
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSub 10 nm CoO nanoparticle-decorated graphitic carbon nitride for solar hydrogen generation via efficient charge separation†
Aniruddha
Mondal
a,
Shubham
Biswas
b,
Srishti
b,
Aditya
Kumar
b,
Jong-Sung
Yu
 *cd and
Apurba
Sinhamahapatra
*b
*cd and
Apurba
Sinhamahapatra
*b
aDepartment of Chemical Engineering and Biotechnology, Tatung University, No. 40, Sec., 3, Chungshan North Rd., Taipei City 104, Taiwan
bDepartment of Chemical Engineering, Indian Institute of Technology (ISM), Dhanbad, Dhanbad-826004, Jharkhand, India. E-mail: apurba@iitism.ac.in
cDepartment of Energy Science and Engineering, DGIST, Daegu, 42988, Republic of Korea. E-mail: jsyu@dgist.ac.kr
dDepartment of Materials Science and Engineering, National Taiwan University of Science and Technology, Taipei 10607, Taiwan
First published on 5th August 2020
Abstract
Solar hydrogen generation is one of the most compelling concepts in modern research to address both the energy and environmental issues simultaneously for the survival of the human race. A Type II heterojunction (CoO–GCN) was fabricated by decorating sub 10 nm CoO nanoparticles (NPs) on the graphitic carbon nitride (GCN) surface. It exhibited improved absorption of UV-VIS light and efficiently separate the photogenerated electrons and holes in opposite directions. A maximum hydrogen generation rate of 9.8 mmol g−1 h−1 was recorded using CoO–GCN from 10% aqueous triethanolamine under simulated sunlight in the presence of 1 wt% Pt. The rate is 3.8 times higher than that of bare GCN. Furthermore, it showed excellent stability for up to five repeated uses. Interestingly, the study also revealed that untreated seawater could replace the deionized water. The cooperative participation of the uniform shape and size of CoO NPs firmly grafted on GCN resulted in remarkable performance for solar hydrogen generation.
Introduction
The search for proficient and clean substitute energy resources is one of the major tasks for future society owing to the reduced fossil-fuel assets and ever-growing environmental pollution.1,2 In this context, hydrogen (H2) is an alternative potential ideal energy carrier compared to traditional fuels, e.g., coal, gasoline, natural gas, and oil.3 H2 is well-known for its benign nature towards the environment and possesses the highest specific energy density compared to any existing commercial fuel.2,4,5 Furthermore, no pollutants such as carbon monoxide (CO), odor, or soot are formed during the combustion process. Although H2 is the best energy carrier, it would be referred to as a green energy carrier only when it is produced via a green path using renewable energy. Several methods for green hydrogen production from water have been developed, such as thermal dissociation of water, electrocatalytic water splitting, photocatalytic water splitting, and hydrolysis of metal hydride. Among the developed processes, photocatalytic water splitting for hydrogen production is the most straightforward, cheap, environment-friendly, and convenient for the further quantification of hydrogen gas. The method needs efficient nanostructured semiconductor-based photocatalysts with a tuned band edge, which can offer the utmost light-harvesting ability and stability in reaction media under light illumination.2,6–8 In recent years, 1D, 2D, and 3D-based nanostructured materials with different molecular architectures have been evolved.9 Among them, 2D based nanoscale materials are widely used owing to their enhanced physicochemical properties and tuned bandgap.In this context, in recent decades, metal-free 2D graphitic carbon nitride (g-C3N4) (GCN) has grown tremendously due to its tunable bandgap towards visible light-assisted photocatalysis.10–18 However, it was observed that the pure GCN photocatalyst system yielded a poor rate of hydrogen generation due to its inefficiency in photogenerated charge separation.11,19–21 Various attempts have been made to develop GCN based nanocomposites for further improvement of the photocatalytic hydrogen generation.22–24 Due to the planned scheme for the formation of the heterostructure along with n-type and p-type semiconductors, the composite will have the following properties: (i) stable oxidation of metal oxides after the formation of the heterostructure, (ii) appropriate band structures, (iii) excellent ability to accelerate the water-splitting reaction, (iv) enhanced visible-light absorption and (v) competent charge separation between the two p–n junctions. Fundamentally, the nanostructured heterojunctions are defined as Type I, Type II, and Type III as per the band positions.25–28 Owing to this, the coupling of two heterostructures such as nanoscale metal oxides and GCN is meaningful to make a potential heterojunction for improved photocatalytic performance towards hydrogen generation. GCN is an n-type semiconductor, and it can form strong non-covalent interactions with multiple classes of p-type semiconductors leading to enhancement in photocatalytic performances.11,22,29–34
Transition metal-based metal oxides have been used as alternative resources to precious metals. They have received tremendous attention due to their environmental benignity, low cost, natural abundance, and improved physicochemical properties.35–41 In particular, Co-oxides, earth-abundant transition metal oxides, substitutes for noble metals, less expensive p-type semiconductors, and less toxic materials have gained remarkable attention due to their attractive physicochemical properties.42–44 Nanostructured Co-oxides, e.g., CoO and Co3O4, have been studied for hydrogen generation. Co3O4 consisting of Co(II) and Co(III) has direct bandgap energy and has sufficient potential to produce H2 gas by the reduction of water molecules.44–46
In comparison, CoO has been less exploited for light-assisted hydrogen generation. The bulk CoO has been found to be inactive towards photocatalytic hydrogen evolution due to their band position.45,47 However, nanostructured CoO, tuned by the quantum confinement effect, possesses a wider bandgap that could be suitable for visible-light absorption. At the nanoscale level of CoO, the bandgap turned out to be increased, and the conduction band (CB) is situated more negative compared to the reduction potential value of H+/H2. The phenomenon is mostly observed when the particle size of CoO is ≤10 nm.45 An average size of 10 nm for CoO NPs was shown to generate a solar to hydrogen efficiency of 5%, but it turned out to be inactive after one-hour of the reaction.45 The stable photocatalytic performance of CoO nanoparticles would be fascinating. In recent years, CoO nanowires with two different sizes (34 nm and 65 nm) constructed with the corresponding NPs revealed multiple positions of the various band edges and displayed unique H2 production via the photocatalytic pathway.46,48,49 However, little effort has been made/reported for the synthesis of nanostructured CoO because of its inflexible synthesis route. CoO at the nanoscale level requires a distinct approach to incorporate cobalt into the lower valence state.45 Due to the presence of a lower oxidation state in the CoO crystal architecture, it had a meager lifetime owing to the further oxidation during the reaction, resulting in further corrosion.50,51 Therefore, it is better for it to be coupled or embedded with other nanoscale materials (e.g., GCN), which can overcome the present limitation by forming a heterojunction.22,25 Mao et al. successfully prepared CoO/GCN NS (NS: nanosheet) nanocomposites via a one-pot method and used them in photocatalytic H2 generation. However, the embedded CoO NPs (size ∼ 30–50 nm) were aggregated, and this resulted in a loss of 17% catalytic activity after three recycling runs.52 Guo et al. also synthesized CoO/GCN NSs via the solvothermal method for overall water splitting and for tetracycline degradation under visible light, which showed improved stability with a sample containing 30 wt% 10–20 nm CoO NPs.53,54 They also suggested high photocatalytic activity of the CoO/GCN due to the rearrangement of band positions in the heterojunction because of charge diffusion for the Fermi level equilibration, which leads to effective separation of photogenerated electrons/holes.54 Wang et al. reported a two-step hydrothermal process followed by pyrolysis for the synthesis of CoO nanorod/C3N4 composites that exhibited improved water splitting activity when they contained 10% CoO.55 Zhu et al. discussed the formation of Type I and II heterojunctions of Co3O4 and CoO, respectively, with GCN nanotubes and a composite content of 7 wt% CoO exhibited the highest rate of hydrogen generation.25 The composite with CoO also exhibited better stability than Co3O4.
