
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous Pt@PtM (M = Co, Ni) cage-bell nanostructures toward methanol electro-oxidation†
Shuli
Yin
,
Ziqiang
Wang
 *,
Chunjie
Li
,
Hongjie
Yu
,
Kai
Deng
,
You
Xu
,
Xiaonian
Li
,
Liang
Wang
* and
Hongjing
Wang
*
*,
Chunjie
Li
,
Hongjie
Yu
,
Kai
Deng
,
You
Xu
,
Xiaonian
Li
,
Liang
Wang
* and
Hongjing
Wang
*
State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: zqwang@zjut.edu.cn; wangliang@zjut.edu.cn; hjw@zjut.edu.cn
First published on 10th February 2020
Abstract
Rational design of Pt-based nanostructures with a controllable morphology and composition is vital for electrocatalysis. Herein, we demonstrate a dual-template strategy to fabricate well-defined cage-bell nanostructures including a Pt core and a mesoporous PtM (M = Co, Ni) bimetallic shell (Pt@mPtM (M = Co, Ni) CBs). Owing to their unique nanostructure and bimetallic properties, Pt@mPtM (M = Co, Ni) CBs show higher catalytic activity, better durability and stronger CO tolerance for the methanol oxidation reaction than commercial Pt/C. This work provides a general method for convenient preparation of cage-bell nanostructures with a mesoporous bimetallic shell, which have high promising potential for application in electrocatalytic fields.
Introduction
Direct methanol fuel cells (DMFCs) as portable energy conversion devices have the characteristics of convenient utilization of methanol, high energy density, low emission of pollutants, and low operating temperature in practical applications.1–4 To commercialize DMFCs, high-efficiency and low-cost electrocatalysts for the methanol oxidation reaction (MOR) are highly desired. Nowadays, platinum (Pt) attracts considerable attention because of its outstanding performance in electrocatalytic applications, such as in the MOR, formic oxidation reaction (FOR), and oxygen reduction reaction (ORR).5–8 Nevertheless, the disadvantages of Pt-based catalysts, such as high price and easy deactivation by CO poisoning, greatly limit their large-scale applications.9–11 To resolve these problems, it is urgent to improve Pt utilization, reduce Pt loading and enhance the activity and durability of Pt-based catalysts by controlling the morphology and composition.An effective way to improve the performance of Pt-based electrocatalysts is to adjust the morphology of nanomaterials.12–14 Abundant catalysts with various morphologies, such as hollow nanostructures, dendritic nanostructures and mesoporous nanostructures, for the enhancement of MOR performance have been reported.15–18 Among them, mesoporous materials have received considerable interest because they can accelerate mass/charge transfer, increase the number of active sites and reduce diffusion resistance. Mesoporous Pt-based nanoarchitectures, which are usually prepared by using a soft or hard template, have been proven to be excellent electrode materials for the MOR.19–21 Currently, mesoporous Pt-based nanomaterials are in the form of mesoporous nanoparticles (NPs) and films, which makes it difficult to make full use of Pt. 22–25 Hollow architectures of Pt-based materials can effectively further improve their intrinsic performance. The combination of mesoporous and hollow structures can greatly improve the utilization of catalysts and significantly enhance the catalytic performance over their solid counterparts. The common fabrication of hollow structures is based on replacement and dealloying methods that have difficulty in shape control.26–29 To make full use of catalysts and effectively prevent their agglomeration, the introduction of nanoparticles into hollow structures is a very interesting strategy. Integrating the structural advantages of nanoparticles, and mesoporous and hollow structures would be highly desired to enhance the activity and stability of Pt-based materials in electrochemical applications.
Alloying Pt with another metal is also considered to be a promising approach. Although marked progress has been achieved by PtRu and PtPd alloys, their price is still very high. Low-cost transition metals (M = Co, Ni, Fe, Cu) have also been widely used for alloying with Pt.30–33 The adsorption of intermediate products and toxic substances on the surface of electrocatalysts can be effectively decreased by the addition of transition metals.34 The formation of such Pt–M alloy structures causes a weak interaction of the Pt–CO bond and the generation of OH species at a lower potential that easily react with intermediates on the surface of catalysts.35,36 Therefore, the incorporation of low-cost transition metals can reduce the Pt loading, enhance resistance against poisoning, and improve catalytic stability.37,38 Inspired by the above ideas, it is highly desired to design and synthesize PtM (M = Co, Ni) alloy catalysts, which are expected to achieve higher catalytic activity, better durability and CO tolerance for the MOR.
Herein, a dual-template approach is rationally designed to prepare Pt core@mesoporous PtM (M = Co, Ni) shell cage-bell nanostructures (Pt@mPtM (M = Co, Ni) CBs). In the process of preparation, SiO2 NPs and Pluronic F127 are used as the hard template and soft template for producing the cage-bell nanostructure. Benefiting from the unique nanoarchitecture and bimetallic composition, these Pt@mPtM (M = Co, Ni) CBs exhibit outstanding catalytic activity, durability and CO tolerance for application in the MOR.
