
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH Transitions and electrochemical behavior during the synthesis of iron oxide nanoparticles with gas-diffusion electrodes†
Rutely C.
Burgos-Castillo
ab,
Arturo
Garcia-Mendoza
c,
Yolanda
Alvarez-Gallego
ad,
Jan
Fransaer
de,
Mika
Sillanpää
b and
Xochitl
Dominguez-Benetton
 *ad
*ad
aSeparation and Conversion Technologies, Flemish Institute for Technological Research (VITO), Boeretang 200, 2400, Mol, Belgium. E-mail: xoch@vito.be; rcbcastillo@gmail.com
bDepartment of Green Chemistry, School of Engineering Science, Lappeenranta University of Technology, Sammonkatu 12, FI-50130 Mikkeli, Finland
cDepartamento de Química Analítica, Facultad de Química, Universidad Nacional Autónoma de México, Av. Universidad 3000, C.U, Mexico City, 04510, Mexico
dSIM vzw, Technologiepark 935, BE-9052 Zwijnaarde, Belgium
eDepartment of Materials Engineering, Katholieke Universiteit Leuven (KU Leuven), Kasteelpark Arenberg 44 - bus 2450, B-3001 Leuven, Belgium
First published on 13th February 2020
Abstract
Gas diffusion electrocrystallization (GDEx) was explored for the synthesis of iron oxide nanoparticles (IONPs). A gas-diffusion cathode was employed to reduce oxygen, producing hydroxyl ions (OH−) and oxidants (H2O2 and HO2−), which acted as reactive intermediates for the formation of stable IONPs. The IONPs were mainly composed of pure magnetite. However, their composition strongly depended on the presence of a weak acid, i.e., ammonium chloride (NH4Cl), and on the applied electrode potential. Pure magnetite was obtained due to the simultaneous action of H2O2 and the buffer capacity of the added NH4Cl. Magnetite and goethite were identified as products under different operating conditions. The presence of NH4Cl facilitated an acid–base reaction and, in some cases, led to cathodic deprotonation, forming a surplus of hydrogen peroxide, while adding the weak acid promoted gradual changes in the pH by slightly enhancing H2O2 production when increasing the applied potential. This also resulted in smaller average crystallite sizes as follows: 20.3 ± 0.6 at −0.350 V, 14.7 ± 2.1 at −0.550 and 12.0 ± 2.0 at −0.750 V. GDEx is also demonstrated to be a green, effective, and efficient cathodic process to recover soluble iron to IONPs, being capable of removing >99% of the iron initially present in the solution.
Introduction
Nanoparticle production methods that are economical, clean, safe, easy-to-implement, and upscalable are an active research subject.1–3 A key disadvantage of most alternatives4–7 is the use of non-aqueous molecular solvents and reducing and capping agents, which are often environmentally hazardous and added in excess.8,9 Moreover, most methods involve several processing steps, often including different upstream and downstream units, which can appear to be suitable at the lab scale but are challenging to implement industrially.5,10–12The development of environmentally friendly synthesis methods has gained particular attention,13–17 intensifying the interest in electrochemistry as a synthetic platform under mild conditions.18–25 In most instances, known electrochemical approaches for the synthesis of nanoparticles26–28 involve the growth of films onto inert substrates, mostly by one-step electrodeposition, at working temperatures between 70° and 90 °C. A compilation of pathways and methods for the electrochemical formation of magnetite nanoparticles is available in the scientific literature.23,26,29–32
In the present study, a new electrochemical method, called gas-diffusion electrocrystallization (GDEx),14 is employed for the formation of iron oxide nanoparticles (IONPs)33 from a soluble iron precursor (Fe2+). A gas diffusion cathode is used for the electroreduction of oxygen (O2) contained in a gas phase (air) which, in turn, drives the precipitation of crystalline IONPs at the electrochemical interface.
The oxygen reduction reaction (ORR) using carbon-based gas-diffusion electrodes has been thoroughly investigated and well demonstrated in acidic and alkaline media. Reactions (1) and (2) show the two-electron ORR of O2 to H2O2 or hydroperoxide ions (HO2−)41 and occur under acidic and alkaline conditions, respectively. Besides, under alkaline conditions, HO2− can be reduced to OH−via reaction (3).
| O2 + 2H+ + 2e− → H2O2 | (1) |
| O2 + H2O + 2e− → HO2− + OH− | (2) |
| HO2− + H2O + 2e− → 3OH− | (3) |
Throughout this work, both chemical species, i.e., H2O2 and HO2−, will be referred to as H2O2, granting that at more alkaline pHs HO2− will be the most abundant species (pKa = 11.65).24,34 The effective dissolution of O2 gas is the limiting step of this process, but with gas-diffusion electrodes this is overcome by the high mass transport rates at the triple-phase boundary.35
The present work differs from previous electrochemical methods in various ways. First, it employs a gas-diffusion cathode to drive the formation of IONPs. Thus, the precipitating agents (OH−) and H2O2 are produced in situ via the ORR. The synthesis takes place in an aqueous solution with a supporting electrolyte containing no other additives or surfactants, over a range of applied potentials between −0.350 V and −0.750 V versus a reference Ag/AgCl (saturated KCl) electrode. Furthermore, our method does not require a high temperature, and it involves only one synthesis step. An understanding of GDEx is incipient. Thus, the present research attempts to reveal relevant phenomenological and mechanistic events to establish it as a prominent green route to fabricating nanoparticles.
The first purpose of this work was to identify pH transitions imposed by the ORR products (i.e., OH− and H2O2) in the catholyte bulk, as well as the electrochemical behaviour (current–potential response) of the system. In addition to investigating the unbuffered system, the effect of adding a weak acid (WA) was studied, hypothesizing that the WA would serve as a co-catalyst for the ORR and, hence, the formation of H2O2. Our key aim was to study this in the presence of a soluble iron precursor, for its recovery to NPs. Furthermore, the correlation between the yield of H2O2 and the recovery rate of iron upon the addition of a WA (i.e., NH4Cl) was studied. Ultimately, the influence of adding a WA on the production of NPs was assessed.
Experimental
Materials and methods
The reference electrode was a Ag/AgCl electrode (satd. KCl, Biologic, model: R-XR300). All potentials measured in this work are reported versus its potential. The anode (counter electrode) was a platinum disc of a 10 cm2 projected surface area, which was laser welded onto a titanium plate. This anode generated O2 or Cl2, which had a negligible influence on the electrochemical (cathodic) process and products of interest. The cathode was a cold-rolled gas-diffusion electrode (VITO CORE®) composed of a hydrophilic active carbon-polytetrafluoroethylene layer (C-PTFE with a C to PTFE weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) pressed in between stainless steel mesh serving as a current collector and a hydrophobic PTFE layer through which oxygen percolated.15 The active carbon was Norit® SX 1G (Cabot, Europe). The projected surface area of the cathode was 10 cm2.15 Air was supplied (as the source of O2 gas) at 200 mL min−1 using a mass flow controller at an overpressure of 30 mbar (g).
