
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic host–guest behavior in halogen-bonded two-dimensional molecular networks investigated by scanning tunneling microscopy at the solid/liquid interface†
Yoshihiro
Kikkawa
 *,
Mayumi
Nagasaki
,
Emiko
Koyama
,
Seiji
Tsuzuki
,
Thierry
Fouquet
and
Kazuhisa
Hiratani
*,
Mayumi
Nagasaki
,
Emiko
Koyama
,
Seiji
Tsuzuki
,
Thierry
Fouquet
and
Kazuhisa
Hiratani
National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: y.kikkawa@aist.go.jp
First published on 21st August 2020
Abstract
The fabrication of supramolecularly engineered two-dimensional (2D) networks using simple molecular building blocks is an effective means for studying host–guest chemistry at surfaces toward the potential application of such systems in nanoelectronics and molecular devices. In this study, halogen-bonded molecular networks were constructed by the combination of linear halogen-bond donor and acceptor ligands, and their 2D structures at the highly oriented pyrolytic graphite/1-phenyloctane interface were studied by scanning tunneling microscopy. The bi-component blend of the molecular building blocks possessing tetradecyloxy chains formed a lozenge structure via halogen bonding. Upon the introduction of an appropriate guest molecule (e.g., coronene) into the system, the 2D structure transformed into a hexagonal array, and the central pore of this array was occupied by the guest molecules. Remarkably, the halogen bonding of the original structure was maintained after the introduction of the guest molecule. Thus, the halogen-bonded molecular networks are applicable for assembling guest species on the substrate without the requirement of the conventional rigid molecular building blocks with C3 symmetry.
Introduction
Supramolecular chemistry of well-ordered two-dimensional (2D) networks at surfaces is of great importance owing to the potential application of such systems in nanoelectronics, molecular devices, and lithographic techniques.1–6 The fine-tuning of the molecular organizations at surfaces has been attempted by the design and preparation of molecular building blocks with functional groups for enabling supramolecular interactions such as hydrogen bonding, metal coordination, dispersion force, and halogen bonding. The halogen bonding is a highly directional interaction of halogen atoms with other nucleophilic atoms carrying lone electron pairs, and its strength is often comparable to that of hydrogen bonding.7–10 Halogen bonds have attracted interest because they can play important roles in the molecular recognition processes, and the formation of supramolecular structures in natural as well as artificial systems.11–15Previously, supramolecularly engineered 2D structures in physisorbed monolayers at solid/liquid interfaces have been investigated by scanning tunneling microscopy (STM) with submolecular resolution.16–23 The formation of 2D structures at the solid/liquid interface is affected by several parameters such as the solvent type, temperature, concentration, alkyl chain length, intermolecular interactions, and electric field. By tuning these parameters, a wide variety of 2D network structures have been constructed.16–23
The 2D porous networks of supramolecular assemblies have attracted much attention because they can capture and immobilize guest molecules in their pores, and they are potentially applicable in molecular devices, nanoelectronics, sensors, catalysis, etc.24–34 There have been many remarkable studies on the host–guest chemistry at surfaces accommodating guest molecules such as coronene and fullerene, allowing the construction of multi-component molecular assemblies.24–34 However, in most of these studies, the 2D porous networks were created via hydrogen bonding and dispersion interactions of alkyl chains using rigid molecular building blocks with C3 symmetry.27–34
In a previous study,35 we prepared simple and linear molecular building blocks with halogen bonding acceptor and donor units (Py-Cn and FI-Cn (n = 18), respectively, in Chart 1). The binary blends of Py-C18 and FI-C18 formed hexagonal and porous networks, as observed by STM, at the highly oriented pyrolytic graphite (HOPG)/1-phenyloctane interface. This result prompted us to apply the halogen-bonded structures to study host–guest chemistry at surfaces. To the best of our knowledge, there are few reports on the host–guest chemistry in halogen-bonded 2D molecular networks.34
 | ||
| Chart 1 Chemical structures of the molecular building blocks used in this study: pyridine (Py-Cn), tetrafluoroiodobenzene-based molecules (FI-Cn), and coronene (COR) (n = 14 or 15). | ||
In this contribution, we present the formation of halogen-bonded molecular networks, which adaptively capture the guest molecule, coronene, resulting in the transformation of the 2D structure. In this study, we prepared halogen-bond donor and acceptor ligands (Py-Cn and FI-Cn: n = 14 and 15; Chart 1) and studied the 2D structures of single and bi-component systems by STM at the HOPG/1-phenyloctane interface. In addition, at this interface, we investigated guest inclusion into the 2D molecular networks.
