DOI:
10.1039/D0NA00491J
(Paper)
Nanoscale Adv., 2020,
2, 4639-4651
Synthesis and catalytic evaluation of PVP–CeO2/rGO as a highly efficient and recyclable heterogeneous catalyst for multicomponent reactions in water†
Received
16th June 2020
, Accepted 24th June 2020
First published on 28th August 2020
Abstract
A highly efficient and eco-friendly route for the reduction of graphene oxide (GO) to reduced graphene oxide (rGO) was developed by using polyvinylpyrrolidone coated CeO2 NPs (PVP–CeO2) as a reducing and stabilizing agent. The resulting carbonaceous material, PVP–CeO2/rGO, was well characterized with different spectroscopic techniques such as Fourier Transform Infrared (FTIR) spectroscopy, Scanning Electron Microscopy (SEM), Energy Dispersive X-ray (EDX), elemental mapping, Transmission Electron Microscopy (TEM), Raman spectroscopy, powder X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET), X-ray Photoelectron Spectroscopy (XPS), and Thermal Gravimetric (TG) analyses. The material exhibited high catalytic potential towards multicomponent reactions for the synthesis of biologically relevant benzodiazepine derivatives in aqueous media. The efficiency of the material for the desired reaction was shown in the form of an excellent product yield (96–98%) and a very short reaction time period (7–10 min). The use of water as solvent and recyclability of the catalyst made the present protocol acceptable from a green perspective.
1. Introduction
Graphene is a 2D system with very strong sp2 hybridized carbon–carbon bonds arranged in a hexagonal manner to form a layered structure with a high aspect ratio and a large π-electronic surface. It has some unique and remarkable properties such as exceptionally high thermal conductivity, a greater surface area, very high carrier mobility at room temperature and excellent mechanical properties.1 Graphene is generally produced by three methods viz. chemical reduction of graphene oxide (GO),2 chemical vapor deposition3 and micro-mechanical stripping.4 The chemical reduction of GO to rGO is a simple and large scale route but it involves hazardous reducing agents such as hydrazine,5 NaBH4,6 dimethyl hydrazine,7 hydroquinone etc.8 Recently, the development of innovative synthetic approaches utilizing non-toxic and cost effective chemicals for the reduction of GO to rGO has attracted prime interest and as a result various environmentally benign methods have been adopted to synthesize rGO.9–12 The incorporation of metal nanoparticles into GO sheets has become a widespread green methodology for the reduction of GO resulting in the formation of metal functionalized rGO such as Au–rGO,13 Ag–rGO,14 Pd–rGO,15 W–rGO,9 Fe–rGO,16 Ti–rGO,17 Mo–rGO,18 Ce–rGO,19etc. Metal functionalized rGO materials have potential applications including stripping analysis of methyl parathion,13 biological activities,14 catalysis,15,18 photoelectrochemical water splitting,16 and sensing.17,20 Focussing on catalytic applications, graphene based materials have been extensively used as catalysts in various organic transformations such as reduction of nitro organics,21 oxidative C–H functionalization of tertiary amines,22 Suzuki and Heck cross-coupling,23 oxidative esterification of alcohols,24 alkene hydrogenation,25 Friedel–Crafts addition,26a Knoevenagel condensation,26betc. To the best of our knowledge, there is no report of using metal functionalized rGO for the synthesis of benzodiazepine derivatives. Cerium oxide nanoparticles (CeO2 NPs) have attracted tremendous interest in the field of science and nanotechnology because of their useful industrial and synthetic applications.20,27–30 CeO2 NPs owing to their acid–base and redox properties have been widely used as an efficient catalyst in various organic transformations such as dehydration of alcohols, alkylation of aromatic compounds, carbamate synthesis from CO2, reduction of carboxylic acid, ketonization of alcohols, etc.31 Different polymeric materials have been used for the stabilization of CeO2 NPs. Polyvinylpyrrolidone (PVP) is a highly cross-linked, non-ionic,32 and non-toxic33 polymer containing both hydrophilic (the pyrrolidone moiety) and hydrophobic components (the alkyl group) in its structure.34 It acts as a stabilizer by inhibiting the aggregation of NPs resulting from the repulsive forces through its hydrophobic carbon chains that extend into solvents and interact with each other.35
Benzodiazepine nuclei are important pharmacophoric scaffolds and constitute a special class of heterocycles with diverse biological activities such as anti-HIV-1,36 antiviral,37 antitumor,38etc. Besides their eye-catching biological properties, benzodiazepines have also been used as valuable synthons for various fused ring compounds such as imidazo,39 triazolo,40 and pyrimido-benzodiazepines.41 Water as a solvent also contributes to green chemistry by featuring many benefits such as simplifying workup procedures, enabling the recycling of the catalyst, improving reactivity and selectivity, and allowing mild reaction conditions and protecting-group free synthesis in addition to being benign itself.42 Taking into account the aforementioned properties of graphene based materials and in continuation of our ongoing research,43 we herein, describe an easy route for the synthesis of PVP–CeO2 coated reduced graphene oxide (PVP–CeO2/rGO). The material was well characterized and used as an efficient heterogeneous catalyst for water compatible synthesis of benzodiazepine derivatives under environmentally benign conditions.
2. Experimental
2.1. Synthesis of graphene oxide (GO)
GO was synthesized using modified Hummers' method.44 In a 250 mL RB flask, 1.2 g of graphite flakes, 0.55 g of NaNO3 and 25 mL conc. H2SO4 were added and the resulting mixture was stirred for 30 min in an ice bath at 0–5 °C to obtain a black paste. To this mixture, 3.2 g of KMnO4 was slowly added (at a rate of 1 g per 15 min) with vigorous stirring and temperature was maintained using an ice bath to avoid explosion. After complete addition of KMnO4, the ice bath was removed and the reaction mixture was stirred at 35 °C for 2 h. After that, 50 mL of de-ionized water was added very slowly to the reaction mixture. Since, it is a highly exothermic process, a huge amount of heat is produced during this process and the temperature reaches around 100 °C and the colour of the reaction mixture turns brownish. After 25 min, 10 mL of 30% H2O2 and 100 mL warm water were added to the reaction flask and the mixture was stirred for 5 min resulting in the formation of a yellowish-brown solution. The resulting solution was filtered, treated with 5% HCl to remove impurities and subsequently washed with a copious amount of double distilled water. As obtained, the product (yellowish-brown) was oven dried at 60 °C for 14 h and re-dispersed in 100 mL double distilled water by ultrasonication for 3 h to obtain a brown colored GO dispersion. The resulting dispersion was filtered and washed thoroughly with deionized water and dried at 60 °C for 6 h to obtain a brown colored solid (GO) (yield: GO = 1.52 g).
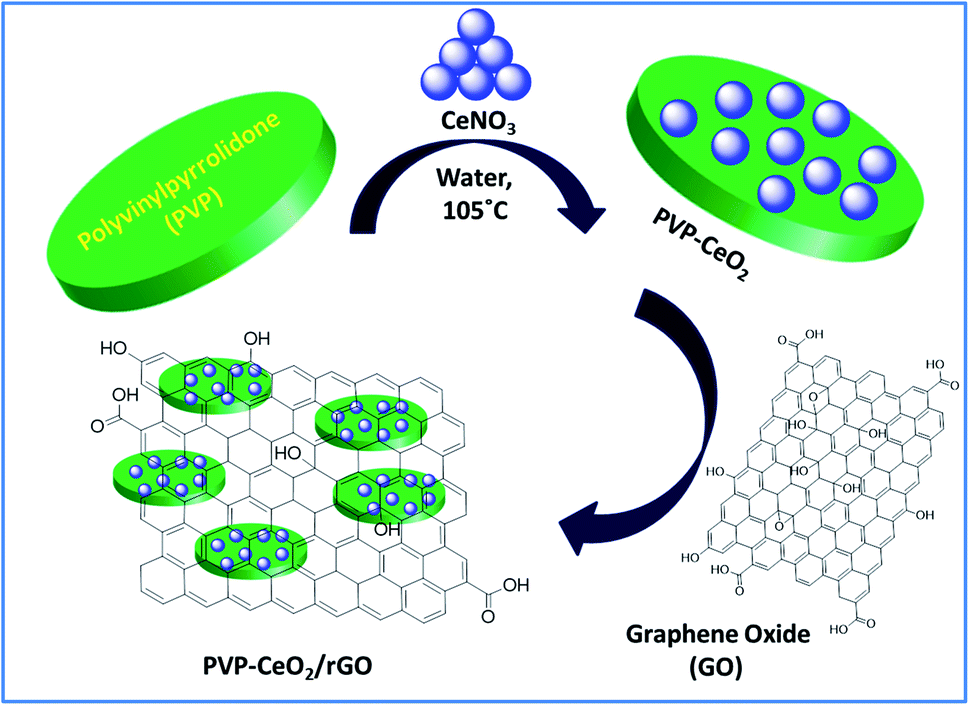
2.2. Synthesis of PVP–CeO2 coated reduced graphene oxide (PVP–CeO2/rGO)
In the first step, PVP coated cerium oxide NPs (PVP–CeO2 NPs) were synthesized by adopting a previously reported procedure with a slight modification.45 In a typical run, CeNO3 (130 mg) was dissolved in 5 mM aqueous solution (50 mL) of PVP and the mixture was heated at 105 °C for 3 h. After heating, the reaction mixture was poured into cold water and acetone (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, acetone/particle suspension) to remove excess PVP. The mixture was then centrifuged at 4000 rpm for 10 min. The yellow pellets (PVP–CeO2 NPs) thus obtained were retained and the excess liquid was discarded (yield: PVP–CeO2 = 0.98 g).
:1 ratio, acetone/particle suspension) to remove excess PVP. The mixture was then centrifuged at 4000 rpm for 10 min. The yellow pellets (PVP–CeO2 NPs) thus obtained were retained and the excess liquid was discarded (yield: PVP–CeO2 = 0.98 g).
In the second step, the pellets were re-suspended in 100 mL double distilled water containing GO (1 g) and the resulting mixture was sonicated for 30 min at 45 °C. The yellowish brown colour of the solution turned black indicating the reduction of GO and formation of PVP–CeO2/rGO. The obtained PVP–CeO2/rGO was separated by centrifugation, washed with double distilled water and dried in a vacuum oven at 60 °C for 24 h (yield: PVP–CeO2/rGO = 1.78 g).
2.3. General procedure for the synthesis of benzodiazepine derivatives (4a–j)
A mixture of 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), aromatic/hetero aromatic aldehydes 2a–e (1 mmol), 1,2-diamines 3a,b (1 mmol) and PVP–CeO2/rGO (30 mg) were taken in a round bottom flask (50 mL) and stirred in water at room temperature for a specified time period. The completion of the reaction was marked by TLC. The reaction mixture was kept at room temperature for a few minutes and ethyl acetate (5 mL) was added to extract the product. The catalyst being insoluble in ethyl acetate was separated by filtration, washed thoroughly with ethyl acetate (2 × 10 mL), dried and reused. The filtrate containing the crude products was washed thoroughly with double distilled water (4 × 10 mL), dried over anhydrous sodium sulphate and evaporated under reduced pressure. The crude products were recrystallized from ethanol to afford pure products 4a–j.
3. Results and discussion
3.1. Characterization of PVP–CeO2/rGO
The material PVP–CeO2/rGO was synthesized as outlined in Scheme 1.