:20) pressed in between stainless steel mesh serving as a current collector and a hydrophobic PTFE layer through which oxygen percolated.15 The active carbon was Norit® SX 1G (Cabot, Europe). The projected surface area of the cathode was 10 cm2.15 Air was supplied (as the source of O2 gas) at 200 mL min−1 using a mass flow controller at an overpressure of 30 mbar (g).
 | (4) |
 | (5) |
Results and discussion
Control electrolytes without Fe2+
The aim of this part of the study was two-fold: (i) to establish the capability of the process to accumulate H2O2 in the absence of metal ions (i.e., Fe2+) and (ii) to identify the consumed charge to transform the pH from acidic (2.7–3) to alkaline conditions (pH 8–12). Three different electrolyte compositions were studied at applied potentials of −0.350 V (a); −0.550 V (b); and −0.750 V (c). The conditions of the control experiments are outlined in Table 1: 0.14 M NaCl (assays I-a, I-b, and I-c), 0.14 M NaCl + 10 mM NH4Cl (assays II-a, II-b, and II-c), 0.14 M NaCl + 30 mM NH4Cl (assays III-a, III-b, and III-c). For comparison purposes between experiments, a one-hour period was chosen to measure the concentration of H2O2, which was then normalized to the amount of charge consumed for each experiment from which the corresponding electrolyte samples were extracted. In all instances, this allowed sufficient evolution to reach the target pH.| Assay | Electrolyte | Applied potential (V) |
|---|---|---|
| I-a | 140 mM NaCl | −0.350 |
| I-b | 140 mM NaCl | −0.550 |
| I-c | 140 mM NaCl | −0.750 |
| II-a | 140 mM NaCl + 10 mM NH4Cl | −0.350 |
| II-b | 140 mM NaCl + 10 mM NH4Cl | −0.550 |
| II-c | 140 mM NaCl + 10 mM NH4Cl | −0.750 |
| III-a | 140 mM NaCl + 30 mM NH4Cl | −0.350 |
| III-b | 140 mM NaCl + 30 mM NH4Cl | −0.550 |
| III-c | 140 mM NaCl + 30 mM NH4Cl | −0.750 |
pH evolution
To assess the evolution of pH and accumulation of H2O2—during the electroreduction of O2—the applied potential was varied for each experiment, as stated above. In addition, two different concentrations of NH4Cl (pKa = 9.26) were tested and compared to the case without the WA. Given the theory of cathodic deprotonation of WAs,37,39,40 it was expected that the addition of NH4Cl would induce a co-catalytic effect on the electroreduction of O2 to H2O2.41–43Fig. 1 depicts the evolution of pH against either time or the charge consumed for the control experiments using NaCl as the background electrolyte, i.e., without addition of the metal precursor. The pH increase is attributed to the generated OH− ions in the active cathode layer during the electrolytic process.29 Such ions diffuse towards the bulk electrolyte, increasing both the local pH at the electrochemical interface and the pH of the bulk liquid. The plots depicted for systems I-a, I-b, and I-c are comparable to the chemical titration of a strong acid, i.e., HCl with a strong base (OH−). The OH− ions formed in the GDE first react with the H3O+ ions generated from the dissociation of the HCl added to adjust the initial pH (∼3) of the solution. Notably, the pH evolved faster as the applied potential increased, i.e., at either −0.55 V or −0.750 V (I-b and I-c), and as the current density increased correspondingly.
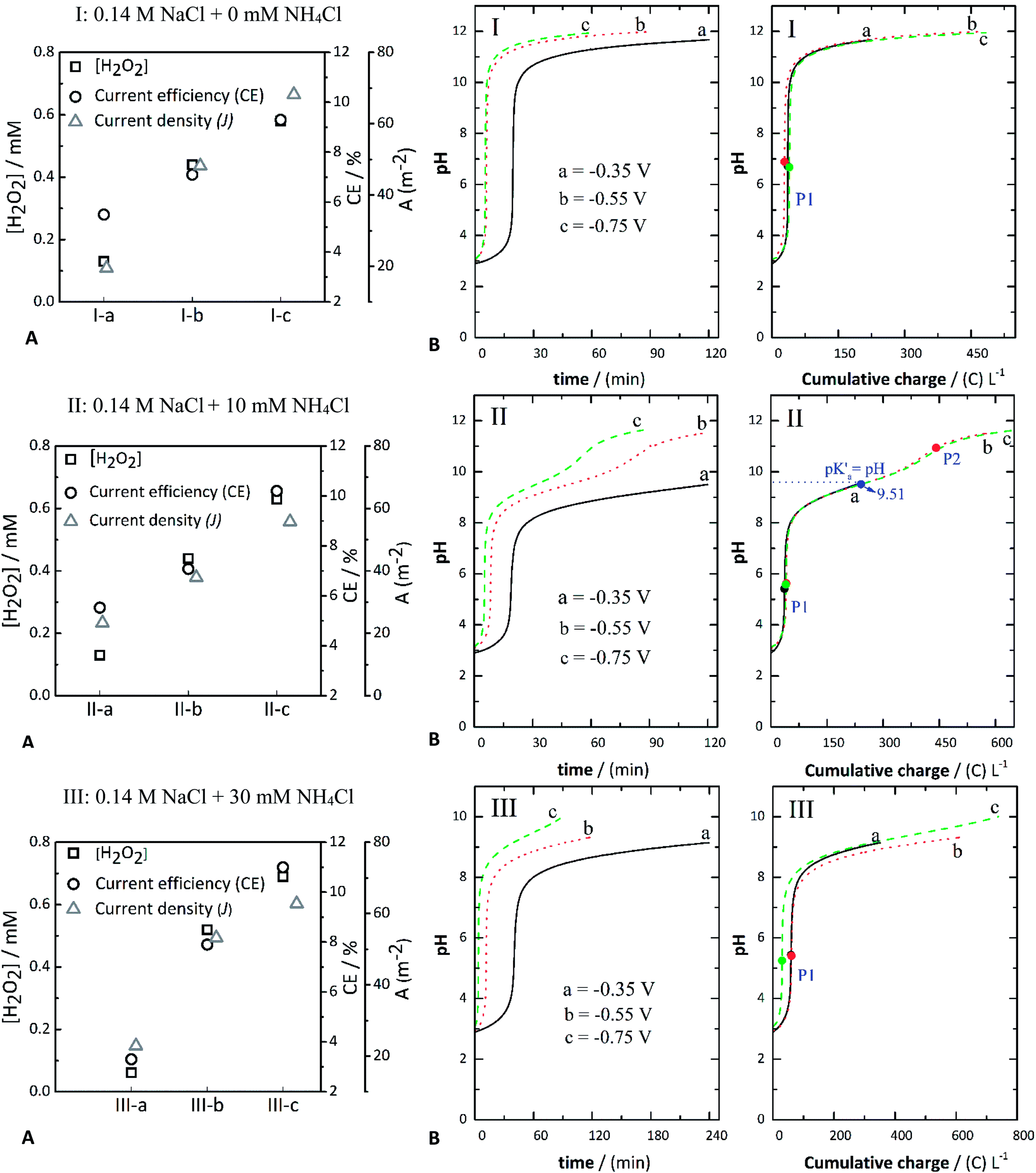 | ||
| Fig. 1 Concentration of H2O2, current efficiency (CE%) and current density (J) (A) and evolution of pH (B) against either time or charge consumed in: 0.14 M NaCl (I-a, I-b, and I-c), 0.14 M NaCl + 10 mM NH4Cl (II-a, II-b, and II-c), and 0.14 M NaCl + 30 mM NH4Cl (III-a, III-b, and III-c) at different applied potentials of −0.350 V (a), −0.550 V (b), and −0.750 V (c). | ||
The presence of NH4Cl influenced the evolution of pH. Its effect can be observed by comparing the systems without (series of assays “I,” Fig. 1(I)) and with NH4Cl (Fig. 2(II) and (III)). When NH4Cl is present, two buffer regions can be differentiated, corresponding to stepwise neutralization of the different protonated species present in the electrolyte (i.e., HCl and NH4Cl). The equivalence point of each region is identified as P1 and P2, respectively, in the plot of pH against charge (Fig. 1). In Fig. 1, the equivalence points are marked with black, red, or green circles. Another characteristic point is the pH value halfway (blue marker circles denoted as pK′a) between the charge consumed to go from P1 to P2, in which the OH− equivalents that have reacted equal one-half of the WA equivalents initially present in the solution. In other words, at this point the concentration of the WA (NH4+) and its conjugate base (NH3) should be equal; thus, the corresponding pH should be the pKa of the neutralized acid, in this case, NH4+.