Experimental
Materials and chemicals
Chloroplatinic acid hexahydrate (H2PtCl6·6H2O), potassium tetrachloroplatinate (K2PtCl4), ascorbic acid (AA), sodium citrate, cobalt chloride hexahydrate (CoCl2·6H2O) and nickel chloride hexahydrate (NiCl2·6H2O) were purchased from Aladdin. Pluronic F127, 3-aminopropyltrimethoxysilane (APTMS) and Nafion 117 solution (5 wt%) were ordered from Sigma-Aldrich. Tetraethylorthosilicate (TEOS), isopropanol (98%), ammonia solution (NH3·H2O), hydrofluoric acid (HF 40%), sulfuric acid (H2SO4) and methanol were brought from Beijing Chemical Reagent. Commercial Pt/C (20 wt% Pt) was obtained from Alfa Aesar Co.Synthesis of Pt nanoparticles (Pt NPs)
Pt NPs were fabricated via a one-step reduction reaction in 4.2 mL of aqueous solution containing H2PtCl6 (0.04 mmol), sodium citrate (0.068 mmol) and AA (0.2 mmol) for 2 h at 308 K under ultrasonication. The Pt NPs were collected by centrifugation at 7000 rpm, and dispersed in 44 mL of H2O for further use.Synthesis of Pt@SiO2 core–shell nanoparticles (Pt@SiO2 NPs)
In brief, 20 mL of isopropanol, 0.06 mL of TEOS, 4 mL of Pt NPs and 0.5 mL of NH3·H2O were mixed under stirring for 12 h at 298 K. The Pt@SiO2 NPs were collected by centrifugation at 7000 rpm, washed three times with ethanol/water, and then dried for 24 h at 323 K.Synthesis of Pt@mPtM (M = Co, Ni) CBs
First, APTMS was used to modify the Pt@SiO2 NPs. Typically, 50 mg of Pt@SiO2 NPs, 0.2 mL of APTMS and 10 mL of isopropanol were evenly mixed and refluxed for 12 h at 353 K. After cooling to room temperature, APTMS-modified Pt@SiO2 NPs were collected by centrifugation at 6500 rpm, washed three times with ethanol/water, and then dried under vacuum at 313 K for 24 h. After this, Pt@SiO2@mPtCo NPs were obtained from the reduction reaction in an aqueous solution (4 mL) containing APTMS-modified Pt@SiO2 (2 mg), K2PtCl4 (0.03 mmol), CoCl2 (0.01 mmol), F127 (20 mg) and AA (0.2 mmol) for 2 h at 308 K under ultrasonication. After centrifugation and washing, the obtained Pt@SiO2@mPtCo NPs were etched in 20 mL of HF (10 wt%) for 12 h. The Pt@mPtCo CBs were collected by centrifugation at 3000 rpm and washed five times with H2O. The synthesis of Pt@mPtNi CBs was similar to the synthesis of Pt@mPtCo CBs, except for using NiCl2 instead of CoCl2.Characterization
The particle size and morphology of the samples were characterized by using a ZEISS SUPRA 55 scanning electron microscope (SEM) operated at 5 kV. Transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and energy-dispersive X-ray spectroscopy (EDS) were performed with a JEOL-2100F operated at 200 kV. The phase and crystallinity of the samples were studied by X-ray diffraction (XRD, PANalytical X'Pert Powder) using Cu Kα radiation.Electrochemical measurements of the MOR
A CHI 660D electrochemical analyzer was used to measure the electrochemical performance. A traditional three-electrode cell including a working electrode (modified glassy carbon electrode), a counter electrode (Pt wire) and a reference electrode (Ag/AgCl electrode (3 M KCl)) was used. For the modification of the working electrode, 10 μg of the catalyst was dropped on the surface of a clean glassy carbon electrode and dried at 323 K. Then 3 μL of Nafion (0.5%) was coated and left to dry at the same temperature. The current densities are normalized by the electrode area (0.071 cm2). The electrochemically active surface area (ECSA) was determined by integrating the charge involved in the hydrogen under potential deposition (HUPD) region in the voltammograms obtained in sulfuric acid after the subtraction of the double layer contribution (ECSA = Q/(0.21 × m), assuming 210 μC cm−2 for the total charge and m (mg) for the Pt loading). MOR investigations were conducted in 0.5 M H2SO4 with 1.0 M methanol electrolyte at a scan rate of 50 mV s−1.Results and discussion
