62 discussed the structure observed in the green luminescence displayed by their samples on the basis of the WGM in micro-nanostructured ZnO optical resonators.
This structured GL band is lost upon Al-doping and the corresponding formation of ZnAl2O4/ZnO heterostructures. Although the related centre may remain optically active, other optical centres that are introduced during mixture with the Al-based salt, and subsequent thermal treatment to dope/form the composite structures, dominate the overall emission. With the introduction of the Al-based salt, the deep level emission shifts to longer wavelengths (Fig. 4(a)). However, this shift does not show a correlation with the amount of the Al-based precursor. At low temperature, the band observed for the 2:0.5 sample peaks at ∼672 nm (∼1.84 eV), while for 2:1 and 2:1.5 samples, the maxima are placed at ∼635 nm (∼1.92 eV) and ∼680 nm (∼1.82 eV), respectively. As will be further discussed, this lack of trend in the shift of the peak position with the ZnO:Al ratio is justified by the presence of multiple recombination channels in the present samples, whose contributions are sample-dependent, not relying solely on the Al content. For the Al-doped ZnO/ZnAl2O4 structures, several effects may account for the luminescence outcome, namely the ZnO phase, the ZnAl2O4 one, Al-doping of the ZnO structures, the formation of the composite and the resultant ZnO/ZnAl2O4 interface, the increase in the surface-to-volume ratio due to the formation of the wrinkled rings (as observed in the SEM images), the existence of additional chemical species adsorbed at the surface of the structures promoted by the Al-based salt or the interaction between defects already present and the newly-introduced ones. In fact, if these new defects are more effective in capturing photogenerated carriers, their recombination will occur mainly from those defect levels, rather than from the ones originally present in the ZnO-T samples. By increasing the surface area of the structures, the influence of their surface on the optical properties could increase and become predominant over bulk-related phenomena, being dependent on how point defects are closely located to the material's surface and how will they interact with the surrounding environment.79
The orange-red luminescence that becomes dominant in the Al-doped ZnAl2O4/ZnO structures has been attributed to defects connected with excess oxygen, particularly with oxygen interstitial defects,29,31 or even to surface defects or interstitial zinc and zinc vacancies.11,29,53,79–83 Previous studies on ZnO structures prepared by the hydrothermal method11,84 suggested the association of the orange-red band observed in those cases with defects present on the surface of the crystals. This hypothesis was raised since PL degradation occurred upon increasing photon illumination, also being dependent on the atmosphere in which the measurements were conducted (air vs. vacuum). On the other hand, Djurišić et al.,29,31 reported that the weak orange-red emission detected in ZnO nanoneedles produced by thermal evaporation was reduced after annealing in argon and enhanced by annealing in air, thus pointing to defects related with excess oxygen as a possible origin for this recombination. Additionally, the Zn vacancy (VZn) has been suggested as a dominant compensating acceptors in n-type ZnO. This is consistent with the results of first-principles calculations that indicate VZn as a deep acceptor, with the lowest formation energy among all native defects in n-type ZnO.42,85,86 According to the studies performed by Wang et al.,42 the VZn− defect acts as a deep acceptor and it is responsible for the red emission (near ∼1.6 eV). Unlike many reports in the literature, these authors claim that such a defect does not participate in green emission. The formation of this defect is more favourable under oxygen-rich conditions,79,87 which connects well with the mentioned assumptions by Djurišić et al.29,31 Moreover, positron annihilation spectroscopy measurements performed by Zubiaga et al.88 demonstrated that the VZn related defects are mostly located near the surface of ZnO and hence provide a higher contribution to the emission when the surface area is increased. Furthermore, being at the surface may result in a more noticeable influence by the measurements conditions, as reported in reference.11,84
Finally, the yellow emission has been previously attributed to the presence of adsorbates at the surface of ZnO, namely OH groups resulting from synthesis processes.29,31,54 Djurišić et al.29 observed that the yellow band redshifted with increasing annealing temperature, being replaced by the orange-red one, thus confirming its assignment to the presence of hydroxyl groups or Zn(OH)2 species. OH groups can also occur after prolonged storage of the samples in air, being affected by the distinctive water adsorption properties of different surfaces of the ZnO crystal.29,31 If such groups are formed during the processing steps with the Al-base salt, they may also contribute to the broad luminescence band. Nevertheless, as stated above, one should bear in mind that all the above discussed hypotheses are sample-dependent and can vary to a great extend depending on the synthesis/growth methods, although some of these explanations seem to agree well with the observation reported here.
Previously studied ZnO:Al samples produced by the same FTS approach and then mixed with Al by a different route (followed by subsequent thermal annealing in air in a furnace at 1150 °C for 5 h3) showed similar PL features as the ones detected here (also with the formation of a secondary ZnAl2O4 phase). In that work, the PL spectra of the ZnO:Al samples were seen to be strongly dependent on the excitation spot, revealing two different emission bands, one peaked in the green and other in the orange-red spectral regions, depending on the probed area. These results indicated that the optically active defects were inhomogeneously distributed in the 3D network, which differs from the case reported here where similar spectra were obtained independently on the probed spot, suggesting a higher uniformity of the ZnAl2O4/ZnO distribution. Indeed, the orange-red luminescence (peaked at ∼1.95 eV) observed in that work resembles the one recorded for 2:0.5 and 2:1.5 samples, with a similar spectral shape and peak position. On the other hand, the GL of the previous ZnO:Al samples matches well the one identified for the reference ZnO-T sample. In addition, Wang et al.89 also reported a red shift of the visible band from 518 nm (∼2.39 eV) to 565 nm (∼2.19 eV) with the increase of Al doping concentrations from 0 to 2.0 at% in samples prepared by sol–gel and annealed at 850 °C. The authors attributed this shift to competition between VO+ and Oi−, since when Al replaces Zn, excess oxygen is introduced at interstitial sites, with its concentration increasing with the Al doping. As mentioned above, interstitial oxygen is frequently associated with the presence of orange-red luminescence; thus by promoting the formation of such defects this band becomes more predominant, shifting the overall peak position towards longer wavelengths.
While ZnO broad visible bands are widely studied and reported, the luminescence features of ZnAl2O4 are significantly less studied, hampering a proper comparison with the literature. Even so, it is reasonable to assume that similar defects as the ones highlighted above may be present in this semiconductor, giving rise to luminescence bands in similar spectral regions. Previous reports on similar composites and doped ZnAl2O4 samples70,90,91 point to the fact that their luminescence properties are strongly dependent on the size of the produced structures, uniform distribution of the secondary phase and/or dopants, morphologies and preparation methods. Motloung et al.90 also observed the presence of both green and orange luminescence features in ZnAl2O4/ZnO samples prepared via the citrate sol–gel method. According to these authors, both bands can be ascribed to intra bandgap defects in both semiconductors, namely vacancies, interstitial defects or even antisites, which makes it rather difficult to identify the material (and defects) for which the optical recombination takes place. Thus, taking all this information into account, the most likely explanation for the PL results presented here is that the overall luminescence should be the result of the contributions from both semiconductors and their interface. The large bandwidth verified for all the Al-doped ZnO/ZnAl2O4 samples is most probably a result from a myriad of defect-related recombinations that include the ones considered here, as well as other unknown origins.