The potential physicochemical properties, including the structural and bandgap profile of the different heterojunctions, depend on their design and synthetic strategies. In this work, sub 10 nm CoO nanoparticles well confined on the surface of the GCN NSs were synthesized via a simple two-step process (see Fig. 1). The prepared CoO/GCN NS heterojunction has shown good activity towards photo-assisted hydrogen generation in the presence of photo-deposited Pt NPs as the co-catalyst. Different characterization techniques have been used to examine the morphology, microstructure properties, band positions, and charge transfer mechanisms. The detailed investigation suggested the formation of Type-II heterojunction.
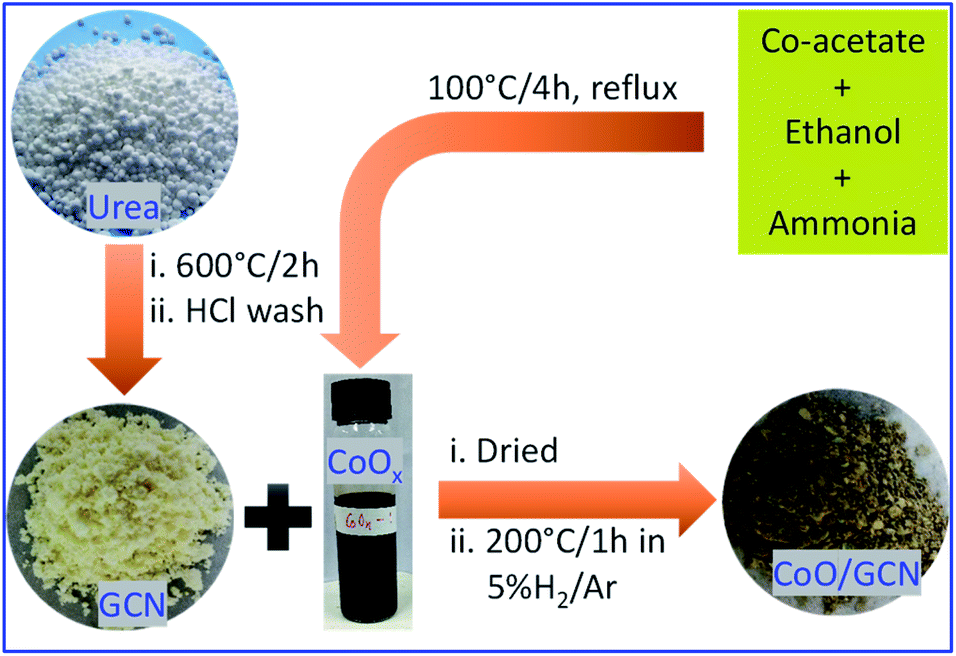 | ||
| Fig. 1 Synthesis: schematic representation regarding the preparation of GCN and CoO/GCN samples. | ||
Experimental
Synthesis of graphitic carbon nitride
In a typical procedure, urea (NH2CONH2), procured from Sigma-Aldrich, was heated at 600 °C for 2 h. The obtained solid was stirred in dilute HCl for 12 h. Then, the product was filtered, washed with water–ethanol, and dried at 70–80 °C for one day to obtain yellow fine GCN powder.Synthesis of the CoOx colloidal solution
In a typical procedure, 500 mg of cobalt acetate (Co(CH3COO)2·4H2O), purchased from Sigma-Aldrich, was dissolved in 25 ml of ethanol, and then 2.5 ml of 25% ammonium hydroxide solution was added under vigorous stirring. After 15 min of stirring, the mixture was refluxed at 100 °C for 4 h in a round bottom flask. After cooling to room temperature, a dark brown solution was obtained with ∼0.08 M concentration of Co2+.Synthesis of sub 10 nm CoO nanoparticle decorated graphitic carbon nitride
In a typical procedure, 1 g of prepared GCN was dispersed in ethanol and the desired amount (1, 2, 4, and 8 ml) of freshly prepared Co(O)x solution was added and stirred for 4 h and then dried at 80 °C for 12 h. The obtained powder was further treated at 200 °C for 1 h in a 5% H2/Ar atmosphere to obtain CoO-decorated GCN. The prepared samples were named CoO/GCN-x, where x is 1, 2, 4, and 8, indicating the volume of the CoOx solution added. The calculated weight percentages (wt%) of CoO in heterojunctions are 0.59, 1.18, 2.36, and 4.72 for CoO/GCN-1, 2, 4, and 8, respectively. The synthesis procedure is schematically represented in Fig. 1.Characterization
Different analytical techniques were used to characterize the prepared materials. A Rigaku Smartlab diffractometer was used to obtain the powder X-ray diffraction patterns of the samples. The diffuse reflectance spectra (DRS) were recorded for the solid samples using an Agilent CARY5000 Ultraviolet-Visible-Near Infrared Spectrophotometer. High-Resolution Transmission Electron Microscopy images of the samples were obtained using a JEOL FE-2010. An ESCALAB 250 XPS System with a monochromated Al Kα (150 W) source was used to collect the X-ray photoelectron spectra of the solid samples. An Agilent Technologies Cary Eclipse Fluorescence Spectrophotometer was used to obtain the photoluminescence spectra.Photocatalytic hydrogen generation
In a typical procedure, 20 mg of CoO/CN-x was dispersed in 10% triethanolamine–water, and a calculated amount of H2PtCl6 solution was added to obtain 1 wt% Pt with respect to the photocatalyst. The mixed solution was kept in a cylindrical quartz reactor. The reactor temperature was maintained at ∼25 °C by using chilled water circulation. The solution was irradiated under a 450 W Xe light (UV-VIS) for 2 h for Pt NP photodeposition on CoO/GCN-x. Then the reactor was degassed again with Ar and studied for hydrogen production under simulated sunlight (1 Sun, AM-1.5 G). The produced hydrogen was measured using a connected gas chromatography (GC) instrument. The performance stability of the heterojunction was studied by storing the solution under dark conditions. The solution was irradiated once every week for up to 5 runs, and the hydrogen generated was recorded. For visible light-assisted hydrogen generation, a cut off filter (λ > 395 nm) was used. For the use of seawater, only distilled water was replaced with untreated filtered seawater keeping all other parameters the same.Results and discussion