The proposed fabrication process of Pt@mPtM (M = Co, Ni) CBs can be briefly summarized as follows. First, Pt@SiO2 core–shell NPs with a Pt core and SiO2 shell are synthesized by the Stöber method (Fig. S1†). Then the SiO2 surfaces are functionalized with amino groups using APTMS. Due to the electrostatic interaction between –NH2+ and PtCl42−, Pt and Co (or Ni) precursors are subsequently reduced to form a mesoporous PtCo (or PtNi) alloy shell on the surface of APTMS-modified Pt@SiO2 NPs with the assistance of F127. Finally, the SiO2 interlayer in Pt@SiO2@mPtM (M = Co, Ni) NPs is etched with HF to obtain Pt@mPtM (M = Co, Ni) CBs. The Pt@mPtM (M = Co, Ni) CBs can provide more catalytically active sites and transfer channels for the MOR.As the starting materials, the monodisperse Pt@SiO2 core–shell NPs with high yield possess a uniform diameter of 150 nm. The particle size of Pt NPs is around 20 nm and the thickness of the SiO2 shell is about 70 nm. Subsequently, Pt and Co species are reduced by AA on the surface of the APTMS-modified Pt@SiO2 NPs with well-developed mesoporous structures (Fig. S2†). After etching the SiO2, Pt@mPtCo CBs are obtained. It can be seen from the SEM images that the high-dispersion Pt@mPtCo CBs have a continuous well-developed mesoporous structure on the surface without obvious changes (Fig. 1a and b). In the high-magnification SEM image, it can be clearly observed that the average particle size of Pt@mPtCo CBs is around 200 nm and the average pore size is mainly distributed at around 20 nm. From the large-scale TEM image of the Pt@mPtCo CBs (Fig. S3a†), it can be seen that the yield is very high. As shown in the high-magnification TEM image (Fig. 1c), the Pt@mPtCo CBs consist of spatially separated Pt NPs and a mesoporous PtCo alloy shell. The thickness of the mesoporous PtCo alloy shell is about 25 nm. The size of Pt@mPtCo CBs is uniform and each PtCo shell is equipped with a Pt NP core. The ring-like selected-area electron diffraction (SAED) pattern of Pt@mPtCo CBs indicates their polycrystalline nature (Fig. S3b†). The HRTEM image shows that the d spacing of the edge for the PtCo shell is measured to be 0.23 nm, which is attributed to the (111) plane of the face centered cubic (fcc) PtCo alloy (Fig. 1d–f). The high-angle annular dark-field scanning TEM (HAADF-STEM), EDS elemental mapping and line-scan profile images clearly confirm the formation of cage-bell mesoporous nanostructures and the alloy state with a Pt and Co atomic ratio of 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fig. 2).
:2 (Fig. 2).
 | ||
| Fig. 1 (a and b) SEM images of Pt@mPtCo CBs. (c) TEM image of a Pt@mPtCo CB. (d) HRTEM image of the Pt@mPtCo CBs. (e) Fourier filtered lattice fringe image of the square area in (d) and (f) the corresponding FFT pattern. | ||
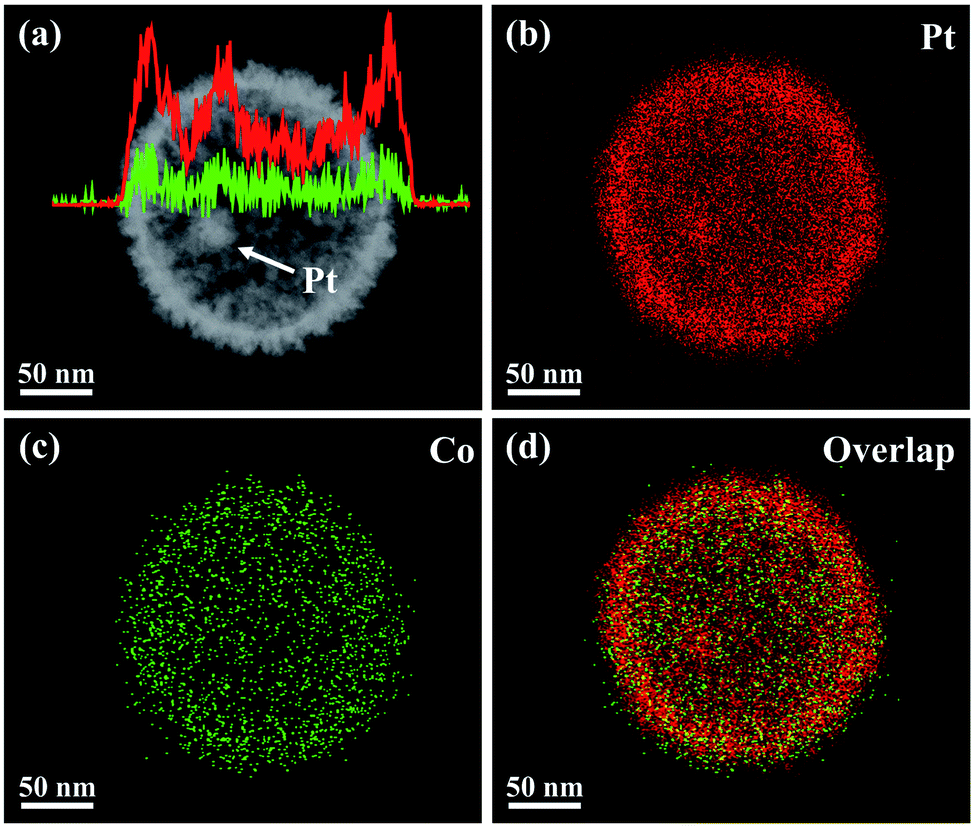 | ||
| Fig. 2 (a) HAADF-STEM image and compositional line profiles, and (b–d) EDS elemental mapping images of a Pt@mPtCo CB. | ||
In order to further confirm the crystal structure of Pt@mPtCo CBs, the XRD pattern of the Pt@mPtCo CBs is obtained. As shown in Fig. 3, the five diffraction peaks in the XRD pattern can be indexed to the (111), (200), (220), (311) and (222) planes of the metallic fcc structure, in accordance with the SAED result. These diffraction peaks deviate slightly from the standard Pt peaks (JCPDS card no. 04-0802), and no other peaks appear, indicating the formation of the PtCo alloy. The shift of diffraction peaks from the standard Pt peaks is probably due to the fact that the incorporation of Co atoms into Pt lattices causes the lattice expansion of Pt. To prove the importance of the inner Pt core, we synthesized PtCo hollow mesoporous nanoparticles (PtCo HMNs) as the comparison sample (Fig. S4†). The synthesis method is similar with the typical synthesis conditions except for replacing Pt@SiO2 NPs with SiO2 NPs as the starting material.
