
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast and surfactant-free synthesis of Sub-3 nm nanoalloys by shear-assisted liquid-metal reduction†
Mengjie
Zhang
,
Wenchang
Zhu
,
Xingzhe
Yang
,
Hao
Chen
and
Hongbin
Feng
 *
*
Institute of Materials for Energy and Environment, School of Materials Science and Engineering, Qingdao University, Qingdao 266071, China. E-mail: fenghongbin@qdu.edu.cn
First published on 31st August 2020
Abstract
The uniform and surfactant-free synthesis of ultrafine alloy nanoparticles (NPs) still remains an ongoing challenge. Here, we developed a general route for synthesizing sub-3 nm nanoalloys on a carbon support via a shear-assisted liquid-metal reduction (SA-LMR) strategy. The utilization of shearing treatment resulted in the rapid release of electrons from the liquid sodium-potassium (NaK) alloy, which enabled the ultrafast reduction of precursor metal ions to metallic nanoalloys at room temperature. As a model system, Pt–Cu NPs with uniform distribution were synthesized and characterized by spectroscopic and microscopic techniques. The size, composition and structure of the Pt–Cu NPs could be tuned by controlling the volume of liquid NaK alloy and the reaction time. To prove the universality and utility of our method, the binary Pt–M (Fe, Co, Ni, and Cu) and ternary Pt–Cu–Fe nanoalloys deposited on a carbon support were prepared as hydrogen evolution reaction (HER) catalysts. This facile, rapid and universal synthesis technique has the potential to be employed in the large-scale production of nanomaterials and suggests a new direction for nanoalloys.
1. Introduction
Nanoalloys (NAs), made by mixture of elements into one form of nanoparticles (NPs), are of great interest in many applications, including catalysis, energy storage, electronics, and sensing applications.1–9 The desire to fabricate metallic nanoalloys with controllable size and composition is often for an enhancement in specific properties due to their high specific surface area, rich diversity of compositions and structures, and their synergistic effects.10–17 Previous studies have shown that Pt-based alloys present good catalytic activity as an ideal catalyst for the hydrogen evolution reaction, oxygen reduction reaction, and ethanol oxidation reaction.18–24 At the same time, the alloying of low-cost transition metals and precious metals can effectively reduce the amount of precious metals and control costs.25 However, current methods of manufacturing alloys are generally complicated and time consuming, and therefore it is very important to develop a convenient, efficient, and rapid method of manufacturing alloys.12,26,27Commonly, the wet chemistry synthesis of well-dispersed alloy NPs needs the assistance of organic surfactants or capping agents, such as cetyltrimethylammonium chloride, ethylene glycol, oleic acid, or oleylamines, to protect them from agglomeration.28–33 Nevertheless, the residual surfactants are often difficult to remove completely, which makes it difficult to obtain nanocrystals with clean surfaces.34 Especially for catalytic applications, the active sites are occupied, which prevents the adsorption of reactants and leads to a decrease in the catalytic activity. So, a general and facile method that can easily control the nucleation and growth of uniform and ultrafine alloy NPs with clean surfaces is highly desirable.35–38 Recently, Sheng Hu et al. reported a method for the synthesis of intermetallic nanoparticles without using surfactants via laser ablation in a solution-galvanic replacement reaction and achieved good electrochemical performance.39–41 As in the general case for nanocrystal synthesis, the speed of the nucleation process is critical for controlling the uniformity of NPs. To obtain uniform and ultrasmall nanocrystals, nucleation must occur rapidly and instantaneously. Fast nucleation is easily achieved by the rapid reduction of metal ions using a strong reductant. However, the ultrafast, uniform, and surfactant-free synthesis of ultrafine nanoalloys in a strong reduction reaction system remains a challenge.
Here we report an ultrafast and surfactant-free approach for the preparation of alloy NPs with controllable size and composition by using a shear-assisted liquid metal reduction (SA-LMR) strategy at room temperature. A very active liquid metal, sodium–potassium (NaK) alloy, was first used as a rapid and effective heterogeneous reductant for the general synthesis of metallic nanoalloys via a redox substitution reaction. The entire reaction process does not require the addition of surfactants and so can avoid the particle agglomeration problem. When the highly active liquid NaK alloy reductant was used, a burst-nucleation was achieved at room temperature, in which the concentration of monomers in the solution rose rapidly to reach the concentration of nucleation, thereby shortening the nucleation time and ensuring that the size of the nanocrystals tended to be uniform. The kinetics became sufficiently fast, and the reaction time was very short (∼30 s), dramatically shorter than in previous reported studies using wet chemical synthesis.29,31 Additionally, with the simplicity of the shear-assisted process, the synthesis could be easily scaled up while maintaining the uniformity of the particle size, as well as homogenizing the reducing agent, precursor, and the carbon supports. This facile, universal, and large-scale potential technique provides a new route for the synthesis of uniform metallic alloy nanoparticles and can shed light on further innovation in nanocatalysts.
2. Experimental
2.1 Synthesis of alloy NPs on a carbon support
The synthesis of Pt–M (Cu, Fe, Co, Ni) alloy NPs on a carbon support was achieved by a one-pot shear-assisted strong reduction process at room temperature. In a typical procedure, the metal salt precursors Cu(acac)2 (0.05 mmol), H2PtCl6 (0.05 mmol), and the carbon support (vxc-72 carbon black, 51.6 mg) were mixed in dimethoxyethane (DME, 100 mL), followed by sonication treatment (5 min) to form a homogeneous suspension. After that, argon gas was introduced into the solution throughout the whole process to avoid the influence of water and oxygen in the air. A rapid shearing process was then applied by using a high-speed shear emulsifier (FLUKO FA25, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). Next, the prepared potassium–sodium alloy (NaK, wt% = 1:3, 160 μL) was added into the as-obtained solution and the reaction time controlled from tens of seconds to a few minutes. The resultant product was centrifuged to remove and recycle the DME. Then, cycles of washing with ethanol and water, sonication, and centrifugation were continuously conducted five times (note that in order to avoid the potential threat of excessive NaK, washing with ethanol was performed first).42 The final as-produced alloy samples were obtained after drying them in a vacuum oven at 60 °C for 8 h. Other Pt-based bimetallic and trimetallic alloys were also synthesized by similar methods. The content of the as-synthesized Pt-based alloy in the catalyst was approximately 20 wt%.