Temperature-dependent PL studies were conducted in order to get a better insight regarding the mechanisms involved in the luminescence processes. The spectra for each sample are shown in Fig. 5. In all cases, a strong reduction in the overall luminescence intensity is observed with increasing temperatures. This reduction is more pronounced in the case of the reference sample, with the RT intensity corresponding to only ∼15% of the intensity measured at 14 K (decrease of ∼85%), while decreases of 30%, 28% and 31% were found for Al-doped ZnO/ZnAl2O4 samples with ZnO:Al ratios of 2:0.5, 2:1 and 2:1.5, respectively, comparing the same range of temperatures. Nevertheless, the most interesting fact was the shift of the PL peak position to shorter wavelengths (higher energies) with increasing temperatures verified for the Al-doped ZnO/ZnAl2O4 structures. While the reference sample maintained the GL visible band maximum unchanged in the analysed temperature range, the samples with ZnAl2O4 exhibited a blueshift. Values of about 27 nm (∼80 meV), 19 nm (∼60 meV) and 36 nm (∼100 meV) were obtained with increasing ZnO:Al ratios. Both 2:0.5 and 2:1.5 samples present an orange-red band peaking at ∼638 nm (∼1.94 eV), while the 2:1 sample exhibits a band centred in the yellow spectral region at ∼595 nm (∼2.08 eV). The reference sample maintained the green luminescence with a maximum at ∼535 nm (∼2.33 eV). The blueshift observed for the Al-doped composites is likely accounted by the presence of multiple optically active defects contributing to the detected emission, as discussed in other studies.3,15 With increasing temperature, the relative intensity of the transitions associated with these defect centres changes, leading to a shift in the position of the band maxima.
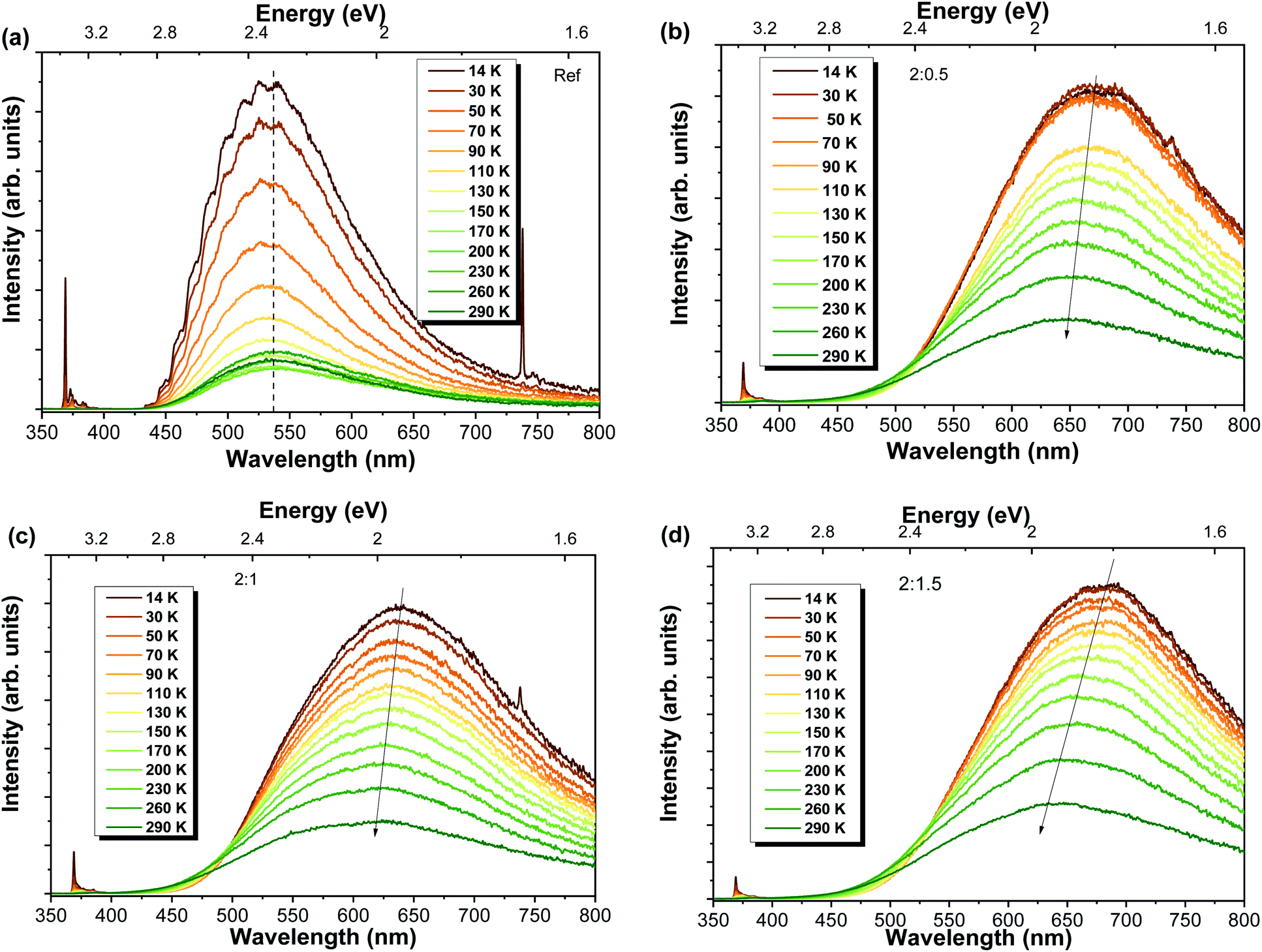 | ||
| Fig. 5 Temperature-dependent PL spectra obtained under 325 nm laser excitation for the Al-doped ZnO/ZnAl2O4 samples: (a) reference sample and samples with different ZnO:Al ratios (b) 2:0.5, (c) 2:1 and (d) 2:1.5. | ||
The presence of more than one recombination channel contributing to the broad visible band of the Al-doped ZnO/ZnAl2O4 composites was corroborated by the effect of the excitation density (at RT). These measurements were carried out by applying neutral density filters to the laser line excitation. Fig. 6 depicts the results obtained for 2:1 and 2:1.5 samples. Even though only a slight shift of the peak position of the visible band was observed, this shift was towards higher energies when the excitation density decreased. A shift of the peak position towards higher energies would be expected with increasing power when centres of DAP nature are involved in the luminescence features. When this type of centre is examined under low excitation densities, typically only a fraction of the donors and acceptors is excited. As the excitation density increases, all the donors and acceptors become excited (saturation condition), leading to an additional contribution from the closer pairs to the recombination spectra92,93 and a blue shift of the DAP emission peak is observed. Yet, the opposite is observed in the present case, which suggests a different origin for this shift and corroborates our previous assumption regarding the existence of multiple recombination channels contributing to the broad luminescence.