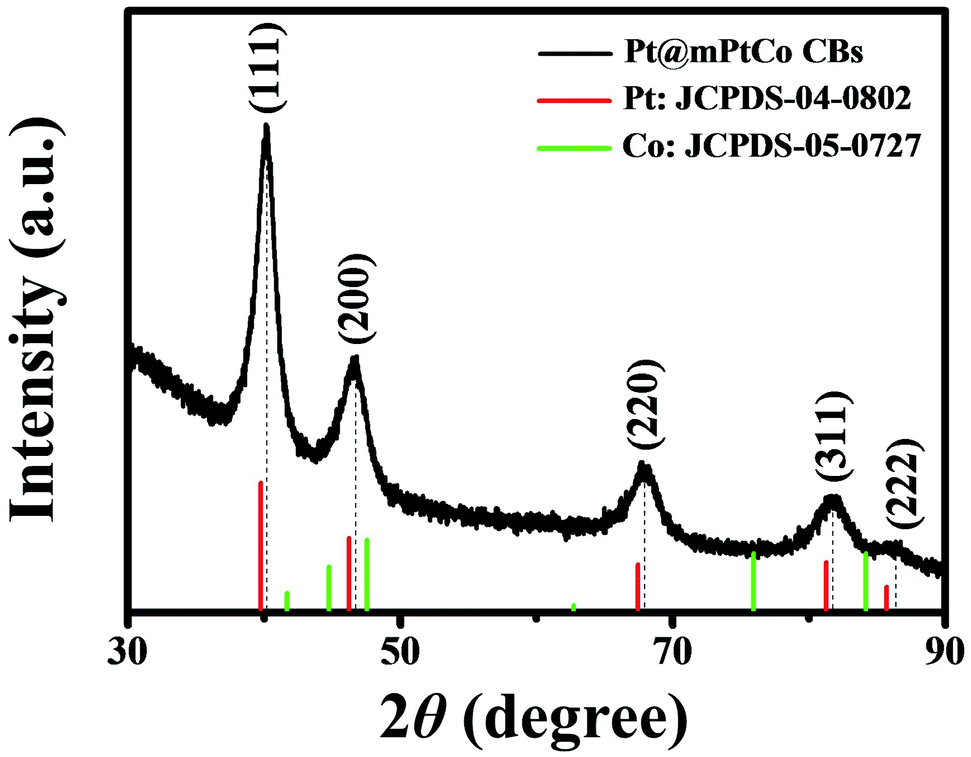 | ||
| Fig. 3 XRD pattern of Pt@mPtCo CBs. | ||
This method is universal for the preparation of cage-bell nanostructures. For instance, Pt@mPtNi CBs are also fabricated by replacing Co species with Ni species. SEM and TEM images of Pt@mPtNi CBs can clearly exhibit the uniform particle size and mesoporous surface structure with a uniform pore size distribution of approximately 20 nm, as well as the existence of the inner Pt core (Fig. S5a–c†). The HRTEM image shows that the d spacing of the PtNi shell is measured to be 0.23 nm, matching with the (111) plane of the face centered cubic (fcc) PtNi alloy (Fig. S5d–f†). From the large-scale TEM image of the Pt@mPtNi CBs, it can be seen that the yield is very high (Fig. S6†). Similar to Pt@mPtCo CBs, the SAED (Fig. S6†) and XRD patterns (Fig. S7†) of Pt@mPtNi CBs indicate the polycrystalline nature and the formation of the PtNi alloy. Additionally, the HAADF-STEM image and the corresponding EDS elemental mapping and line-scan profile images also confirm that the Pt@mPtNi CBs possess the cage-bell mesoporous nanostructure with a Pt and Ni atomic ratio of 98:2 (Fig. S8†).
Based on the above results, we would speculate the formation process of Pt@mPtM (M = Co, Ni) CBs. The Pt NPs are first synthesized via a simple reduction reaction and they are coated with SiO2 to form Pt@SiO2 core–shell NPs. After modification of Pt@SiO2 NPs with APTMS, rich amino groups are formed on the surface of SiO2 NPs. The NH2-modified Pt@SiO2 NPs are mixed with K2PtCl4 aqueous solution, CoCl2 (or NiCl2) aqueous solution, Pluronic F127 and AA. Due to the higher concentration of F127 than the critical micelle concentration at the reaction temperature, spherical molecules can be formed in the solutions. The hydrophilic groups in F127 can bind with the amino groups on the surface of Pt@SiO2 NPs via hydrogen bonds. F127 micelles have hydrophilic ethylene oxide groups at each end, which can adsorb the metal precursors.39 Pt and transition metal species are reduced by AA, and Pt-based alloys are coated on the Pt@SiO2 surfaces. F127 is removed by washing with water to form mesoporous structures. Finally, HF is applied to etch the SiO2 interlayer to synthesize the final product Pt@mPtM (M = Co, Ni) CBs.
