
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrowth mechanism of CH3NH3I in a vacuum processed perovskite†
Beom-Soo
Kim
a,
Yoonjay
Han
a and
Jang-Joo
Kim
 *ab
*ab
aDepartment of Materials Science and Engineering, Seoul National University, Seoul, 151-742, South Korea. E-mail: jjkim@snu.ac.kr
bResearch Institute of Advanced Materials (RIAM), Seoul National University, Seoul, 151-744, South Korea
First published on 24th July 2020
Abstract
In the field of halide perovskite research, the growth of high quality films has been a critical issue. Among the reported growth methods, vacuum processes have attracted much attention due to their accurate controllability and high reproducibility, as proven in the manufacture of vacuum deposited organic-light-emitting-diode industry. In a vacuum process, the major difficulty for growing a perovskite film is control of a precursor, methylammonium iodide (MAI), originating from its uncontrollable behavior i.e., a high working pressure and poor adsorption characteristics. Thus, it is crucial to understand the growth mechanism of MAI vapor for the successful application of vacuum processes in the growth of halide perovskite films. In this paper, we report the growth mechanism and deposition kinetics of MAI in a vacuum. Unlike that of conventional materials evaporated in a vacuum, the deposition rate of MAI was found to be much faster on the reactive surface, PbI2, compared to other non-reactive materials. Surprisingly, a very thin (2 nm-thick) PbI2 layer increased the initial growth rate of MAI 2.7-fold. Based on the real-time monitored data from a quartz microbalance and surface study, we suggest dipole-induced adsorption as the MAI growth mechanism on PbI2 and the perovskite in the vacuum process. We believe that this work will provide meaningful insight into film growth in vacuum processed perovskites.
Introduction
MAPbI3-based perovskites (MA = CH3NH3+) have attracted considerable attention over the past several years, resulting in solar cells and light-emitting diodes with high efficiency. One of the critical issues in this field is reproducible fabrication of high-quality perovskite films. After Kojima et al. (2009) reported a perovskite solar cell produced using a one-step spin-coating process,1 various methods have been introduced to improve film quality, including sequential deposition,2,3 solvent engineering,4,5 vacuum processes,6–10 hybrid deposition,11–13 direct contact of methylammonium iodide (MAI) hot powders on lead halides,14–16 and growth under humid conditions.17,18 The power conversion efficiency of perovskite solar cells has improved from 3.8% in early studies to over 20% through optimization of the growth conditions, thus demonstrating the importance of perovskite film quality for optoelectronic device applications.1,4,19–26 A vacuum process is particularly promising and has distinct characteristics from solution processes. For example, high-purity materials can be easily deposited on large-area substrates, eliminating atmospheric effects and solvent exposure for good reproducibility. Additionally, a conformal morphology on textured Si solar cells can readily be obtained to realize efficient perovskite/silicon tandem solar cells.27–30 Following Liu's successful application of vacuum processes to perovskite solar cells, several groups have reported perovskite solar cell fabrication using vacuum co-deposition processes;6–10,31–46 however, controlling and/or calibrating the deposition of MAI is a known to be a difficult problem7,8,10,31,32,36,40,47–50 and has been attributed to the gas-like behavior of MAI. Several studies have been reported to explain the origin of behaviour of MAI in a vacuum, indicating that the decomposition of MAI, impurities, and variations in adhesion property of MAI can be attributed to the uncontrollable behaviour of MAI in a vacuum process.51–54 In this regard, accurate monitoring and control over MAI deposition in vacuum processes require a better understanding of the growth mechanism in a vacuum, which is closely related to deposition parameters and perovskite film formation.In this paper, we report the growth mechanism and resulting deposition kinetics of MAI vapour in a vacuum, which directly affects the deposition parameters and film formation in the vacuum process. MAI showed surface-dependent growth characteristics, which varied for the reactive surface, PbI2. On the PbI2 layer, MAI growth is affected by reaction, diffusion, and dipole effects, as evidenced in quartz crystal microbalance (QCM), surface potential microscopy (SPM), and atomic force microscopy (AFM) measurements. Our results suggest that the surface dipoles of the perovskite formed by reacting with the adsorbed MAI trigger the adsorption of polar molecules such as MAI which in turn increases the adsorption rate for film formation in a vacuum.