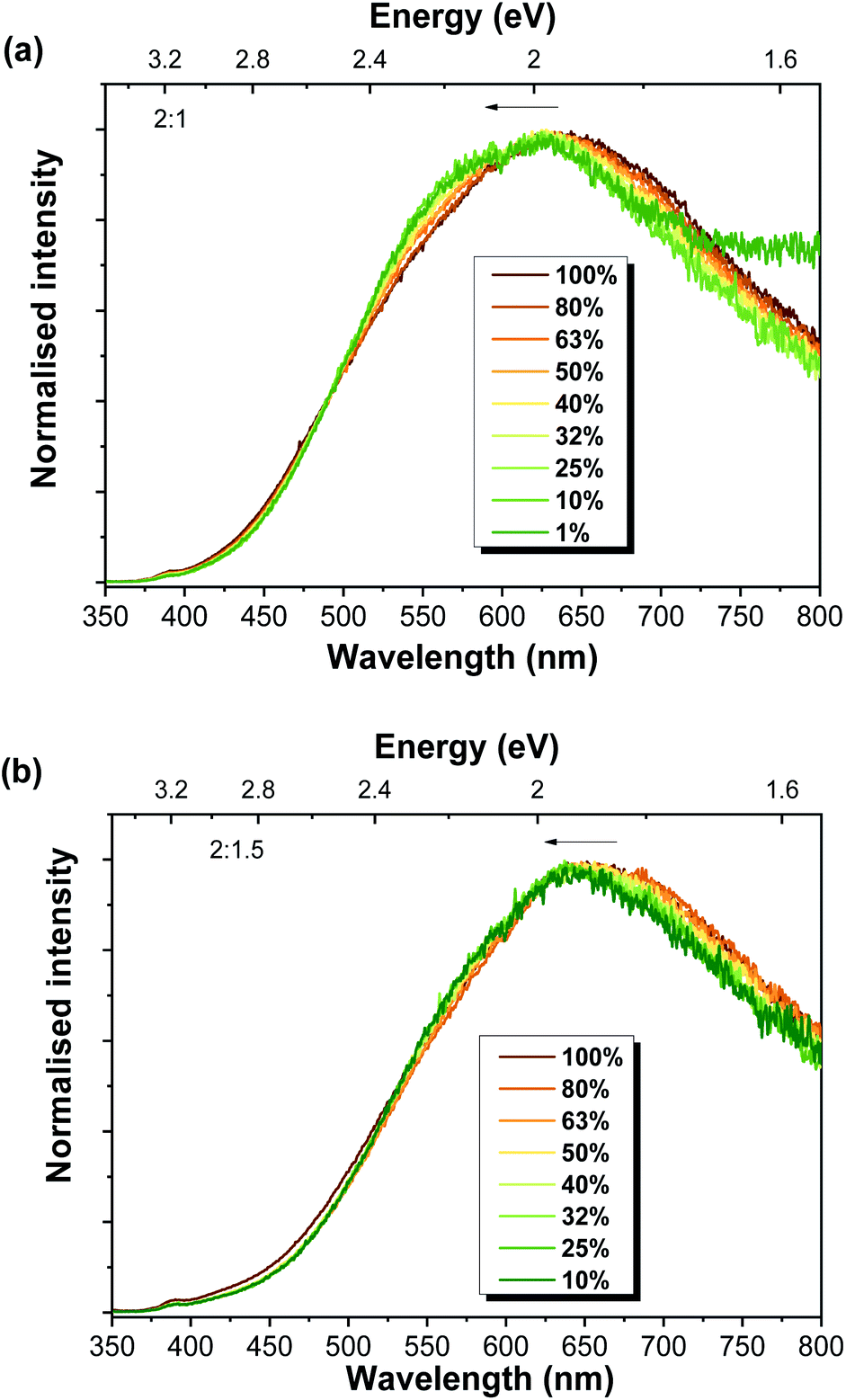 | ||
| Fig. 6 RT power-dependent PL spectra for the Al-doped ZnO/ZnAl2O4 samples with ZnO:Al ratios of (a) 2:1 and (b) 2:1.5. | ||
To acquire a better understanding on the phenomena involved in the visible band luminescence and to further confirm the presence of the different recombination channels, RT time-resolved measurements were carried out, as demonstrated in Fig. 7. It is important to take into account that at such temperatures some of the contributions may no longer be assessed due to thermal dissociation of the emitting defects. Thus, only the contributions that remain at RT could be evaluated. In all cases, the spectra were recorded under 325 nm excitation for increasing time delays. For the Al-doped ZnO/ZnAl2O4 samples, increasing the delay after flash, and thus reducing the components with faster decays, reveal the presence of a slower additional band in the red spectral region, whose predominance increases with the increasing Al content. Indeed, after 1 ms it becomes the dominant emission for the 2:1.5 sample, suggesting that the presence of the defects that originate this red emission are enhanced by the presence of a higher concentration of ZnAl2O4 at the surface of the Al-doped ZnO tetrapodal network. In the case of the reference sample, the red emission appears to overlap with the green component, even after 1 ms of delay after flash, indicating a much lower concentration of this defect. For this sample, the overall emission becomes broader with increasing time delays due to the strong reduction of the faster green component, whose intensity is reduced by about 3 orders of magnitude after a delay of 1 ms. Considering the 2:0.5 sample, identification of the presence of at least 2 components was clearly observed with the increasing delays. It is seen that the yellow/orange component (peaked at ∼620 nm/∼2 eV) disappears after ∼1 ms, while the red (∼720 nm/∼1.71 eV) one is still present until ∼3 ms, evidencing a longer lifetime. In the case of 2:1 and 2:1.5 samples, two bands are present in the green/yellow and red spectral regions, with an additional shoulder at ∼630–640 nm, which becomes more evident in the case of the 2:1.5 sample. For the latter, the yellow emission (with maximum intensity at ∼560 nm/∼2.21 eV) almost vanishes after 1 ms, while the red component (∼725 nm/∼1.71 eV) is still visible until ∼10 ms.