The prepared samples were first characterized by recording their absorption spectra to examine the changes in optical properties (Fig. 2a). The prepared GCN exhibited a steep absorption edge at 442 nm. The CoO-NP-decorated GCN samples showed advancement in the absorption of visible light along with a prominent shift of the absorption peak towards a higher wavelength. Moreover, light absorption in the visible region was increased with the increasing number of CoO nanoparticles. The pattern of CoO–GCN's spectrum was dissimilar compared to that of the bare GCN. It exhibited two new adsorption edges in the visible region due to the presence of the CoO NPs. A slope up to 550 nm and a broad hump centered above 700 nm were observed due to ligand (OII) to metal (CoII) charge transfer and d–d transition in CoII, respectively. The advancement in visible light absorption occurred due to the synergistic effect of GCN and CoO NPs. Furthermore, the absorption edge possesses a slight redshift, and the shifting increased with an increase of the CoO amount, suggested the formation of a heterojunction between CoO and GCN. It should also be noted that the UV absorption region also increased in the composite samples compared to pure GCN due to the presence of the CoO NPs. For further analysis, the samples were studied by photoluminescence (PL) spectroscopy to understand the effect of CoO NPs on charge separation between CoO and GCN (Fig. 2b). The PL spectra of the samples revealed a prominent peak at 465 nm at an excitation wavelength of 350 nm, and the intensity gradually decreased with the increasing CoO amount in the CoO–GCN samples. The optical energy of the emission band has a close resemblance to that for GCN, indicating intrinsic emission ascribed to direct electron–hole recombination of the band shift. It was also seen that the CoO/GCN composite material shows a weaker emission profile compared to the bare GCN counterpart. The presence of non-covalent interactions between CoO and GCN triggered the faster migration of charge transporters. The decrease in intensity demonstrates the improvement of the charge separation efficiency in the composite compared to the bare GCN. The PL results gave an overall photochemical sketch about the recombination rate of the photogenerated charge transporters, which is immensely controlled by the well-defined heterojunction between CoO and GCN.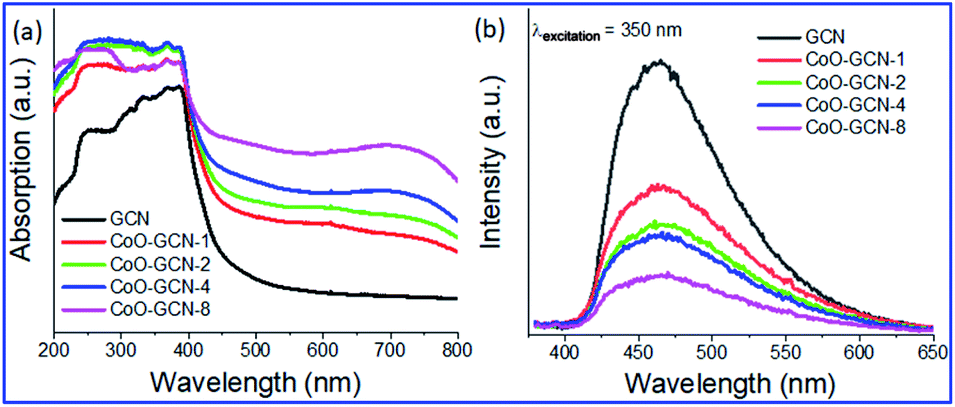 | ||
| Fig. 2 Optical properties: (a) UV-VIS DRS spectra and (b) PL spectra for the as-prepared GCN and CoO/GCN samples. | ||
After the confirmation of improvement in optical properties, the materials were investigated for morphological analysis via microscopic techniques. Fig. 3 represents the high-resolution transmission microscope (HR-TEM) images of GCN and CoO–GCN-1. The pure GCN appeared to have a wrapped nanosheet type morphology (Fig. 3a) with an average diameter of around 70 nm. The arrangement of the wrapped nanosheets was irregular. In Fig. 3b–d and S1a–d,† the TEM images of CoO–GCN-1 reveal that the small spherical particles are uniformly well-distributed throughout the surface of the GCN. Additionally, the HR-TEM images further revealed a narrow size distribution centered near <10 nm for the CoO NPs. The observed d-spacing of 0.265 nm was indexed to the (002) plane of CoO.56 The SAED patterns shown in Fig. S2† indicate the amorphous nature of the synthesized samples. However, some diffuse rings appeared in the case of CoO–GCN due to the presence of CoO (Fig. S2b†). The elemental mapping images and EDS spectra of the materials (GCN and Co–GCN-1) are shown in Fig. S3 and S4.† Homogeneous distribution of C and N atoms in the CN sample was seen with a C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N atomic ratio of 1:0.71, suggesting that N vacancies might occur probably due to preparation from urea at a high temperature of 600 °C. N vacancies in GCN boosted the photocatalytic activity as reported earlier.57 The C:N ratio for CoO–GCN-1 changes to 1:0.58. The additional carbon would have come from the acetate used in the preparation of CoO NPs. Furthermore, the mapping analysis revealed the homogeneous distribution of Co and O, also indicating the monodispersed small CoO NPs. The atomic ratio of Co:O is found to be 1:0.95, which further evidenced the formation of cobalt monoxide. The results are strong evidence in favor of sub 10 nm CoO NP-decorated graphitic carbon nitride nanosheets.