000 rpm). Next, the prepared potassium–sodium alloy (NaK, wt% = 1:3, 160 μL) was added into the as-obtained solution and the reaction time controlled from tens of seconds to a few minutes. The resultant product was centrifuged to remove and recycle the DME. Then, cycles of washing with ethanol and water, sonication, and centrifugation were continuously conducted five times (note that in order to avoid the potential threat of excessive NaK, washing with ethanol was performed first).42 The final as-produced alloy samples were obtained after drying them in a vacuum oven at 60 °C for 8 h. Other Pt-based bimetallic and trimetallic alloys were also synthesized by similar methods. The content of the as-synthesized Pt-based alloy in the catalyst was approximately 20 wt%.
2.2 Characterization
X-ray diffraction (XRD) patterns were collected on a Rigaku Ultima IV X-ray diffractometer equipped with Cu Kα radiation (λ = 0.15418 nm). XPS characterization was carried out on a Thermo Escalab 250Xi electron spectrometer system with a monochromatized Al Ka standard X-ray source. TEM and high-resolution TEM (HRTEM) images of the alloy nanoparticles (NPs) were observed by a transmission electron microscope (JEOL JEM-2100). The Pt L3 edge XAFS spectra were recorded at the Taiwan National Synchrotron Radiation Research Center. Pt foil was used as a reference sample and measured in the transmission mode. The data were normalized and analyzed using Athena.2.3 Electrocatalytic measurements
Electrocatalyst inks were prepared by dispersing the synthesized catalyst samples (4 mg) in a mixed solution (1 mL) containing ethanol and water (1:3) and 5 wt% Nafion solution (10 μL), followed by ultrasonication for 30 min to form a uniform suspension. Then, 5 μL of the uniform catalyst ink was deposited onto a freshly polished glass carbon disk electrode (3 mm). After drying under a baking lamp, the working electrode with uniform catalyst loading was successfully prepared. All the electrochemical performance tests were carried out in a standard three-electrode system with a modified glassy carbon electrode as the working electrode, a carbon rod as the counter electrode, and Ag/AgCl electrode as the reference electrode using 0.5 M sulfuric acid solution as the electrolyte. In order to facilitate the evaluation of the performance of the catalyst, all the potentials were calibrated to a reversible hydrogen electrode. All the electrochemical tests were performed using an electrochemical workstation (CHI 660E, Shanghai, Chenghua Co.) at room temperature. Linear sweep voltammetry (LSV) with a scan rate of 5 mV s−1 was performed on a 0.5 M H2SO4 electrolyte and all the polarization curves were iR compensated.
3. Results
Fig. 1 illustrates a schematic diagram of this ultrafast and surfactant-free synthesis of metallic nanoalloys by the SA-LMR strategy. As a proof of concept, the Pt–Cu alloy NPs were formed by this shear-assisted solution-based coreduction of their precursor Cu(acac)2 and H2PtCl6 using a liquid sodium–potassium (NaK) alloy as the reducing agent and dimethoxyethane (DME) as the solvent. First, two precursor and carbon support were added to the reaction system for sufficient mixing via shearing treatment. Second, the reducing agent NaK alloy was injected into the solution and promptly downsized into tiny droplets by the action of shearing force. Then, the precursor metal ions were instantaneously reduced to produce enough monomers corresponding to their metals ions in the system, and the monomer concentration rapidly rose to reach the nucleation concentration. According to LaMer's theory,43,44 a two-step synthesis mechanism, that is instantaneous-nucleation and ultrafast-growth, was presumed in our formation process. When the liquid NaK alloy was added, instantaneous nucleation occurred due to its strong reducibility. Also, the crystal nucleus grew immediately in a short time (from ten seconds to several minutes) until the precursor metal ions or reductants were depleted. Therefore, the size of the alloy NPs could be controlled as sub-3 nm by controlling the reaction time and the amount of reducing agent. Moreover, the explosively rapid nucleation enabled the nanocrystals to grow at the same time, which ensured that the particle size of the alloy NPs was relatively uniform.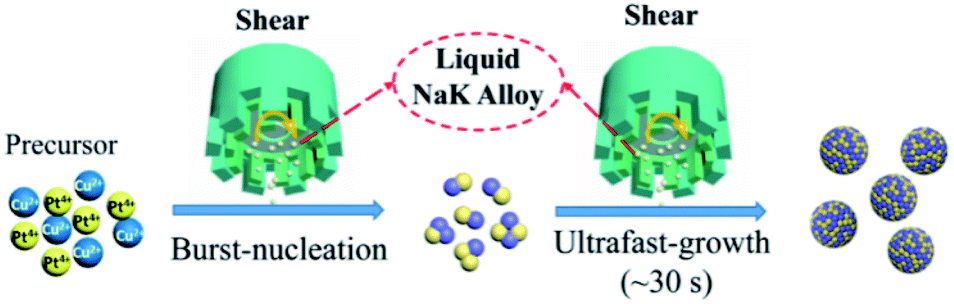 | ||
| Fig. 1 Scheme for the synthesis of ultrasmall Pt–Cu nanoparticles in the strong reduction non-aqueous system by a shear-assisted liquid NaK alloy reduction process. | ||
In order to reveal the nucleation and growth process of the alloy NPs in our system, taking Pt–Cu nanoalloys as an example, we synthesized a series of Pt–Cu alloy NPs on the carbon support (Pt–Cu-xvc72) with different sizes by regulating the reaction time (tR) and the amount of liquid NaK reducing agent (VR). The transmission electron micrograph (TEM) images (Fig. 2a–i and S1†) showed the morphologies and the size changes of a series of Pt–Cu alloy NPs on carbon black that were related to the VR and tR in the reaction system. When the tR and other conditions were kept constant, the more the VR added to the system, the larger the size of the Pt–Cu NPs formed by the reaction. Fig. 2a–e display the TEM images of Pt–Cu NPs and their particle-size variation along with the changes of VR from 10, 20, 40, and 80 μL to 160 μL, respectively, when the tR was constant at 10 s. As shown in the insert images in Fig. 2a–i, on the basis of the TEM observation, we measured the size of different 100 nanoparticles (NPs) in each of the samples and plotted the particle-size distribution.