Due to the outstanding performance of Pt-based catalysts in electrocatalysis, their controllable synthesis has attracted intensive attention. To date, incorporation of transition metals into Pt electrocatalysts with different nanostructures has been widely reported.40–42 However, most PtM catalysts are intact or smooth, and the inner catalysts cannot be utilized. For example, Shen et al.43 reported deeply excavated Pt3Co nanocubes prepared by a solvothermal method in oleylamine at high temperature (150 °C). Chen and co-workers44 prepared cyanide (CN−)-functionalized PtNi hollow nanospheres using a cyanogel reduction method. Lee's group45 reported the synthesis of Pt–Ni multiframes in the oil phase at 270 °C. Many PtM nanocatalysts with other morphologies such as core–shell NPs, nanowires, and nanoflowers, have also been synthesized.46–48 However, these catalysts usually suffer from harsh synthetic conditions and/or irregular morphology. In our case, we employ a versatile route to construct yolk–shell catalysts with mesoporous structures on the surface. The mesoporous nanostructures can provide sufficient active sites and facile mass/charge transfer channels for adsorption and activation of target species. Meanwhile, the hollow structure allows the accessibility of species to the internal active sites and inner Pt core through the mesoporous structure and prevents the agglomeration of Pt nanoparticles, leading to high Pt utilization and enhanced performance.
As is well known, Pt-based materials are considered to be excellent electrocatalysts for fuel cells. Therefore, the MOR is chosen to explore the performance of Pt@mPtM (M = Co, Ni) CBs in acidic media. PtCo HMNs and commercial Pt/C are selected to measure MOR performance for comparison. Cyclic voltammograms (CVs) are measured in a N2-saturated H2SO4 (0.5 M) aqueous solution at a scan rate of 50 mV s−1. The hydrogen adsorption and desorption peaks on the CVs can be used to calculate the ECSA (Fig. S9†). The ECSAs are calculated to be 31.6, 28.5, 26.1 and 43.0 m2 g−1 for Pt@mPtCo CBs, Pt@mPtNi CBs, PtCo HMNs and Pt/C, respectively. The ECSA of Pt@mPtCo CBs is higher than that of the Pt@mPtNi CBs, indicating that more electrochemically active sites can be generated when Pt alloys with Co. The advantages of inner Pt NPs can be illustrated by the comparison of ECSA for Pt@mPtCo CBs and PtCo HMNs. The MOR performance of these catalysts is estimated from CVs in H2SO4 (0.5 M) and methanol (1.0 M) solution at a scan rate of 50 mV s−1. For all catalysts, the peak of methanol electro-oxidation in the positive sweep is located at around 0.7 V. The reactivation of oxidized Pt species in the negative sweep occurs at about 0.4 V. Compared with the other three catalysts, Pt@mPtCo CBs exhibit higher current density for oxygen adsorption and desorption due to their structural and compositional advantages. We employ specific activity that is defined as the ECSA-normalized current to investigate the intrinsic activity (Fig. 4a). As we can see from the CVs, the specific activity of Pt@mPtCo CBs at the peak potential in the positive sweep is 1.84 mA cm−2, which is 1.16, 1.30 and 4.18 times that of the Pt@mPtNi CBs (1.58 mA cm−2), PtCo HMNs (1.41 mA cm−2) and Pt/C (0.44 mA cm−2) (Fig. 4c). Simultaneously, we measure mass activity that is the Pt mass-normalized current to study the Pt utilization (Fig. 4b). The mass activity of Pt@mPtCo CBs at the peak potential in the positive sweep is 0.58 mA μgPt−1, which is 1.29, 1.57 and 3.05 times that of the Pt@mPtNi CBs (0.45 mA μgPt−1), PtCo HMNs (0.37 mA μgPt−1) and Pt/C (0.19 mA μgPt−1) (Fig. 4c). These results demonstrate that the Pt@mPtCo CBs can achieve excellent MOR activity with reduced usage of Pt due to the cage-bell mesoporous structures of Pt cores and bimetallic PtCo shells.