:N atomic ratio of 1:0.71, suggesting that N vacancies might occur probably due to preparation from urea at a high temperature of 600 °C. N vacancies in GCN boosted the photocatalytic activity as reported earlier.57 The C:N ratio for CoO–GCN-1 changes to 1:0.58. The additional carbon would have come from the acetate used in the preparation of CoO NPs. Furthermore, the mapping analysis revealed the homogeneous distribution of Co and O, also indicating the monodispersed small CoO NPs. The atomic ratio of Co:O is found to be 1:0.95, which further evidenced the formation of cobalt monoxide. The results are strong evidence in favor of sub 10 nm CoO NP-decorated graphitic carbon nitride nanosheets.
 | ||
| Fig. 3 Morphology: HR-TEM images of (a) GCN and (b–d) CoO/GCN. | ||
For further evidence of the phases, composition, and structural features of the synthesized nanostructured materials, powdered XRD and XPS analysis were performed. Fig. S5† displays the XRD patterns of GCN and CoO–CNs. The pristine GCN nanosheets exhibited two major XRD peaks around 2θ = 13° (100) and 27° (002). The former peak indicated the presence of in-plane structural packing, while the latter peak emerged due to the interlayer stacking of the conjugated aromatic system.58 The two corresponding peaks were observed in all the synthesized samples, which indicated that the typical structure of GCN did not change much upon decoration with CoO NPs. The peaks for the CoO NPs were not observed well when the amount was low; however, they started to appear when increasing the CoO amount and suggested a hexagonal crystal system with the P63mc (186) space group (PDF Card no. 01-089-2803). The XPS data are presented in Fig. 4. The XPS survey spectrum (Fig. 4a) of the as-synthesized materials, GCN and CoO–GCN-1, confirmed the presence of C and N for GCN with a low concentration of oxygen. The high-resolution Co 2p, C 1s, O 1s, and N 1s spectra for CoO–GCN are shown in Fig. 4b–e. The C:N atomic ratios were found to be 1:0.72 and 1:0.65 for the GCN and CoO–GCN-1, respectively, which followed a similar trend to that of the TEM-EDS results. The core-level C 1s spectra of the GCN and CoO–GCN in Fig. 4b exhibited two peaks at 284.5 and 287.9 eV corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and N–CN groups. The core-level N 1s XPS peak (Fig. 4c) appeared at 398.6 eV with extended tails towards higher binding energies corresponding to CN–C, N(C)3, and C–NHx groups. Another small peak appeared at 404.0 eV due to the π excitations. In Fig. 4d, the core-level Co 2p XPS spectrum exhibited two significant peaks at 781.3 and 796.6 eV for p3/2 and p1/2, respectively, along with two corresponding shake-up satellite peaks at 785.9 and 802.8 eV. A split spin–orbit component of 15.3 eV was calculated, indicating the presence of Co2+. The shake-up satellite peaks are often used to characterize the phase purity of Co oxides, which appears due to ligand to metal charge transfer during photoemission. The peak position and satellite features confirmed the formation of phase pure CoO59–61 in line with the XRD and TEM results. In Fig. 4e, the core-level O 1s XPS spectra of the as-prepared GCN and Co–GCN-1 are presented. The O 1s peak for GCN appeared at 532.4 eV, indicating the presence of a small amount of oxygen. A comparatively strong peak at 531.6 eV in the case of the CoO/GCN was observed due to the grafted CoO.
C and N–CN groups. The core-level N 1s XPS peak (Fig. 4c) appeared at 398.6 eV with extended tails towards higher binding energies corresponding to CN–C, N(C)3, and C–NHx groups. Another small peak appeared at 404.0 eV due to the π excitations. In Fig. 4d, the core-level Co 2p XPS spectrum exhibited two significant peaks at 781.3 and 796.6 eV for p3/2 and p1/2, respectively, along with two corresponding shake-up satellite peaks at 785.9 and 802.8 eV. A split spin–orbit component of 15.3 eV was calculated, indicating the presence of Co2+. The shake-up satellite peaks are often used to characterize the phase purity of Co oxides, which appears due to ligand to metal charge transfer during photoemission. The peak position and satellite features confirmed the formation of phase pure CoO59–61 in line with the XRD and TEM results. In Fig. 4e, the core-level O 1s XPS spectra of the as-prepared GCN and Co–GCN-1 are presented. The O 1s peak for GCN appeared at 532.4 eV, indicating the presence of a small amount of oxygen. A comparatively strong peak at 531.6 eV in the case of the CoO/GCN was observed due to the grafted CoO.