Results and discussion
Table 1 displays the thicknesses of vacuum-deposited MAI on different substrates in the same batch for 60 min at a pressure of 7 × 10−5 Torr. The thicknesses are measured by using a profilometer after scratching the film. Interestingly, the thickness varied remarkably depending on the presence or absence of a PbI2 layer, with relatively small variations on other substrates. The MAI on the PbI2 layer is ca. 2.1–2.5 times thicker than on substrates without a PbI2 sub-layer. Here, we speculate that the variation of MAI thickness on different substrates may explain why it is difficult to accurately calibrate MAI thickness or control MAI and/or MAPbI3 deposition in vacuum processes, as reported previously.7,8,10,31,32,36,40,47–50| a MAI: methylammonium iodide (MA = CH3NH3+); NPB: N,N′-di(naphthalene-1-yl)-N,N′-diphenyl-benzidine; C60: fullerene; ITO: indium tin oxide. | |||||
|---|---|---|---|---|---|
| Substrate | ITO | NPB | C60 | Au | PbI2 |
| Thickness of MAI (nm) | 72 | 76 | 85 | 86 | 184 |
The deposited film thickness also depends on the thickness of the initial PbI2 layers, as shown in Fig. 1. The initial thickness of the PbI2 layer varied from 0, on the indium tin oxide (ITO) surface, to 200 nm; the substrates were exposed to MAI for 60 min at a pressure of 7 × 10−5 Torr (in the same batch). The black line indicates the measured thickness of the film (MAI + PbI2) and the red line represents the thickness change, which is the thickness obtained by subtracting the initial thickness of the PbI2 from the thickness indicated by the black line (MAI + PbI2). Surprisingly, only a 2 nm-thick PbI2 layer resulted in more than twice the MAI film thickness compared to the ITO surface without a PbI2 layer; specifically, the MAI film thickness was 70 nm on ITO and 155 nm on 2 nm-thick PbI2. The MAI thickness showed little change as the PbI2 layer thickness increased from 2 to 200 nm. Deposition of MAI transformed the PbI2 layer into a perovskite film, as shown in Fig. 1b, consistent with the findings of previous reports.6,7,10,40 X-ray diffraction measurements showed a perovskite (110) peak at 14.2° for 20, 100, and 200 nm-thick PbI2 layers. Notably, the MAI/PbI2 2 nm-thick film exhibited MAPbI3 perovskite (310) peaks with low intensity at 31.7°. A strong (310) peak is often observed for thin perovskites, such as for perovskite quantum dots,55 indicating the formation of a thin perovskite layer on the 2 nm-thick PbI2 film; however, the origin of this preferred orientation is not yet clear. The MAI film deposited on ITO showed the same peaks as the films grown on PbI2 layers, but with slightly lower crystallinity. The 200 nm-PbI2 substrate exhibited a strong PbI2 (001) peak at 12.6°, indicating the substantial presence of unreacted PbI2.
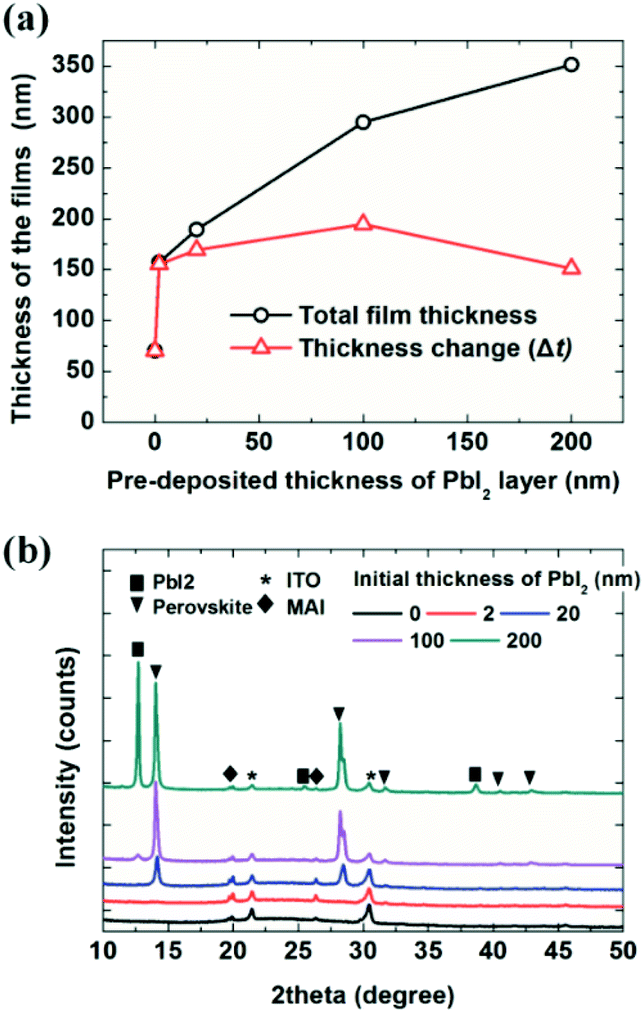 | ||
| Fig. 1 (a) Thickness of MAI/PbI2 films with respect to the initial PbI2 thickness after a 60 min deposition under a constant MAI working pressure. (b) X-ray diffraction patterns of the MAI layer deposited on PbI2 films of different thicknesses. | ||
To analyze the deposition kinetics, we devised an experiment using two QCMs in a vacuum, as shown in Fig. 2. The two QCM sensors were located side-by-side (only 3 cm apart), approximately 40 cm above the source in the vacuum chamber (Fig. 2a). Only one of the sensors was covered with PbI2 (x nm) via vacuum deposition before MAI exposure. Both sensors were then exposed to MAI at a constant working pressure of 7 × 10−5 Torr for 1 h. Fig. 2b shows the monitored weight increase with exposure time on the PbI2 (x nm) QCM. The final deposited weights were 19.6, 34.9, 48.3, 64.4, and 70.5 μg cm−2, as the PbI2 layer thickness increased from 0 to 2, 20, 100, and 200 nm, respectively. It is interesting to note that the weight gain upon exposure to MAI increased with the initial thickness of PbI2, which differed from the thickness change tendencies shown in Fig. 1a. Again, a much larger amount (∼1.8 times) of MAI was deposited on 2 nm-thick PbI2 compared to the bare ITO substrate. Fig. 2c shows the variation in the deposition rate and working pressure with exposure time. The deposition rates increased with the working pressure in the early stages of deposition, reaching a peak value as the working pressure approached the set point of 7 × 10−5 Torr. The rates increased more rapidly to higher peak values with the initial PbI2 layer, and then slowed gradually under the constant working pressure. The deposition rates for the different QCMs reached the same value after ∼40 min. Taken together, understanding the dependence of the deposition rates of MAI on PbI2 layers is a crucial step in resolving the growth mechanism of MAI in a vacuum. We speculate that the deposition characteristics are related to adsorption and diffusion processes of MAI into PbI2 to form MAxPbI2+x, or the deposition of MAI if the reaction does not take place, e.g., on ITO or after completion of the conversion from PbI2 to MAPbI3. A much higher deposition rate on the thin 2 nm-thick PbI2 layer compared to the bare ITO substrate suggests an attractive force for MAI adsorption on the PbI2 surface.