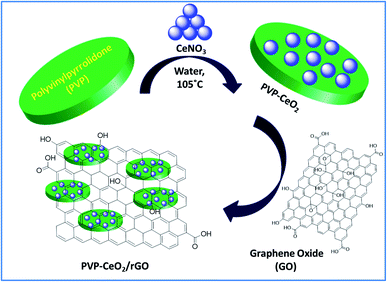 |
| | Scheme 1 Synthesis of PVP–CeO2/rGO. | |
The synthesized material (PVP–CeO2/rGO) combines the properties of PVP, CeO2 and rGO. CeO2 NPs provided sufficient acidity to the catalyst, PVP acted as a stabilizing agent for NPs and graphene sheets provided a surface for the catalytic activity. The material was characterized by various techniques such as FTIR, SEM, EDX, elemental mapping, TEM, Raman spectroscopy, XRD, BET, XPS and TG analyses.
3.1.1 FTIR/SEM analyses.
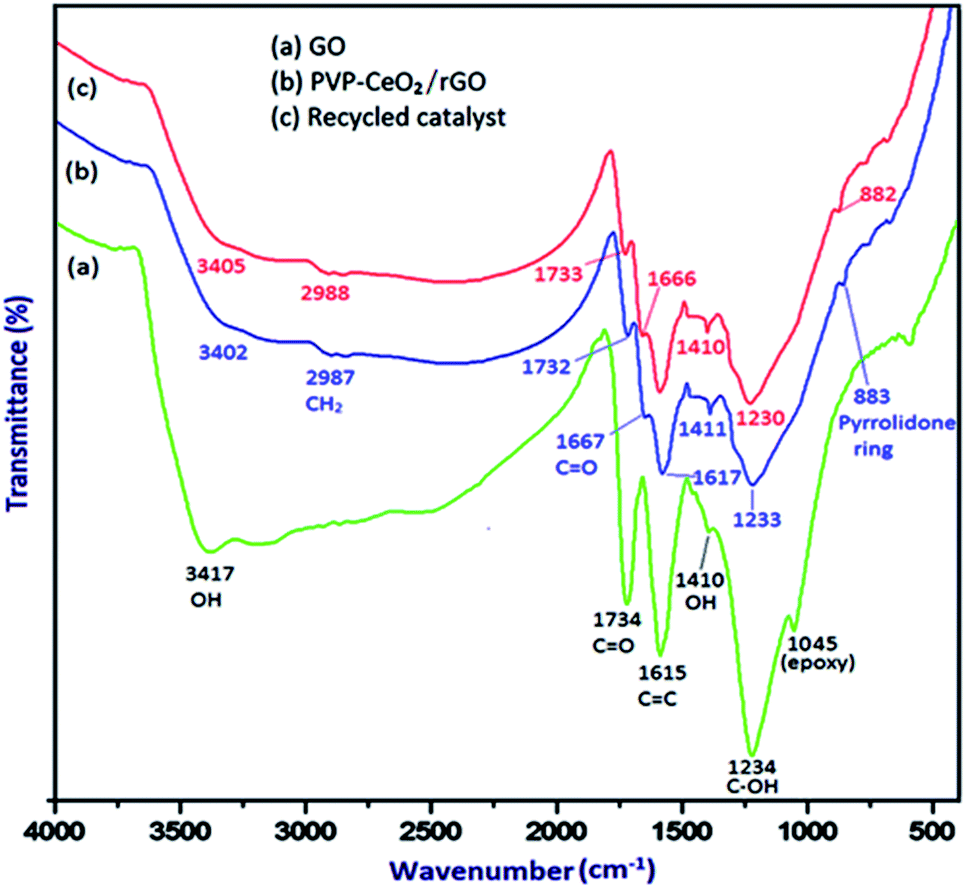
The FTIR spectrum of GO (Fig. 1a) displayed the characteristic bands at 3417 cm−1, 1734 cm−1, 1615 cm−1, 1410 cm−1, 1234 cm−1 and 1045 cm−1 for the stretching vibration of the –OH group, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, CC stretching vibration of aromatic rings, –OH deformation, C–OH stretching and C–O (epoxy) stretching respectively.46 When PVP–CeO2 was introduced into GO sheets, the intensity of CO and C–OH bands decreased significantly whereas the band at 1045 cm−1 disappeared completely indicating the reduction of GO leading to the formation of PVP–CeO2/rGO (Fig. 1b). The characteristic bands of PVP appeared at 2987 cm−1, 1667 cm−1 and 883 cm−1 due to the stretching vibration of CH2 of alkyl chain, CO stretching and breathing vibration of the pyrrolidone ring respectively (Fig. 1b).47 The surface morphology of GO (Fig. 2a) showed a sheet like structure, whereas the uniform distribution of PVP–CeO2 NPs over the rGO sheets was clearly discernible in PVP–CeO2/rGO (Fig. 2b and c).
O stretching vibration, CC stretching vibration of aromatic rings, –OH deformation, C–OH stretching and C–O (epoxy) stretching respectively.46 When PVP–CeO2 was introduced into GO sheets, the intensity of CO and C–OH bands decreased significantly whereas the band at 1045 cm−1 disappeared completely indicating the reduction of GO leading to the formation of PVP–CeO2/rGO (Fig. 1b). The characteristic bands of PVP appeared at 2987 cm−1, 1667 cm−1 and 883 cm−1 due to the stretching vibration of CH2 of alkyl chain, CO stretching and breathing vibration of the pyrrolidone ring respectively (Fig. 1b).47 The surface morphology of GO (Fig. 2a) showed a sheet like structure, whereas the uniform distribution of PVP–CeO2 NPs over the rGO sheets was clearly discernible in PVP–CeO2/rGO (Fig. 2b and c).
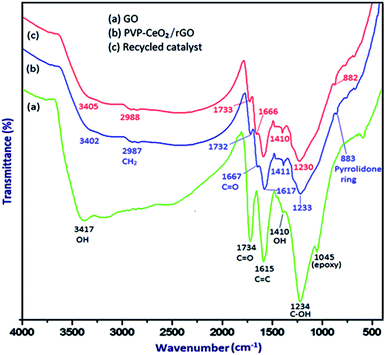 |
| | Fig. 1 FTIR spectrum of (a) GO, (b) PVP–CeO2/rGO and (c) the recycled catalyst after six runs. | |
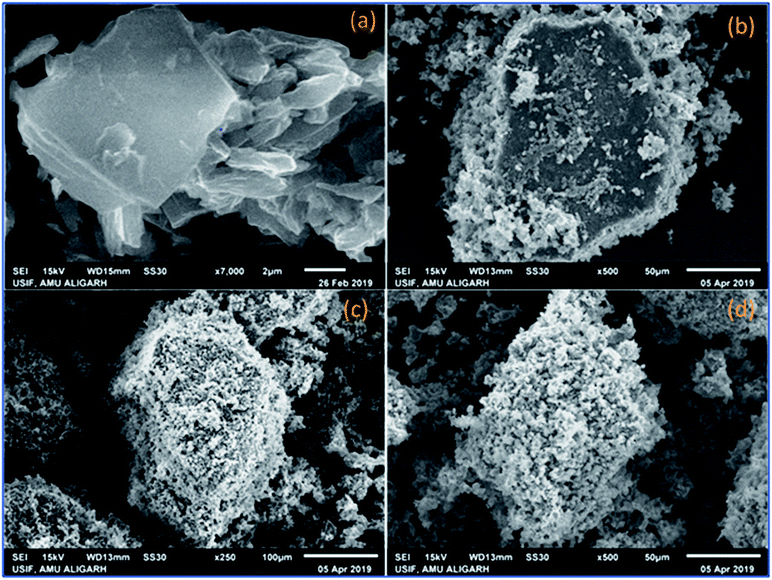 |
| | Fig. 2 SEM analysis of (a) GO, (b) PVP–CeO2/rGO at 50 μm, (c) PVP–CeO2/rGO at 100 μm and (d) the recycled catalyst after six runs. | |
3.1.2 EDX/elemental mapping analyses.
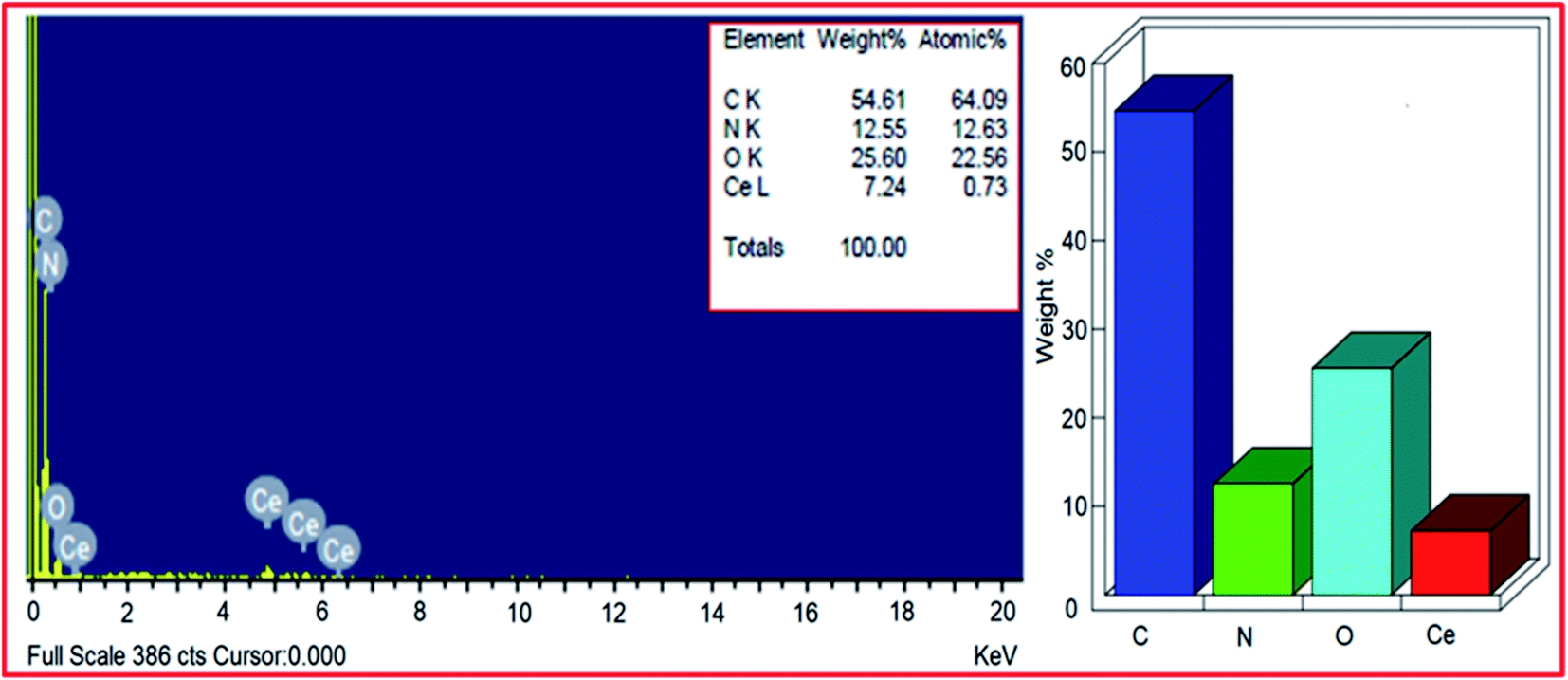
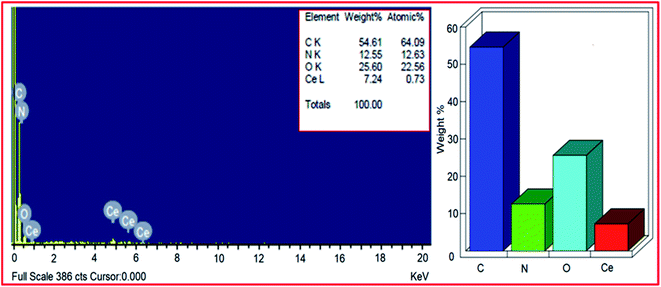
The elemental composition of PVP–CeO2/rGO was studied by EDX analysis and revealed the presence of carbon, nitrogen, oxygen and cerium elements (Fig. 3). From the EDX pattern, the weight% of carbon, nitrogen, oxygen and cerium was found to be 54.61%, 12.55%, 25.60% and 7.24% respectively. In general, the carbon to oxygen ratio in GO is found to be approximately 3:148 and in rGO most of the oxygen functionalities were removed. But, from the EDX pattern of PVP–CeO2/rGO, the carbon:oxygen ratio was obtained to be approximately 3:1.5. This showed the presence of excess oxygen in PVP–CeO2/rGO due to the incorporation of CeO2 NPs. From the EDX pattern, the atomic% of nitrogen was obtained to be 12.63% which has come from the PVP moiety present in the catalyst. Elemental mapping analysis of PVP–CeO2/rGO was performed to observe the distribution of the elements in the matrix. The elemental mapping image of the PVP–CeO2/rGO material (Fig. 4a) confirmed the uniform distribution of carbon (Fig. 4b), oxygen (Fig. 4c) cerium (Fig. 4d) and nitrogen (Fig. 4e) elements in the catalyst.