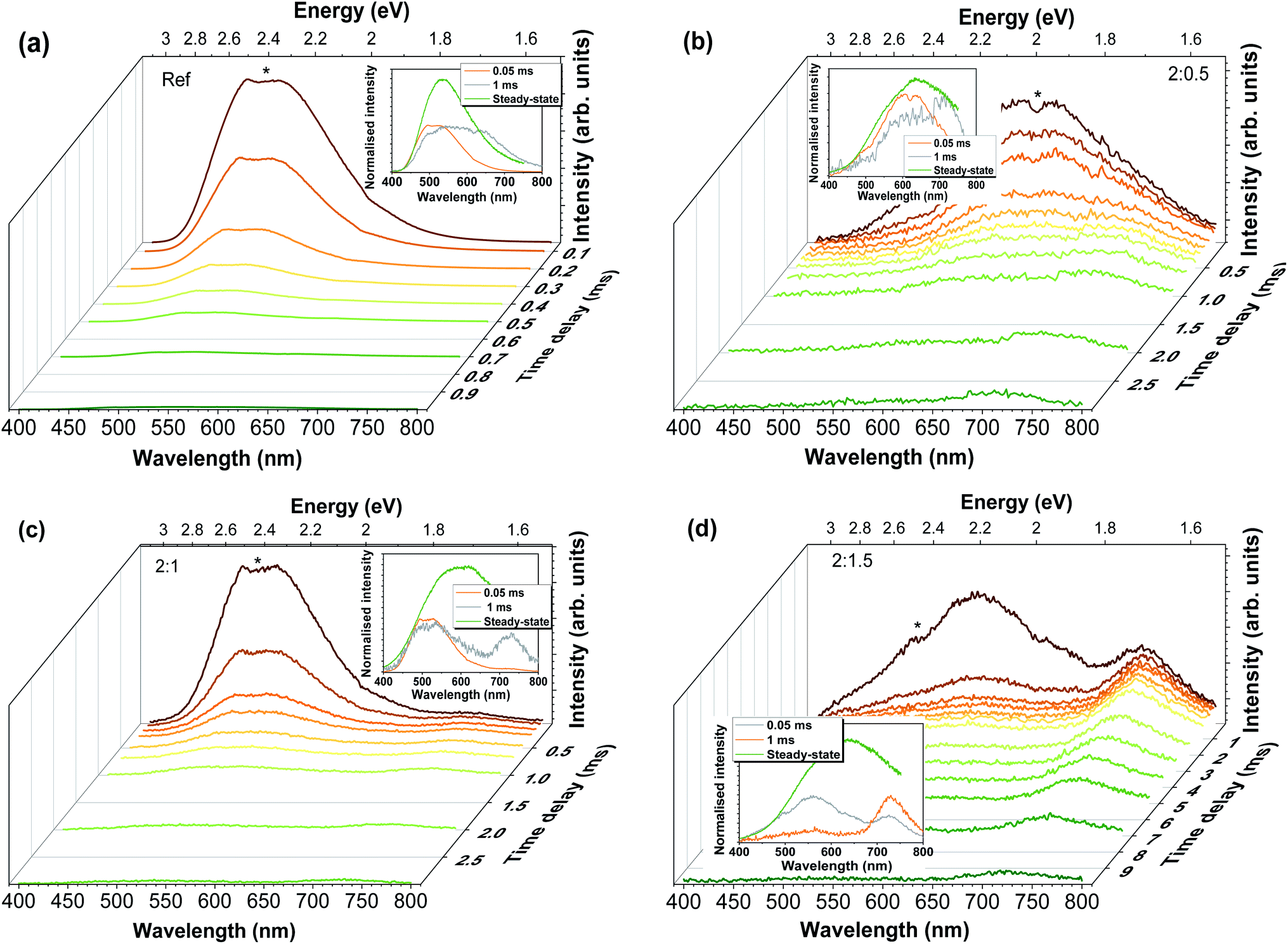 | ||
| Fig. 7 RT TRPL spectra obtained after 325 nm pulsed excitation for increasing delay times (time window of 10 ms) for (a) the reference sample, and Al-doped ZnO/ZnAl2O4 samples with different ZnO:Al ratios (b) 2:0.5, (c) 2:1 and (d) 2:1.5. Inset: normalised time-resolved spectra for 2 different delays, 0.05 ms and 1 ms, and the steady-state spectrum, evidencing a change in the spectral shape with increasing delay after flash. The small signal depression denoted with an asterisk corresponds to the blaze of the detection grating. | ||
Finally, considering the data provided by the previous measurements, the preferential excitation pathways for the present samples were assessed via PLE, as depicted in Fig. 8. The RT PLE spectra were acquired by monitoring the PL emission at the maximum of the broad band for each sample. Again, clear differences are observed between the reference sample and the Al-doped ZnO/ZnAl2O4 (Fig. 8(a)). In the first case, the energy position for the excitation associated with the exciton is peaked at ∼377 nm (∼3.29 eV), while this value shifts towards higher energies in the presence of ZnAl2O4 particles (∼3.38 eV), corresponding to a blue shift of ∼90 meV (better seen in Fig. 8(b)). Besides, an increase in the full width at half maximum of the line also occurs. It is interesting to note that the same energy shift was measured for all Al-doped samples. Additionally, when the excitation spectra are monitored at longer wavelengths of the broad bands (∼630 nm), an increase in the band tail states (states at lower energies than the ZnO bandgap) is observed, indicating that below bandgap population pathways occur. The PL/PLE results obtained for the present set of samples are similar with the ones found for the previous ZnO:Al samples produced by FTS in which the additional phase of ZnAl2O4 was also present.3
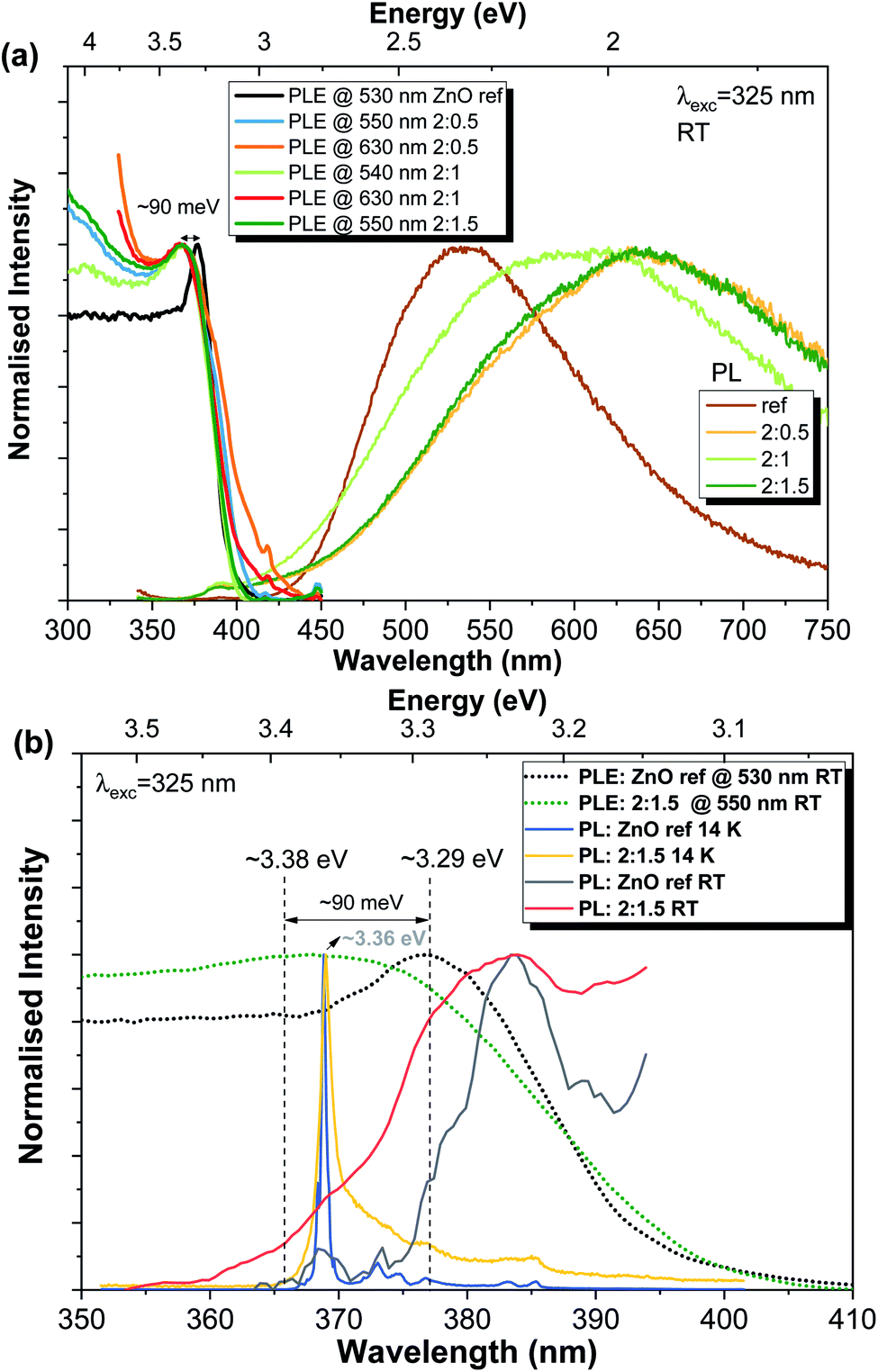 | ||
| Fig. 8 (a) PL/PLE normalised intensity spectra for all analysed samples. (b) Enlarged view of normalised PL/PLE spectra in the high-energy region for the reference and 2:1.5 samples. 14 K and RT PL spectra are displayed for comparison. | ||
The ∼3.38 eV energy value measured for the Al-doped ZnO/ZnAl2O4 samples does not correspond to the value expected for the bandgap energy of the additional ZnAl2O4 phase (Eg ∼ 3.8 eV3,70). Instead, the observed energy shift (ΔEg ∼ 90 meV) should be related to the Burstein–Moss effect, which is commonly observed in heavily doped semiconductors.48,50,94
In n-type semiconductors, this effect occurs when the electrons occupy shallow donor states spread into the conduction band, raising the Fermi level above its minimum. Since the Pauli principle is still obeyed, optical transitions can only occur for higher photon energies to assure the vertical transitions from the valence band to above the Fermi level, which is located inside the conduction band. Thus, a blue-shift of the optical bandgap of the material is observed.48,50 The energy bandgap broadening (ΔEg-MB) is related to the carrier (electron) concentration (ne) and, in the parabolic band approximation, it can be described by the following expression:48–50
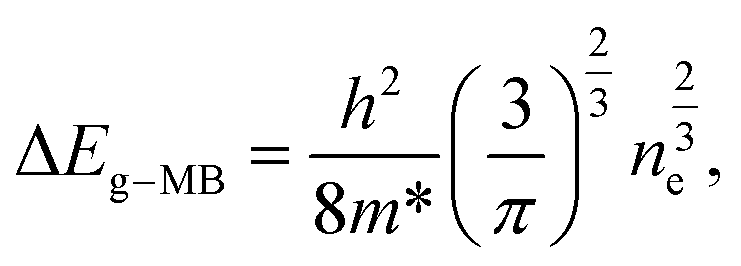 | (1) |