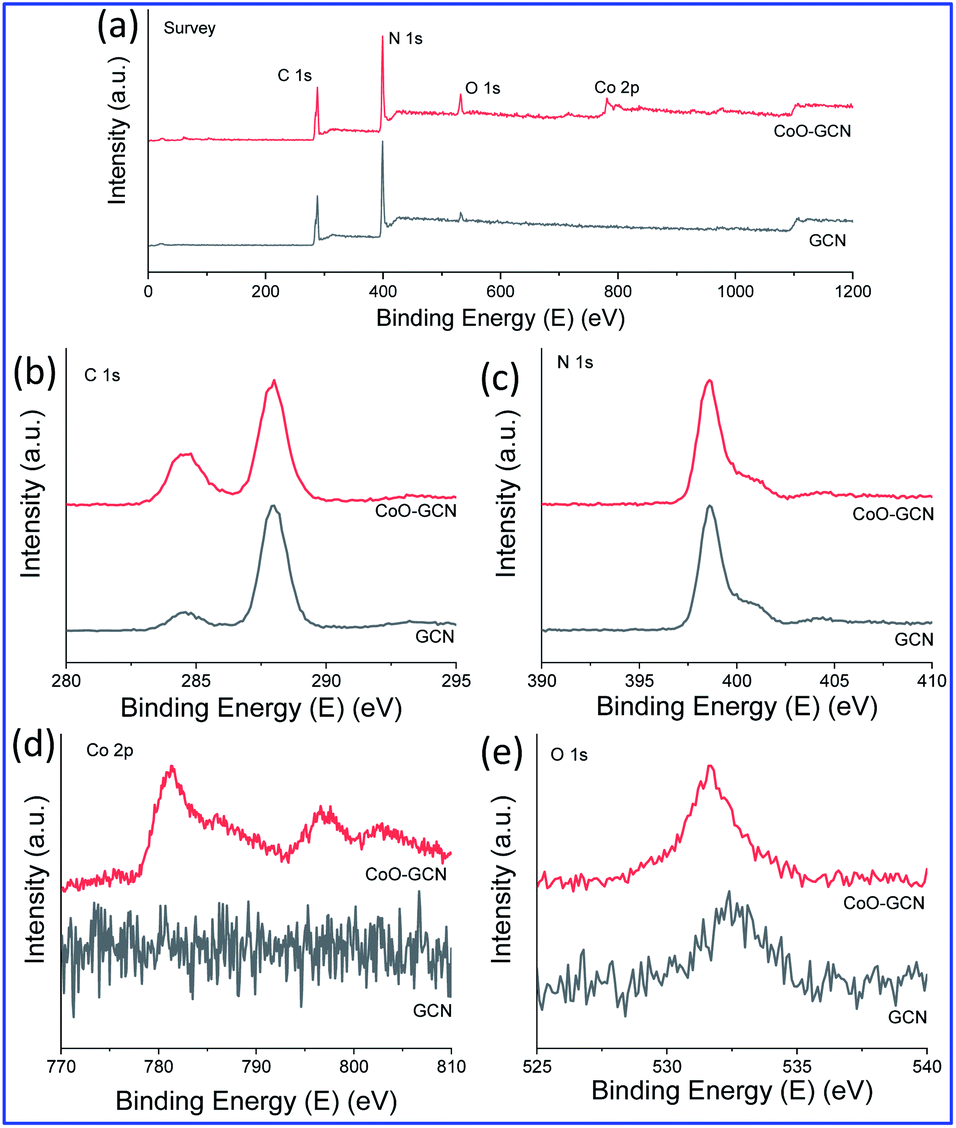 | ||
| Fig. 4 Elemental composition: XPS spectra of (a) survey, (b) C 1s, (c) N 1s, (d) Co 2p and (e) O 1s for the as-prepared GCN and CoO/GCN. | ||
Photo assisted hydrogen generation
The as-prepared CoO-decorated GCN samples were studied for photo-assisted hydrogen production from 10% triethanolamine-water under simulated sunlight (1 Sun, AM-1.5 G) using the full solar wavelength range of light in the presence of photo-deposited Pt as the co-catalyst. The non-stop hydrogen production profile and the rate of H2 production obtained from different samples are shown in Fig. 5a and b, respectively. The hydrogen production rate for bare GCN was 2.6 mmol h−1 g−1, and it increased with the addition of CoO NPs and further increased with the increasing CoO amount. Finally, a maximum rate of 9.8 mmol h−1 g−1 was obtained for CoO–GCN-4, which was ∼3.8 times higher than that for the bare GCN. A further increase in the CoO amount beyond 2.4 wt% did not increase the rate of hydrogen production. A possible reason for the reduced photocatalytic activity might be that the overloading of CoO hindered the active sites on the surface of g-C3N4.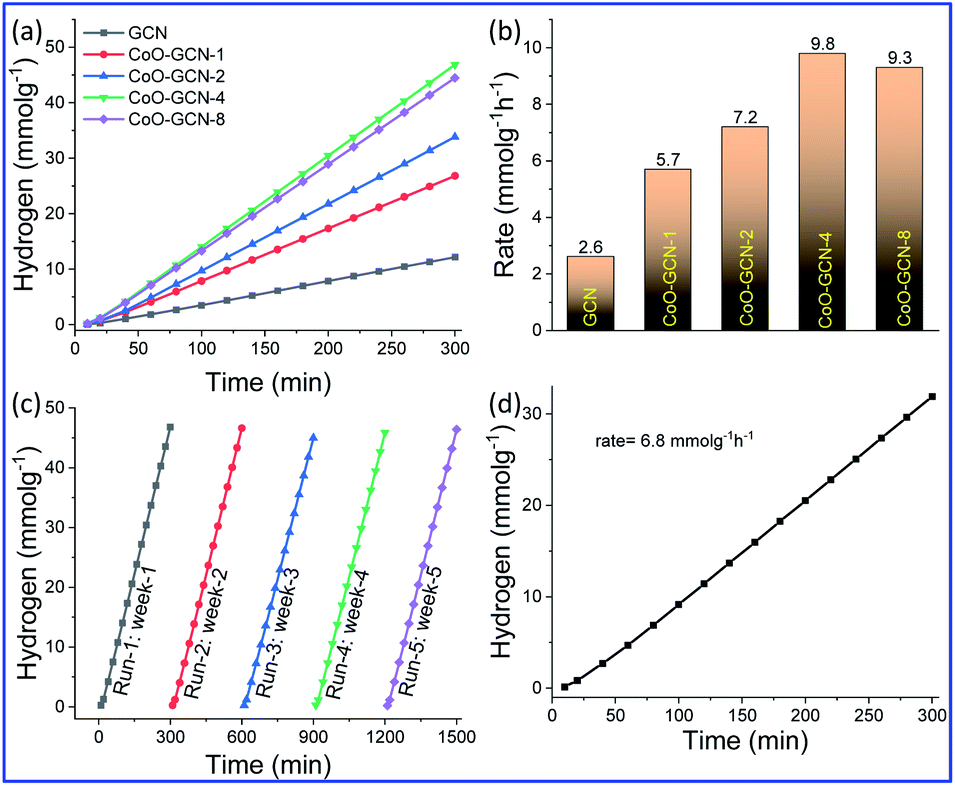 | ||
| Fig. 5 Photo-assisted hydrogen generation: (a) continuous hydrogen generation profile and (b) rate of hydrogen generation for GCN and CoO–GCN. (c) Stability performance in terms of hydrogen generation for CoO–GCN-4. (d) Hydrogen generation from seawater. Reaction conditions: light: 1 Sun, AM 1.5 G, solution: 10% triethanolamine–water, photocatalyst: 20 mg, co-catalyst: 1% photodeposited Pt, Ar atmosphere. | ||
Therefore, the increment in the hydrogen generation rate was prominent after the decoration of sub 10 nm CoO NPs on the GCN. It was observed that on introduction of CoO, the charge transfer of the photogenerated electrons and holes is reasonably increased compared to that in the bare GCN, as observed in the PL results (Fig. 1c). Further, the activity of GCN and CoO–GCN-4 was examined in the absence of the co-catalyst (Pt). The obtained rates were 1.04 mmol g−1 h−1 for CoO–GCN-4 and 0.34 mmol g−1 h−1 for GCN (Fig. S6†). The results indicated satisfactory improvement in photoactivity of the prepared heterojunction compared to GCN because of an increase in charge transfer. The Pt co-catalyst further accelerated the process.
It is also important that the increase in the light absorption in both the UV and visible region was observed in the DRS results (Fig. 1a), which also contributed to catalysts' efficiency advancement. Hydrogen generation was also performed using only visible light (Fig. S7†). Hydrogen generation rates of 0.49 and 1.58 mmol g−1 h−1 were obtained for GCN and CoO–GCN-4, respectively. The results suggested a reasonable increase of visible light activity for the prepared heterojunction due to the enhanced visible light absorption and faster charge separation. As discussed earlier, the minimal size of CoO triggered the photocatalytic activity for the efficient production of hydrogen. This was further confirmed by preparing a photocatalyst system comprising bulk CoO and GCN, which exhibited a lower rate of hydrogen generation than bare GCN (Fig. S8†). Although it is not worth comparing different catalyst systems unless studied under identical conditions, a comparison including many aspects for the various catalysts is provided in Table 1. In comparison to the reported CoO–g-C3N4 heterojunction systems, our developed one demonstrated reasonable advancement. The rate of hydrogen production is remarkably higher than those for the reported composites. After the effective production of H2 using CoO–GCN-4, it is essential to check the stability of the corresponding catalyst (Fig. 5c). The hydrogen generation experiment was performed by storing the same solution in the absence of light. In the investigation, no significant decrease was observed for up to 5 cycles in different weeks. The stable photocatalytic performance of the heterojunction also suggested no alteration in the CoO–GCN during reaction. The production of H2 using CoO/GCN-4 from seawater (Fig. 5d) was also examined. The observed rate of hydrogen production was 6.8 mmol g−1 h−1.