 |
| | Fig. 3 Energy dispersive X-ray analysis of PVP–CeO2/rGO. | |
 |
| | Fig. 4 Elemental mapping images of (a) PVP–CeO2/rGO, showing the presence of (b) carbon, (c) oxygen, (d) cerium and (e) nitrogen. | |
3.1.3 TEM analysis.
TEM analysis of GO showed a folded sheet like structure (Fig. 5a), whereas PVP–CeO2/rGO displayed the uniform distribution of PVP–CeO2 nanoparticles over the rGO sheets (Fig. 5b).
 |
| | Fig. 5 TEM analysis of (a) GO, (b) PVP–CeO2/rGO and (c) the recycled catalyst after six runs. | |
3.1.4 Raman analysis.
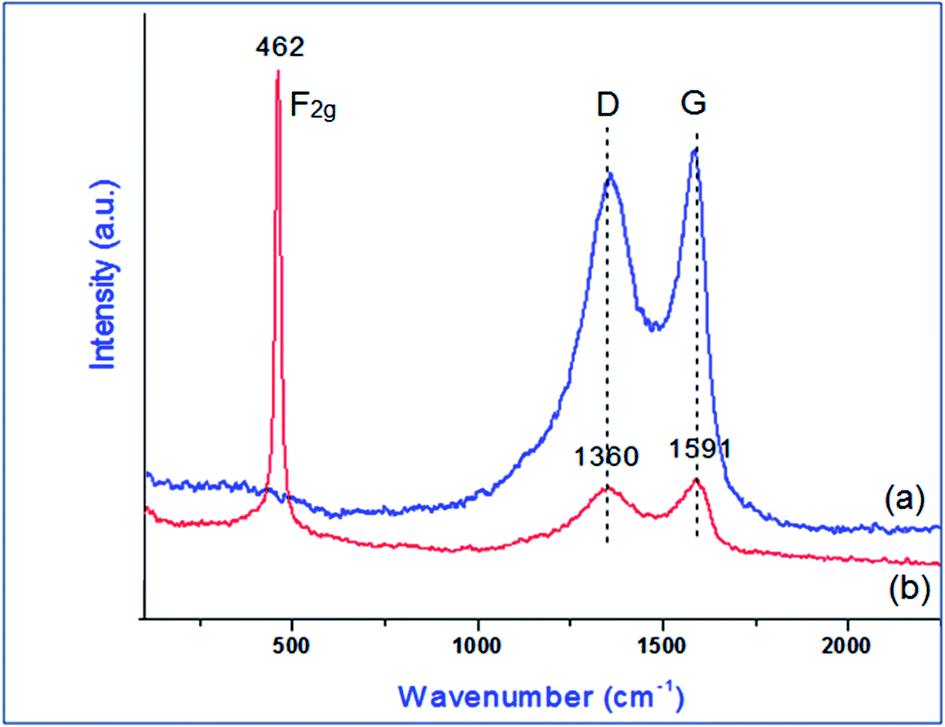
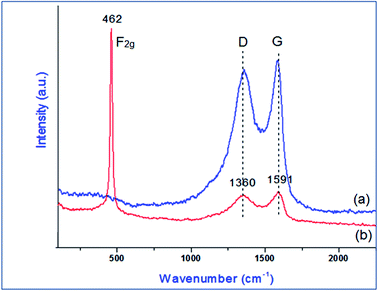
Raman analysis was performed to understand the structural properties of GO and PVP–CeO2/rGO. The presence of two characteristic broad peaks at 1360 cm−1 and 1591 cm−1 corresponds to the D and G bands of the carbon skeleton of the GO sheets (Fig. 6a).49 After the functionalization of GO with PVP–CeO2, a sharp characteristic peak at 462 cm−1 appeared due to the F2g band of CeO2 NPs50 (Fig. 6b). The intensity of bands at 1360 cm−1 and 1591 cm−1 corresponding to the D and G bands of the carbon skeleton of GO was reduced indicating the formation of rGO (Fig. 6b).
 |
| | Fig. 6 Raman spectrum of (a) GO and (b) PVP–CeO2/rGO. | |
3.1.5 XRD analysis.
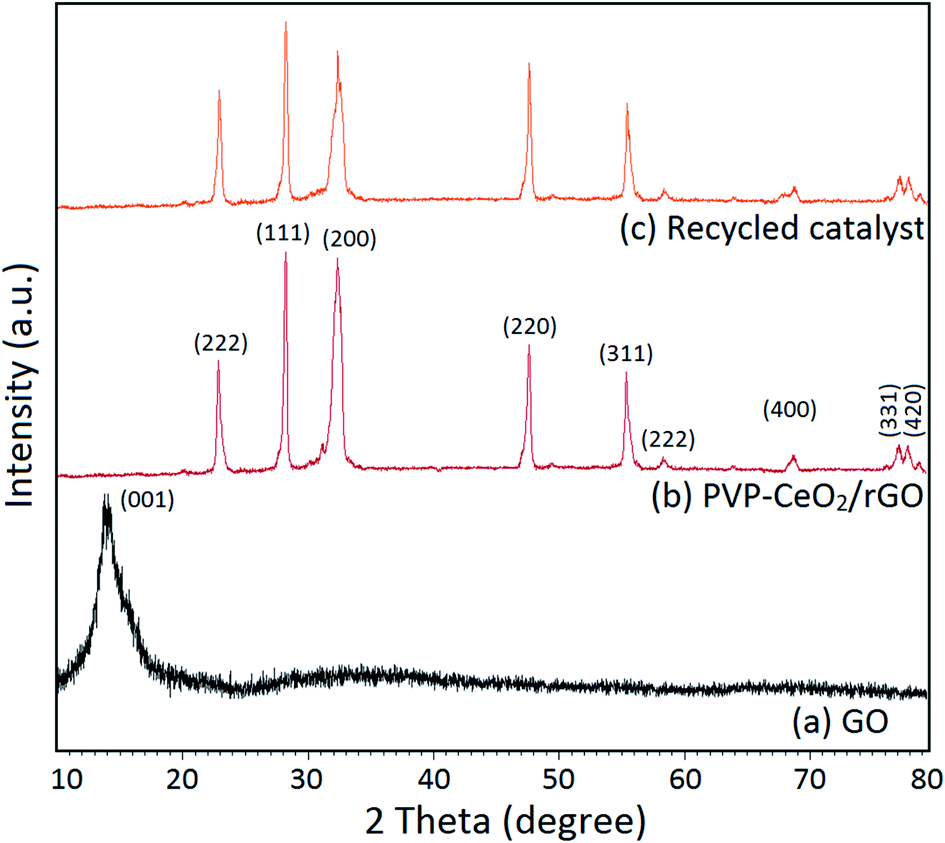
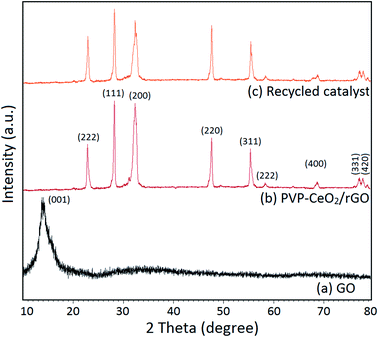
The characteristic peak for the GO material appeared at 2θ = 13.3° corresponding to the (001) plane (Fig. 7a). This indicated the exfoliation of the GO sample and intercalation of water molecules in graphite galleries which generated oxygen functional groups in between the graphite layers upon oxidation.51 After the functionalization of GO with PVP–CeO2, most of the oxygen functional groups present in the GO were removed which was confirmed by the disappearance of the peak at 2θ = 13.3°. A strong diffraction peak at 2θ = 23.0° corresponding to the (222) reflection plane was due to the presence of rGO in the catalyst.52 The diffraction bands at 2θ = 28.1, 32.1, 47.7, 55.4, 58.3, 68.8, 78.0, and 78.9 corresponding to (111), (200), (220), (311), (222), (400), (331) and (420) planes respectively were due to the presence of CeO2 nanoparticles.53 The presence of characteristic peaks for both rGO and CeO2 indicates the successful formation of the desired catalytic system.
 |
| | Fig. 7 XRD pattern of (a) GO, (b) PVP–CeO2/rGO and (c) the recycled catalyst after six runs. | |