 | ||
| Fig. 2 TEM images and average particle-size statistics for Pt–Cu-vxc72 synthesized by different amounts of reducing agent (VR) and reaction times (tR). (a) VR = 10 μL, tR = 10 s (The inset shows the structure of Pt–Cu-vxc72); (b) VR = 20 μL, tR = 10 s; (c) VR = 40 μL, tR = 10 s; (d) VR = 80 μL, tR = 10 s; (e) VR = 160 μL, tR = 10 s; (f) VR = 160, μL tR = 30 s; (g) VR = 160 μL, tR = 2 min; (h) VR = 160 μL, tR = 5 min; (i) VR = 160 μL, tR = 10 min. (j) Three-dimensional statistical image of the average particle size with the reducing agent volume (VR) and reaction time (tR). (k) The statistical curves of the average sizes of the samples with more or less reducing agent as the reaction time went on. | ||
As shown in the fitted curve, the particle-size distribution substantially conformed to the Gaussian distribution mode, and the particle-size distribution of the particles in each sample was narrow. The statistical chart also showed that the as-synthesized samples had a relatively uniform particle size, and as the VR added increased, the average particle size of the NPs also gradually increased from 1.57, 1.99, 2.03, 2.14 nm to 2.42 nm, respectively. On the other hand, in the case when the VR of the liquid NaK alloy remained constant with an excessive mole number over the stoichiometric precursor metal ions, the particle diameter of the alloy NPs also increased as the reaction time went on. We fixed VR at 160 μL, and changed the tR from 10 s, 30 s, 2 min, and 5 min to 10 min, respectively. We found the particle-size distribution of the samples was also in a narrow range, and the average sizes of the Pt–Cu NPs gradually increased as tR increased, with the sizes being 2.42, 2.5, 2.6, 2.7, and 3.2 nm, respectively. A three-dimensional map was plotted and is shown in Fig. 2j, in which the variation of the average sizes of the as-synthesized Pt–Cu NPs with VR and tR can be clearly observed. Meanwhile, another statistical curve in Fig. 2k shows that the average sizes of the samples synthesized using more reducing agent had a larger increase than the smaller ones as the reaction time went on. These results indicated that it was easy to control the synthesis of sub-3 nm alloy NPs by changing the amount of reducing agent and the reaction time at room temperature.
Fig. 3a and b further display the detailed morphology of the Pt–Cu NPs formed on vxc-72 through the TEM and HRTEM images, which clearly show the uniform and dense dispersion of Pt–Cu NPs on the surface of the carbon supports. The average particle diameter of the alloy NPs was about 3 nm. Also, essentially, there were no separate alloy particles that were not supported on the carbon, which demonstrated that heterogeneous nucleation in the reduction system is advantageous in the presence of a carbon support. Fig. 3b shows a high-resolution transmission electron microscope (HRTEM) image of the Pt–Cu alloy NPs. The interplanar spacing was measured as 0.22 nm, which corresponded well to the (111) plane of the Pt–Cu crystallite. The formation of the alloy and the elemental surface fraction can be disclosed by the elemental mapping images of Pt, Cu and C in Fig. 3d, e and S2 in the ESI.† As expected, Pt, Cu atoms were evenly distributed on the alloy particles. X-ray adsorption near edge structure (XANES) and extended X-ray adsorption fine structure (EXAFS) spectroscopy were performed for the PtCu/C sample and reference Pt foil. The Pt L3-edge XANES results of the PtCu/C and the Pt foil are shown in Fig. 3f. The white line intensities in the normalized XANES spectra reflect the oxidation state of Pt. As shown in Fig. 3f, the white line intensity of PtCu/C is lower than that of the Pt foil, revealing a decrease in the oxidation state of Pt within PtCu/C. Fig. 3g shows the R-space Pt K edge of PtCu/C and the Pt foil, respectively. It can be seen from the elemental distribution map and Pt L3 edge XAFS spectra that the reduction method we used successfully reduced the added precursor to the alloy NPs.