 | ||
| Fig. 2 (a) Schematic figure of a comparative deposition experiment on quartz crystal microbalances (QCMs). (b) The monitored deposited weight of MAI on QCM/PbI2 (x nm). (c) The monitored deposition rate of MAI on QCM/PbI2 (x nm) and working pressures. | ||
A measurement mode that combined AFM with scanning Kelvin probe microscopy (SKPM) was applied to examine the growth characteristics of the films. Thermally evaporated PbI2 on ITO formed smooth films covering the ITO surface, with the exception of the 200 nm-thick PbI2 film, which showed a slightly rougher surface than the thinner layers (Fig. 3a and S2–S4†). Deposition of MAI on the ITO substrate gradually increased the surface roughness, which is relevant to the intrinsic properties of the MAI film deposited on a non-reactive surface. MAI deposition on 2 nm-thick PbI2 resulted in a rapid increase in roughness after 10 min of exposure. In contrast, MAI deposition for 10 min on 20, 100, and 200 nm-thick PbI2 layers did not increase the roughness significantly, and even reduced the roughness of the 200 nm-thick PbI2 layer. This indicates that MAI deposited on PbI2 diffuses into PbI2 to form a perovskite in the early stages. Exposure times longer than 20–30 min increased the roughness, depending on the initial PbI2 thickness, indicating that MAI grows on the perovskite films after a certain time. Since PbI2 remains after exposure of 100 and 200 nm-thick PbI2 films to MAI (Fig. 1b) for 60 min, the composition of MAI (x value) in MAxPbI(2+x) gradually changed from 0 (PbI2) at the ITO/PbI2 interface to 1 (MAPbI3), followed by deposition of MAI on top of MAPbI3. The transition from perovskite formation to MAI growth on the surface was evidenced by the change in density of the deposited films, as shown in Fig. 3b, which was calculated according to the thickness and weight change of the deposited films (Fig. S5†) for each time interval. The density of MAI, PbI2, and MAPbI3 is 2.22, 4.15, and 6.16 g cm−3, respectively.36,56,57Fig. 3b shows that the density of the deposited film decreased gradually, approaching the density of MAI over time. During the first 10 min, the perovskite with x < 1 (or a mixture of perovskite and PbI2) formed for films thicker than 20 nm; the x value decreased with increasing PbI2 thickness. However, MAI was mostly deposited after 30 min, even on the 200 nm-thick PbI2 film, indicating the growth of MAI on MAPbI3. The change in the surface potential of the films as a function of exposure time (Fig. 3c) also provides information on the growth of the perovskite, MAI, or MAI on the perovskite over time. The surface potential is expressed as Vs = (ϕtip − ϕsample)/e, where Vs is the external voltage applied to the tip to nullify the displacement current, ϕtip and ϕsample are the work functions of the tip (Cr/Pt coated tip with a work function of 5.0 eV (ref. 58)) and the sample, respectively, and e is the electronic charge. Measurements of ITO and the PbI2 film before MAI deposition indicated work functions of 4.9 and 5.7 eV, respectively, approximately 0.3 eV higher than the work function obtained by ultraviolet photoelectron spectroscopy (UPS); UPS measures secondary electrons from the Fermi edge with a low-energy cut-off.59,60 The surface potential of the 2 nm-thick PbI2 film measured by SKPM was 0.1 to 0.2 V higher than that of the thick films. This was attributed to electron transfer from ITO to PbI2, resulting in the Fermi level of PbI2 being closer to the conduction band, thus lowering the work function of PbI2 near the ITO/PbI2 interface. The surface potential of the MAI films on the ITO substrate shifted gradually to 0.4 V, which corresponds to a work function of 4.6 eV for MAI.60 On thin (2 to 20 nm-thick) PbI2 films, the potential shifted rapidly to 0.2 V during the first 20 min, corresponding to that of MAI, and then followed the same growth trend as that on ITO. The surface potential of MAI-deposited films grown on 100 and 200 nm-thick PbI2 films gradually shifted to ∼0.1 V during the first 30 min and then slowly increased to reach 0.3 V at 60 min. Given a reported surface potential of −0.1 V for MAPbI3 using SKPM,61,62 it can be deduced that the potential change on thick PbI2 involves a change in the surface from PbI2 to the perovskite to MAI, based on roughness and density measurements (Fig. 3a and b).