3.1.6 XPS analysis.
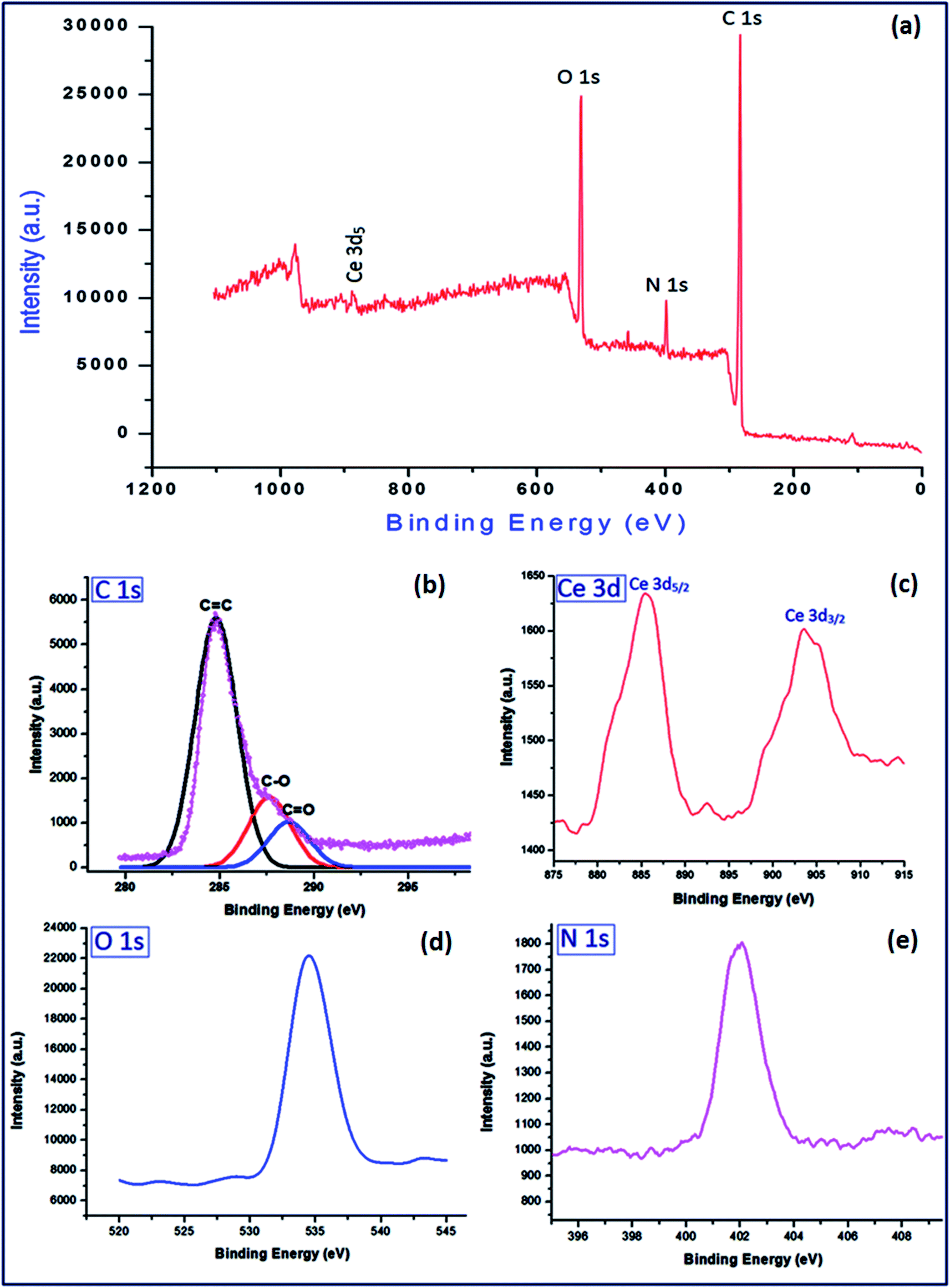
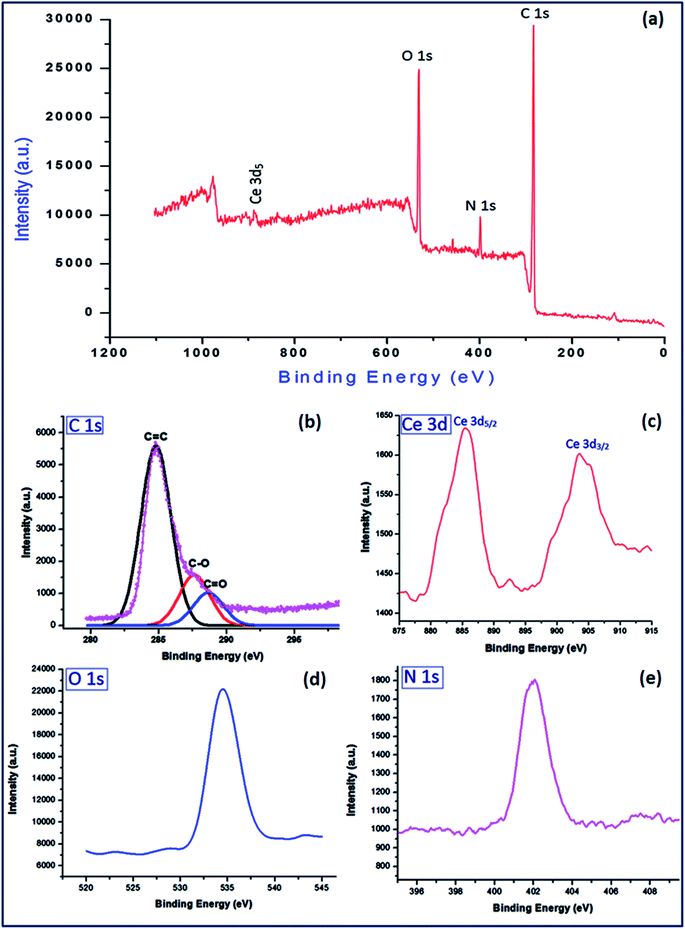
To investigate the surface composition of different elements of the synthesized material (PVP–CeO2/rGO), XPS analysis was carried out and the results are shown in Fig. 8. The high resolution XPS spectrum of C 1s, Ce 3d, O 1s and N 1s were also obtained (Fig. 8a–d). The C 1s showed atomic configurations of different functional groups CC, C–O and CO at binding energies 284.8 eV, 287.6 eV and 288.3 eV respectively2 (Fig. 8a). The Ce 3d displayed two sharp peaks for 3d5/2 and 3d3/2 at 885.4 eV and 903.2 eV respectively54 (Fig. 8b). The sharp bands at binding energies 534.6 eV and 402.1 eV were assigned to O 1s (Fig. 8c) and N 1s (Fig. 8d) respectively. The cerium content was found to be 7.34% which corresponds to 0.523 mmol g−1 of the catalyst.
 |
| | Fig. 8 XPS analysis of (a) PVP–CeO2/rGO; survey spectrum of (b) C 1s, (c) Ce 3d, (d) O 1s and (e) N 1s. | |
3.1.7 BET analysis.
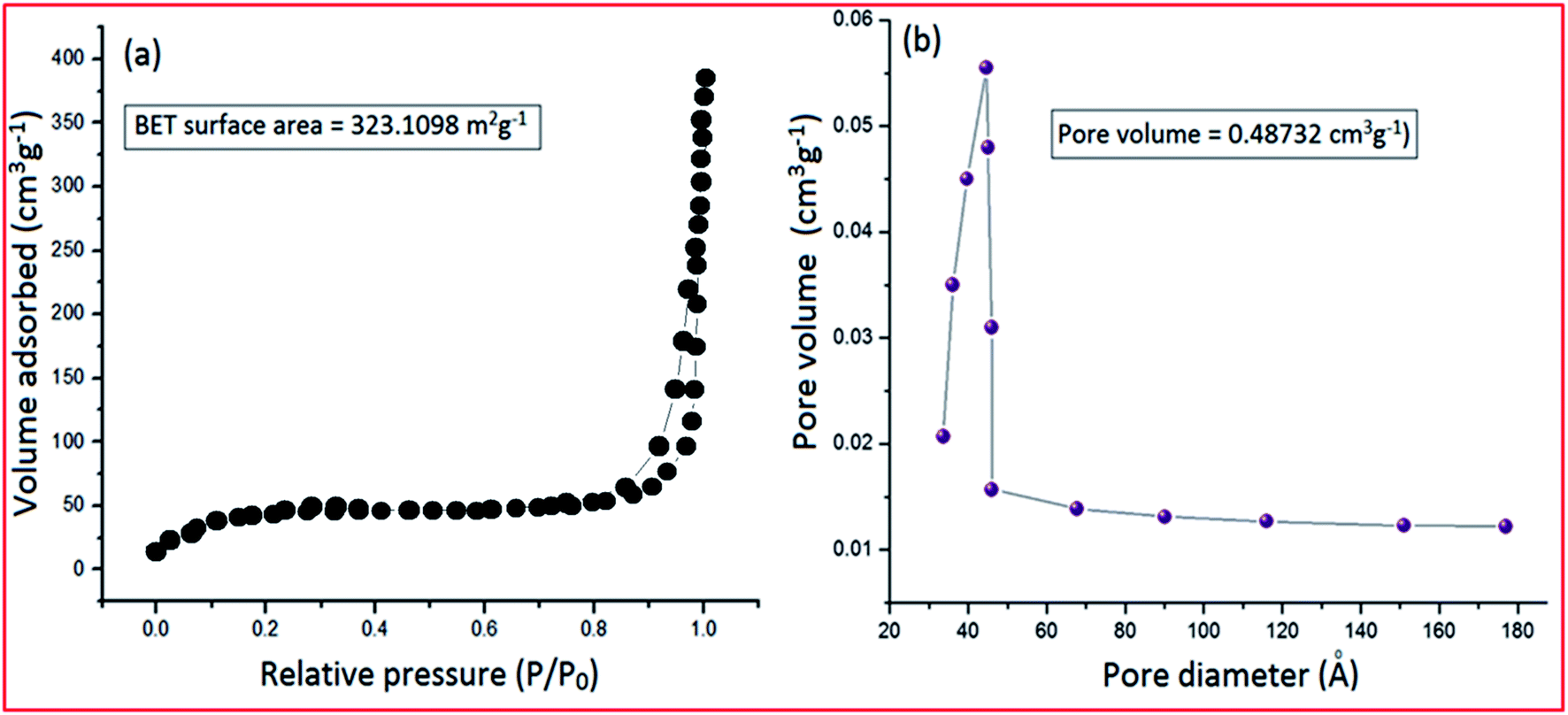
Most of the catalytic properties of nano-materials are generally associated with the surface to volume ratio; the higher the ratio, the better the catalytic activity. The measurement of the surface area and porosity of PVP–CeO2/rGO was conducted by an adsorption/desorption experiment using BET analysis under a N2 atmosphere (Fig. 9). The N2 adsorption/desorption curve of PVP–CeO2/rGO showed a surface area of 323.1098 m2 g−1 confirming the porous nature of the material, and thus, providing more active sites for the catalytic activity (Fig. 9a). The pore size distribution curve exhibited a large pore volume (0.48732 cm3 g−1), which further benefited the exposure of active sites (Fig. 9b).
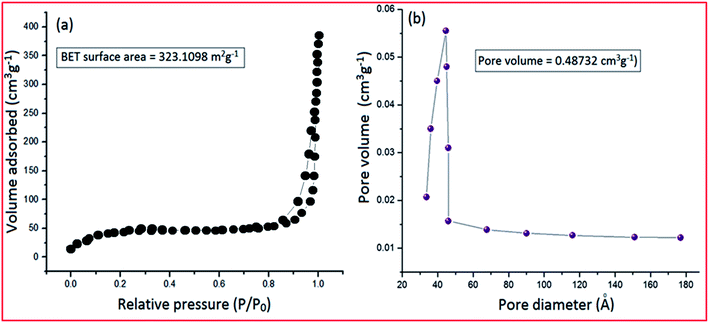 |
| | Fig. 9 (a) N2 adsorption isotherm of PVP–CeO2/rGO and (b) pore size distribution of PVP–CeO2/rGO. | |