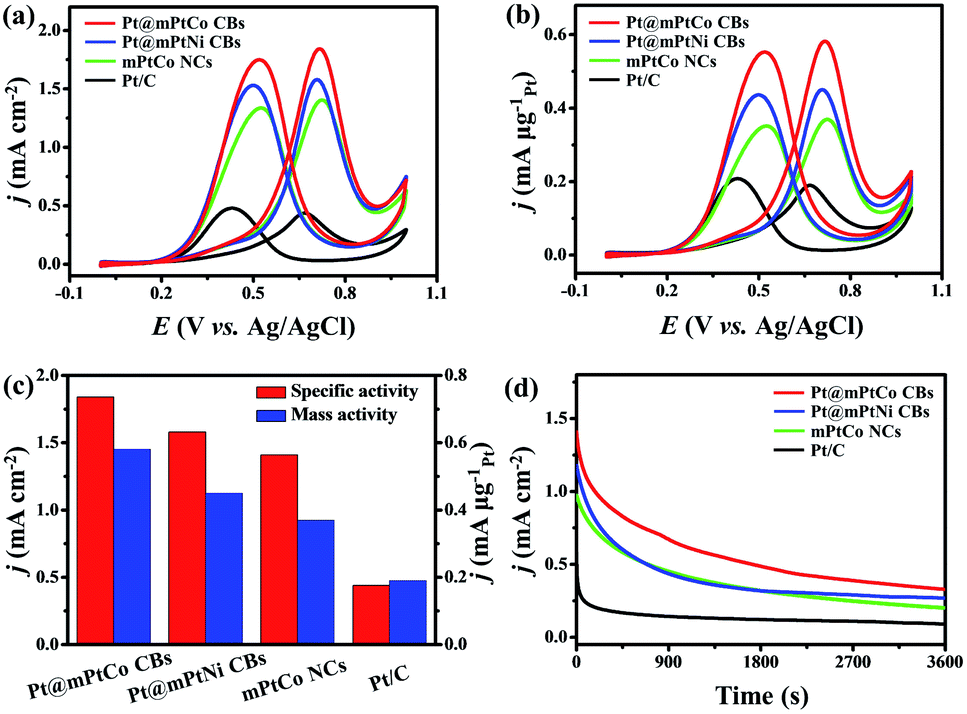 | ||
| Fig. 4 (a) ECSA-normalized and (b) mass-normalized CV curves of Pt@mPtCo CBs, Pt@mPtNi CBs, mPtCo NCs, and Pt/C in 0.5 M H2SO4 + 1 M CH3OH at a scan rate of 50 mV s−1. (c) The comparisons of the mass activities and specific activities. (d) Chronoamperometric curves (recorded at 0.65 V) obtained in 0.5 M H2SO4 + 1 M CH3OH. The currents densities were normalized by the ECSAs. | ||
The stability, CO tolerance and durability of catalysts are critical for their practical applications. To investigate the stability of different catalysts in the MOR, long-term catalytic performances are measured. Chronoamperometric curves are recorded at 0.65 V for 3600 s in H2SO4 (0.5 M) and methanol (1.0 M) solution (Fig. 4d). At the initial stage, the current density of all catalysts decreases sharply. As the reaction proceeds, the methanol concentration decreases and the catalyst is poisoned, resulting in the decrease of the current density. During the entire reaction process, the current density of the Pt@mPtCo CBs is always higher than that of the contrast samples, indicating the superior durability of Pt@mPtCo CBs for the MOR. Moreover, CO stripping tests of the Pt@mPtCo CBs, Pt@mPtNi CBs, PtCo HMNs and Pt/C are performed to investigate the CO tolerance of these catalysts. CO stripping cyclic voltammetry is conducted in CO-saturated H2SO4 (0.5 M) at a scan rate of 50 mV s−1 (Fig. 5). Because CO is easily adsorbed on the catalyst surface, no hydrogen desorption peak appears during the first positive sweep. The CO oxidation peak of the Pt@mPtCo CBs appears at 0.67 V (Fig. 5a), which is more negative than that of the Pt@mPtNi CBs (0.69 V), PtCo HMNs (0.69 V) and Pt/C (0.71 V) (Fig. 5b–d). The onset potential of CO oxidation in Pt@mPtCo CBs is more negative, indicating that Pt@mPtCo CBs are more resistant to CO than other catalysts. It can be seen from these results that Pt@mPtCo CBs have higher CO oxidation activity and better CO tolerance.
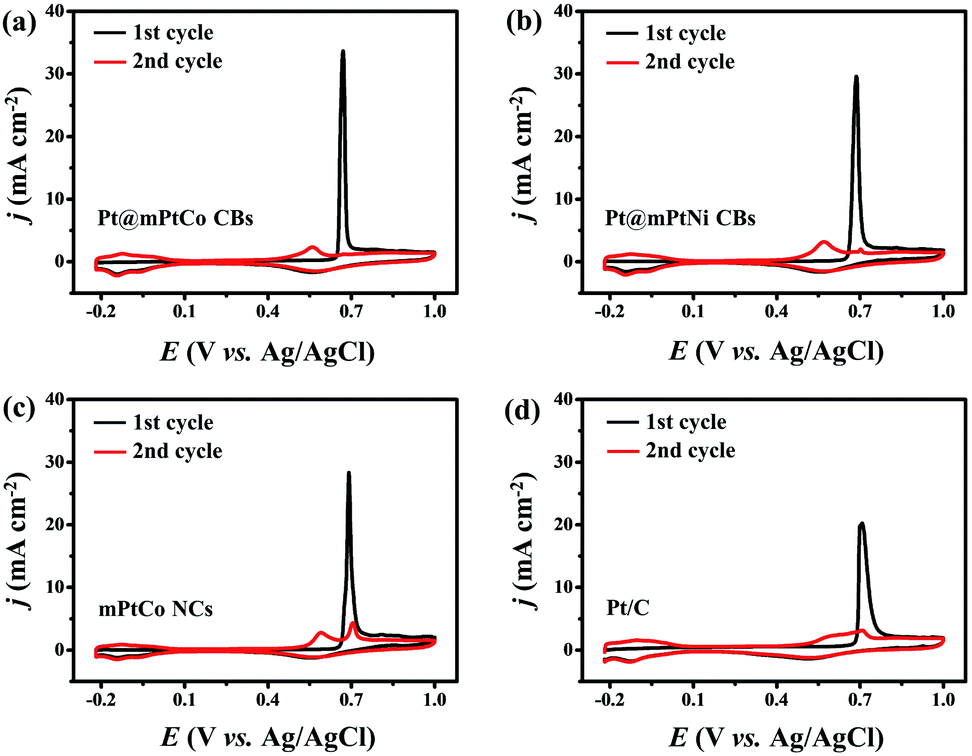 | ||
| Fig. 5 CO stripping voltammograms of (a) Pt@mPtCo CBs, (b) Pt@mPtNi CBs, (c) mPtCo NCs and (d) Pt/C. | ||
Conclusions
In conclusion, we have successfully designed a universal dual-template method for synthesizing well-defined cage-bell nanostructures including a Pt core and a mesoporous PtM (M = Co, Ni) bimetallic shell with uniform morphology and high yield. These catalysts possess several advantages including all-metallic cage-bell, mesoporous, hollow structure, and bimetallic composition, which are beneficial for the MOR. Therefore, the unique Pt@mPtM (M = Co, Ni) CB nanoarchitecture exhibits outstanding catalytic activity, durability and CO tolerance for the MOR. More significantly, this synthesis strategy can be extended to other materials to form cage-bell mesoporous structures by changing metal precursors for application in various electrocatalytic fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21601154, 21776255, 21972126, 21978264, and 21905250).References