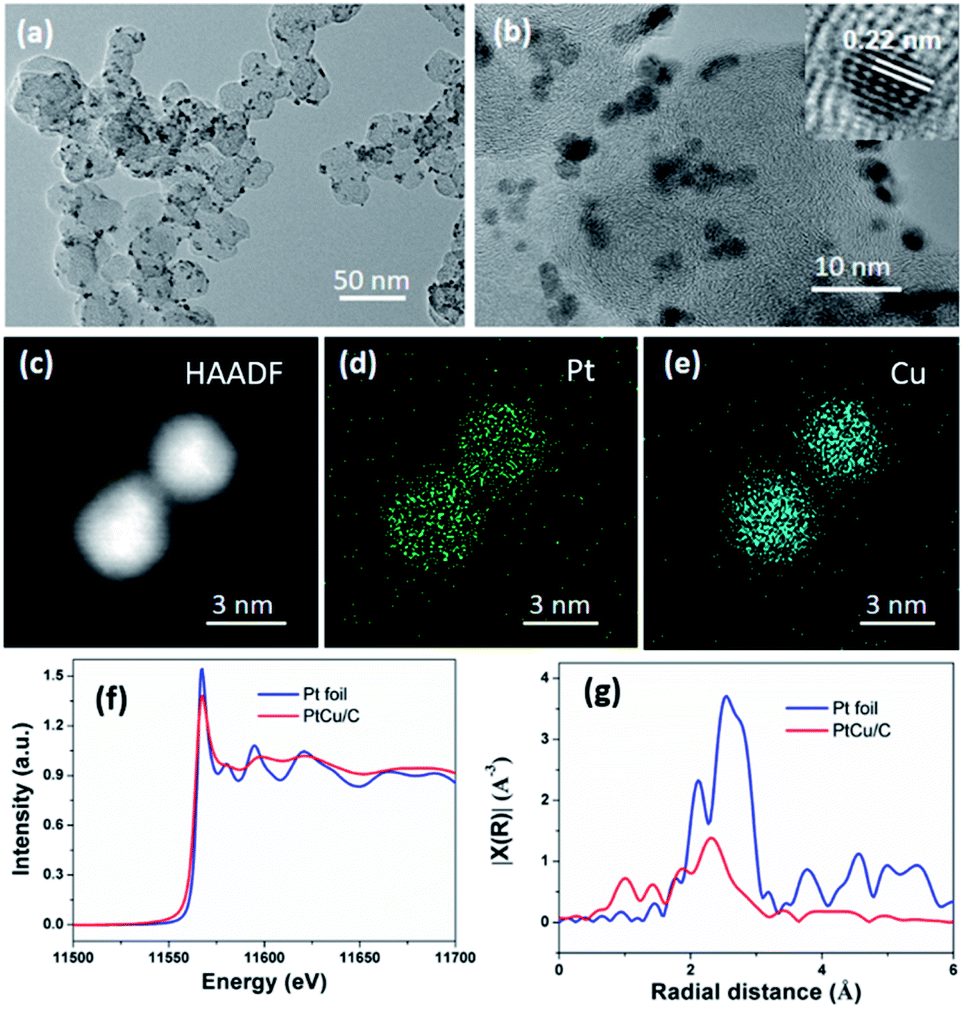 | ||
| Fig. 3 (a) TEM, (b) HRTEM images and (c–e) HAADF-STEM images and the corresponding EDS elemental maps of Pt–Cu NPs. (f) Pt L3-edge XANES for all the samples. (g) The k3-weighted R-space Fourier-transform EXAFS spectra of different catalysts and reference samples. | ||
Further, a series of ex situ X-ray diffraction (XRD) and ex situ X-ray photoelectron spectra (XPS) measurements were performed on the Pt–Cu nanoalloys to track the changes of their structure, composition, and valence states during the different reaction conditions. Fig. 4a shows the XRD patterns of the Pt–Cu alloy NPs with a face-centered cubic (FCC) structure and a lattice constant greater than copper and less than platinum. The peak appearing at 26° is the (002) crystal plane of the carbon support.45 The peaks appearing at 41° and 47° correspond to the (111) and (200) crystal planes of the Pt–Cu alloy NPs, respectively.34 On the basis of the XRD data shown in Fig. 4a, a clear transformation feature of the crystal lattice of the Pt–Cu nanoalloys during the different reaction conditions can be seen. When the reaction time was short (tR = 10 s) and the amount of the NaK alloy added was low (VR = 10 and 20 μL), it is most likely that Cu existed predominantly in the metallic alloy state. While, with increasing the reducing agent or extending the reaction time, the XRD peaks were shifted to the low angle direction, but close to that of the Pt–Cu alloy, indicating the formation of the Pt–Cu alloys. There were no separate characteristic peaks corresponding to Pt and Cu in the sample, which reflected that the as-synthesized alloy NPs appeared to be atomically mixed Pt–Cu alloy phases.
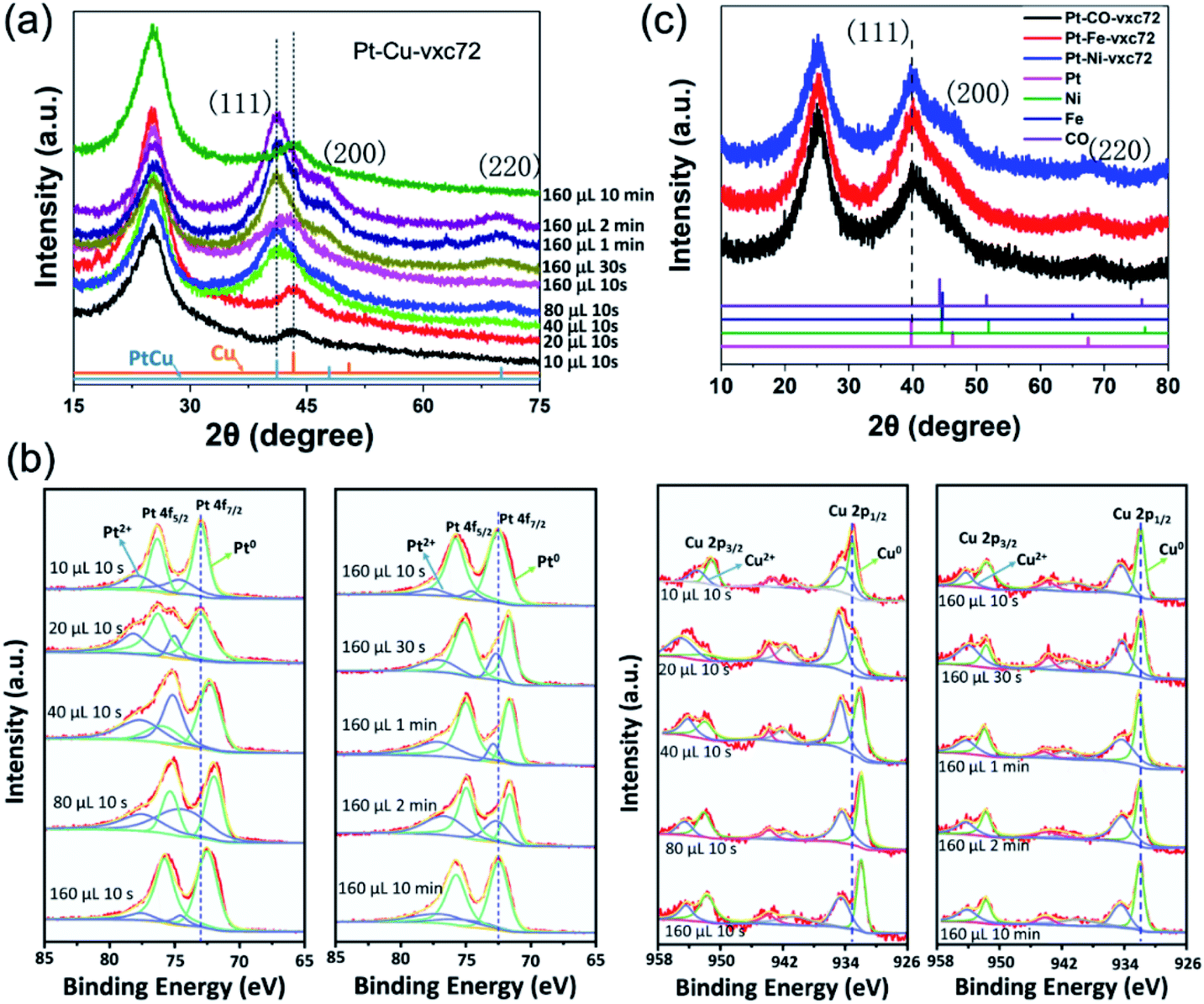 | ||
| Fig. 4 XRD patterns (a) and the Pt 4f regions of the XPS spectra (b) of a series of Pt–Cu-vxc72 samples at different conditions by changing the amount of reducing agent (VR) and the reaction time (tR). When fixing tR = 10 s, VR was changed from 10 μL to 160 μL; when fixing VR = 160 μL, tR changed from 10 s to 10 min. (c) XRD patterns of Pt–M-vxc72 (M = Fe, Co, Ni, VR = 160 μL, tR = 1 min). | ||
Fig. 4b shows the XPS spectra of a series of Pt–Cu nanocrystals on carbon supports. The relatively strong two peaks at 72.9 and 76.3 eV correspond to the metal Pt0, and the weaker peaks are derived from Pt2+. As the reducing agent or the reaction time increased, Pt 4f was shifted to a lower binding energy (BE). Thus, the lower-energy shift of Pt 4f indicated the formation of Pt–Cu alloys, which was in agreement with the XRD results. Likewise, the peaking of the Cu 2p orbital with the two peaks at 932.6 and 952.5 eV correspond to zero-valent copper. The low-shifting tendency of the Cu 2p peak can be clearly seen in the left graph in Fig. 4b, which is consistent with the trend of variation of Pt 4f. This negative shift phenomenon may be caused by an initial state effect, for example, charge transfer from a metal to a carrier, due to the small particle size, lattice strain, and many atoms having electron defects.33,46