Therefore, the value of pH at this point should coincide with the theoretical pKa of NH4+ (∼9.2). However, the experimental value lies around pK′a = 9.51 (Fig. 1(II)). These pK′a values vary mainly due to reactions that occur at the same time as the equilibrium of NH4+, e.g., reactions (7) and (8). The pH versus cumulative charge curves for the case when 30 mM NH4Cl is added are shown in Fig. 1(III). As more NH4Cl is present, the buffer region becomes larger. In contrast, the plateau length decreases when increasing the potential (from −0.35 V and −0.75 V) at a given concentration of NH4Cl. It should be noted that the pH values at P1 seem to be influenced by the applied potential, decreasing at more negative applied potentials. The more negative potentials increased the production of OH− ions. The pH versus cumulative charge curves for the case when 30 mM NH4Cl is added are shown in Fig. 1(III). As more NH4Cl is present, the buffer region becomes larger. In contrast, the plateau length decreases when increasing the potential (from −0.35 V and −0.75 V) at a given concentration of NH4Cl. It should be noted that the pH values at P1 seem to be influenced by the applied potential, decreasing at more negative applied potentials.
The experimental pH curves versus time were fitted using a polynomial model to elucidate the reactions occurring in the cathodic compartment and that are likely to explain the titration curves presented in Fig. 1. These fitted curves are depicted in Fig. S4 (ESI†). As a result, it was confirmed that reaction (6) is involved in the reduction of oxygen at the cathode.
| O2 + 2H2O + 2e− ⇌ 2HO− + H2O2 | (6) |
| H3O+ + OH− ⇌ 2H2O | (7) |
| NH4+ + OH− ⇌ NH3 + H2O | (8) |
From the experimental data, the following phenomena can be highlighted: (1) Reactions (7) and (8) occurred in sequence during the electrolytic ORR when NH4Cl was added. In its absence, only the ORR of reaction (6) took place, resulting in an OH− titration. Therefore in the assays containing only NaCl as the background electrolyte (Fig. 1(I)), only one equivalence point was expected (circle markers in Fig. 1(I)), denoted as P1. The pH value was thus established by the action of water acting as an amphoteric molecular solvent, meaning that at P1 the number of moles of H3O+ and of OH− ions is equal. (2) The pH value expected in the absence of NH4Cl was around seven, in close agreement with the values calculated from the experimental pH data described in Fig. 1, being 6.72 at −0.350 V (assay I-a), 6.89 at −0.550 V (assay I-b), and 6.67 at −0.750 V (assay I-c). (3) In the assays, including NaCl + NH4Cl, as previously discussed, two equivalence points were identified and marked as P1 and P2 (Fig. 1(II)). (4) The pH values expected at P1 (around 5.63) when adding 10 mM NH4Cl (Fig. 1(II)) are somewhat in agreement with the values calculated, being around 5.42 ± 0.01 at −0.35 V, 5.63 ± 0.02 at −0.55 V, and 5.58 ± 0.11 at −0.75 V. These values are only slightly divergent from the values expected. (5) The pH values fitted for the different experiments shown in Fig. 1(III) were also in close agreement. The pH anticipated at P1 was 5.39, and the values obtained from the experimental curves were 5.44 ± 0.01 at −0.35 V, 5.41 ± 0.02 at −0.55 V, and 5.25 ± 0.11 at −0.75 V.
| H2O2 + 2H+ + 2e− → 2H2O | (9) |
Overall, there was a slight increase in the H2O2 concentration, with an increasing concentration of the WA at a constant applied potential that also corresponded to a small increase in the current density. For instance, assays with 10 mM NH4Cl (II-b and III-b) achieved a CE% of 7.1% and 7.9% and a current density of around 38 and 53 A m−2, respectively. The CE% for assays with 30 mM NH4Cl (I-c, II-c, and III-c) was around 9%, 10%, and 11%, whereas the current density was approximately 68, 56, and 63 A m−2, respectively. These results show the co-catalytic effect of NH4Cl via the cathodic deprotonation process, resulting in a higher production rate and concentration of hydrogen peroxide (Fig. 1A).