| Photocatalyst | Synthetic approach | Photo-assisted reaction conditions | Results | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Carbon nitride | CoO composite | Solution | Amount (mg) | Co-catalyst | Light source | Rate (mmol g−1 h−1) | Improvement | Stability | ||
| a The values are calculated from the results presented in the article. b No values are provided. | ||||||||||
| g-C3N4/CoO nanocomposites with 0.5 wt% CoO | Prepared at 550 °C for 6 h from urea | Preparation of Co-species loaded C3N4 followed by sintering at 400 °C for 4 h in Ar | 10% triethylamine–water, 100 ml | 100 | Photodeposited Pt (3 wt%) | 300 W xenon lamp/400 nm cutoff filter/100 mW cm−2 | H2: 0.651 | 2.2 times compared to bare g-C3N4a | Run: 3, total time: 12 h, activity retention: 83%a | Mao et al., 201752 |
| CoO/g-C3N4 heterojunction with 30 wt% CoO | Prepared at 550 °C for 3 h from urea | Solvothermal treatment at 220 °C for 4 h | Ultrapure water, 20 ml | 50 | No | White light-emitting diode (LED) (λ > 400 nm) | H2: 0.00251,a O2: 0.00139a | 17.2 times compared to CoOa | Run: 15, total time: 360 h, activity retention: goodb | Guo et al. 201853 |
| CoO nanorod/C3N4 heterostructure with 10 wt% CoO | Prepared at 550 °C from urea | Hydrothermal treatment at 90 °C for 10 h, followed by thermal annealing at 450 °C for 4 h in Ar | Water, 80 ml | 30 | Photodeposited Pt (3 wt%) | 300W xenon lamp (λ > 400 nm) | H2: ∼3,a O2: ∼1.5a | Run: 3, total time: b, activity retention: decreasingb | Wang et al., 201855 | |
| Heterojunction of CoO nanoparticles confined in g-C3N4 nanotubes with 7 wt% CoO | Prepared at 550 °C for 3 h from urea | Preparation of CoOx decorated on C3N4 followed by annealing at 400 °C for 4 h under vacuum | 10% triethylamine–water, 40 ml | 40 | No | 300 W xenon lamp/420 nm cutoff filter | H2: 0.263a | 2.2 times compared to bare g-C3N4a | Run: 4, Total time: 12 h, activity retention: 94% | Zhu et al., 201925 |
| 10 nm CoO nanoparticle decorated graphitic carbon nitride (GCN) with 2.36 wt% CoO | Prepared at 600 °C for 2 h from urea | Preparation of Co(O)x followed by grafting on carbon nitride via drying and annealing at 200 °C for 1 h in 5%H2/Ar | 10% triethylamine–water, 80 ml | 20 | Photodeposited Pt (1 wt%) | Simulated sunlight (1 Sun, AM-1.5 G), only visible light (λ > 395 nm) | H2: 9.8, H2: 1.58 | 3.8 times compared to GCN, 3.2 times compared to GCN | Run: 5, total time: 25 h, activity retention: 98% | Present study |
Besides the excellent performance of the developed heterojunction for solar hydrogen generation, it is also necessary to study the designed heterojunction in depth. The DRS data were computed to obtain the Tauc plot (Fig. 6a). The Tauc plot of CoO–GCN revealed two different light absorption edges at 2.84 and 2.02 eV. The former absorption edge corresponds to the GCN and the latter to CoO in the heterojunction. Therefore, the bandgaps of GCN and CoO were 2.84 and 2.02 eV in the heterojunction. The bandgap of bare GCN was found to be 2.83 eV (Fig. S9†), which was not compromised during the formation of the heterojunction. A bandgap of 2.02 eV for nanoscale CoO was observed due to quantum confinement of small sized (<10 nm) CoO NPs.45 Next, valence band (VB) XPS (Fig. 6b) was performed to find out the VB top of the two semiconductors in the heterojunction. As the VBs of GCN and CoO are generally constructed from the molecular orbitals of nitrogen and oxygen, respectively, the VBs possibly observe at different bind energies in the VB XPS of CoO–GCN. The VB XPS results indicated the VB top at 2.69 and 1.00 eV for GCN and CoO, respectively. Subtraction of the valance band top position from the bandgap leads to the CB bottom. In Fig. 6c, a schematic representation of the heterojunction (in potential vs. the NHE scale) is depicted based on the above results, which revealed the formation of a Type II (staggered) heterojunction. Light was absorbed in the UV-VIS region by both semiconductors and charges are generated at the VB and CB. For effective charge separation, the generated holes migrated to CoO's VB, whereas electrons migrated to the CB of GCN. Oxidation takes place at the VB of CoO to produce H+ and other oxidized products. The H+ migrated to the surface of the Pt co-catalyst attached to the CB of GCN and was reduced by the electrons to produce H2. The evidence for effective charge separation can be further realized by photocurrent measurements.62,63 The photocurrent was measured for GCN and CoO–GCN-4 (Fig. S10†). A distinct increase in photocurrent for CoO–GCN was observed compared to GCN, which further confirmed the effective charge separation in the prepared heterojunction.