To prove the universality of our synthesized strategy, a series of binary Pt–M (M = Fe, Co, Ni) and ternary Pt–Cu–Fe nanoalloys were deposited on a carbon support (vxc72), and were noted as Pt–Cu-vxc72, Pt–Co-vxc72, Pt–Ni-vxc72, Pt–Fe-vxc72, and Pt–Cu–Fe-vxc72. Fig. S2f–h† show the HAADF-STEM images and the corresponding EDS maps of Pt–Co NPs. Fig. 4c presents the XRD patterns of a series of as-synthesized binary alloys, which had an FCC structure with a lattice parameter closer to that of platinum. After comparison with the standard XRD spectra of Fe, Co, and Ni, it was found that no single peak of the corresponding metal appeared in the sample, and so it can be judged that the synthesized sample contained substantially no single metal particles and these had formed an alloy.
The overall morphology of the ternary Pt–Cu–Fe-vxc72 sample synthesized by the same method (VR = 160 μL, tR = 2 min) is shown in Fig. 5. The as-produced ternary alloy NPs were uniformly grown on carbon black. Within the entire larger display range, a relatively uniform and good dispersibility of the nanoparticles were demonstrated and no obvious agglomeration was observed. All of these topographical features contribute to further improving the catalytic performance of the material. The inside illustration is an HRTEM image of a Pt–Cu–Fe NPs, from which the crystal lattice of the alloy particles can be clearly shown. It could be calculated that the interplanar spacing of the nanoparticles was 0.23 nm, which corresponded to the (111) crystal plane of the alloy. Fig. 5c–f present the elemental distribution of the sample, showing the distribution of the four elements Pt, Cu, Fe, and C. The three metal elements Pt, Cu, and Fe were distributed in the nanoparticles, which further proved that we had successfully synthesized the ternary alloy. The valence state of the metal in the Pt–Cu–Fe-vxc72 sample could be analyzed by XPS, as shown in Fig. S3.† The survey XPS spectrum showed that the Pt–Cu–Fe-vxc72 contained elements such as Pt, Cu, Fe, C, and O. Among these, the O element may be derived from the partial oxidation of the sample when it was placed in the air.33,47
 | ||
| Fig. 5 TEM (a), HRTEM (b), BF-STEM (c), and HAADF-STEM (d) images and (e–h) the corresponding EDS elemental maps of Pt–Cu–Fe-vxc72. | ||
To prove the utility of the nanoalloys, the hydrogen evolution reaction (HER) performance of a series of Pt–Cu-vxc72 samples was performed and the results are shown in Fig. 6. The polarization curves were obtained in a 0.5 M sulfuric acid electrolyte at a sweep rate of 5 mV s−1 (Fig. 6a). It can be seen that the different reducing agents and reaction time factors had a great influence on the electrocatalytic HER performance. When the amount of the reducing agent added was 160 μL and the reaction time was 10 min, the best hydrogen evolution performance of Pt–Cu-vxc72 was obtained, that is, the overpotential was 17 mV at a current density of 10 mA cm−2. In addition, for a more intuitive comparison of the sample performance, we calculated the overpotential of each material at a current density of 10 mA cm−2 and prepared a columnar chart (Fig. 6c).