Current–potential curves
Current–potential curves were recorded by LSV at the beginning (Fig. 2) and the end (data not shown here) of every experiment to investigate the effect of NH4Cl addition. The inset in Fig. 2 shows the full current–potential patterns. The shape of these curves had the same pattern when the experiments were repeated. During the LSV experiments (Fig. 2), the polarization potential was varied from 0 V to −0.75 V using the following background electrolytes: 0.14 M NaCl (black solid line), 0.14 M NaCl + 10 mM NH4Cl (red solid line), and 0.14 M NaCl + 30 mM NH4Cl (green solid line). In this context, J1, J2, and J3 denote the cathodic current density measured for 0 mM, 10 mM, and 30 mM NH4Cl in NaCl solutions, respectively. These LSV results recorded at the start of the experiments suggest that the addition of NH4Cl resulted in a consistent small increase in cathodic current density. This can be associated with an increase in the rate of the electrochemical reactions taking place, which in this case led to a higher production of H2O2 in some assays (II-b, III-b, I-c, II-c, and III-c), which is in good agreement with the slightly increasing concentration of H2O2 reported in Fig. 1A.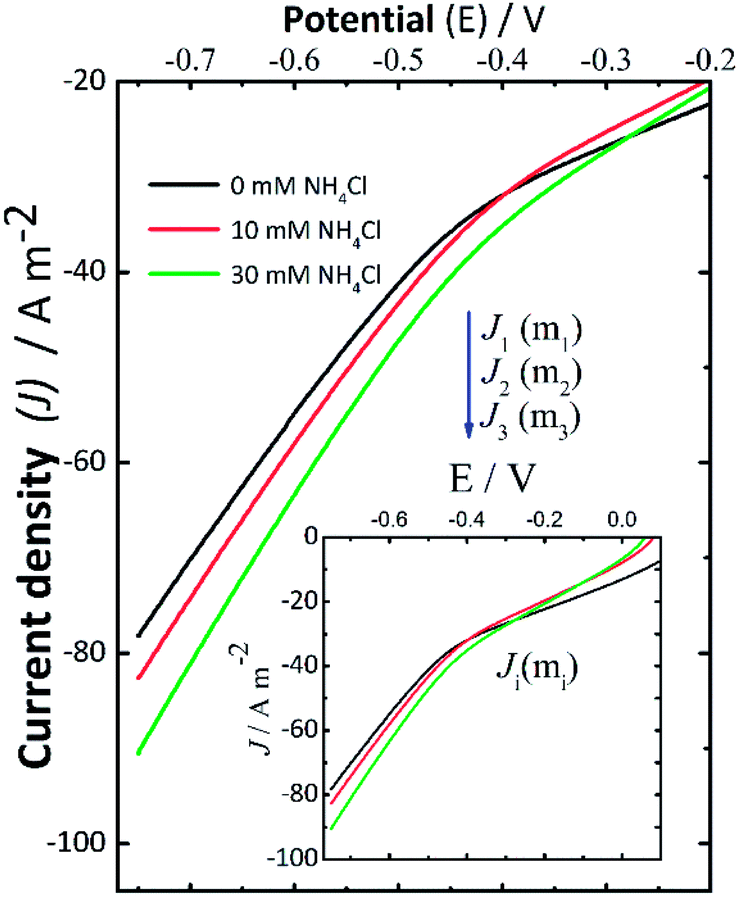 | ||
| Fig. 2 Current–potential curves recorded before starting the electrolysis by linear scan voltammetry (LSV) at a scan rate of 10 mV s−1 varying the polarization potential from open circuit potential to −0.75 V vs. Ag/AgCl (satd. KCl) in different background electrolytes: 0.14 M NaCl (black solid line), 0.14 M NaCl + 10 mM NH4Cl (red solid line), and 0.14 M NaCl + 30 mM NH4Cl (green solid line). The inset shows the full current–potential curves. | ||
When the pH reached a given alkaline pH value, the increase of current was not obvious as in the LSV recorded before applying the desired potential to start the electrolyses. Therefore, these changes in the behavior of the LSV curves suggest the occurrence of cathodic deprotonation. Thus, it could be inferred from the LSV curves (Fig. 2) recorded at the start of the experiments that NH4+ ions should be responsible for the current increase as the potential in the LSV curves became more negative during these assays. This effect is not present in LSV curves recorded at the end of experiments (not shown here) wherein the pH is high, and thus protonated species are presumably depleted. The increase in H2O2 formation thus could be the consequence of the cathodic deprotonation process, as hypothesized. In view of this theory, the protonated species of a buffer (e.g., NH4Cl) will lose protons, causing a dependence of the produced current on the concentration of the WA. It should be noted that the electroactive species, electrochemically reduced, are not protons themselves but protonated species, which in turn depend on the dissociation constant of the species and the pH of the solution.40,42 Under these experimental conditions, the cathodic deprotonation of NH4+ ions occurs mostly below and up to a pH close to the pKa of NH4Cl.
On the other hand, from the modelling of the pH curves presented in Fig. 1 (shown in Fig. S4, ESI†), the amount of NH4Cl that seemed to be involved in reaction (8) was lower than the added concentration of either 10 mM or 30 mM in assays II-b, II-c, and III-c. This parameter was only determined in those assays where oxygen was reduced for a sufficient time to effectively induce the reaction between NH4+ added and the OH− ions formed, to produce the second equivalence point (P2).
Formation of IONPs
In general, irrespective of the approach used to form the iron oxides of interest, a supersaturation condition is a prerequisite for the precipitation of solids. In turn, such a condition can tailor their size, phase, and other properties.28 Thus, a series of electrolysis experiments was performed to prove that the products from the two-electron pathway of the ORR could be used to establish the supersaturation of the solution, its alkalinity, and the necessary redox conditions to obtain stable crystals of iron oxides like magnetite. Besides, the composition of the electrolyte can also influence the final products when they contain inorganic species like chloride ions.28 FeCl2 was used as the source of Fe2+ ions in 0.14 M NaCl to investigate the behaviour of this process, following our previous work.33 In addition, in separate experiments, the effect of two different concentrations of NH4Cl i.e., 10 mM and 30 mM, respectively, was investigated. These assays are listed in Table 2 and depicted in Fig. 3. The rationale behind this is that the presence of the WA can accelerate the production of H2O2via the cathodic deprotonation mechanism. As shown in our previous research, this should also result in the formation of smaller particles.44| Assay | Electrolyte | Applied potential (V) |