3.1.8 TG analysis.
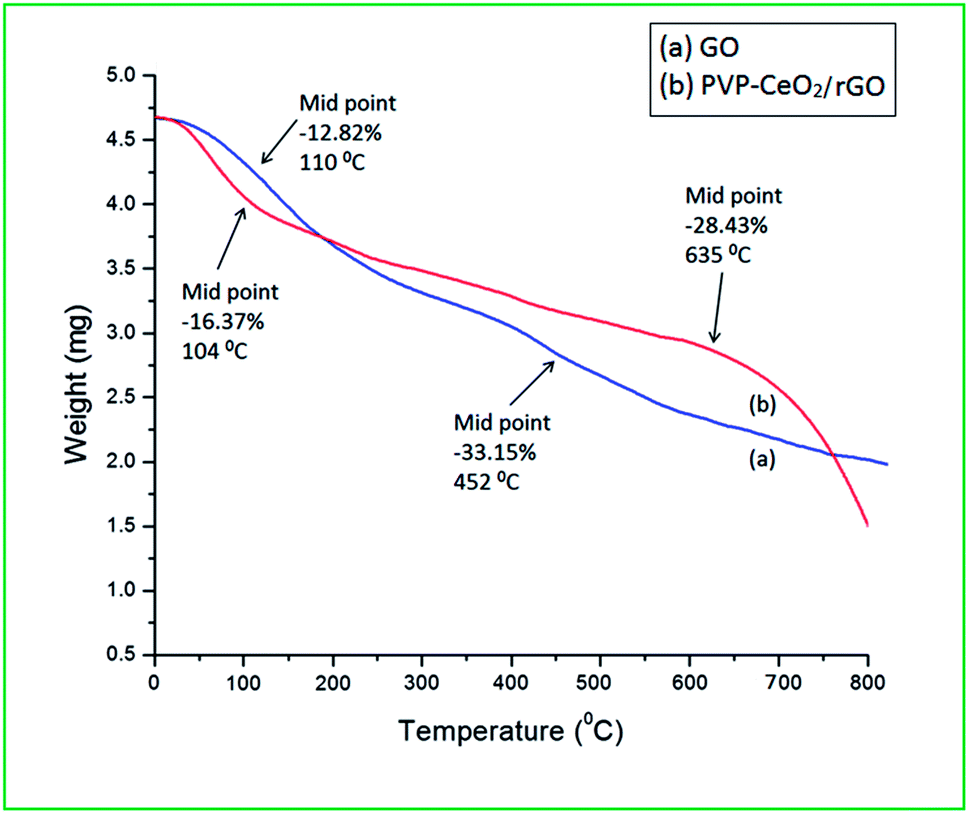
The TG curves of GO and PVP–CeO2/rGO are shown in Fig. 10. The TG curve of GO (Fig. 10a) showed two stage weight loss at 110 °C (12.82%) and 452 °C (33.15%) due to the removal of absorbed water molecules from the GO material and decomposition of oxygen-containing functional groups respectively.55 In the TG curve of PVP–CeO2/rGO (Fig. 10b), the corresponding weight loss occurred at a relatively higher temperature of 635 °C (28.43%) indicating higher thermal stability of the material as compared to GO.
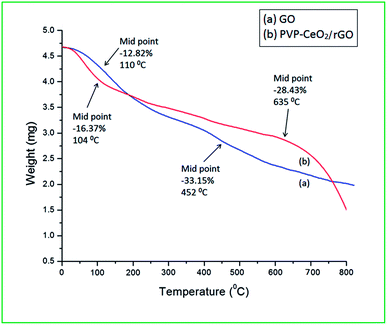 |
| | Fig. 10 TG analysis of (a) GO and (b) PVP–CeO2/rGO. | |
3.2. Catalytic evaluation
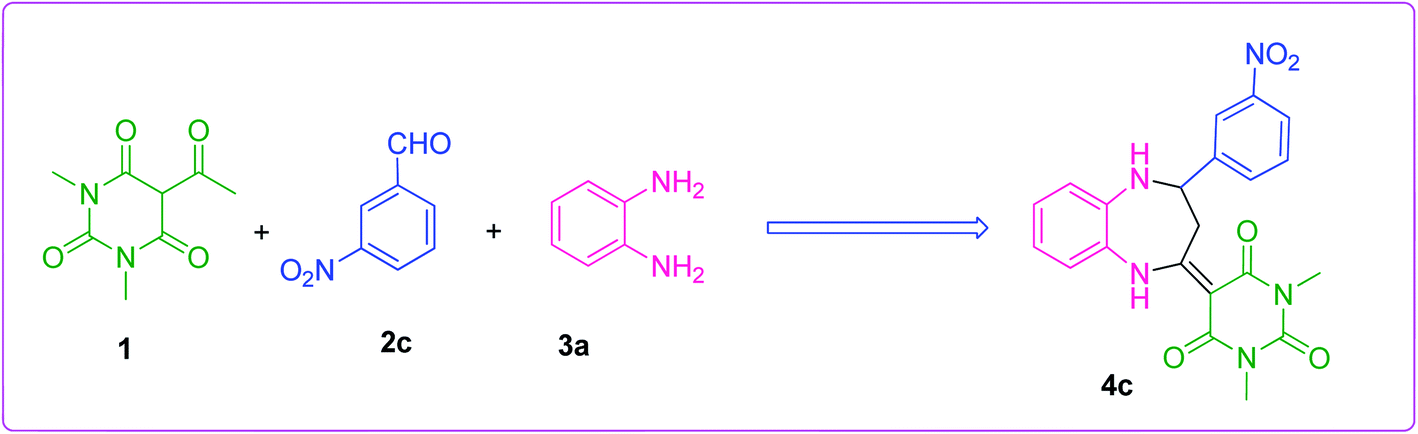
The efficiency of the synthesized material as a catalyst was tested by synthesizing benzodiazepine derivatives. Several parameters such as the effect of different catalysts, solvents, catalyst loading and temperatures on a model reaction were taken into consideration. A reaction between 5-acetyl-1,3-dimethylbarbituric acid 1, 3-nitrobenzaldehyde 2c and o-phenylenediamine 3a to produce substituted benzodiazepine 4c was chosen as the model reaction (Scheme 2). The components 1 and 3a had no substituents and 2c was functionalized with a m-NO2 substituent which had the minimum effect out of all aldehydes used for the synthesis of benzodiazepine derivatives. Therefore, Scheme 2, was the best composition of reactants to see the effect of different parameters.
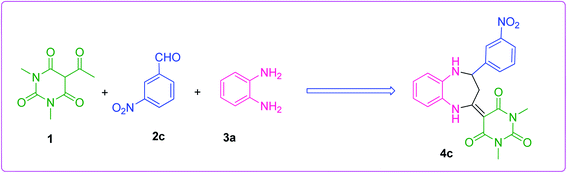 |
| | Scheme 2 Model reaction. | |
3.2.1 Effect of different catalysts.
In order to establish the superiority of our synthesized catalyst (PVP–CeO2/rGO) for the desired transformation, different catalysts such metal oxide NPs, polymer modified metal oxide NPs and supported polymer modified metal oxide NPs were employed in the model reaction (Table 1). First, the model reaction was carried out without the catalyst and the reaction afforded no product even after 24 h (Table 1, Entry 1). Then the investigation was carried out using different metal NPs as the catalyst and the reaction afforded low to moderate yields of the product in a longer reaction time period (Table 1, Entries 2–7). Among the different metal NPs tested, CeO2 NPs showed the best activity affording a good yield in a comparatively less reaction time period (Table 1, Entry 7). After that, the model reaction was investigated by employing different polymer modified CeO2 NPs and the reaction showed improved results in terms of product yield and reaction time (Table 1, Entries 8–12). PVP–CeO2 was chosen among different polymer modified CeO2 catalysts for producing the best result (Table 1, Entry 10). Finally, PVP–CeO2 was supported on different support materials such as Al2O3, ZrO2, SiO2 and rGO for the purpose of enhancing the catalytic activity (Table 1, Entries 13–16). It was observed that among the different support materials, rGO proved to be the best support material making PVP–CeO2/rGO the best catalyst for affording the maximum yield (98%) of the product in a minimum reaction time period (7 min) (Table 1, Entry 16).
Table 1 Effect of different catalysts on the model reactiona
| Entry |
Catalyst |
Timeb |
Yieldc (%) |
|
Reaction conditions: 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), 3-nitrobenzaldehyde 2c (1 mmol), o-phenylenediamine 3a (1 mmol), different catalysts (30 mg), water, r.t. stirring.
Reaction progress monitored by TLC.
Isolated yield.
|
| 1 |
— |
24 h |
— |
| 2 |
ZnO NPs |
5 h |
40 |
| 3 |
CaO NPs |
4.2 h |
42 |
| 4 |
CuO NPs |
4 h |
45 |
| 5 |
TiO2 NPs |
3.5 h |
48 |
| 6 |
Fe2O3 NPs |
3 h |
51 |
| 7 |
CeO2 NPs |
2 h |
54 |
| 8 |
PPy–CeO2 |
1.8 h |
56 |
| 9 |
PANI–CeO2 |
1.5 |
59 |
| 10 |
PVA–CeO2 |
1.2 h |
61 |
| 11 |
Chitosan–CeO2 |
1 h |
64 |
| 12 |
PVP–CeO2 |
50 min |
70 |
| 13 |
PVP–CeO2/Al2O3 |
28 min |
73 |
| 14 |
PVP–CeO2/ZrO2 |
25 min |
76 |
| 15 |
PVP–CeO2/SiO2 |
20 min |
78 |
| 16 |
PVP–CeO2/rGO |
7 min |
98 |
3.2.2 Effect of solvents.
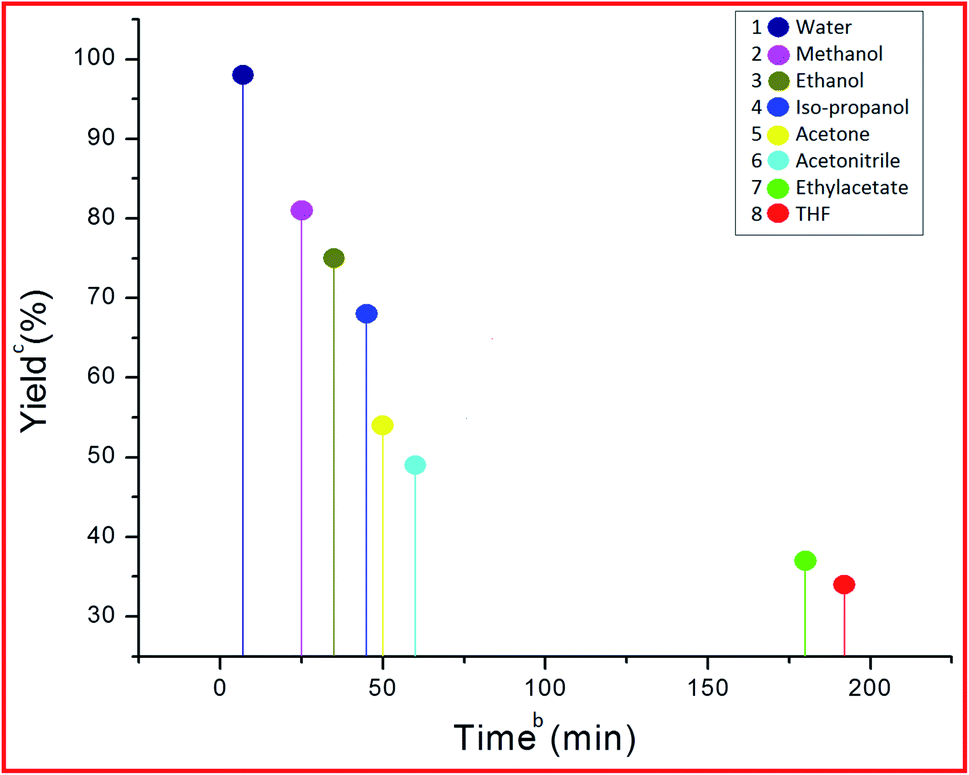
After finding out the most suitable catalyst, investigation was carried out to select the best solvent for the model reaction. In this effort, different protic (iso-propanol, ethanol, methanol, and water) as well as aprotic (THF, ethyl acetate, acetonitrile, and acetone) solvents (Fig. 11) were employed in the model reaction. It was observed that aprotic solvents afforded a poor yield of the product in a longer reaction time period (Fig. 11, Entries 1–6), whereas protic solvents gave a satisfactory product yield (68–96%) (Fig. 11, Entries 7–10). Among different protic solvents, water proved to be the best solvent for the desired reaction affording an excellent yield (98%) of the product in minimum reaction time period (7 min) (Fig. 11, Entry 10).