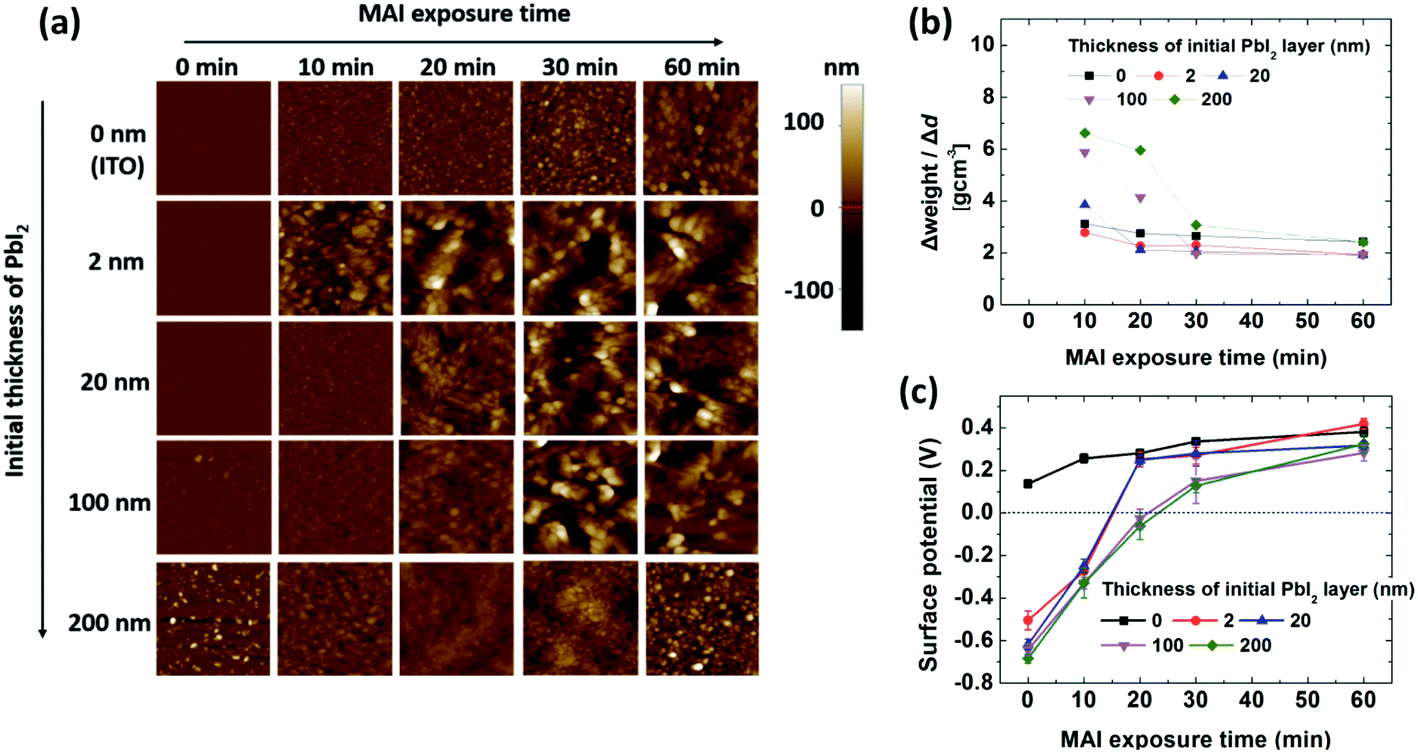 | ||
| Fig. 3 (a) Topographic images (5 × 5 μm2) and (b) average density of the deposited films calculated according to the weight gain from the QCM and thickness increase from the surface profiler at the given times. (c) Surface potential of the films, as measured by scanning Kelvin probe microscopy (SKPM). Samples of PbI2 on ITO substrates (PbI2 thickness: 0, 2, 20, 100, and 200 nm) were exposed to MAI for 0, 10, 20, 30 and 60 min. | ||
Interpretation
The growth of MAI on PbI2 in a vacuum can be described in terms of the diffusion of MAI forming MAxPbI2+x with moving boundaries, up to a certain point in time, where this depends on the initial thickness of PbI2, in which x decreases gradually from the surface toward the film interior, as explained schematically in Fig. 4a. In the early stages of film deposition, the diffusion of MAI into PbI2 is faster than the deposition of MAI to form MAxPbI2+x at the surface. However, the diffusion of MAI into PbI2 slows down over time and MAI eventually begins to grow on MAxPbI2+x when x ≅ 1 or slightly higher than 1; in this case, the deposition rate of MAI is higher than the diffusion rate at the perovskite surface. The growth of the perovskite via MAI diffusion into thick PbI2 layers (100 and 200 nm-thick films) results in changes in density and surface potential over time, as shown in Fig. 3b and c, respectively. However, this observation does not hold for very thin PbI2 layers, e.g., a 2 nm-thick layer, in which MAI growth is expected within a short time (<10 min). Importantly, therefore, the effects of interdiffusion of MAI into PbI2 can be excluded to explain this enhanced growth rate, where the surface is fully covered with MAI after converting all of the 2 nm PbI2. The surface potential shown in Fig. 3c indicates that, however, the MAI potential is negative (−0.3 V) at 10 minutes exposure on the 2 nm PbI2 substrate. Afterwards, the potential becomes 0.3–0.4 V eventually. This