|---|---|---|
| IV-a | 140 mM NaCl + 0 mM NH4Cl + 2.25 mM Fe(II) | −0.350 |
| IV-b | 140 mM NaCl + 0 mM NH4Cl + 2.25 mM Fe(II) | −0.550 |
| IV-c | 140 mM NaCl + 0 mM NH4Cl + 2.25 mM Fe(II) | −0.750 |
| V-a | 140 mM NaCl + 10 mM NH4Cl + 2.25 mM Fe(II) | −0.350 |
| V-b | 140 mM NaCl + 10 mM NH4Cl + 2.25 mM Fe(II) | −0.550 |
| V-c | 140 mM NaCl + 10 mM NH4Cl + 2.25 mM Fe(II) | −0.750 |
| VI-a | 140 mM NaCl + 30 mM NH4Cl + 2.25 mM Fe(II) | −0.350 |
| VI-b | 140 mM NaCl + 30 mM NH4Cl + 2.25 mM Fe(II) | −0.550 |
| VI-c | 140 mM NaCl + 30 mM NH4Cl + 2.25 mM Fe(II) | −0.750 |
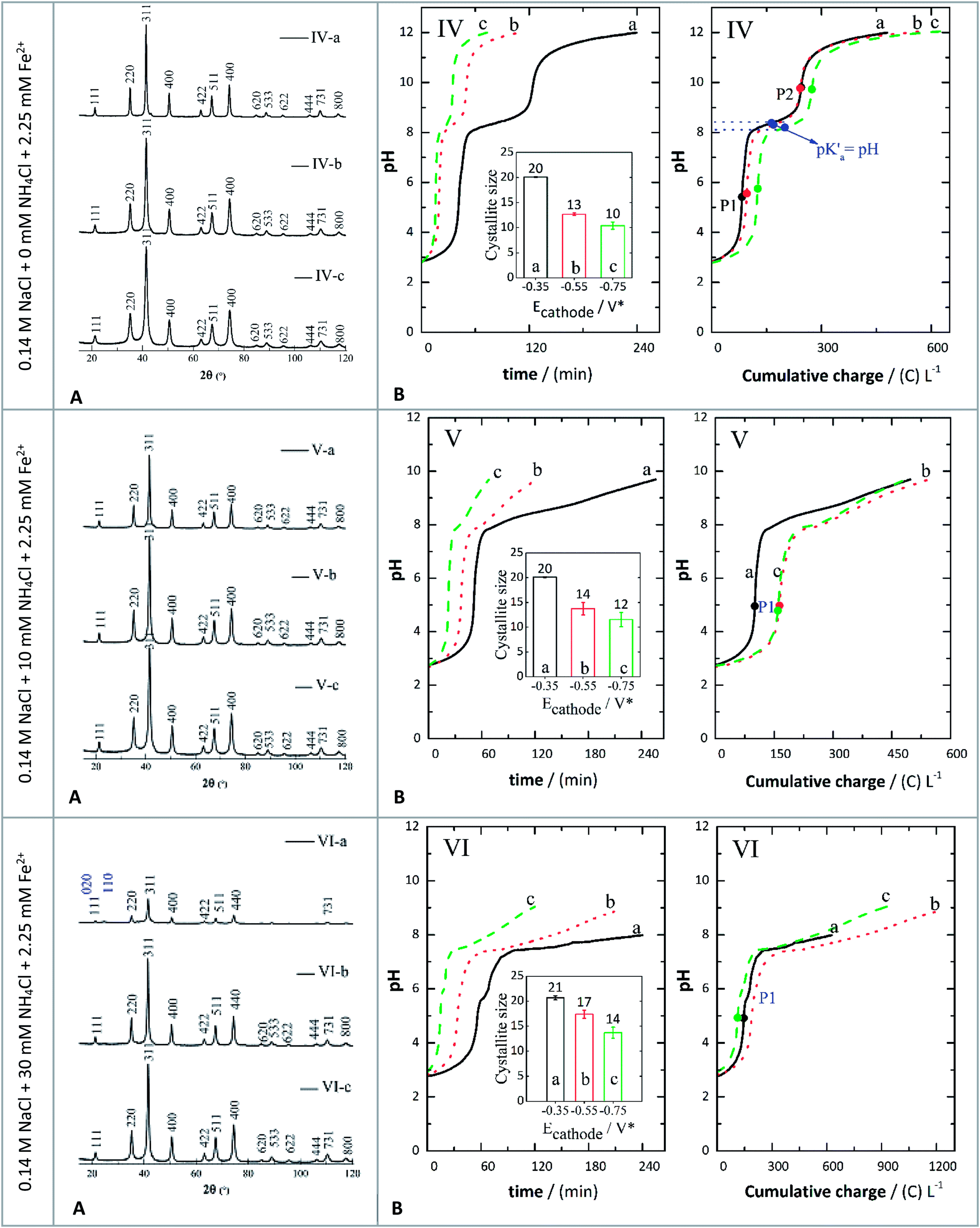 | ||
| Fig. 3 XRD patterns (A) and evolution of pH (B) against either time or charge consumed in 0.14 M NaCl + 2.25 mM Fe2+ (IV-a, IV-b, and IV-c), 0.14 M NaCl + 2.25 mM Fe2+ + 10 mM NH4Cl (V-a, V-b, and V-c), and 0.14 M NaCl + 30 mM NH4Cl + 2.25 mM Fe2+ (VI-a, VI-b, and VI-c) at different applied potentials of −0.350 V (a), −0.550 V (b), and −0.750 V (c). The insets show the crystallite sizes determined by XRD. | ||
XRD analysis of precipitates
The crystal structures of the prepared samples were characterized by XRD and the results are shown in Fig. 3(A). The peaks indexed (Fig. 3(A)) correspond to crystallographic planes in the following order: (111), (220), (311), (400), (422), (511), (440), (620), (533), (622), (444), (731) and (800). This identification was performed by comparing the XRD diffractograms with reference patterns from the International Centre for Diffraction Data (ICDD) database, and these peak indexes were matched with cards 01-088-0315 (magnetite) and 00-029-0713 (goethite). The XRD patterns showed sharp and intense peaks, suggesting the presence of well-defined crystalline phases, identified mostly as magnetite, except for assay VI-a (Fig. 3) which contained a magnetite-goethite mixture. The latter was identified by the low-angle peaks (020) and (110) marked in Fig. 3(A), corresponding to assay VI-a. Consequently, when the GDEx process was performed by adding 30 mM NH4Cl at −0.350 V, crystals of goethite were detected. This is also consistent with the findings of Prato et al.33The average crystal size distribution (insets in Fig. 4B) obtained for the IONPs from XRD was between 12 nm and 20 nm. Specifically, without NH4Cl the average sizes were around 20 nm (assay IV-a), 13 nm (assay IV-b) and 10 nm (assay IV-c). When adding NH4Cl the average crystal sizes were 20 nm (assay V-a), 14 nm (assay V-b), 12 nm (assay V-c), 21 nm (assay VI-a), 17 nm (assay VI-b) and 14 nm (assay VI-c). It is important to recall here that the indexes a, b and c, correspond to the applied potentials of −0.350 V, −0.550 V, and −0.750 V, respectively. It seems that there is a higher correlation of the crystal size with the applied polarization potential than with the presence or absence of NH4Cl. Yet, the influence of adding the WA can still be appreciated. The bigger crystals were on average 20 nm at −0.350 V, irrespective of the addition of WA, but at −0.550 V and −0.750 V slightly smaller crystals were obtained.
 | ||
| Fig. 4 Current–potential curves recorded before starting the electrolysis by linear scan voltammetry (LSV) at a scan rate of 10 mV s−1 varying the polarization potential from open circuit potential to −0.75 V vs. Ag/AgCl (satd. KCl) in different background electrolytes: 0.14 M NaCl + 2.25 mM Fe2+ (black solid line), 0.14 M NaCl + 10 mM NH4Cl + 2.25 mM Fe2+ (red solid line), and 0.14 M NaCl + 30 mM NH4Cl + 2.25 mM Fe2+ (green solid line). The inset shows the full current–potential curves. | ||
This effect was in disagreement with previous research reported where the mechanism through which the crystal size is influenced is as follows: higher supersaturation leads to higher nucleation rates, which in turn generate smaller crystals.45,46 This is because when adding the WA the gradual changes in pH presumably favoured the generation of slightly larger crystals.
Magnetite (Fe3O4) was the only pure phase present at the end of almost all the experiments, except for assay VI-a, as explained before (Fig. 4A). The formation of crystallite domains of Fe3O4 in this work can be possibly explained by the concomitant formation of oxidising and reducing species like H2O2. The likely partial oxidation of H2O2 (ref. 47) via either homogeneous Fenton or heterogeneous Fenton-like processes could play an important role in the early stages of formation of the predominant phase of the iron oxide identified at the end of the assays. However, GDEx differs from Fenton or electro-Fenton processes as the latter takes place under acidic conditions (<4), wherein the precipitated products observed in this work would not remain stable.