 | ||
| Fig. 6 (a) Linear sweep voltammograms of Pt–Cu-vxc72 at different reaction times and amounts of reducing agent. (b) Overpotentials and Tafel slopes of Pt–Cu-vxc72 synthesized at different times and at different amounts of reducing agent. (c) Tafel slope diagram of Pt–Cu-vxc72 samples synthesized with different reducing agents at 10 s. (d) Tafel slope diagram of Pt–Cu-vxc72 samples synthesized with different reducing times with 160 μL reducing agent. | ||
When the amount of the reducing agent was insufficient (10 μL) and the reaction time was short (10 s), the sample had poor HER performance, and the overpotential at a current density of 10 mA cm−2 was as high as 165 mV. Fig. S4† shows the HER stability of the sample in 0.5 M H2SO4 (160 μL 1 min). The Tafel slope of each sample can be obtained from Fig. 6c and d. It can be seen that the sample prepared by using 10 μL reducing agent and a reaction time of 10 s had a relatively poor performance, with the largest Tafel slope of about 68 mV dec−1. The best HER performance of the Pt–Cu-vxc72 sample (160 μL 10 min) had the smallest Tafel slope, about 22 mV dec−1.
The HER performances of Pt-based binary Pt–M (M = Fe, Co, Ni) alloys NPs grown on carbon supports are shown in Fig. 7a. The Pt–Co-vxc72 sample had the best catalytic performance with only 3 mV overpotential to achieve a current density of 10 mA cm−2 in these samples, which was better than Pt–Fe-vxc72 and Pt–Ni-vxc72. At a current density of 60 mA cm−2, Pt–Co-vxc72 required a minimum overpotential of 42 mV, followed by Pt–Ni-vxc72 at 56 mV, while Pt–Fe-vxc72 required the maximum overpotential (59 mV). The Tafel slopes corresponding to the three samples can be observed in Fig. 7b. The Pt–Co-vxc72 sample had the smallest Tafel slope with only 30 mV dec−1. The Tafel slope can reflect information about the reaction rate control steps, which can be used to analyze the kinetic factors of electrochemical reactions.20 Previous studies have shown that there are three main steps in the hydrogen evolution reaction in an acidic solution. The smaller Tafel slope indicated that in our synthetic samples, the HER process occurred via the Volmer–Heyrovsky mechanism.48 A small Tafel slope means that the current density increases rapidly with the increase of the overpotential, so it can be judged that Pt–Co-vxc72 had the best catalytic performance among the three. Although our synthetic carbon-supported binary alloys had excellent catalytic properties, their stability in acidic media was not satisfactory (Fig. S4†). Therefore, we added Fe(acac)3 as a precursor of iron during the synthesis, and synthesized ternary Pt–Cu–Fe nanoalloys.
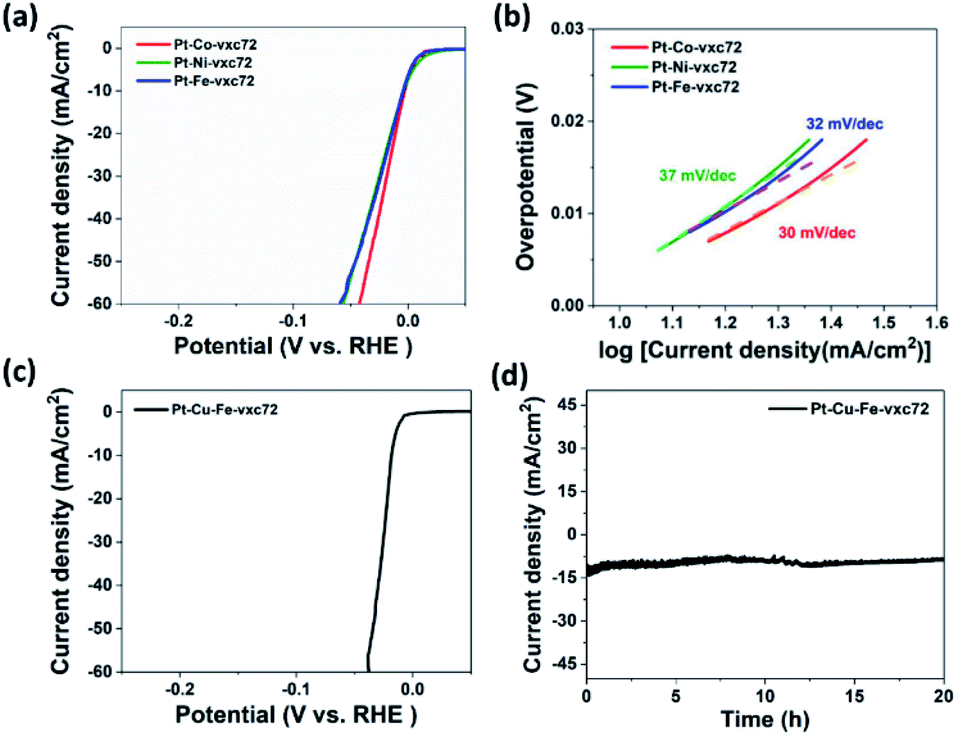 | ||
| Fig. 7 (a) Linear sweep voltammograms and (b) the corresponding Tafel plots of Pt–Co-vxc72, Pt–Ni-vxc72, and Pt–Fe-vxc72. (c) Linear sweep voltammograms of Pt–Cu–Fe-vxc72. (d) The current–time plot of the Pt–Cu–Fe-vxc72 in 0.5 M H2SO4. | ||
Fig. 7c shows the polarization curve of Pt–Cu–Fe-vxc72 in 0.5 M H2SO4 electrolyte. Although the amount of noble metal platinum was further reduced by doping the iron precursor to form a ternary alloy, the catalyst still maintained large activity in the hydrogen evolution reaction. Only 6 mV overpotential was needed to reach a current density of 10 mA cm−2. A current density of 60 mA cm−2 could be achieved with an overpotential of 16 mV. In addition, Fig. 7d shows the I-T curve of the material tested in the same electrolyte at the overpotential of 32 V vs. RHE, indicating that the ternary alloy formed by the addition of iron had good stability in the acidic environment. The current density was maintained for 20 h without significant attenuation, indicating that the activity of the catalyst was well maintained.