change of surface potential upon the same material, MAI, deposition may originate from the orientation change of polar MAI on the ferroelectric surface, MAPbI3, as described schematically in Fig. 4b.63–66 The ordering of MAI decreases gradually with increasing MAI thickness, such that the surface potential of MAI converges to that of bulk MAI. This explains the much higher deposition rate of MAI on very thin 2 nm-thick PbI2 compared to bare ITO (Fig. 1). Specifically, the surface polarization of ferroelectric MAPbI3 attracts MAI molecules via a Coulomb interaction or dipole-induced absorption. This observation can be supported by similar reports on enhanced adsorption of gas molecules such as CH3OH, CO2 and CO on an oxide perovskite.67,68 In these regards, we can conclude that the surface potential changes in Fig. 3c are related to the conversion of PbI2 to MAxPbI2+x, combined with the orientation of MAI molecules on the ferroelectric substrate of MAxPbI2+x associated with dipole-induced absorption.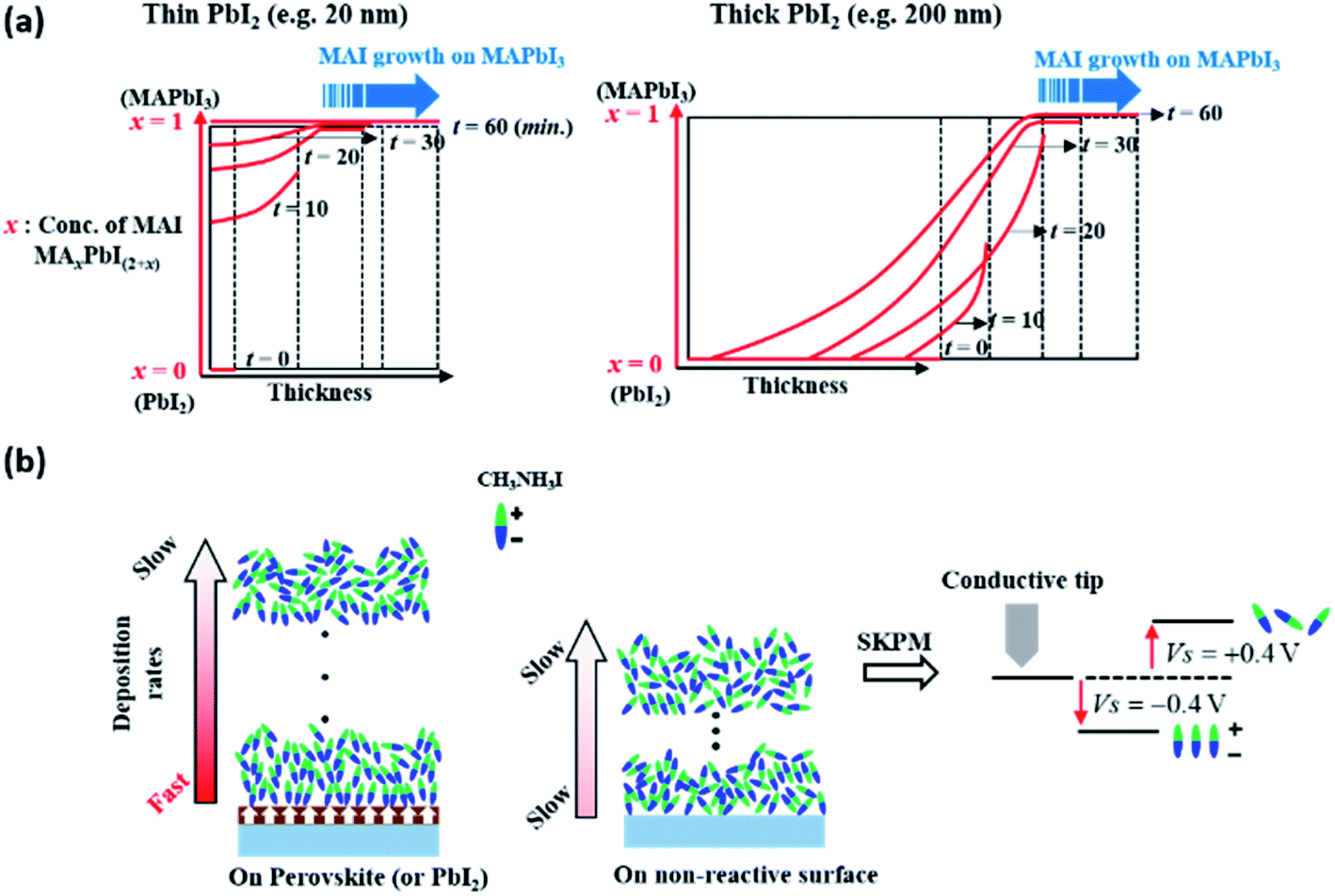 | ||
| Fig. 4 Schematic illustration of (a) MAI growth with respect to the PbI2 layer thickness and (b) an interpretation of the change in the surface potential of MAI shown by SKPM. | ||
Conclusion
We investigated the growth mechanism of MAI in a vacuum process. MAI showed surface-dependent deposition characteristics that varied significantly in the presence of a reactive surface, PbI2 layer. As the thickness of the PbI2 layer increased, the deposition rate of MAI increased rapidly initially and then decreased to a similar value after a certain time, depending on the initial PbI2 thickness under a constant working pressure. We suggest a dipole-induced adsorption mechanism for MAI in vacuum processes on PbI2 or perovskite. This directly affects the film thickness and the thickness monitor, QCM, which are closely related to deposition parameters of the film fabrication process. Understanding the surface-dependent growth characteristics