pH evolution when adding a metal precursor
In Fig. 3, the pH evolution curves against either time or charge consumed are depicted for the following electrolytes: (IV) 0.14 M NaCl + 10 mM NH4Cl + 2.25 mM Fe2+, (V) 0.14 M NaCl + 10 mM NH4Cl + 2.25 mM Fe2+, and (VI) 0.14 M NaCl + 30 mM NH4Cl + 2.25 mM Fe2+ at the same polarization potentials of −0.350 V (a), −0.550 V (b), and −0.750 V (c) as previously described. At the beginning of each constant polarization experiment, the solutions were colorless and progressively became yellowish. When small precipitates were noticeable, the color transitioned to orange-yellow, ultimately turning into brown-green. The greenish colour may indicate the presence of Fe(OH)2.48 It then shifted to brown-orange and finally to black, indicating the formation of magnetite (Fe3O4) (Fig S1, ESI†). However, in some cases the final solution was brown-black, suggesting the formation of more than one phase (Table S2, ESI†).Because OH− ions were generated continuously, the pH of the electrolyte increased and led to the precipitation of iron hydroxides via reactions (10) and (11). In particular, both reactions (10) and (11) are driven by the pH increase near the cathode due to reaction (6).49,50 In this regard, the Fe3+ ions formed could react with OH− ions to form orange-yellow hydroxides in the solution via reaction (10),23 whereas Fe2+ hydroxides (i.e., Fe(OH)2 may completely precipitate at a pH higher than 8.5.51 In these experiments, when the electrolyte reached a final pH of 8, in the obtained particles, a mixture of goethite and magnetite was evidenced from the XRD patterns (assay VI-a, Fig. 3(A)). At a final pH around 9 (assays VI-b and VI-c) the prepared crystals were identified as magnetite. Similarly, at a final pH of 9.7 (assays V-a, V-b, and V-c) and pH around 12 (assays IV-a, IV-b, and IV-c), in the dried samples, a single phase corresponding to magnetite was observed.
| Fe3+ + 3OH− → Fe(OH)3(s) | (10) |
| Fe2+ + 2OH− ⇄ Fe(OH)2(s) | (11) |
As reaction (2) took place continuously, the electrogeneration of H2O2 and OH− also created the necessary supersaturation conditions to transform goethite and Fe(OH)2 into magnetite. Reaction (11) was determined to be one of the equilibrium processes depicted in Fig. 3(IV), established between P1 and P2. This is because the solubility product of Fe(OH)2 is higher than that of Fe(OH)3; thus the partial dehydroxylation of Fe(OH)2 is favoured as the concentration of OH− ions increased. Furthermore, the pH condition marked as P2 represents the condition of pH established to form magnetite as a pure phase under the conditions of these assays, which seems to be around a pH of 9.6. In addition, the equivalence points P1 and P2 for assays IV-a, IV-b, and IV-c (Fig. 3) were determined using the first and second derivative of the experimental pH data versus either time or charge. From the half-point (corresponding to the pKa) between P1 and P2, it was inferred that the equilibrium process of reaction (11) had an average pKa of 8.28 ± 0.1 in the systems without the supplemented WA (Fig. 3B-IV).
On the other hand, adjusted curves (shown in Fig. S4, ESI†) to the results in Fig. 3 indicate that the formation of magnetite likely followed the chemical steps in a similar way to what was previously discussed by Lozano et al.29 Reaction (11) is expressed as reversible to point out that this is the reaction involved in the acid-base equilibrium depicted in Fig. 4B(IV–VI). This equilibrium results from reactions between products of the reduction of oxygen at the cathode as presented at the beginning of the manuscript and the iron species formed, whereas reactions (12) and (13) are likely the chemical steps occurring in the bulk of the electrolyte whose titration curves are depicted in Fig. 3B.
Lozano et al.29 stated that Fe(OH)2, under alkaline conditions and in the presence of air, can be transformed into γ-FeOOH (lepidocrocite phase) that finally reacted with additional Fe(OH)2 to form magnetite via a topotactic process.29 This pathway seemed to be promoted by the presence of a high concentration of Fe2+ ions, as their study involved the anodic dissolution of a sacrificial iron anode. Nonetheless, in this research, the concentration of Fe2+ ions was limited by an initial amount added to the electrolytes as shown in Fig. 3. This lack of excess Fe2+ ions likely promoted an alternative mechanism to form magnetite, which did not involve lepidocrocite as the main intermediate as proposed by Lozano et al.29 Instead, goethite (α-FeOOH(s)) was identified by XRD as a precursor in our systems as discussed before. This phase has been obtained by direct transformation of Fe(OH)2 under highly alkaline conditions at pH values between 12.8 and 12.3.52 It has also been previously obtained through GDEx under a specific set of experimental conditions, mainly related to the concentration of Fe2+ in solution and the charge applied in GDEx.33 In this research the main transformations were observed at pH values lower than those reported by Gilbert et al.52 Hence, the formation of goethite is inferred to be the result of the slow hydrolysis of Fe3+ hydroxy species.53 These species could act as seeds to direct the transformation of FeOOH(s) formed via partial oxidation of Fe(OH)2 by H2O2, as shown in reaction (12). Finally, α-FeOOH(s) and Fe(OH)2 could react in a topotactic process to form magnetite via reaction (13) as previously discussed for other FeOOH polymorphs.29,52,54,55
| Fe(OH)2(s) + H2O2 → FeOOH(s) + H2O | (12) |
| 2α-FeOOH(s) + Fe(OH)2(s) → Fe3O4(s) + 2H2O | (13) |
Current–potential curves
Current–potential curves were recorded before starting and at the end of each electrolysis by LSV (Fig. 4). The aim was to investigate the effect of adding Fe2+ ions compared to the cathodic deprotonation effects previously presented for the ORR to H2O2 described in Fig. 3, from 0 V to −0.75 V. The insets correspond to the full current–potential curves using the following background electrolytes: 0.14 M NaCl + 2.25 mM Fe2+, 0.14 M NaCl + 10 mM NH4Cl + 2.25 mM Fe2+, and 0.14 M NaCl + 30 mM NH4Cl + 2.25 mM Fe2+. These LSV curves show the same changes when adding NH4Cl, either with or without Fe2+. The open-circuit potential for these curves was around −0.24 ± 0.01 V. As mentioned before, J1, J2, and J3 denote the cathodic current density measured for the assays described in Table 2, using NaCl as the background electrolyte (Fig. 4). The LSV curves recorded showed a similar behavior to those in Fig. 2, where the addition of NH4Cl (from 10 mM to 30 mM) resulted in a consistent small increase in cathodic current density. As discussed earlier, this is a relevant outcome and has favourable implications for the formation of the crystallite domains of iron because the gradual changes in pH can allow conditions for the slow oxidation of intermediates to form pure phases like magnetite. However, it is important to bear in mind that appropriate selection of the added WA should be accompanied by the analysis of the chemical nature of the species of interest to ultimately produce suitable supersaturation conditions (i.e., temperature, applied potential or current density).Recovery efficiencies of iron (η).The recovery efficiencies from the assays described in Fig. 3 were determined by monitoring the concentration of total iron dissolved in the solution at the beginning and at the end of each assay by ICP-MS. From these analyses, it was established that an average recovery of 99.9 ± 0.2% of the total iron added was obtained in the aqueous electrolyte. Thus, GDEx provides an effective route to recovering soluble iron species to IONPs.
Conclusions
The buffering ability of NH4Cl in aqueous solutions of 0.1 M NaCl enhanced the generation of H2O2via the cathodic deprotonation process. The applied polarization potential had a greater influence on the size of the crystals of iron measured. This effect was exceptionally noticeable in almost all instances where magnetite (Fe3O4) was identified at a final pH higher than 8.5. Otherwise, a mixture of magnetite-goethite was evidenced at lower pH values. The chemical reactions occurring to continuously form H2O2 and OH− facilitated the necessary conditions of supersaturation, leading to the production of smaller crystals. This study demonstrates the feasibility of the method in preparing nano-sized magnetite particles, with in situ generated oxidizing species in aqueous solutions. Moreover, it establishes GDEx as an effective strategy to completely deplete iron ions in the solution, with prospective applications in metal recovery and remediation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
XDB, YAG, and JF acknowledge the support from the Flemish SIM MaRes programme under grant agreement no. 150626 (Get-A-Met project). We also acknowledge the support given by the Finnish Funding Agency for Innovation. XDB and JF acknowledge the funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 654100 (CHPM2030 project). We sincerely acknowledge Myrjam Mertens for XRD measurements.References
- M. Ma, Y. Zhang, Z. Guo and G. Ning, Facile synthesis of ultrathin magnetic iron oxide nanoplates by Schikorr reaction, Nanoscale Res. Lett., 2013, 8, 16 CrossRef PubMed.