Experimental
Preparation and characterization of molecular building blocks
The chemicals were purchased from Sigma-Aldrich, Kanto Chemical, Kishida Chemical, or Tokyo Chemical Industry, and they were used without further purification. Except for the change in the length of the alkyl chains, the building blocks of Py-Cn and FI-Cn (n = 14 and 15) were prepared as reported previously.35Briefly, for the preparation of Py-Cn, the alkylation of catechol with an alkyl bromide followed by iodination was performed to yield 1,2-bis(alkyloxy)-4-iodobenzene. Then, the Sonogashira coupling of 3-ethynylpyridine with 1,2-bis(alkyloxy)-4-iodobenzene was performed in dry tetrahydrofuran with a Pd(PPh3)4 catalyst, CuI co-catalyst, and triethylamine. The flask was cooled, degassed, charged with N2 gas, and stirred at 60 °C for 7 days. The crude product of Py-Cn was purified by silica gel column chromatography with CHCl3 as the eluent.
For the preparation of FI-Cn, methyl 3,5-dihydroxybenzoate was alkylated, and the resultant compound was reduced with LiAlH4 to obtain 3,5-bis(alkyloxy)benzylalcohol. Then, 3,5-bis(alkyloxy)benzylalcohol in a small amount of dry N,N-dimethylformamide was reacted with pentafluoroiodobenzene in the presence of Cs2CO3 for 15 h. The crude product of FI-Cn was purified by silica gel column chromatography using CHCl3/n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent.
:1) as the eluent.
The prepared compounds were characterized by 1H and 13C NMR spectroscopy (Bruker Avance 400 or 500 NMR spectrometer) using tetramethylsilane as the internal standard and Fourier-transform infrared spectroscopy (FTIR; JASCO FT/IR 420) by the KBr pellet method. The PhCF3 signal at −62.61 ppm (ref. 36) was used as the internal standard for 19F NMR measurements. Matrix-assisted laser desorption/ionization high-resolution mass spectrometry (MALDI-HRMS) was conducted on a JMS-S3000 spiralTOF mass spectrometer (JEOL, Tokyo, Japan). Trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile was used as the matrix and silver trifluoroacetate was used as the cationizing agent. The prepared compounds were mixed with the matrix and the cationizing agent in a solvent-free sample preparation (the solid chemicals were ground using a mortar and pestle for a few minutes) and the powdery mixture with an approximate matrix:cation:sample molar ratio of 50:1:1 was pressed onto a MALDI target using a spatula to form a thin film.
STM observations
STM was performed with a Nanoscope IIIa system (Digital Instruments). The compounds were dissolved in 1-phenyloctane at different concentrations, and the solutions were deposited on freshly cleaved HOPG substrates (ZYB grade, Momentive Performance Materials Quartz, Inc.) to obtain physisorbed monolayers. A Pt–Ir wire (90/10; 0.25 mm ϕ) was mechanically cut to prepare STM tips, and STM was carried out at the 1-phenyloctane/HOPG interface. The captured STM images were calibrated and corrected with reference to the HOPG lattice underneath the physisorbed monolayer using the SPIP software.The solutions of Py-Cn and FI-Cn (n = 14 and 15) in 1-phenyloctane were prepared, and STM was carried out before and after blending them at a molar ratio of 1:1. The guest, coronene (COR), was introduced with the coadsorption of molecular building blocks: a saturated 1-phenyloctane solution of COR was mixed with that of the bi-component blend (Py-Cn/FI-Cn) at a 1:1 ratio, and the relative ratio between COR and Py-Cn/FI-Cn was tuned. The pre-mixed solution of Py-Cn/FI-Cn/COR deposited on the HOPG surface was stored for 15 h before performing STM on the host–guest system.