of MAI is necessary to better control the perovskite film growth in vacuum processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Midcareer Research Program (2018018455) through a National Research Foundation (NRF) grant funded by the Ministry of Science, Information, and Communication Technology (ICT) and Future Planning (MSIP).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed
- D. Bi, S.-J. Moon, L. Häggman, G. Boschloo, L. Yang, E. M. J. Johansson, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, RSC Adv., 2013, 3, 18762–18766 RSC
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed
- O. Malinkiewicz, A. Yella, Y. H. Lee, G. M. Espallargas, M. Graetzel, M. K. Nazeeruddin and H. J. Bolink, Nat. Photonics, 2013, 8, 128–132 CrossRef
- L. E. Polander, P. Pahner, M. Schwarze, M. Saalfrank, C. Koerner and K. Leo, APL Mater., 2014, 2, 081503 CrossRef
- A. S. Subbiah, A. Halder, S. Ghosh, N. Mahuli, G. Hodes and S. K. Sarkar, J. Phys. Chem. Lett., 2014, 5, 1748–1753 CrossRef CAS PubMed
- B.-S. Kim, T.-M. Kim, M.-S. Choi, H.-S. Shim and J.-J. Kim, Org. Electron., 2015, 17, 102–106 CrossRef CAS
- L. K. Ono, S. Wang, Y. Kato, S. R. Raga and Y. Qi, Energy Environ. Sci., 2014, 7, 3989–3993 RSC
- M. R. Leyden, L. K. Ono, S. R. Raga, Y. Kato, S. Wang and Y. Qi, J. Mater. Chem. A, 2014, 2, 18742–18745 RSC
- Y. Peng, G. Jing and T. A. Cui, J. Mater. Chem. A, 2015, 3, 12436–12442 RSC
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622–625 CrossRef CAS PubMed
- J. Zhang, Y. Zhao, D. Yang, C. Li and S. Liu, RSC Adv., 2016, 6, 93525–93531 RSC
- X. Zhu, D. Yang, R. Yang, B. Yang, Z. Yang, X. Ren, J. Zhang, J. Niu, J. Feng and S. Liu, Nanoscale, 2017, 9, 12316–12323 RSC
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed
- G. D. Niu, W. Z. Li, F. Q. Meng, L. D. Wang, H. P. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705–710 RSC
- National renewable energy laboratory, Best research-cell efficiency chart, https://www.nrel.gov/pv/cell-efficiency.html.