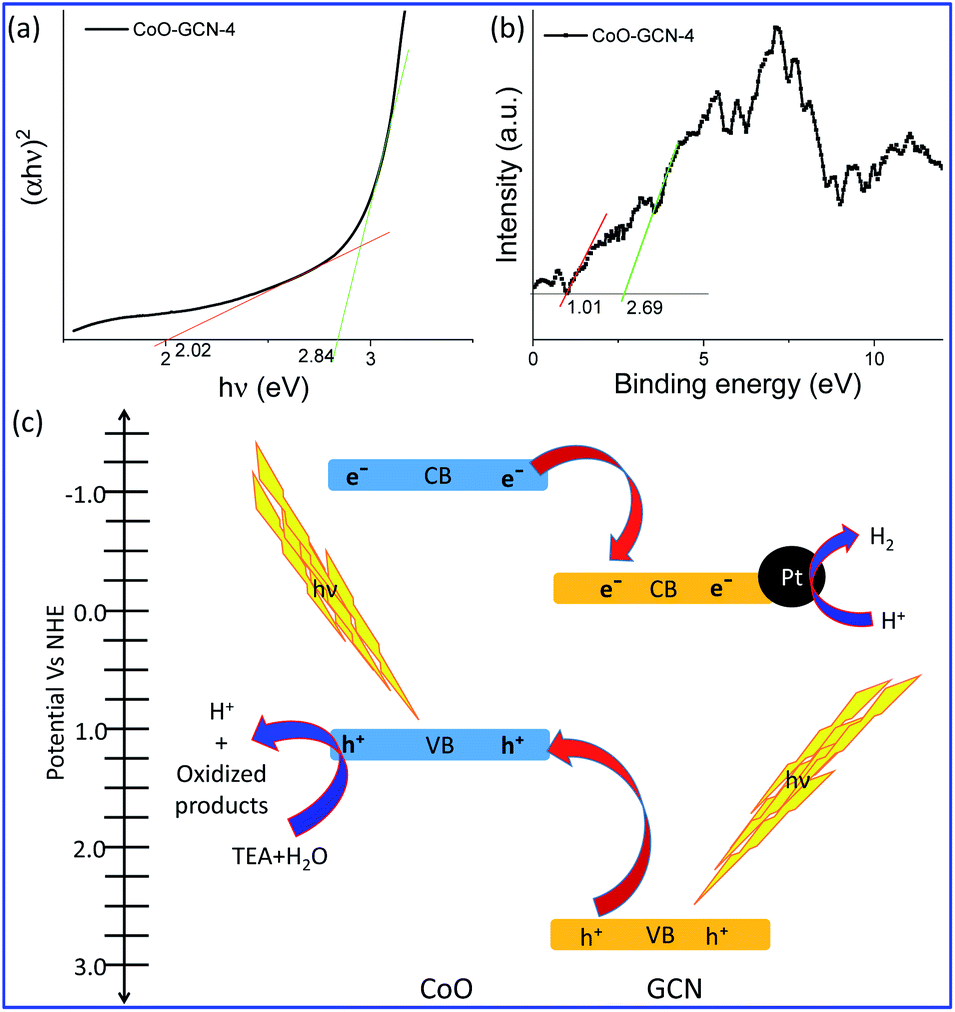 | ||
| Fig. 6 (a) Tauc plot calculated from the DRS data and (b) valence band XPS of CoO–GCN-4. (c) Schematic representation of hydrogen generation over the Type II heterojunction of CoO–GCN-4 elucidated from the data presented in (a) and (b). | ||
Conclusion
In conclusion, a photoactive heterojunction made of sub 10 nm CoO nanoparticles implanted on GCN nanosheets was prepared. The CoO nanoparticles with a uniform shape and size were regularly dispersed over GCN. The study revealed the formation of a Type II heterojunction capable of UV-VIS light absorption and faster charge migration in the opposite direction. The heterojunction exhibited a distinct activity and stability for hydrogen production under simulated solar light. The achieved rate of hydrogen generation was 9.8 mmol g−1 h−1, and 98% activity was retained after five different series of cycles performed in a gap of one week. The results are reasonably improved relative to those for the previously reported CoO/C3N4 heterojunction. The developed photocatalyst system was also found to be proficient at producing hydrogen with a reasonable rate of 6.8 mmol g−1 h−1 when deionized water is replaced with untreated seawater. The study takes the research one step forward to the feasibility of the commercial production of solar hydrogen as a clean and efficient energy carrier.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Department of Science and Technology (DST) – Ministry of Science and Technology, India for the funding and the DST Inspire faculty scheme (Grant No. IFA17-MS107). JSYU acknowledges the National Research Foundation (2019R1A2C2086770) funded by the Korea Ministry of Science, ICT & Future Planning for funding.Notes and references
- Y.-P. Yuan, L.-W. Ruan, J. Barber, S. C. Joachim Loo and C. Xue, Energy Environ. Sci., 2014, 7, 3934–3951 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- A. Mondal, A. Paul, D. N. Srivastava and A. B. Panda, ACS Appl. Nano Mater., 2018, 1, 4622–4632 CrossRef CAS.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- A. Mondal, K. Sinha, A. Paul, D. N. Srivastava and A. B. Panda, Int. J. Hydrogen Energy, 2020, 45, 18623–18634 CrossRef CAS.
- C. Xu, P. Ravi Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868–3902 RSC.
- A. Sinhamahapatra, H. Y. Lee, S. H. Shen, S. S. Mao and J. S. Yu, Appl. Catal., B, 2018, 237, 613–621 CrossRef CAS.
- A. Saha, A. Sinhamahapatra, T. H. Kang, S. C. Ghosh, J. S. Yu and A. B. Panda, Nanoscale, 2017, 9, 17029–17036 RSC.
- X. Zhang, X. Yuan, L. Jiang, J. Zhang, H. Yu, H. Wang and G. Zeng, Chem. Eng. J., 2020, 390, 124475 CrossRef CAS.
- X. Gao, J. Feng, D. Su, Y. Ma, G. Wang, H. Ma and J. Zhang, Nano Energy, 2019, 59, 598–609 CrossRef CAS.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- D. Huang, Z. Li, G. Zeng, C. Zhou, W. Xue, X. Gong, X. Yan, S. Chen, W. Wang and M. Cheng, Appl. Catal., B, 2019, 240, 153–173 CrossRef CAS.
- A. Akhundi, A. Badiei, G. M. Ziarani, A. Habibi-Yangjeh, M. J. Muñoz-Batista and R. Luque, Mol. Catal., 2020, 488, 110902 CrossRef CAS.
- W. Shi, M. Li, X. Huang, H. Ren, C. Yan and F. Guo, Chem. Eng. J., 2020, 382, 122960 CrossRef.
- L. Wang, Y. Hong, E. Liu, Z. Wang, J. Chen, S. Yang, J. Wang, X. Lin and J. Shi, Int. J. Hydrogen Energy, 2020, 45, 6425–6436 CrossRef CAS.
- Y. Hong, L. Wang, E. Liu, J. Chen, Z. Wang, S. Zhang, X. Lin, X. Duan and J. Shi, Inorg. Chem. Front., 2020, 7, 347–355 RSC.
- M. S. Nasir, G. Yang, I. Ayub, S. Wang, L. Wang, X. Wang, W. Yan, S. Peng and S. Ramakarishna, Appl. Catal., B, 2019, 257, 117855 CrossRef CAS.
- Q. Wang, S. Yu, W. Qin and X. Wu, Nanoscale Adv., 2020, 2, 274–285 RSC.
- F. Guo, M. Li, H. Ren, X. Huang, K. Shu, W. Shi and C. Lu, Sep. Purif. Technol., 2019, 228, 115770 CrossRef CAS.
- M. Z. Rahman and C. B. Mullins, Acc. Chem. Res., 2019, 52, 248–257 CrossRef CAS PubMed.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391(part B), 72–123 CrossRef CAS.