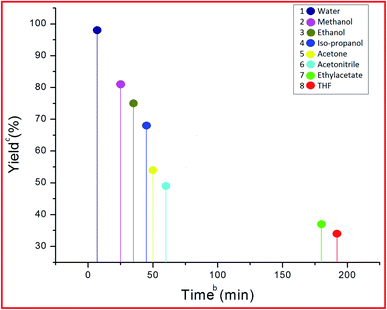 |
| | Fig. 11 Effect of different solvents on the model reaction.a aReaction conditions: 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), 3-nitrobenzaldehyde 2c (1 mmol), o-phenylenediamine 3a (1 mmol), PVP–CeO2/rGO (30 mg), solvents, r.t. stirring. bReaction progress monitored by TLC. cIsolated yield. | |
3.2.3 Effect of catalyst loading.
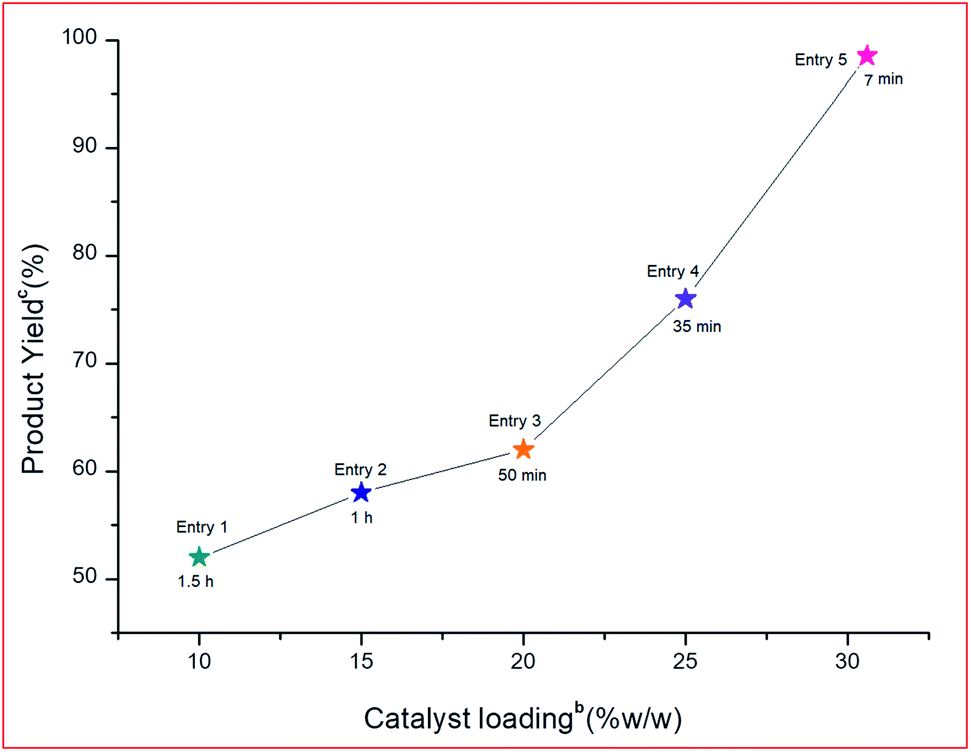
To determine the effect of different PVP–CeO2 loadings on rGO, different loading amounts such as 10, 15, 20, 25 and 30% w/w were employed in the model reaction. It was observed that the rate of reaction increased linearly with an increased loading amount from 10–30% w/w (Fig. 12, Entries 1–5).
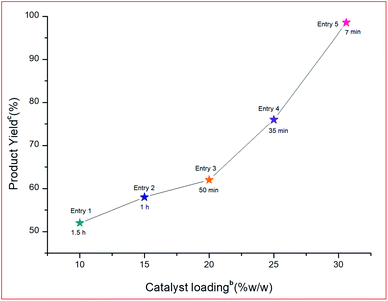 |
| | Fig. 12 Effect of different PVP–CeO2 loadings on the support for the model reaction.a aReaction conditions: 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), 3-nitrobenzaldehyde 2c (1 mmol), o-phenylenediamine 3a (1 mmol), PVP–CeO2/rGO (30 mg), different PVP–CeO2 loadings on support, r.t. stirring. bReaction progress monitored by TLC. cIsolated yield. | |
3.2.4 Effect of the amount of catalyst.
In order to choose the optimum amount of the catalyst, the model reaction was investigated with different amounts (10, 20, 25, 30, and 40 mg) of PVP–CeO2/rGO (Table 2, Entries 1–5). It was observed that on increasing the amount of catalyst (from 10 mg to 30 mg), the yield of the product and turnover frequency (TOF) increased from 43% to 98% and 54.81 h−1 to 535.37 h−1 respectively (Table 2, Entries 1–4). This showed that the efficiency of the catalyst improved on increasing the catalyst amount. Further increasing the catalyst amount did not affect the reaction rate and hence, 30 mg of PVP–CeO2/rGO (producing the highest TOF = 535.37 h−1) was found to be the optimum amount for the desired reaction affording the maximum yield of the product (98%) in minimum reaction time period (7 min) (Table 2, Entry 4).
Table 2 Effect of different amounts of the catalyst on model reactiona
| Entry |
Catalyst loading |
Timeb (min) |
Yieldc (%) |
TON |
TOF (h−1) |
|
Reaction conditions: 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), 3-nitrobenzaldehyde 2c (1 mmol), o-phenylenediamine 3a (1 mmol), PVP–CeO2/rGO, different amount of catalyst, r.t. stirring.
Reaction progress monitored by TLC.
Isolated yield.
|
| 1 |
10 mg |
90 |
43 |
82.21 |
54.81 |
| 2 |
20 mg |
45 |
62 |
59.27 |
79.03 |
| 3 |
25 mg |
25 |
81 |
61.95 |
148.68 |
| 4 |
30 mg
|
7
|
98
|
62.46
|
535.37
|
| 5 |
40 mg |
7 |
98 |
46.84 |
401.52 |
3.3. Catalytic reaction
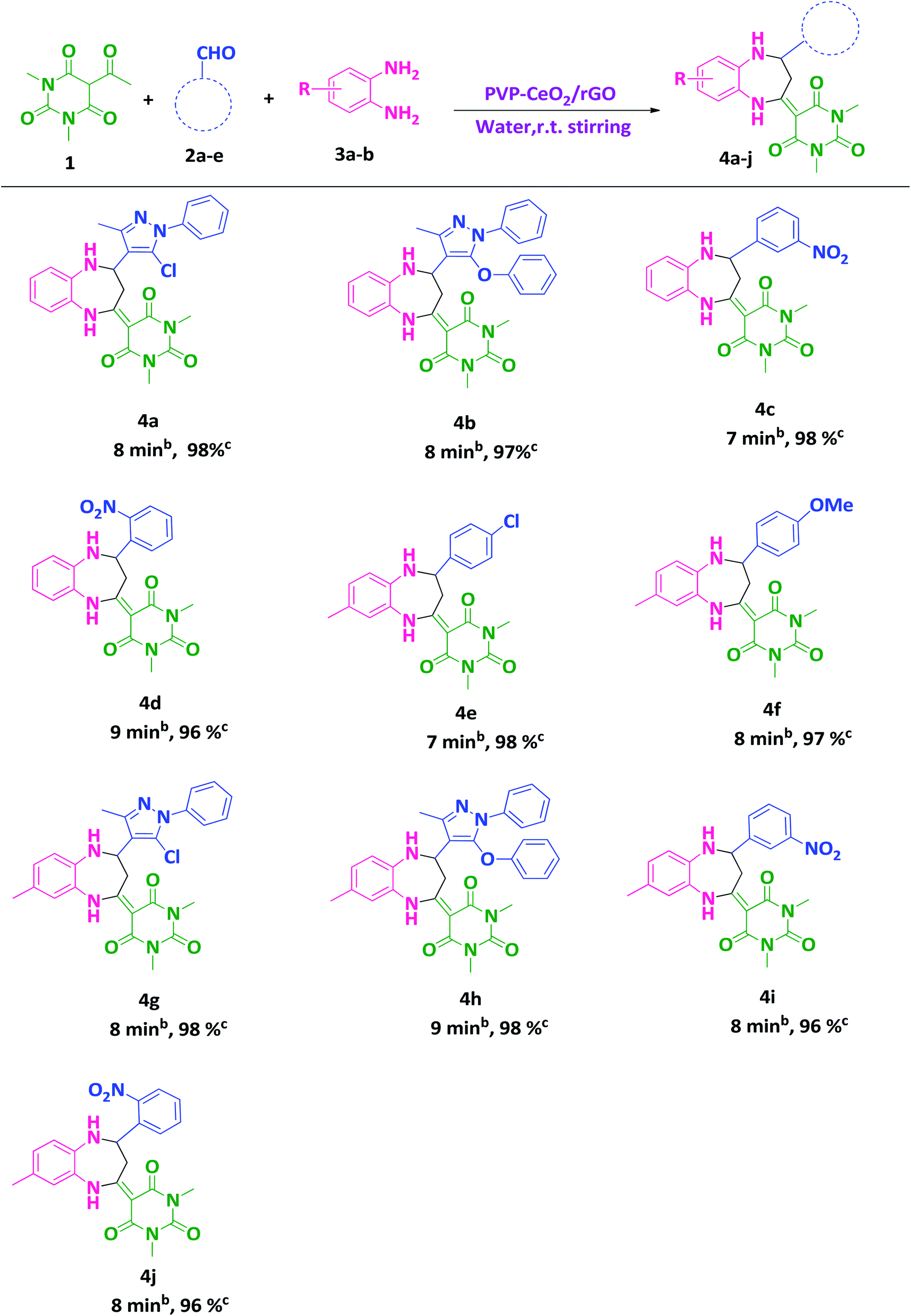
After finding out the most suitable reaction conditions, substituted benzodiazepine derivatives were synthesized by carrying out the reaction between 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), hetero-aromatic/aromatic aldehydes 2a–e and 1,2-diamines 3a,b (1 mmol) in the presence of PVP–CeO2/rGO at room temperature stirring in water for a specified time period (Scheme 3). The turnover number (TON) and turnover frequency (TOF) of the catalyst were calculated to show the efficiency of PVP–CeO2/rGO. The high TONs and TOFs confirmed the excellent performance of PVP–CeO2/rGO for the synthesis of benzodiazepines (Table 3).