- X. Zeng, T. Zhou, C. Leng, Z. Zang, M. Wang, W. Hu, X. Tang, S. Lu, L. Fang and M. Zhou, J. Mater. Chem. A, 2017, 5, 17499–17505 RSC
- M. Wang, H. Wang, W. Li, X. Hu, K. Sun and Z. Zang, J. Mater. Chem. A, 2019, 7, 26421–26428 RSC
- T. Zhou, M. Wang, Z. Zang and L. Fang, Adv. Energy Mater., 2019, 9, 1900664 CrossRef
- M. Wang, Z. Zang, B. Yang, X. Hu, K. Sun and L. Sun, Sol. Energy Mater. Sol. Cells, 2018, 185, 117–123 CrossRef CAS
- X. Liu, M. Wang, F. Wang, T. Xu, Y. Li, X. Peng, H. Wei, Z. Guan and Z. Zang, IEEE Electron Device Lett., 2020, 41, 1044–1047 Search PubMed
- H. Ji, Z. Shi, X. Sun, Y. Li, S. Li, L. Lei, D. Wu, T. Xu, X. Li and G. Du, ACS Appl. Mater. Interfaces, 2017, 9, 42893–42904 CrossRef CAS PubMed
- Z. Shi, L. Lei, Y. Li, F. Zhang, Z. Ma, X. Li, D. Wu, T. Xu, Y. Tian, B. Zhang, Z. Yao and G. Du, ACS Appl. Mater. Interfaces, 2018, 10, 32289 CrossRef CAS PubMed
- J. Ávila, C. Momblona, P. P. Boix, M. Sessolo and H. J. Bolink, Joule, 2017, 1, 431–442 CrossRef
- P.-S. Shen, Y.-H. Chiang, M.-H. Li, T.-F. Guo and P. Chen, APL Mater., 2016, 4, 091509 CrossRef
- H. Kim, K.-G. Lim and T.-W. Lee, Energy Environ. Sci., 2016, 9, 12–30 RSC
- L. K. Ono, M. R. Leyden, S. Wang and Y. Qi, J. Mater. Chem. A, 2016, 4, 6693–6713 RSC
- Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2014, 9, 106–112 CrossRef
- T.-W. Ng, C.-Y. Chan, M.-F. Lo, Z. Q. Guan and C.-S. Lee, J. Mater. Chem. A, 2015, 3, 9081–9085 RSC
- C. Gao, J. Liu, C. Liao, Q. Ye, Y. Zhang, X. He, X. Guo, J. Mei and W. Lau, RSC Adv., 2015, 5, 26175 RSC
- D. T. C. Tran, Y. Liu, E. S. Thibau, A. Llanos and Z.-H. Lu, AIP Adv., 2015, 5, 087185 CrossRef
- J. Borchert, H. Boht, W. Fränzel, R. Csuk, R. Scheer and P. Pistor, J. Mater. Chem. A, 2015, 3, 19842 RSC
- J. Teuscher, A. Ulianov, O. Müntener, M. Grätzel and N. Tetreault, ChemSusChem, 2015, 8, 3847–3852 CrossRef CAS PubMed
- Z. Su, F. Hou, F. Jin, L. Wang, Y. Li, J. Zhu, B. Chu and W. Li, Org. Electron., 2015, 26, 104–108 CrossRef CAS
- W. Ke, D. Zhao, C. R. Grice, A. J. Cimaroli, G. Fang and Y. Yan, J. Mater. Chem. A, 2015, 3, 23888–23894 RSC
- C. Momblona, L. Gil-Escrig, E. Bandiello, E. M. Hutter, M. Sessolo, K. Lederer, J. Blochwitz-Nimoth and H. J. Bolink, Energy Environ. Sci., 2016, 9, 3456–3463 RSC
- B.-S. Kim, M.-H. Choi, M.-S. Choi and J.-J. Kim, J. Mater. Chem. A, 2016, 4, 5663–5668 RSC
- D. Zhao, W. Ke, C. R. Grice, A. J. Cimaroli, X. Tan, M. Yang, R. W. Collins, H. Zhang, K. Zhu and Y. Yan, Nano Energy, 2016, 19, 88–97 CrossRef CAS
- L. Calió, C. Momblona, L. Gil-Escrig, S. Kazim, M. Sessolo, Á. Sastre-Santos, H. J. Bolink and A. Shahzada, Sol. Energy Mater. Sol. Cells, 2017, 163, 237–241 CrossRef
- H.-J. Song, H. Lee, S. Ahn, H. Song and C. Lee, Jpn. J. Appl. Phys., 2018, 57, 102303 CrossRef
- V. Arivazhagan, J. Xie, Z. Yang, P. Hang, M. M. Parvathi, K. Xiao, C. Cui, D. Yang and X. Yu, Sol. Energy, 2019, 181, 339–344 CrossRef CAS
- H. Xu, F. Xu, W. Wang, Y. Zhu, Z. Fang, B. Yao, F. Hong, J. Cui, F. Xu, R. Xu, C.-C. Chen and L. Wang, Org. Electron., 2019, 69, 329–335 CrossRef CAS