- G. Mirabello, J. J. M. Lenders and N. A. J. M. Sommerdijk, Bioinspired synthesis of magnetite nanoparticles, Chem. Soc. Rev., 2016, 45(18), 5085–5106 RSC.
- H. Duan, D. Wang and Y. Li, Green chemistry for nanoparticle synthesis, Chem. Soc. Rev., 2015, 44(16), 5778–5792 RSC.
- T.-D. Nguyen, From formation mechanisms to synthetic methods toward shape-controlled oxide nanoparticles, Nanoscale, 2013, 5(20), 9455–9482 RSC.
- R. Koirala, S. E. Pratsinis and A. Baiker, Synthesis of catalytic materials in flames: opportunities and challenges, Chem. Soc. Rev., 2016, 45(11), 3053–3068 RSC.
- D. Lisjak and A. Mertelj, Anisotropic magnetic nanoparticles: A review of their properties, syntheses and potential applications, Prog. Mater. Sci., 2018, 95, 286–328 CrossRef CAS.
- J. Xiao, P. Liu, C. X. Wang and G. W. Yang, External field-assisted laser ablation in liquid: An efficient strategy for nanocrystal synthesis and nanostructure assembly, Prog. Mater. Sci., 2017, 87, 140–220 CrossRef CAS.
- J. M. Patete, X. Peng, C. Koenigsmann, Y. Xu, B. Karn and S. S. Wong, Viable methodologies for the synthesis of high-quality nanostructures, Green Chem., 2011, 13(3), 482–519 RSC.
- Y. Min, J. Kwak, A. Soon and U. Jeong, Nonstoichiometric Nucleation and Growth of Multicomponent Nanocrystals in Solution, Acc. Chem. Res., 2014, 47(10), 2887–2893 CrossRef CAS PubMed.
- T. R. Kline, M. Tian, J. Wang, A. Sen, M. W. H. Chan and T. E. Mallouk, Template-Grown Metal Nanowires, Inorg. Chem., 2006, 45(19), 7555–7565 CrossRef CAS PubMed.
- Y. Liu, J. Goebl and Y. Yin, Templated Synthesis of Nanostructured Materials, Chem. Soc. Rev., 2013, 42(7), 2610–2653 RSC.
- K. Woo, J. Hong, S. Choi, H.-W. Lee, J.-P. Ahn and C. S. Kim, et al., Easy Synthesis and Magnetic Properties of Iron Oxide Nanoparticles, Chem. Mater., 2004, 16(14), 2814–2818 CrossRef CAS.
- R. Zboril, M. Mashlan and D. Petridis, Iron(III) Oxides from Thermal Processes Synthesis, Structural and Magnetic Properties, Mössbauer Spectroscopy Characterization, and Applications, Chem. Mater., 2002, 14(3), 969–982 CrossRef CAS.
- X. Dominguez-Benetton, Y. Alvarez-Gallego, C. Porto-Carrero, K. Gijbels and S. Rajamani, An Electrochemical Process for Preparing a Compound Comprising a Metal or Metalloid and a Peroxide, Ionic or Radical Species, WO 2016/110597 Al, 2016.
- Y. Alvarez-Gallego, X. Dominguez-Benetton, D. Pant, L. Diels, K. Vanbroekhoven and I. Genné, et al., Development of Gas Diffusion Electrodes for Cogeneration of Chemicals and Electricity, Electrochim. Acta, 2012, 82, 415–426 CrossRef CAS.
- L. Machala, J. Tucek and R. Zboril, Polymorphous Transformations of Nanometric Iron(III) Oxide: A Review, Chem. Mater., 2011, 23(14), 3255–3272 CrossRef CAS.
- B. K. Poudel, K.-O. Doh and J. H. Byeon, Green and Continuous Route to Assemble Lateral Nanodimensional Graphitic Oxide Composites without Process Interruption, Green Chem., 2018, 20(13), 2984–2989 RSC.
- M. D. Kärkäs, Electrochemical Strategies for C–H Functionalization and C–N Bond Formation, Chem. Soc. Rev., 2018, 47(15), 5786–5865 RSC.
- D. Nick, B. Tom, F. J. Vankelecom Ivo and P. Pescarmona Paolo, Influence of the Composition and Preparation of the Rotating Disk Electrode on the Performance of Mesoporous Electrocatalysts in the Alkaline Oxygen Reduction Reaction, ChemElectroChem, 2017, 5(1), 119–128 Search PubMed.
- R. Karlsson, Theoretical and Experimental Studies of Electrode and Electrolyte Processes in Industrial Electrosynthesis, 2015 Search PubMed.
- N. Elgrishi, B. D. McCarthy, E. S. Rountree and J. L. Dempsey, Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes, ACS Catal., 2016, 6(6), 3644–3659 CrossRef CAS.
- B. Horstmann, T. Danner and W. G. Bessler, Precipitation in Aqueous Lithium-Oxygen Batteries: A Model-based Analysis, Energy Environ. Sci., 2013, 6(4), 1299–1314 RSC.
- S. Mosivand and I. Kazeminezhad, Functionalization and Characterization of Electrocrystallized Iron Oxide Nanoparticles in the Presence of β-cyclodextrin, CrystEngComm, 2016, 18(3), 417–426 RSC.
- J. F. Carneiro, R. S. Rocha, P. Hammer, R. Bertazzoli and M. R. V. Lanza, Hydrogen Peroxide Electrogeneration in Gas Diffusion Electrode Nanostructured with Ta2O5, Appl. Catal., A, 2016, 517, 161–167 CrossRef CAS.
- W. R. P. Barros, T. Ereno, A. C. Tavares and M. R. V. Lanza, In Situ Electrochemical Generation of Hydrogen Peroxide in Alkaline Aqueous Solution by using an Unmodified Gas Diffusion Electrode, ChemElectroChem, 2015, 2(5), 714–719 CrossRef CAS.
- D. Carlier, C. Terrier, C. Arm and J.-P. Ansermet, Preparation and Magnetic Properties of Fe3O4 Nanostructures Grown by Electrodeposition, Electrochem. Solid-State Lett., 2005, 8, C43 CrossRef CAS.
- T. Yousefi, R. Davarkhah, A. Nozad Golikand, M. Hossein Mashhadizadeh and A. Abhari, Facile Cathodic Electrosynthesis and Characterization of Iron Oxide Nano-particles, Prog. Nat. Sci.: Mater. Int., 2013, 23(1), 51–54 CrossRef.
- L. Martinez, D. Leinen, F. Martin, M. Gabas, J. Ramos-Barrado and E. Quagliata, et al., Electrochemical Growth of Diverse Iron Oxide Fe3O4,-FeOOH, and -FeOOH Thin Films by Electrodeposition Potential Tuning, 154, 2007, D126.