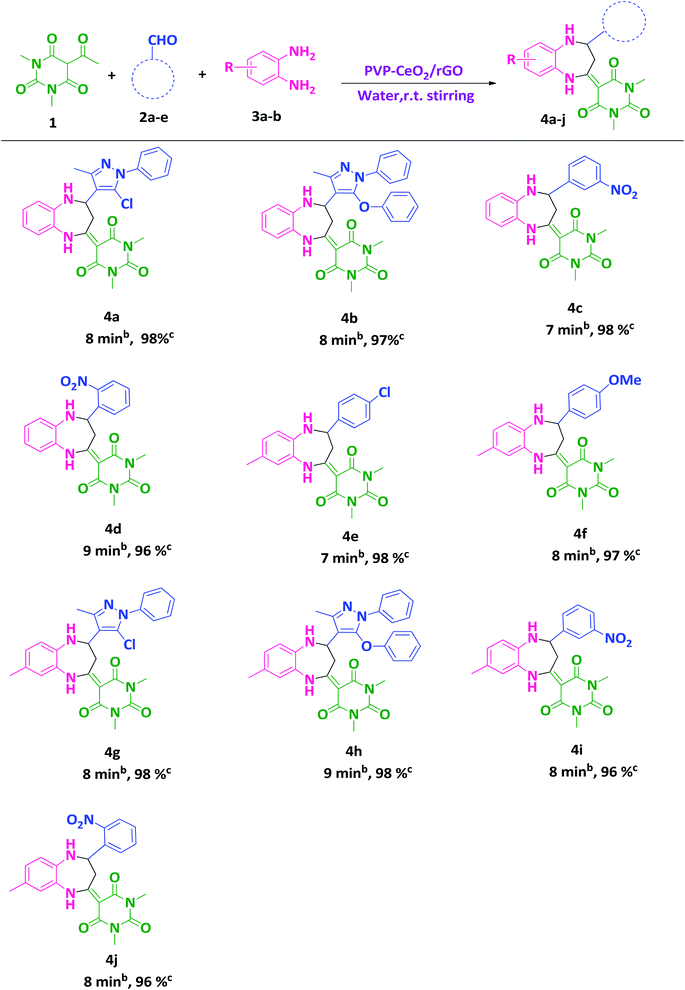 |
| | Scheme 3 Water mediated synthesis of substituted benzodiazepine derivatives.a aReaction conditions: 1 (1 mmol), 2a–e (1 mmol), 3a,b (1 mmol), PVP–CeO2/rGO (30 mg), water, r.t. stirring. bReaction progress monitored by TLC. cIsolated yield. | |
Table 3 Calculation of the TON and TOF of PVP–CeO2/rGO
| Entry |
Product |
Time (min) |
Yield (%) |
TON |
TOF (h−1) |
| 1 |
4a
|
8 |
98 |
62.46 |
468.45 |
| 2 |
4b
|
8 |
97 |
61.82 |
463.65 |
| 3 |
4c
|
7 |
98 |
62.46 |
535.37 |
| 4 |
4d
|
9 |
96 |
61.18 |
407.86 |
| 5 |
4e
|
7 |
98 |
62.46 |
535.37 |
| 6 |
4f
|
8 |
97 |
61.82 |
463.65 |
| 7 |
4g
|
8 |
98 |
62.46 |
468.45 |
| 8 |
4h
|
9 |
98 |
62.46 |
416.40 |
| 9 |
4i
|
8 |
96 |
61.18 |
458.85 |
| 10 |
4j
|
8 |
96 |
61.18 |
458.85 |
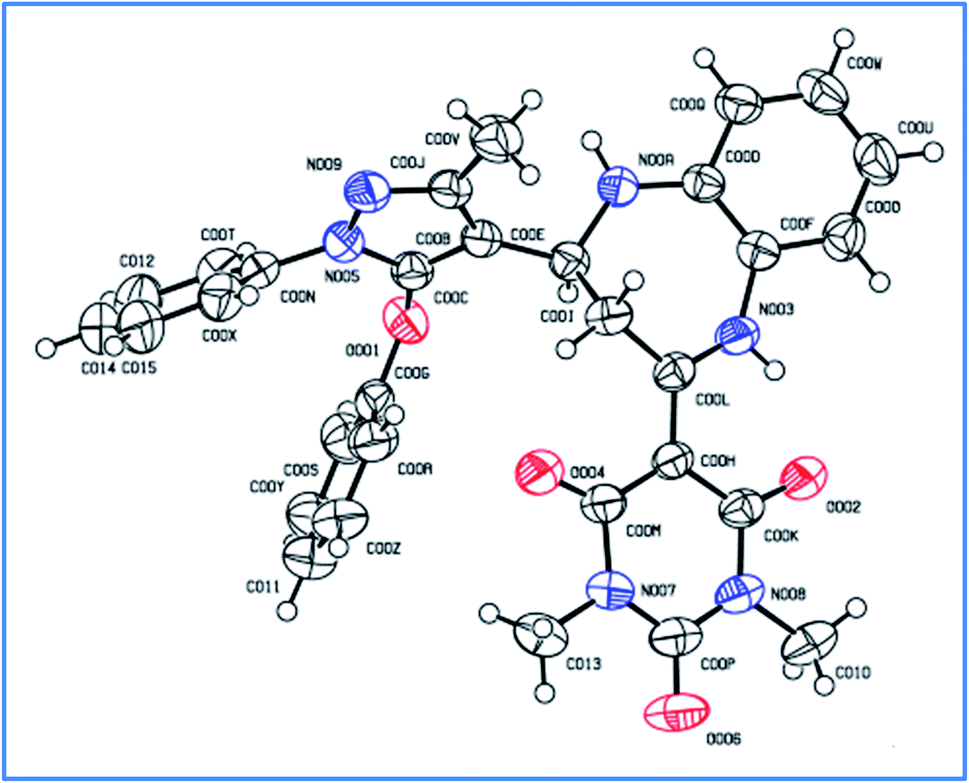
The single crystal structure of one of the synthesized compounds 4b is shown in Fig. 13. The relevant details are provided in Table 4.
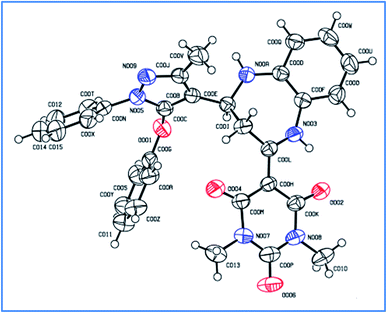 |
| | Fig. 13 ORTEP diagram of compound 4b (ellipsoids are drawn at the 50% probability level). | |
Table 4 Single crystal data for compound 4b
| Identification code |
4b
|
| Empirical formula |
C31H28N6O4 |
| Formula weight |
548.59 |
| Temperature/K |
273.15 |
| Crystal system |
Monoclinic |
| Space group |
P21/n |
|
a/Å |
18.836(3) |
|
b/Å |
7.2533(10) |
|
c/Å |
19.682(3) |
|
α/° |
90 |
|
β/° |
92.081(4) |
|
γ/° |
90 |
| Volume/Å3 |
2687.3(6) |
|
Z
|
4 |
|
ρ
calc/g cm−3 |
1.356 |
|
μ/mm−1 |
0.093 |
|
F(000) |
1152.0 |
| Crystal size/mm3 |
0.52 × 0.25 × 0.08 |
| Radiation |
MoKα (λ = 0.71073) |
| 2Θ range for data collection/° |
5.882 to 49.998 |
| Index ranges |
−22 ≤ h ≤ 22, −8 ≤ k ≤ 8, −23 ≤ l ≤ 23 |
| Reflections collected |
29871 |
| Independent reflections |
4723 [Rint = 0.0593, Rsigma = 0.0407] |
| Data/restraints/parameters |
4723/0/377 |
| Goodness-of-fit on F2 |
1.033 |
| Final R indices I ≥ 2σ(I) |
R
1 = 0.0514, wR2 = 0.1151 |
| Final R indices all data |
R
1 = 0.0943, wR2 = 0.1372 |
| Largest diff. peak/hole/e Å−3 |
0.16/−0.24 |
The green perspective of the present protocol was also discussed in terms of atom economy (AE), reaction mass efficiency (RME), overall efficiency (OE), carbon efficiency (CE), process mass intensity (PMI), E-factor and solvent intensity (SI). The details of the green metric calculations of these parameters are provided in the ESI.† The values of all parameters showed that the present protocol was fitting suitably in the range of sustainability as confirmed by an earlier study.56 The results of the green metric parameters are given in Table 5.
Table 5 The Green metric analysis of the synthesized compounds (4a–j)h,i
| Compound |
% yield |
% AEa |
% CEb |
% RMEc |
% OEd |
PMIe |
SIf |
E-factorg |
|
% AE: percentage atom economy.
% CE: percentage carbon efficiency.
% RME: percentage reaction mass efficiency.
% OE: percentage overall efficiency.
PMI: process mass intensity.
SI: solvent intensity.
E-factor.
Catalyst amount was not used in the calculations because of its reusability.57
Calculations up to the crude product in all cases.
|
|
4a
|
98 |
93.15 |
100.00 |
91.29 |
98.00 |
5.25 |
3.99 |
4.25 |
|
4b
|
97 |
93.83 |
100.00 |
91.01 |
96.99 |
4.86 |
3.53 |
3.86 |
|
4c
|
98 |
92.92 |
100.00 |
90.27 |
97.14 |
5.95 |
4.65 |
4.95 |
|
4d
|
96 |
92.92 |
100.00 |
88.43 |
95.16 |
6.08 |
4.55 |
5.08 |
|
4e
|
98 |
92.17 |
100.00 |
90.33 |
98.00 |
5.92 |
4.62 |
4.92 |
|
4f
|
97 |
92.10 |
100.00 |
89.34 |
97.00 |
6.03 |
4.61 |
5.03 |
|
4g
|
98 |
93.33 |
100.00 |
91.96 |
98.53 |
5.14 |
3.88 |
4.14 |
|
4h
|
98 |
93.96 |
100.00 |
92.11 |
98.03 |
4.71 |
3.48 |
3.71 |
|
4i
|
96 |
92.35 |
100.00 |
88.66 |
96.00 |
5.91 |
4.41 |
4.91 |
|
4j
|
96 |
92.35 |
100.00 |
88.66 |
96.00 |
5.91 |
4.41 |
4.91 |
3.4. Recyclability of the catalyst
The reusability of the catalyst was checked by carrying out the recycling experiment on the model reaction (ESI†). It was observed that the catalyst was efficient up to six subsequent cycles with minor loss in catalytic activity after the sixth run (Table 6 and Fig. 1S†).
Table 6 Recycling study of the catalyst for the model reactiona
| Catalytic cycle |
Timeb (min) |
Yieldc (%) |
|
Reaction conditions: 5-acetyl-1,3-dimethylbarbituric acid 1 (1 mmol), 3-nitrobenzaldehyde 2c (1 mmol), o-phenylenediamine 3a (1 mmol), PVP–CeO2/rGO(30 mg), water, r.t. stirring.
Reaction progress monitored by TLC.
Isolated yield.
|
| 1 |
7 |
98 |
| 2 |
7 |
98 |
| 3 |
7 |
98 |
| 4 |
7 |
98 |
| 5 |
7 |
98 |
| 6 |
7 |
98 |
| 7 |
7 |
90 |
The structural integrity of the recovered catalyst after six runs was established by FTIR, SEM, TEM and powder XRD analyses. In the FTIR spectrum of the recycled catalyst, characteristic peaks were observed in approximately the same region as for the fresh catalyst (Fig. 1c). The SEM and TEM images of the recovered catalyst showed a similar morphology (Fig. 2d and 5c) and characteristic peaks remained the same in the XRD pattern also (Fig. 7c).
3.5. Comparison of the activity of PVP–CeO2/rGO with some previously reported catalysts
In order to determine the advantage of PVP–CeO2/rGO over previously reported catalysts for the synthesis of benzodiazepines, a comparative study was carried out (Table 7). It was observed that the present methodology (Table 7, Entry 5) is better as compared with other reported methods (Table 7, Entries 1–4) in terms of product yield and time. The use of water as a solvent and room temperature synthesis make it environment friendly as well.