For ZnO, the reported effective mass for electrons is 0.28 m0, where m0 corresponds to the electron mass at rest.50 Considering the energy shift measured in the present Al-doped samples, and assuming that the Burstein–Moss effect is the only phenomenon ruling the bandgap shift, the carrier concentration was estimated and a value of ∼1.82 ×1019 cm−3 was obtained. Since the same energy shift was observed for all the Al-doped samples, it is fair to assume that the carrier concentration is analogous in all of them. The estimated value for the carrier concentration is in line with that in previous reports for Al-doped samples48–50 where the shift in the bandgap energy was seen to correlate well with the measured carrier concentration by only taking into account the Burstein–Moss effect.
Besides the shift in the bandgap energy, Fig. 8(b) also evidences the mirror-like shape of the RT NBE emission when compared with the PLE spectra for both reference and 2:1.5 samples. Moreover, in the case of the ZnO reference sample, the difference between the maxima of the PLE and PL spectra (at RT) agrees well with the value expected for the exciton binding energy (∼60 meV).
4 Conclusions
Al-doped ZnO samples decorated with ZnAl2O4 nanoparticles were produced by mixing ZnO tetrapods grown by flame transport synthesis with an Al-based salt. Al-doping was confirmed by the broadening and shift of the peak position of the NBE recombination at low temperature. For the sample prepared with the highest mixing-ratio (2:1.5), the NBE is peaked at ∼3.36 eV, which is in good agreement with the position expected for the exciton bound to Al (I6 line). Additionally, the peak position of the visible luminescence band shifted towards longer wavelengths (lower energy) with the Al-doping and composite formation. These bands arose from overlapping of multiple recombination channels that may originate from ZnO, ZnAl2O4 and/or from the interface between the two materials. This assumption was confirmed by temperature-dependent PL studies, where a blueshift of the broad band with increasing temperature was identified in the Al-doped ZnO samples decorated with ZnAl2O4, while for the reference sample the band maximum remained unchanged. A different density of the defect centres in each sample, and thus different contributions to the overall visible band with the temperature increase, is the most probable explanation for such behaviour. Moreover, the time-resolved measurements unambiguously revealed the existence of more than one emitting centre overlapped under the broad band observed under steady-state PL conditions, indicating that the broad emission band exhibited distinct spectral components. Upon monitoring all Al-doped samples at the maximum of the visible PL band, PLE spectra revealed a high-energy shift of ∼90 meV of the bandgap when compared with the ZnO reference sample. This shift was associated with the Burstein–Moss effect that occurs in heavily doped semiconductors. Considering the parabolic band approximation and that this effect is the only one ruling the measured shift, a value of ∼1.82 ×1019 cm−3 was estimated for the carrier concentration of the doped ZnO samples.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was developed within the scope of the project i3N, UID/CTM/50025/2019, UIDB/50025/2020 & UIDP/50025/2020, financed by national funds through the FCT/MEC and financially supported by FEDER funds through the COMPETE 2020 Programme and National Funds through FCT – Portuguese Foundation for Science and Technology under project POCI-01-0145-FEDER-028755 The Kiel authors acknowledge the German Research Foundation for the financial support under scheme SFB 677 (C14). LK and NW acknowledge funding by the DFG under scheme CRC1261 and thank Prof. Dr Bettina Lotsch for additional TEM time.References
- R. Niepelt, U. C. Schröder, J. Sommerfeld, I. Slowik, B. Rudolph, R. Möller, B. Seise, A. Csaki, W. Fritzsche and C. Ronning, Nanoscale Res. Lett., 2011, 6, 511 CrossRef PubMed.