- I. Lozano, N. Casillas, C. P. de León, F. C. Walsh and P. Herrasti, New Insights into the Electrochemical Formation of Magnetite Nanoparticles, J. Electrochem. Soc., 2017, 164(4), D184–D191 CrossRef CAS.
- M. Y. Nassar, I. S. Ahmed and H. S. Hendy, A facile one-pot hydrothermal synthesis of hematite (α-Fe2O3) nanostructures and cephalexin antibiotic sorptive removal from polluted aqueous media, J. Mol. Liq., 2018, 271, 844–856 CrossRef CAS.
- M. Y. Nassar and S. Abdallah, Facile controllable hydrothermal route for a porous CoMn2O4 nanostructure: synthesis, characterization, and textile dye removal from aqueous media, RSC Adv., 2016, 6(87), 84050–84067 RSC.
- T. A. Sorenson, S. A. Morton, G. D. Waddill and J. A. Switzer, Epitaxial Electrodeposition of Fe3O4 Thin Films on the Low-Index Planes of Gold, J. Am. Chem. Soc., 2002, 124(25), 7604–7609 CrossRef CAS PubMed.
- R. A. Prato, V. Van Vught, S. Eggermont, G. Pozo, P. Marin, J. Fransaer and X. Dominguez-Benetton, Gas Diffusion Electrodes on the Electrosynthesis of Controllable Iron Oxide Nanoparticles., Sci. Rep., 2019, 9, 15370 CrossRef PubMed.
- F. Alcaide, E. Brillas, P.-L. Cabot and J. Casado, Electrogeneration of Hydroperoxide Ion Using an Alkaline Fuel Cell, J. Electrochem. Soc., 1998, 145(10), 3444–3449 CrossRef CAS.
- H.-H. Yang and R. L. McCreery, Elucidation of the Mechanism of Dioxygen Reduction on Metal-Free Carbon Electrodes, J. Electrochem. Soc., 2000, 147(9), 3420–3428 CrossRef CAS.
- Ph Vermeiren, W. Adriansens, J. P. Moreels and R. Leysen, Evaluation of the Zirfon® separator for use in alkaline water electrolysis and Ni-H2 batteries, Int. J. Hydrogen Energy, 1998, 23(5), 321–324 CrossRef CAS.
- K. Claudius, D. W. Bahnemann and M. R. Hoffmann, Photocatalytic production of hydrogen peroxides and organic peroxides in aqueous suspensions of titanium dioxide, zinc oxide, and desert sand, Environ. Sci. Technol., 1988, 22(7), 798–806 CrossRef PubMed.
- A. Özcan, Y. Şahin, A. Savaş Koparal and M. A. Oturan, Carbon sponge as a new cathode material for the electro-Fenton process: Comparison with carbon felt cathode and application to degradation of synthetic dye basic blue 3 in aqueous medium, J. Electroanal. Chem., 2008, 616(1–2), 71–78 CrossRef.
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, Hydrogen Peroxide Synthesis via Enhanced Two-Electron Oxygen Reduction Pathway on Carbon-Coated Pt Surface, J. Phys. Chem. C, 2014, 118(51), 30063–30070 CrossRef CAS.
- S. Da Silva, R. Basséguy and A. Bergel, Electrochemical deprotonation of phosphate on stainless steel, Electrochim. Acta, 2004, 49(26), 4553–4561 CrossRef CAS.
- X. Dominguez-Benetton and Y. Alvarez-Gallego, A device and method for the production of hydrogen peroxide, EP3177754A1, 2017.
- L. D. Munoz, B. Erable, L. Etcheverry, J. Riess, R. Basséguy and A. Bergel, Combining phosphate species and stainless steel cathode to enhance hydrogen evolution in microbial electrolysis cell (MEC), Electrochem. Commun., 2010, 12(2), 183–186 CrossRef CAS.
- S. Da Silva, R. Basséguy and A. Bergel, Electron transfer between hydrogenase and 316L stainless steel: identification of a hydrogenase-catalyzed cathodic reaction in anaerobic mic, J. Electroanal. Chem., 2004, 561, 93–102 CrossRef CAS.
- S. G. F. Eggermont, A. Rua-Ibarz, K. Tirez, X. Dominguez-Benetton and J. Fransaer, Oxidation-assisted alkaline precipitation: the effect of H2O2 on the size of CuO and FeOOH nanoparticles, RSC Adv., 2019, 9(51), 29902–29908 RSC.
- F. H. Scholes, A. E. Hughes, S. G. Hardin, P. Lynch and P. R. Miller, Influence of Hydrogen Peroxide in the Preparation of Nanocrystalline Ceria, Chem. Mater., 2007, 19(9), 2321–2328 CrossRef CAS.
- B. Djuričić and S. Pickering, Nanostructured cerium oxide: preparation and properties of weakly-agglomerated powders, J. Eur. Ceram. Soc., 1999, 19(11), 1925–1934 CrossRef.
- R. C. Burgos-Castillo, J.-M. Fontmorin, W. Z. Tang, X. Dominguez-Benetton and M. Sillanpaa, Towards reliable quantification of hydroxyl radicals in the Fenton reaction using chemical probes, RSC Adv., 2018, 8(10), 5321–5330 RSC.
- R. W. Revie and H. H. Uhlig, Thermodynamics: Pourbaix Diagrams, in Corrosion and Corrosion Control, Wiley-Blackwell, 2008, pp. 43–51 Search PubMed.
- M. Ibrahim, K. G. Serrano, L. Noe, C. Garcia and M. Verelst, Electro-precipitation of magnetite nanoparticles: An electrochemical study, Electrochim. Acta, 2009, 55(1), 155–158 CrossRef CAS.
- L. Cabrera, S. Gutierrez, N. Menendez, M. P. Morales and P. Herrasti, Magnetite nanoparticles: Electrochemical synthesis and characterization, Electrochim. Acta, 2008, 53(8), 3436–3441 CrossRef CAS.
- X. Wei, R. Viadero and K. Buzby, Recovery of Iron and Aluminum from Acid Mine Drainage by Selective Precipitation, Environ. Eng. Sci., 2005, 22, 745–755 CrossRef CAS.
- F. Gilbert, P. Refait, F. Lévêque, C. Remazeilles and E. Conforto, Synthesis of goethite from Fe(OH)2 precipitates: Influence of Fe(II) concentration and stirring speed, J. Phys. Chem. Solids, 2008, 69(8), 2124–2130 CrossRef CAS.
- A. C. Scheinost, Metal Oxides, in Encyclopedia of Soils in the Environment, ed. D. Hillel, Elsevier, Oxford, 2005, pp. 428–38 Search PubMed.
- M. Usman, J. M. Byrne, A. Chaudhary, S. Orsetti, K. Hanna and C. Ruby, et al., Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals, Chem. Rev., 2018, 118(7), 3251–3304 CrossRef CAS PubMed.
- Y. Cudennec and A. Lecerf, Topotactic transformations of goethite and lepidocrocite into hematite and maghemite, Solid State Sci., 2005, 7(5), 520–529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00738e |
| This journal is © The Royal Society of Chemistry 2020 |