Overall, the as-produced metallic alloys exhibited excellent electrocatalytic hydrogen evolution performance, which could be further demonstrated by the CV test and the ECSA values calculated in Fig. S5† and the comparison of the mass specific activity among the Pt/C, Pt–Cu-vxc72, and Pt–Cu–Fe-vxc72 samples in Fig. S6.† The reasons for the excellent electrocatalytic performance are as follows. The absence of a surfactant in the reaction allowed for intimate contact between the Pt–Cu nanocrystals and the carbon support, which facilitated the catalysis of charge transfer in the HER. The ultrafine alloy NPs grown on carbon supports with uniform dispersion and high surface exposure provided a high density of catalytically active sites. The dispersibility between the particles was relatively good, so that the surface of the alloy NPs could be sufficiently exposed, which was favorable for the improvement of the electrocatalytic performance.24
4. Discussion and conclusions
The surfactant-free syntheses of carbon-supported binary metallic Pt–M (Cu, Fe, Co, Ni) alloys and ternary metallic Pt–Cu–Fe alloy were carried out via a liquid NaK alloy reductant at room temperature. This shear-assisted liquid metal reduction (SA-LMR) method provides an excellent platform for synthesis of multimetallic alloy NPs on carbon supports with the following features: rapid preparation rate, uniform, well-dispersed, and controllably sized nanoparticles. The size and surface composition of the Pt–Cu alloys deposited on carbon supports were tuned along with the variation of the reaction time and volume of the reducing agent. The transformation process of Pt–Cu alloy was characterized by ex situ XRD and XPS, which are helpful to reveal the nucleation-growth of nanoalloys during the ultrafast and strong reduction reaction process. The transformation between the Pt–Cu alloy dominant phase and single metal Cu dominant phase structures was clearly observed during the reaction.The synthetic technique also presents several advantages as follows. (I) The K (−2.925 V) and Na (−2.712 V) metals have more negative reduction potentials than most metals, which ensure that this method can create nanoalloys from nearly any metallic combination. Especially, the metallic alloys that are difficult to synthesize by conventional methods in aqueous solution can be synthesized in a non-aqueous strong reduction system by NaK alloy. (II) The particle size, composition as well as the final structure can be handily and effectively tuned by controlling the reaction conditions (shearing time and volume of the reducing agent), which benefits the observation and optimization of new nanocatalysts. The uniform alloying of the Pt atoms in the as-formed PtCu NPs is proved in Fig. S7.† The shearing force generated by the rotor accelerates the homogenization process of the reducing agent with the reaction precursor and the carrier carbon black, so that the reduction reaction can occur rapidly and uniformly throughout the reaction system. (III) The rapid (synthesis in tens of seconds), low energy cost (at room temperature), and scalable (through a shearing treatment) synthetic procedure could enable the highly efficient production of alloy NPs. (IV) Moreover, the synthesis process does not require the addition of a surfactant to inhibit the growth of crystal grains. Instead, a strong reducing agent is used to achieve a process of explosive nucleation in a very short period of time. After the addition of the reducing agent, the concentration of the atoms in the solution rises rapidly to reach the concentration of nucleation, which greatly shortens the nucleation time and ensures that the growth grain size of the nanocrystals tends to be uniform. Meanwhile, a general method for synthesizing alloy nanocrystals without a surfactant at room temperature was proposed, which paves the way for exploring the application of tuning the size and composition of alloy nanocrystals to obtain the desired properties. The SA-LMR method is universal and can synthesize a series of bimetallic catalysts easily and quickly. The utilization of the shear-assisted liquid metal reduction approach to obtain ultrafine alloy NPs opens up new avenues for designing highly efficient nanocatalysts. This method greatly improves the synthesis rate of this type of catalyst, and provides a prospect for large-scale mass production and industrialization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21501172), the Key R & D programs in Shandong (2018GGX102038), the Applied Basic Research Program Project of Qingdao (No. 17-1-1-69-jch), the World-Class University and Discipline Program of Shandong Province and the Taishan Scholars Advantageous and Distinctive Discipline Program for supporting the research team of energy storage materials of Shandong Province, P. R. China.References
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS
.