- K. M. Alam, P. Kumar, P. Kar, U. K. Thakur, S. Zeng, K. Cui and K. Shankar, Nanoscale Adv., 2019, 1, 1460–1471 RSC.
- Y. Zhu, T. Wan, X. Wen, D. Chu and Y. Jiang, Appl. Catal., B, 2019, 244, 814–822 CrossRef CAS.
- F. Opoku, K. K. Govender, C. G. C. E. van Sittert and P. P. Govender, Adv. Sustainable Syst., 2017, 1, 1700006 CrossRef.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- F. Dong, Z. Ni, P. Li and Z. Wu, New J. Chem., 2015, 39, 4737–4744 RSC.
- F. Xu, Z. Mo, J. Yan, J. Fu, Y. Song, W. El-Alami, X. Wu, H. Li and H. Xu, J. Colloid Interface Sci., 2020, 560, 555–564 CrossRef CAS PubMed.
- C. Jin, M. Wang, Z. Li, J. Kang, Y. Zhao, J. Han and Z. Wu, Chem. Eng. J., 2020, 398, 125569 CrossRef CAS.
- P. Mishra, A. Behera, D. Kandi and K. Parida, Nanoscale Adv., 2019, 1, 1864–1879 RSC.
- W. Shi, C. Liu, M. Li, X. Lin, F. Guo and J. Shi, J. Hazard. Mater., 2020, 389, 121907 CrossRef CAS PubMed.
- J. Wang, C. Liu, S. Yang, X. Lin and W. Shi, J. Phys. Chem. Solids, 2020, 136, 109164 CrossRef CAS.
- S. Yang, C. Liu, J. Wang, X. Lin, Y. Hong, F. Guo and J. Shi, J. Solid State Chem., 2020, 287, 121347 CrossRef CAS.
- T. Jafari, E. Moharreri, S. A. Amin, R. Miao, W. Song and L. S. Suib, Molecules, 2016, 21 Search PubMed.
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC.
- A. Okashimo, Y. Sakata, N. Urao, H. Otsuka and H. Imamura, in Studies in Surface Science and Catalysis, ed. E. M. Gaigneaux, M. Devillers, D. E. De Vos, S. Hermans, P. A. Jacobs, J. A. Martens and P. Ruiz, Elsevier, 2006, vol. 162, pp. 1001–1008 Search PubMed.
- A. Zaleska-Medynska, Metal Oxide-Based Photocatalysis: Fundamentals and Prospects for Application, Elsevier, 2018 Search PubMed.
- A. Sinhamahapatra, J. P. Jeon, J. Kang, B. Han and J. S. Yu, Sci. Rep., 2016, 6, 27218 CrossRef CAS PubMed.
- A. Sinhamahapatra, D. Bhattacharjya and J. S. Yu, RSC Adv., 2015, 5, 37721–37728 RSC.
- B. K. Tudu, V. Gupta, A. Kumar and A. Sinhamahapatra, J. Colloid Interface Sci., 2020, 566, 183–193 CrossRef CAS PubMed.
- P. A. Mangrulkar, M. M. Joshi, S. N. Tijare, V. Polshettiwar, N. K. Labhsetwar and S. S. Rayalu, Int. J. Hydrogen Energy, 2012, 37, 10462–10466 CrossRef CAS.
- J. Y. Choi, C. K. Lim, B. Park, M. Kim, A. Jamal and H. Song, J. Mater. Chem. A, 2019, 7, 15068–15072 RSC.
- S. Qiao, J. Guo, D. Wang, L. Zhang, A. Hassan, T. Chen, C. Feng, Y. Zhang and J. Wang, Int. J. Hydrogen Energy, 2020, 45, 1629–1639 CrossRef CAS.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- Z. Lin, C. Du, B. Yan and G. Yang, J. Catal., 2019, 372, 299–310 CrossRef CAS.
- K. M. Nam, W. S. Seo, H. Song and J. T. Park, NPG Asia Mater., 2017, 9, e364 CrossRef CAS.
- X. Zhan, Z. Wang, F. Wang, Z. Cheng, K. Xu, Q. Wang, M. Safdar and J. He, Appl. Phys. Lett., 2014, 105, 153903 CrossRef.
- K.-W. Park and A. M. Kolpak, J. Mater. Chem. A, 2019, 7, 16176–16189 RSC.
- L. Soriano, M. Abbate, A. Fernández, A. R. González-Elipe, F. Sirotti and J. M. Sanz, J. Phys. Chem. B, 1999, 103, 6676–6679 CrossRef CAS.
- A. Indra, P. W. Menezes, C. Das, C. Göbel, M. Tallarida, D. Schmeiβer and M. Driess, J. Mater. Chem. A, 2017, 5, 5171–5177 RSC.
- Z. Mao, J. Chen, Y. Yang, D. Wang, L. Bie and B. D. Fahlman, ACS Appl. Mater. Interfaces, 2017, 9, 12427–12435 CrossRef CAS PubMed.
- F. Guo, W. Shi, C. Zhu, H. Li and Z. Kang, Appl. Catal., B, 2018, 226, 412–420 CrossRef CAS.
- F. Guo, W. Shi, H. Wang, M. Han, H. Li, H. Huang, Y. Liu and Z. Kang, Catal. Sci. Technol., 2017, 7, 3325–3331 RSC.
- N. Wang and X. Li, Inorg. Chem. Commun., 2018, 92, 14–17 CrossRef CAS.
- X. Deng, D. Yang, G. Tan, X. Li, J. Zhang, Q. Liu, H. Zhang, N. J. Mellors, D. Xue and Y. Peng, Nanoscale, 2014, 6, 13710–13718 RSC.
- J. Huang, Y. Cao, H. Wang, H. Yu, F. Peng, H. Zou and Z. Liu, Chem. Eng. J., 2019, 373, 687–699 CrossRef CAS.
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS.
- R. Dedryvère, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J. M. Tarascon, Chem. Mater., 2004, 16, 1056–1061 CrossRef.
- N. Weidler, S. Paulus, J. Schuch, J. Klett, S. Hoch, P. Stenner, A. Maljusch, J. Brötz, C. Wittich, B. Kaiser and W. Jaegermann, Phys. Chem. Chem. Phys., 2016, 18, 10708–10718 RSC.
- M. M. Natile and A. Glisenti, Chem. Mater., 2002, 14, 3090–3099 CrossRef CAS.
- F. Guo, M. Li, H. Ren, X. Huang, W. Hou, C. Wang, W. Shi and C. Lu, Appl. Surf. Sci., 2019, 491, 88–94 CrossRef CAS.
- W. Shi, H. Ren, M. Li, K. Shu, Y. Xu, C. Yan and Y. Tang, Chem. Eng. J., 2020, 382, 122876 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00508h |
| This journal is © The Royal Society of Chemistry 2020 |