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- L. Huang, X. Zhang, Y. Han, Q. Wang, Y. Fang and S. Dong, Chem. Mater., 2017, 29, 4557–4562 CrossRef CAS.
- Y. Feng, H. Liu and J. Yang, Sci. Adv., 2017, 3, e1700580 CrossRef PubMed.
- W. Zhao, B. Ni, Q. Yuan, P. He, Y. Gong, L. Gu and X. Wang, Adv. Energy Mater., 2017, 7, 1601593 CrossRef.
- L. Han, P. Cui, H. He, H. Liu, Z. Peng and J. Yang, J. Power Sources, 2015, 286, 488–494 CrossRef CAS.
- F. Li, Y. Ding, X. Xiao, S. Yin, M. Hu, S. Lia and Y. Chen, J. Mater. Chem. A, 2018, 6, 17164–17170 RSC.
- L. Zeng, X. Cui and J. Shi, Sci. China Mater., 2018, 61, 1557–1566 CrossRef CAS.
- C. Zhang, Y. Dai, H. Chen, Y. Ma, B. Jing, Z. Cai, Y. Duan, B. Tang and J. Zou, J. Mater. Chem. A, 2018, 6, 22636–22644 RSC.
- S. Yin, R. Kumar, H. Yu, C. Li, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 14867–14873 CrossRef CAS.
- L. Luo, F. Zhu, R. Tian, L. Li, S. Shen, X. Yan and J. Zhang, ACS Catal., 2017, 7, 5420–5430 CrossRef CAS.
- H. Wang, Y. Wu, X. Luo, L. Jiao, X. Wei, W. Gu, D. Du, Y. Lin and C. Zhu, Nanoscale, 2019, 11, 10575–10580 RSC.
- S. Yin, H. Wang, K. Deng, Z. Dai, Z. Wang, Y. Xu, X. Li, H. Xue and L. Wang, Chem.–Eur. J., 2019, 25, 5316–5321 CrossRef CAS PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- M. Gong, Z. Deng, D. Xiao, L. Han, T. Zhao, Y. Lu, T. Shen, X. Liu, R. Lin, T. Huang, G. Zhou, H. Xin and D. Wang, ACS Catal., 2019, 9, 4488–4494 CrossRef CAS.
- E. D. Goodman, S. Dai, A. Yang, C. J. Wrasman, A. Gallo, S. R. Bare, A. S. Hoffman, T. F. Jaramillo, G. W. Graham, X. Pan and M. Cargnello, ACS Catal., 2017, 7, 4372–4380 CrossRef CAS.
- R. Wu, Y. Chong, G. Fang, X. Jiang, Y. Pan, C. Chen, J. Yin and C. Ge, Adv. Funct. Mater., 2018, 28, 1801484 CrossRef.
- X. Yan, S. Yu, Y. Tang, D. Sun, L. Xu and C. Xue, Nanoscale, 2018, 10, 2231–2235 RSC.
- C. Li, Y. Xu, D. Yang, X. Qian, X. Chai, Z. Wang, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9709–9716 CrossRef CAS.
- B. Jiang, C. Li, H. Qian, M. S. A. Hossain, V. Malgras and Y. Yamauchi, Angew. Chem., Int. Ed., 2017, 56, 7836–7841 CrossRef CAS PubMed.
- H. Ataee-Esfahani, Y. Nemoto, L. Wang and Y. Yamauchi, Chem. Commun., 2011, 47, 3885–3887 RSC.
- C. Li, M. Iqbal, B. Jiang, Z. Wang, J. Kim, A. K. Nanjundan, A. E. Whitten, K. Wood and Y. Yamauchi, Chem. Sci., 2019, 10, 4054–4061 RSC.