- Z. Zhao, W. Lei, X. Zhang, B. Wang and H. Jiang, Sensors, 2010, 10, 1216–1231 CrossRef CAS PubMed.
- O. Lupan, V. Postica, J. Gröttrup, A. K. Mishra, N. H. de Leeuw, J. F. C. Carreira, J. Rodrigues, N. Ben Sedrine, M. R. Correia, T. Monteiro, V. Cretu, I. Tiginyanu, D. Smazna, Y. K. Mishra and R. Adelung, ACS Appl. Mater. Interfaces, 2017, 9, 4084–4099 CrossRef CAS PubMed.
- O. Lupan, F. Schütt, V. Postica, D. Smazna, Y. K. Mishra and R. Adelung, Sci. Rep., 2017, 7, 14715 CrossRef PubMed.
- J. Rodrigues, N. Ben Sedrine, M. R. Correia and T. Monteiro, Mater. Today Chem., 2020, 16, 100243 CrossRef CAS.
- N. R. Shanmugam, S. Muthukumar, A. P. Selvam and S. Prasad, Nanomedicine, 2016, 11, 1345–1358 CrossRef CAS PubMed.
- S. S. Bhat, A. Qurashi and F. A. Khanday, TrAC, Trends Anal. Chem., 2017, 86, 1–13 CrossRef CAS.
- A. Wei, L. Pan and W. Huang, Mater. Sci. Eng., B, 2011, 176, 1409–1421 CrossRef CAS.
- A. Tereshchenko, M. Bechelany, R. Viter, V. Khranovskyy, V. Smyntyna, N. Starodub and R. Yakimova, Sens. Actuators, B, 2016, 229, 664–677 CrossRef CAS.
- J. Rodrigues, A. Pimentel, E. Fortunato, T. Monteiro and F. M. Costa, Phys. Status Solidi, 2018, 215, 1800155 CrossRef.
- A. Pimentel, J. Rodrigues, P. Duarte, D. Nunes, F. M. Costa, T. Monteiro, R. Martins and E. Fortunato, J. Mater. Sci., 2015, 50, 5777–5787 CrossRef CAS.
- C. Karunakaran, V. Rajeswari and P. Gomathisankar, J. Alloys Compd., 2010, 508, 587–591 CrossRef CAS.
- G. Singh, A. Choudhary, D. Haranath, A. G. Joshi, N. Singh, S. Singh and R. Pasricha, Carbon, 2012, 50, 385–394 CrossRef CAS.
- X. Yang, H. Li, T. Li, Z. Li, W. Wu, C. Zhou, P. Sun, F. Liu, X. Yan, Y. Gao, X. Liang and G. Lu, Sens. Actuators, B, 2019, 282, 339–346 CrossRef CAS.
- V. Postica, J. Gröttrup, R. Adelung, O. Lupan, A. K. Mishra, N. H. de Leeuw, N. Ababii, J. F. C. Carreira, J. Rodrigues, N. Ben Sedrine, M. R. Correia, T. Monteiro, V. Sontea and Y. K. Mishra, Adv. Funct. Mater., 2017, 27, 1604676 CrossRef.
- X. Yang, S. Zhang, Q. Yu, L. Zhao, P. Sun, T. Wang, F. Liu, X. Yan, Y. Gao, X. Liang, S. Zhang and G. Lu, Sens. Actuators, B, 2019, 281, 415–423 CrossRef CAS.
- J. Cui, L. Shi, T. Xie, D. Wang and Y. Lin, Sens. Actuators, B, 2016, 227, 220–226 CrossRef CAS.
- G. Lu, J. Xu, J. Sun, Y. Yu, Y. Zhang and F. Liu, Sens. Actuators, B, 2012, 162, 82–88 CrossRef CAS.
- X. Liu, Y. Sun, M. Yu, Y. Yin, B. Du, W. Tang, T. Jiang, B. Yang, W. Cao and M. N. R. Ashfold, Sens. Actuators, B, 2018, 255, 3384–3390 CrossRef CAS.
- J. Rodrigues, D. Smazna, N. Ben Sedrine, E. Nogales, R. Adelung, Y. K. Mishra, B. Mendez, M. R. Correia and T. Monteiro, Nanoscale Adv., 2019, 1, 1516–1526 RSC.
- D. Smazna, J. Rodrigues, S. Shree, V. Postica, G. Neubüser, A. F. Martins, N. Ben Sedrine, N. K. Jena, L. Siebert, F. Schütt, O. Lupan, R. Ahuja, M. R. Correia, T. Monteiro, L. Kienle, Y. Yang, R. Adelung and Y. K. Mishra, Nanoscale, 2018, 10, 10050–10062 RSC.
- T. Gao, Q. Li and T. Wang, Chem. Mater., 2005, 17, 887–892 CrossRef CAS.
- B. K. Meyer, H. Alves, D. M. Hofmann, W. Kriegseis, D. Forster, F. Bertram, J. Christen, A. Hoffmann, M. Straßburg, M. Dworzak, U. Haboeck and A. V. Rodina, Phys. Status Solidi, 2004, 241, 231–260 CrossRef CAS.
- U. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S.-J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- C. F. Klingshirn, A. Waag, A. Hoffmann and J. Geurts, Zinc Oxide: From Fundamental Properties Towards Novel Applications, Springer, 1st edn, 2010 Search PubMed.
- Y. Gu, I. L. Kuskovsky, M. Yin, S. O'Brien and G. F. Neumark, Appl. Phys. Lett., 2004, 85, 3833–3835 CrossRef CAS.
- M. Yin, Y. Gu and I. Kuskovsky, J. Am. Chem. Soc., 2004, 126, 6206–6207 CrossRef CAS PubMed.