- R. Kottokkaran, H. A. Gaonkar, H. A. Abbas, M. Noack and V. Dalal, J. Mater. Sci.: Mater. Electron., 2019, 30, 5487–5494 CrossRef CAS
- C. W. Chen, H. W. Kang, S. Y. Hsiao, P. F. Yang, K. M. Chiang and H.-W. Lin, Adv. Mater., 2014, 26, 6647–6652 CrossRef CAS PubMed
- D. Yang, Z. Yang, W. Qin, Y. Zhang, S. Liu and C. Li, J. Mater. Chem. A, 2015, 3, 9401–9405 RSC
- S.-Y. Hsiao, H.-L. Lin, W.-H. Lee, W.-L. Tsai, K.-M. Chiang, W.-Y. Liao, C.-Z. Ren-Wu, C.-Y. Chen and H.-W. Lin, Adv. Mater., 2016, 28, 7013–7019 CrossRef CAS PubMed
- S. Wang, L. K. Ono, M. R. Leyden, Y. Kato, S. R. Raga, M. V. Lee and Y. Qi, J. Mater. Chem. A, 2015, 3, 14631–14641 RSC
- L. Liu, J. A. McLeod, R. Wang, P. Shen and S. Duhm, Appl. Phys. Lett., 2015, 107, 061904 CrossRef
- S. Olthof and K. Meerholz, Sci. Rep., 2017, 7, 40267 CrossRef CAS PubMed
- J. Borchert, I. Levchuk, L. C. Snoek, M. U. Rothmann, R. Haver, H. J. Snaith, C. J. Brabec, L. M. Herz and M. B. Johnston, ACS Appl. Mater. Interfaces, 2019, 11, 28851 CrossRef CAS PubMed
- K. B. Lohmann, J. B. Patel, M. U. Rothmann, C. Q. Xia, R. D. J. Oliver, L. M. Herz, H. J. Snaith and M. B. Johnston, ACS Energy Lett., 2020, 5, 710–717 CrossRef CAS PubMed
- J. H. Im, C. R. Lee, J. W. Lee, S. W. Park and N. G. Park, Nanoscale, 2011, 3, 4088–4093 RSC
- O. Yamamuro, M. Oguni, T. Matsuo and H. J. Suga, Chem. Thermodyn., 1986, 18, 939 CrossRef CAS
- J. Huang, K. Jiang, X. Cui, Q. Zhang, M. Gao, M. Su, L. Yang and Y. Song, Sci. Rep., 2015, 5, 15889 CrossRef CAS PubMed
- R. Vidyasagar, B. Camargo, E. Pelegova, K. Romanyuk and A. L. Kholkin, Ferroelectrics, 2017, 508, 115 CrossRef CAS
- J. S. Kim, B. Lagel, E. Moons, N. Johansson, I. D. Baikie, W. R. Salaneck, R. H. Friend and F. Cacialli, Synth. Met., 2000, 111, 311 CrossRef
- T.-W. Ng, H. Thachoth Chandran, C.-Y. Chan, M.-F. Lo and C.-S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 20280–20284 CrossRef CAS PubMed
- J. R. Harwell, T. K. Baikie, I. D. Baikie, J. L. Payne, C. Ni, J. T. S. Irvine, G. A. Turnbull and I. D. W. Samuel, Phys. Chem. Chem. Phys., 2016, 18, 19738–19745 RSC
- K. Song, Y. Wu, X. Chen, Y. He, L. Liu, G. Chen and R. Liu, AIP Adv., 2018, 8, 035114 CrossRef
- Y. Li, M. Behtash, J. Wong and K. Yang, J. Phys. Chem. C, 2018, 122, 177–184 CrossRef CAS
- J. Lahnsteiner, G. Kresse, A. Kumar, D. D. Sarma, C. Franchini and M. Bokdam, Phys. Rev. B, 2016, 94, 214114 CrossRef
- Y. Rakita, O. Bar-Elli, E. Meirzadeh, H. Kaslasi, Y. Peleg, G. Hodes, I. Lubomirsky, D. Oron, D. Ehre and D. Cahen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5504 CrossRef CAS PubMed
- S. Duhm, G. Heimel, I. Salzmann, H. Glowatzki, R. L. Johnson, A. Vollmer, J. P. Rabe and N. Koch, Nat. Mater., 2008, 7, 326 CrossRef CAS PubMed
- D. Li, M. H. Zhao, J. Garra, A. M. Kolpak, A. M. Rappe, D. A. Bonnell and J. M. Vohs, Nat. Mater., 2008, 7, 473–477 CrossRef CAS PubMed
- L. C. Tănase, N. G. Apostol, L. E. Abramiuc, C. A. Tache, L. Hrib, L. Trupină, L. Pintilie and C. M. Teodorescu, Sci. Rep., 2016, 6, 35301 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00466a |
| This journal is © The Royal Society of Chemistry 2020 |