- H. Wang, H. Yu, Z. Wang, Y. Li, Y. Xu, X. Li, H. Xue and L. Wang, Small, 2019, 15, 1804769 CrossRef PubMed.
- H. Wang, H. Yu, S. Yin, Y. Xu, X. Li, Y. Yamauchi, H. Xue and L. Wang, J. Mater. Chem. A, 2018, 6, 12744–12750 RSC.
- C. Li, H. Tan, J. Lin, X. Luo, S. Wang, J. You, Y.-M. Kang, Y. Bando, Y. Yamauchi and J. Kim, Nano Today, 2018, 21, 91–105 CrossRef CAS.
- C. Li, M. Iqbal, J. Lin, X. Luo, B. Jiang, V. Malgras, K. C. W. Wu, J. Kim and Y. Yamauchi, Acc. Chem. Res., 2018, 51, 1764–1773 CrossRef CAS PubMed.
- M. Zhao, A. O. Elnabawy, M. Vara, L. Xu, Z. D. Hood, X. Yang, K. D. Gilroy, L. Figueroa-Cosme, M. Chi, M. Mavrikakis and Y. Xia, Chem. Mater., 2017, 29, 9227–9237 CrossRef CAS.
- K. Zhang, H. Xu, B. Yan, J. Wang, Y. Du and Q. Liu, Electrochim. Acta, 2018, 268, 383–391 CrossRef CAS.
- Q. Shi, Y. Song, C. Zhu, H. Yang, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 24288–24295 CrossRef CAS PubMed.
- P. Cui, H. He, H. Liu, S. Zhang and J. Yang, J. Power Sources, 2016, 327, 432–437 CrossRef CAS.
- S. Guo, S. Zhang, D. Su and S. Sun, J. Am. Chem. Soc., 2013, 135, 13879–13884 CrossRef CAS PubMed.
- C. Zhang, R. Zhang, X. Li and W. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 29623–29632 CrossRef CAS PubMed.
- Q. Lu, L. Sun, X. Zhao, J. Huang, C. Han and X. Yang, Nano Res., 2018, 11, 2562–2572 CrossRef CAS.
- T. Xia, J. Liu, S. Wang, C. Wang, Y. Sun and R. Wang, Sci. China Mater., 2017, 60, 57–67 CrossRef CAS.
- J. Li, X. Fu, Z. Mao, Y. Yang, T. Qiu and Q. Wu, Nanoscale Res. Lett., 2016, 11, 3 CrossRef PubMed.
- P. Yang, X. Yuan, H. Hu, Y. Liu, H. Zheng, D. Yang, L. Chen, M. Cao, Y. Xu, Y. Min, Y. Li and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1704774 CrossRef.
- H. J. Qiu, X. Shen, J. Q. Wang, A. Hirata, T. Fujita, Y. Wang and M. W. Chen, ACS Catal., 2015, 5, 3779–3785 CrossRef CAS.
- Z. Zhu, Y. Zhai and S. Dong, ACS Appl. Mater. Interfaces, 2014, 6, 16721–16726 CrossRef CAS PubMed.
- C. Wang, Y. Zhang, Y. Zhang, P. Xu, C. Feng, T. Chen, T. Guo, F. Yang, Q. Wang, J. Wang, M. Shi, L. Fan and S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 9444–9450 CrossRef CAS PubMed.
- H. Ataee-Esfahani, J. Liu, M. Hu, N. Miyamoto, S. Tominaka, K. C. W. Wu and Y. Yamauchi, Small, 2013, 9, 1047–1051 CrossRef CAS PubMed.
- R. Chattot, T. Asset, J. Drnec, P. Bordet, J. Nelayah, L. Dubau and F. Maillard, Nano Lett., 2017, 17, 2447–2453 CrossRef CAS PubMed.
- S. Bai, L. Bu, Q. Shao, X. Zhu and X. Huang, J. Am. Chem. Soc., 2018, 140, 8384–8387 CrossRef CAS PubMed.
- S. Yin, Z. Wang, X. Qian, D. Yang, Y. Xu, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 7960–7968 CrossRef CAS.
- H. Du, S. Luo, K. Wang, M. Tang, R. Sriphathoorat, Y. Jin and P. Shen, Chem. Mater., 2017, 29, 9613–9617 CrossRef CAS.
- H. Liu, X. Liu, Y. Li, Y. Jia, Y. Tang and Y. Chen, Nano Res., 2016, 9, 3494–3503 CrossRef CAS.
- H. Kwon, M. Kabiraz, J. Park, A. Oh, H. Baik, S. Choi and K. Lee, Nano Lett., 2018, 18, 2930–2936 CrossRef CAS PubMed.
- L. Wang, W. Gao, Z. Liu, Z. Zeng, Y. Liu, M. Giroux, M. Chi, G. Wang, J. Greeley, X. Pan and C. Wang, ACS Catal., 2018, 8, 35–42 CrossRef CAS.
- P. Wang, Q. Shao, X. Cui, X. Zhu and X. Huang, Adv. Funct. Mater., 2018, 28, 1705918 CrossRef.
- L. Jiang, X. Lin, A. Wang, J. Yuan, J. Feng and X. Li, Electrochim. Acta, 2017, 225, 525–532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00020e |
| This journal is © The Royal Society of Chemistry 2020 |