- V. M. Harutunian, H. L. Margarian, V. A. Melicksetian and J. R. Panossian, J. Phys.: Condens. Matter, 1989, 1, 847–854 CrossRef CAS.
- A. B. Djurišić, Y. H. Leung, K. H. Tam, Y. F. Hsu, L. Ding, W. K. Ge, Y. C. Zhong, K. S. Wong, W. K. Chan, H. L. Tam, K. W. Cheah, W. M. Kwok and D. L. Phillips, Nanotechnology, 2007, 18, 095702 CrossRef.
- D. Li, Y. H. Leung, A. B. Djurišić, Z. T. Liu, M. H. Xie, S. L. Shi, S. J. Xu and W. K. Chan, Appl. Phys. Lett., 2004, 85, 1601–1603 CrossRef CAS.
- A. B. Djurišić, Y. H. Leung, K. H. Tam, L. Ding, W. K. Ge, H. Y. Chen and S. Gwo, Appl. Phys. Lett., 2006, 88, 103107 CrossRef.
- J. Fallert, R. Hauschild, F. Stelzl, A. Urban, M. Wissinger, H. Zhou, C. Klingshirn and H. Kalt, J. Appl. Phys., 2007, 101, 073506 CrossRef.
- M. Biswas, Y. S. Jung, H. K. Kim, K. Kumar, G. J. Hughes, S. Newcomb, M. O. Henry and E. McGlynn, Phys. Rev. B, 2011, 83, 235320 CrossRef.
- M. Hoppe, O. Lupan, V. Postica, N. Wolff, V. Duppel, L. Kienle, I. Tiginyanu and R. Adelung, Phys. Status Solidi A, 2018, 215, 1700772 CrossRef.
- Y. K. Mishra, G. Modi, V. Cretu, V. Postica, O. Lupan, T. Reimer, I. Paulowicz, V. Hrkac, W. Benecke, L. Kienle and R. Adelung, ACS Appl. Mater. Interfaces, 2015, 7, 14303–14316 CrossRef CAS PubMed.
- T. Lu, Z. Ma, C. Du, Y. Fang, H. Wu, Y. Jiang, L. Wang, L. Dai, H. Jia, W. Liu and H. Chen, Sci. Rep., 2014, 4, 6131 CrossRef CAS PubMed.
- T. P. Bartel, M. R. Wagner, U. Haboeck, A. Hoffmann, C. Neumann, S. Lautenschläger, J. Sann and B. K. Meyer, in Zinc Oxide Materials and Devices III, San Jose, CA, USA, 2008, vol. 6895, p. 689502 Search PubMed.
- C. Klingshirn, Phys. Status Solidi, 2007, 244, 3027–3073 CrossRef CAS.
- T. Monteiro, C. Boemare, M. J. Soares, E. Rita and E. Alves, J. Appl. Phys., 2003, 93, 8995 CrossRef CAS.
- T. Monteiro, M. J. Soares, A. Neves, S. Pereira, M. R. Correia, M. Peres, E. Alves, D. Rogers, F. Teherani, V. Munoz-SanJose, T. Trindade and A. Pereira, J. Non-Cryst. Solids, 2006, 352, 1453–1456 CrossRef CAS.
- A. Janotti and C. G. Van De Walle, J. Cryst. Growth, 2006, 287, 58–65 CrossRef CAS.
- X. J. Wang, L. S. Vlasenko, S. J. Pearton, W. M. Chen and I. A. Buyanova, J. Phys. D: Appl. Phys., 2009, 42, 175411 CrossRef.
- R. Dingle, Phys. Rev. Lett., 1969, 23, 579–581 CrossRef CAS.
- N. Y. Garces, L. Wang, L. Bai, N. C. Giles, L. E. Halliburton and G. Cantwell, Appl. Phys. Lett., 2002, 81, 622–624 CrossRef CAS.
- M. Liu, A. H. Kitai and P. Mascher, J. Lumin., 1992, 54, 35–42 CrossRef CAS.
- M. A. Reshchikov, J. Q. Xie, B. Hertog and A. Osinsky, J. Appl. Phys., 2008, 103, 103514 CrossRef.
- Y. N. Chen, S. J. Xu, C. C. Zheng, J. Q. Ning, F. C. C. Ling, W. Anwand, G. Brauer and W. Skorupa, Appl. Phys. Lett., 2014, 105, 041912 CrossRef.
- B. E. Sernelius, K.-F. Berggren, Z.-C. Jin, I. Hamberg and C. G. Granqvist, Phys. Rev. B, 1988, 37, 10244–10248 CrossRef CAS PubMed.
- H. Lee, S. Lau, Y. Wang, K. Tse, H. Hng and B. Tay, J. Cryst. Growth, 2004, 268, 596–601 CrossRef CAS.
- J. G. Lu, S. Fujita, T. Kawaharamura, H. Nishinaka, Y. Kamada, T. Ohshima, Z. Z. Ye, Y. J. Zeng, Y. Z. Zhang, L. P. Zhu, H. P. He and B. H. Zhao, J. Appl. Phys., 2007, 101, 083705 CrossRef.
- A. Mohanta, J. G. Simmons, G. Shen, S. M. Kim, P. Kung and H. O. Everitt, J. Lumin., 2019, 211, 264–270 CrossRef CAS.
- J. Rodrigues, T. Holz, R. Fath Allah, D. Gonzalez, T. Ben, M. R. Correira, T. Monteiro and F. M. Costa, Sci. Rep., 2015, 5, 10783 CrossRef CAS PubMed.
- K. H. Tam, C. K. Cheung, Y. H. Leung, A. B. Djurišić, C. C. Ling, C. D. Beling, S. Fung, W. M. Kwok, W. K. Chan, D. L. Phillips, L. Ding and W. K. Ge, J. Phys. Chem. B, 2006, 110, 20865–20871 CrossRef CAS PubMed.
- A. B. Djurisić and Y. H. Leung, Small, 2006, 2, 944–961 CrossRef PubMed.
- D. Tainoff, B. Masenelli, P. Melinon, A. Belsky, G. Ledoux, D. Amans, C. Dujardin, N. Fedorov and P. Martin, J. Lumin., 2009, 129, 1798–1801 CrossRef CAS.
- J. Grabowska, A. Meaney, K. K. Nanda, J.-P. Mosnier, M. O. Henry, J.-R. Duclère and E. McGlynn, Phys. Rev. B, 2005, 71, 115439 CrossRef.
- J. Li, S. Srinivasan, G. N. He, J. Y. Kang, S. T. Wu and F. A. Ponce, J. Cryst. Growth, 2008, 310, 599–603 CrossRef CAS.
- V. A. L. Roy, A. B. Djurišić, W. K. Chan, J. Gao, H. F. Lui and C. Surya, Appl. Phys. Lett., 2003, 83, 141–143 CrossRef CAS.