- H. Shi, Z. Fang, X. Zhang, F. Li, Y. Tang, Y. Zhou, P. Wu and G. Yu, Nano Lett., 2018, 18, 3193–3198 CrossRef CAS
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS
- Q. Wang, Z. L. Zhao, Z. Zhang, T. Feng, R. Zhong, H. Xu, S. T. Pantelides and M. Gu, Adv. Sci., 2019, 6, 1901279 Search PubMed
- X. Tian, X. Zhao, Y. Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS
- H. Fang, J. Yang, M. Wen and Q. Wu, Adv. Mater., 2018, 30, 1705698 CrossRef
- J. P. Camden, J. A. Dieringer, J. Zhao and R. P. Van Duyne, Acc. Chem. Res., 2008, 41, 1653–1661 CrossRef CAS
- X. Huang, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan and T. Mueller, Science, 2015, 348, 1230–1234 CrossRef CAS
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC
- X. Tan, S. Prabhudev, A. Kohandehghan, D. Karpuzov, G. A. Botton and D. Mitlin, ACS Catal., 2015, 5, 1513–1524 CrossRef CAS
- W. Tu, W. Luo, C. Chen, K. Chen, E. Zhu, Z. Zhao, Z. Wang, T. Hu, H. Zai, X. Ke, M. Sui, P. Chen, Q. Zhang, Q. Chen, Y. Li and Y. Huang, Adv. Funct. Mater., 2019, 29, 1908230 CrossRef
- S. Xu, G. Zhong, C. Chen, M. Zhou, D. J. Kline, R. J. Jacob, H. Xie, S. He, Z. Huang, J. Dai, A. H. Brozena, R. Shahbazian-Yassar, M. R. Zachariah, S. M. Anlage and L. Hu, Matter, 2019, 1, 759–769 CrossRef
- K. Jiang, P. Wang, S. Guo, X. Zhang, X. Shen, G. Lu, D. Su and X. Huang, Angew. Chem., Int. Ed., 2016, 55, 9030–9035 CrossRef CAS
- Z. Zhang, Y. Gong, D. Wu, Z. Li, Q. Li, L. Zheng, W. Chen, W. Yuan and L. Y. Zhang, Int. J. Hydrogen Energy, 2019, 44, 2731–2740 CrossRef CAS
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS
- J. Chen, Y. Yang, J. Su, P. Jiang, G. Xia and Q. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 3596–3601 CrossRef CAS
- M. Li, H. Wang, W. Zhu, W. Li, C. Wang and X. Lu, Adv. Sci., 2019, 6, 1901833 Search PubMed
- M. Lao, K. Rui, G. Zhao, P. Cui, X. Zheng, S. X. Dou and W. Sun, Angew. Chem., Int. Ed., 2019, 58, 5432–5437 CrossRef CAS
- Y. Wang and A. S. Hall, ACS Energy Lett., 2020, 5, 17 CrossRef CAS
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS
- Z. Zhao, C. Chen, Z. Liu, J. Huang, M. Wu, H. Liu, Y. Li and Y. Huang, Adv. Mater., 2019, 31, 1808115 CrossRef
- A. Dutta and J. Ouyang, ACS Catal., 2015, 5, 1371–1380 CrossRef CAS
- F. Wu, D. Zhang, M. Peng, Z. Yu, X. Wang, G. Guo and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 4952–4956 CrossRef CAS
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782 CrossRef CAS
- B. N. Wanjala, B. Fang, S. Shan, V. Petkov, P. Zhu, R. Loukrakpam, Y. Chen, J. Luo, J. Yin, L. Yang, M. Shao and C. J. Zhong, Chem. Mater., 2012, 24, 4283–4293 CrossRef CAS
- A. Mondal and N. R. Jana, ACS Catal., 2014, 4, 593–599 CrossRef CAS
- S. Zhang, Ö. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS
- Y. Yu, W. Yang, X. Sun, W. Zhu, X. Z. Li, D. J. Sellmyer and S. Sun, Nano Lett., 2014, 14, 2778–2782 CrossRef CAS
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef CAS
- W. Y. Zhao, B. Ni, Q. Yuan, P. L. He, Y. Gong, L. Gu and X. Wang, Adv. Energy Mater., 2017, 7, 1601593 CrossRef
- X. Y. Huang, L. X. You, X. F. Zhang, J. J. Feng, L. Zhang and A. J. Wang, Electrochim. Acta, 2019, 299, 89–97 CrossRef CAS
- C. Wang, D. Van Der Vliet, K. L. More, N. J. Zaluzec, S. Peng, S. Sun, H. Daimon, G. Wang, J. Greeley, J. Pearson, A. P. Paulikas, G. Karapetrov, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2011, 11, 919–926 CrossRef CAS
- Q. L. Zhu, D. C. Zhong, U. B. Demirci and Q. Xu, ACS Catal., 2014, 4, 4261–4268 CrossRef CAS
- Z. Sun, J. Masa, W. Xia, D. König, A. Ludwig, Z. A. Li, M. Farle, W. Schuhmann and M. Muhler, ACS Catal., 2012, 2, 1647–1653 CrossRef CAS
- Y. Yao, Z. Huang, P. Xie, T. Li, S. D. Lacey, M. Jiao, H. Xie, K. K. Fu, R. J. Jacob, D. J. Kline, Y. Yang, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar and L. Hu, ACS Appl. Mater. Interfaces, 2019, 11, 29773–29779 CrossRef CAS
- H. Chen, D. Wang, Y. Yu, K. A. Newton, D. A. Muller, H. Abruña and F. J. Disalvo, J. Am. Chem. Soc., 2012, 134, 18453–18459 CrossRef CAS
- S. Hu, M. Tian, E. L. Ribeiro, G. Duscher and D. Mukherjee, J. Power Sources, 2016, 306, 413–423 CrossRef CAS
- S. Hu, K. M. Cheng, E. L. Ribeiro, K. Park, B. Khomami and D. Mukherjee, Catal. Sci. Technol., 2017, 7, 2074–2086 RSC
- S. Hu, G. Goenaga, C. Melton, T. A. Zawodzinski and D. Mukherjee, Appl. Catal., B, 2016, 182, 286–296 CrossRef CAS
- M. Feng, M. Zhang, H. Zhang, X. Liu and H. Feng, Carbon, 2019, 153, 217–224 CrossRef CAS
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS
- Z. Mingshan, C. Xiaoyan, F. Mamoru, Z. Junying and M. Tetsuro, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef
- B. Zhang, H. Zhu, M. L. Zou, X. R. Liu, H. Yang, M. Zhang, W. W. Wu, J. M. Yao and M. L. Du, J. Mater. Sci., 2017, 52, 8207–8218 CrossRef CAS
- G. Zhu, J. Liu, S. Li, Y. Zuo, D. Li and H. Han, ACS Appl. Energy Mater., 2019, 2, 2862–2869 CrossRef CAS
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 1–21 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00412j |
| This journal is © The Royal Society of Chemistry 2020 |