Table 7 Comparison of the catalytic performance of PVP–CeO2/rGO with reported catalysts
| Entry |
Catalyst |
Solvent |
Temperature |
Time |
% yield (maximum) |
Ref. |
| 1 |
[H-NMP][HSO4] |
EtOH |
100 °C |
15 min |
85 |
58
|
| 2 |
FeCl3–SiO2 |
EtOH |
0 °C |
2 h |
95 |
59
|
| 3 |
Nano-γ-Fe2O3–SO3H |
Solvent-free |
90 °C |
2 h |
90 |
60
|
| 4 |
NiO–SiO2 NCs |
EtOH |
70 °C |
10 min |
98 |
61
|
| 5 |
PVP–CeO
2
/rGO
|
Water
|
r.t.
|
7 min
|
98
|
This work
|
4. Conclusion
We have developed a simple and easy route for the reduction of graphene oxide with the help of PVP–CeO2 NPs. The carbonaceous material, PVP–CeO2/rGO, was used as a heterogeneous catalyst for the multicomponent synthesis of benzodiazepines. The high catalytic performance of the carbon material was verified by its excellent product yield (96–98%), short reaction time period (7–10 min) and recyclability. The methodology involved use of non-toxic chemicals and solvents for the synthesis and therefore can be considered as eco-friendly.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
The authors would like to acknowledge DRS-II New Delhi for providing necessary research facilities and USIF, A.M.U. for facilitating SEM/EDX, elemental mapping, and TEM analyses and SAIF, IIT-Patna for single crystal X-ray crystallographic analysis.
References
- K. S. Novoselov, V. I. Fal, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192 Search PubMed.
- Q. Lyu, H. Yan, L. Li, Z. Chen, H. Yao and Y. Nie, Polymers, 2017, 9, 447 Search PubMed.
- K. S. Novoselov, A. K. Geim, S. V Morozov, D. Jiang, Y. Zhang, S. V Dubonos, I. V Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 Search PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706 Search PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 Search PubMed.
- H. Shin, K. K. Kim, A. Benayad, S. Yoon, H. K. Park, I. Jung, M. H. Jin, H. Jeong, J. M. Kim and J. Choi, Adv. Funct. Mater., 2009, 19, 1987–1992 Search PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 Search PubMed.
- G. Wang, J. Yang, J. Park, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 Search PubMed.
- R. Yin, P. Shen and Z. Lu, J. Colloid Interface Sci., 2019, 550, 110–116 Search PubMed.
- R. Wijaya, G. Andersan, S. P. Santoso and W. Irawaty, Sci. Rep., 2020, 10, 1–9 Search PubMed.
- R. Sha and S. Badhulika, J. Electroanal. Chem., 2018, 816, 30–37 Search PubMed.
- L. Gan, B. Li, Y. Chen, B. Yu and Z. Chen, Chemosphere, 2019, 219, 148–154 Search PubMed.
- W. Zhu, W. Liu, T. Li, X. Yue, T. Liu, W. Zhang, S. Yu, D. Zhang and J. Wang, Electrochim. Acta, 2014, 146, 419–428 Search PubMed.
- D. Zhang, X. Liu and X. Wang, J. Inorg. Biochem., 2011, 105, 1181–1186 Search PubMed.
- A. H. Al-Marri, M. Khan, M. R. Shaik, N. Mohri, S. F. Adil, M. Kuniyil, H. Z. Alkhathlan, A. Al-Warthan, W. Tremel and M. N. Tahir, Arabian J. Chem., 2016, 9, 835–845 Search PubMed.
- B. Amanulla, S. Palanisamy, S.-M. Chen, V. Velusamy, T.-W. Chiu, T.-W. Chen and S. K. Ramaraj, J. Colloid Interface Sci., 2017, 487, 370–377 Search PubMed.
- F.-Y. Kong, S.-X. Gu, J.-Y. Wang, H.-L. Fang and W. Wang, Sens. Actuators, B, 2015, 213, 397–403 Search PubMed.
- N. Meng, J. Cheng, Y. Zhou, W. Nie and P. Chen, Appl. Surf. Sci., 2017, 396, 310–318 Search PubMed.
- N. Pachauri, K. Dave, A. Dinda and P. R. Solanki, J. Mater. Chem. B, 2018, 6, 3000–3012 Search PubMed.
- C. R. Stanek, A. H. H. Tan, S. L. Owens and R. W. Grimes, J. Mater. Sci., 2008, 43, 4157–4162 Search PubMed.
- S. Liu, Z. Chen, N. Zhang, Z.-R. Tang and Y.-J. Xu, J. Phys. Chem. C, 2013, 117, 8251–8261 Search PubMed.
- Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341–3344 Search PubMed.
- H. A. Elazab, A. R. Siamaki, S. Moussa, B. F. Gupton and M. S. El-Shall, Appl. Catal., A, 2015, 491, 58–69 Search PubMed.
- S. Verma, D. Verma, A. K. Sinha and S. L. Jain, Appl. Catal., A, 2015, 489, 17–23 Search PubMed.
- Q. M. Kainz, R. Linhardt, R. N. Grass, G. Vilé, J. Pérez-Ramírez, W. J. Stark and O. Reiser, Adv. Funct. Mater., 2014, 24, 2020–2027 Search PubMed.
-
(a) A. V. Kumar and K. R. Rao, Tetrahedron Lett., 2011, 52, 5188–5191 Search PubMed;
(b) T. Wu, X. Wang, H. Qiu, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2012, 22, 4772–4779 Search PubMed.
- T. Hoshino, Y. Kurata, Y. Terasaki and K. Susa, J. Non-Cryst. Solids, 2001, 283, 129–136 Search PubMed.
- P. B. Zantye, A. Kumar and A. K. Sikder, Mater. Sci. Eng., R, 2004, 45, 89–220 Search PubMed.
- M. J. Cumbo, D. Fairhurst, S. D. Jacobs and B. E. Puchebner, Appl. Opt., 1995, 34, 3743–3755 Search PubMed.
- V. Sajith, C. B. Sobhan and G. P. Peterson, Adv. Mech. Eng., 2010, 2, 581407 Search PubMed.
-
(a) L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 Search PubMed;
(b) M. Tamura, K. Shimizu and A. Satsuma, Chem. Lett., 2012, 41, 1397–1405 Search PubMed.
- S. V Jadhav, D. S. Nikam, V. M. Khot, N. D. Thorat, M. R. Phadatare, R. S. Ningthoujam, A. B. Salunkhe and S. H. Pawar, New J. Chem., 2013, 37, 3121–3130 Search PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han and X. Liu, Nat. Chem., 2012, 4, 310 Search PubMed.
-
M. Bahadory, PhD thesis, Drexler University, USA, 2008.
- R. Si, Y.-W. Zhang, L.-P. You and C.-H. Yan, J. Phys. Chem. B, 2006, 110, 5994–6000 Search PubMed.
- M. Di Braccio, G. C. Grossi, G. Roma, L. Vargiu, M. Mura and M. E. Marongiu, Eur. J. Med. Chem., 2001, 36, 935–949 Search PubMed.
- J. R. Kavali and B. V. Badami, Il Farmaco, 2000, 55, 406–409 Search PubMed.
- A. Kamal, N. Shankaraiah, S. Prabhakar, C. R. Reddy, N. Markandeya, K. Laxma and X. Devaiah, Bioorg. Med. Chem. Lett., 2008, 18, 2434–2439 Search PubMed.
- L. Kosychova, L. Pleckaitiene, Z. Staniulyte, R. Janciene, A. Palaima and B. D. Puodziunaite, Arkivoc, 2006,(xiii), 158–164 Search PubMed.
- L. Kosychova, Z. Stumbreviciute, L. Pleckaitiene, R. Janciene and B. D. Puodziunaite, Chem. Heterocycl. Compd., 2004, 40, 811–815 Search PubMed.
- X. Che, L. Zheng, Q. Dang and X. Bai, Tetrahedron, 2006, 62, 2563–2568 Search PubMed.
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 Search PubMed.
-
(a) S. Siddiqui, M. U. Khan and Z. N. Siddiqui, ACS Sustainable Chem. Eng., 2017, 5, 7932–7941 Search PubMed;
(b) S. Siddiqui and Z. N. Siddiqui, Catal. Lett., 2018, 148, 3628–3645 Search PubMed;
(c) S. Siddiqui and Z. N. Siddiqui, Appl. Organomet. Chem., 2019, 33, e5161 Search PubMed;
(d) M. U. Khan and Z. N. Siddiqui, ACS Omega, 2018, 3, 10357–10364 Search PubMed;
(e) M. U. Khan, S. Siddiqui and Z. N. Siddiqui, ACS Omega, 2019, 4, 7586–7595 Search PubMed;
(f) R. A. Rather and Z. N. Siddiqui, J. Organomet. Chem., 2018, 868, 164–174 Search PubMed;
(g) R. A. Rather and Z. N. Siddiqui, RSC Adv., 2019, 9, 15749–15762 Search PubMed.
- M. Kooti and M. Afshari, Mater. Res. Bull., 2012, 47, 3473–3478 Search PubMed.
- R. C. Merrifield, Z. W. Wang, R. E. Palmer and J. R. Lead, Environ. Sci. Technol., 2013, 47, 12426–12433 Search PubMed.
- S. Lv, Q. Zhou, Y. Cui, W. Yang and Y. Li, Arabian J. Chem., 2019, 12, 3028–3037 Search PubMed.
- Y.-J. Song, M. Wang, X.-Y. Zhang, J.-Y. Wu and T. Zhang, Nanoscale Res. Lett., 2014, 9, 17 Search PubMed.
-
I. Tiginyanu, V. Ursaki and V. Popa, Ultra-thin membranes for sensor applications, in Nanocoatings and Ultra-Thin Films, Woodhead Publishing, 2011, pp. 330–354 Search PubMed.
- I. Boukhoubza, M. Khenfouch, M. Achehboune, L. Leontie, A. Carlescu, C. Doroftei, B. M. Mothudi, I. Zorkani and A. Jorio, J. Alloys Compd., 2020, 154874 Search PubMed.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. M. Reddy, RSC Adv., 2014, 4, 11322–11330 Search PubMed.
- P. Chamoli, M. K. Das and K. K. Kar, J. Phys. Chem. Solids, 2018, 113, 17–25 Search PubMed.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car and R. K. Prud'homme, Chem. Mater., 2007, 19, 4396–4404 Search PubMed.
- S. Wang, F. Gao, Y. Zhao, N. Liu, T. Tan and X. Wang, Nanoscale Res. Lett., 2018, 13, 377 Search PubMed.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264–267 Search PubMed.
- P. Chamoli, M. K. Das and K. K. Kar, Phys. E, 2017, 90, 76–84 Search PubMed.
- M. M. Khan, S. Khan, S. Shareef and S. C. Sahoo, RSC Adv., 2018, 8, 41892–41903 Search PubMed.
- J. Safari, S. H. Banitaba and S. D. Khalili, J. Mol. Catal. A: Chem., 2011, 335, 46–50 Search PubMed.
- H. Naeimi and H. Foroughi, Chin. J. Catal., 2015, 36, 734–741 Search PubMed.
- Y. S. An, X. Q. Li, X. R. An and L. Z. Wang, Monatsh. Chem., 2015, 146, 165–172 Search PubMed.
- A. Amoozadeh, M. Malmir, N. Koukabi and S. Otokesh, J. Chem. Res., 2015, 39, 694–697 Search PubMed.
- Z. Nasir, A. Ali, M. Shakir, R. Wahab, Shamsuzzaman and Lutfullah, New J. Chem., 2016, 40, 4223–4227 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, green metrics calculation, recycling experiment and spectral data of synthesized compounds. CCDC 1958389. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0na00491j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Zeba N.
Siddiqui
*
and
Zeba N.
Siddiqui
*