- J. Rodrigues, A. J. S. Fernandes, T. Monteiro and F. M. Costa, CrystEngComm, 2019, 21, 1071–1090 RSC.
- C. Jagadish and S. Pearton, Zinc Oxide Bulk, Thin Films and Nanostructures, Elsevier, 2006 Search PubMed.
- Y. K. Mishra, S. Kaps, A. Schuchardt, I. Paulowicz, X. Jin, D. Gedamu, S. Freitag, M. Claus, S. Wille, A. Kovalev, S. N. Gorb and R. Adelung, Part. Part. Syst. Charact., 2013, 30, 775–783 CrossRef CAS.
- T. Reimer, I. Paulowicz, R. Röder, S. Kaps, O. Lupan, S. Chemnitz, W. Benecke, C. Ronning, R. Adelung and Y. K. Mishra, ACS Appl. Mater. Interfaces, 2014, 6, 7806–7815 CrossRef CAS PubMed.
- J. Gröttrup, I. Paulowicz, A. Schuchardt, V. Kaidas, S. Kaps, O. Lupan, R. Adelung and Y. K. Mishra, Ceram. Int., 2016, 42, 8664–8676 CrossRef.
- P. A. Midgley and A. S. Eggeman, IUCrJ, 2015, 2, 126–136 CrossRef CAS PubMed.
- D. Levy, A. Pavese, A. Sani and V. Pischedda, Phys. Chem. Miner., 2001, 28, 612–618 CrossRef CAS.
- C. J. Sun, J. W. Yang, Q. Chen, M. Asif Khan, T. George, P. Chang-Chien and S. Mahajan, Appl. Phys. Lett., 1996, 68, 1129–1131 CrossRef CAS.
- R. Cuscó, E. Alarcón-Lladó, J. Ibáñez, L. Artús, J. Jiménez, B. Wang and M. Callahan, Phys. Rev. B, 2007, 75, 165202 CrossRef.
- S. López, A. H. Romero, P. Rodríguez-Hernández and A. Muñoz, Phys. Rev. B, 2009, 79, 214103 CrossRef.
- A. Chopelas and A. M. Hofmeister, Phys. Chem. Miner., 1991, 18, 279–293 CrossRef CAS.
- X. Zhao, L. Wang, X. Xu, X. Lei, S. Xu and F. Zhang, AIChE J., 2012, 58, 573–582 CrossRef CAS.
- M. Strassburg, A. Rodina, M. Dworzak, U. Haboeck, I. L. Krestnikov, A. Hoffmann, O. Gelhausen, M. R. Phillips, H. R. Alves, A. Zeuner, D. M. Hofmann and B. K. Meyer, Phys. Status Solidi, 2004, 241, 607–611 CrossRef CAS.
- S. L. Shi, G. Q. Li, S. J. Xu, Y. Zhao and G. H. Chen, J. Phys. Chem. B, 2006, 110, 10475–10478 CrossRef CAS PubMed.
- H. Zeng, G. Duan, Y. Li, S. Yang, X. Xu and W. Cai, Adv. Funct. Mater., 2010, 20, 561–572 CrossRef CAS.
- M. A. Reshchikov, V. Avrutin, N. Izyumskaya, R. Shimada, H. Morkoç and S. W. Novak, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct., 2009, 27, 1749 CrossRef CAS.
- D. Byrne, F. Herklotz, M. O. Henry and E. McGlynn, J. Phys.: Condens. Matter, 2012, 24, 215802 CrossRef CAS PubMed.
- P. Dahan, V. Fleurov, P. Thurian, R. Heitz, A. Hoffmann and I. Broser, J. Phys.: Condens. Matter, 1998, 10, 2007–2019 CrossRef CAS.
- Y. K. Mishra and R. Adelung, Mater. Today, 2017, 21, 631–651 CrossRef.
- T. Nobis, E. M. Kaidashev, A. Rahm, M. Lorenz and M. Grundmann, Phys. Rev. Lett., 2004, 93, 103903 CrossRef PubMed.
- S. S. Kurbanov, S. Z. Urolov, Z. Shaymardanov and T. W. Kang, J. Lumin., 2018, 197, 159–163 CrossRef CAS.
- R. B. M. Cross, M. M. De Souza and E. M. Sankara Narayanan, Nanotechnology, 2005, 16, 2188–2192 CrossRef CAS PubMed.
- S. A. Studenikin, N. Golego and M. Cocivera, J. Appl. Phys., 1998, 84, 2287 CrossRef CAS.
- M. Gomi, N. Oohira, K. Ozaki and M. Koyano, Jpn. J. Appl. Phys., Part 1, 2003, 42, 481–485 CrossRef CAS.
- H. J. Fan, R. Scholz, F. M. Kolb, M. Zacharias, U. Gosele, F. Heyroth, C. Eisenschmidt, T. Hempel and J. Christen, Appl. Phys. A, 2004, 79, 1895–1900 CrossRef CAS.
- A. Pimentel, D. Nunes, P. Duarte, J. Rodrigues, F. M. Costa, T. Monteiro, R. Martins and E. Fortunato, J. Phys. Chem. C, 2014, 118, 14629–14639 CrossRef CAS.
- A. Janotti and C. G. Van De Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165202 CrossRef.
- P. Erhart, K. Albe and A. Klein, Phys. Rev. B, 2006, 73, 205203 CrossRef.
- A. Janotti and C. G. Van De Walle, Rep. Prog. Phys., 2009, 72, 126501 CrossRef.
- A. Zubiaga, F. Tuomisto, F. Plazaola, K. Saarinen, J. A. Garcia, J. F. Rommeluere, J. Zuñiga-Pérez and V. Muñoz-Sanjosé, Appl. Phys. Lett., 2005, 86, 042103 CrossRef.
- M. Wang, K. E. Lee, S. H. Hahn, E. J. Kim, S. Kim, J. S. Chung, E. W. Shin and C. Park, Mater. Lett., 2007, 61, 1118–1121 CrossRef CAS.
- S. V. Motloung, P. Kumari, L. F. Koao, T. E. Motaung, T. T. Hlatshwayo and M. J. Mochane, Mater. Today Commun., 2018, 14, 294–301 CrossRef CAS.
- S. F. Wang, F. Gu, M. K. Lü, X. F. Cheng, W. G. Zou, G. J. Zhou, S. M. Wang and Y. Y. Zhou, J. Alloys Compd., 2005, 394, 255–258 CrossRef CAS.
- P. Y. Yu and M. Cardona, Fundamentals of Semiconductors, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005 Search PubMed.
- J. I. Pankove, Optical Processes in Semiconductors, Dover Publication, Inc., 1971 Search PubMed.
- E. Burstein, Phys. Rev., 1954, 93, 632–633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00730j |
| This journal is © The Royal Society of Chemistry 2020 |