DFT calculations
The Gaussian 16 program37 was employed for density functional theory (DFT) calculations using the 6-311G** basis set. The DGDZVP basis set38 was used for iodides. The B3LYP functional39 was used with Grimme's D3 dispersion correction.40 The geometry of the complex was optimized under imposing C2v symmetry. BSSE41 was corrected by the counterpoise method42 in the calculations of the intermolecular interaction energies.Results and discussion
Single component system
The 2D structures of the single-component system at the HOPG/1-phenyloctane interface were studied by STM. Fig. 1A and S1A (ESI)† show the 2D structure of Py-C14. As shown in Fig. 1A, the π-conjugated moieties (bright contrast in the image) are oriented in a face-to-face manner to form a double-columnar structure. The bright column is periodically separated by the dark region with a width of L1 = 1.77 ± 0.10 nm, which is identical to the length of the tetradecyloxy chain (1.77 nm). The molecular model in Fig. 1C is proposed based on the STM image in Fig. 1A. | ||
| Fig. 1 STM images of Py-C14 (A) and FI-C14 (B) physisorbed at the HOPG/1-phenyloctane interface. The concentrations of Py-C14 and FI-C14 in 1-phenyloctane were set to 30 mM and 25 mM, respectively. A set of arrows indicates the HOPG lattice directions. Proposed molecular models in (C and D) are drawn based on the STM images in (A and B), respectively. Tunneling conditions: (A) I = 25 pA, V = −1076 mV; (B) I = 25 pA, V = −676 mV. | ||
In contrast, FI-C14 showed a single-lined columnar structure (Fig. 1B and S1B (ESI)†), where the 3,5-tetradecyloxy chains with L2 = 1.75 ± 0.09 nm and L3 = 1.78 ± 0.10 nm exist in an expanded conformation (with an angle of 121 ± 2°) to follow the HOPG lattice with a herringbone structure; the direction of the iodine atom alternated in neighboring columns, as shown in the proposed molecular model in Fig. 1D. The 2D molecular arrangements in Py-C14 and FI-C14 are almost identical to those of the previously reported Py-C18 and FI-C18 building blocks, respectively,35 except for the column intervals. A higher concentration of Py-C14 and FI-C14 in 1-phenyloctane was necessary for stable STM investigations as compared to that used for studying Py-C18 and FI-C18. A possible reason for this is that the molecules with shorter alkyl chains (n = 14) have less intermolecular and molecule–substrate interactions compared to those with longer chains (n = 18). The lattice constants of the 2D structures of Py-C14 and FI-C14 are summarized in Table 1.
| a (nm) | b (nm) | γ (°) | |
|---|---|---|---|
| Py-C14 | 1.41 ± 0.02 | 3.43 ± 0.04 | 89 ± 1 |
| FI-C14 | 1.41 ± 0.11 | 4.37 ± 0.06 | 88 ± 1 |
Bi-component system
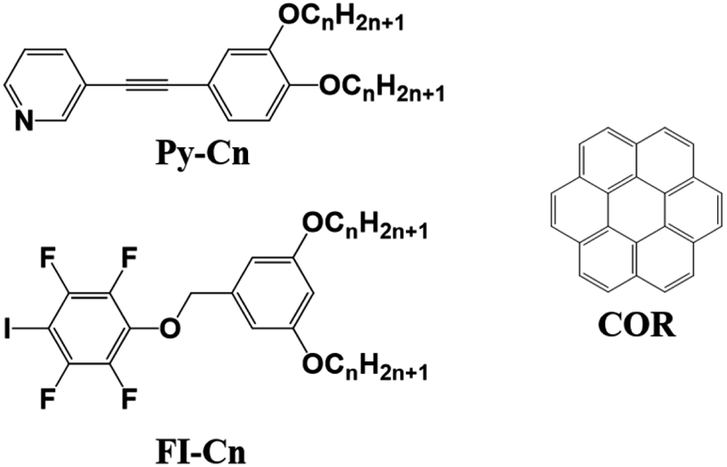
Next, the binary blend of Py-C14 and FI-C14 was studied, expecting the formation of a halogen-bonded 2D molecular network. Fig. 2A and B show the STM images of a binary blend of Py-C14/FI-C14 (1:1 ratio) at the HOPG/1-phenyloctane interface. Bright spots with cross-shaped assemblies are periodically arranged to form a lozenge-shaped lattice, and the lattice constants (Table 2) are different from those of the single component systems (Py-C14 and FI-C14).
 | ||
| Fig. 2 (A, B) STM images of the bi-component blends of Py-C14/FI-C14 (25 mM) physisorbed at the HOPG/1-phenyloctane interface. A set of arrows indicates the HOPG lattice directions. (C) Proposed molecular model of the lozenge structure is depicted based on the STM image in (B). The building blocks in a part of the repeating units for Py-C14 and FI-C14 are highlighted in green and blue, respectively in (B) and (C). The lengths of L4, L5, and L6 were measured to be 1.76 ± 0.08 nm; they are almost identical to that of the tetradecyloxy chain (1.77 nm). (D) Optimized structures of the complex for the head groups in Py-C14 (pyridine) and FI-C14 (2,3,5,6-tetrafluoro-1-iodo-4-methoxybenzene) obtained by DFT calculation. I⋯N halogen bonding and Ar–F⋯H–Py interactions are represented as red and cyan dotted lines, respectively. Tunneling conditions: (A) I = 25 pA, V = −805 mV; (B) I = 25 pA, V = −813 mV. | ||
| a (nm) | b (nm) | γ (°) | |
|---|---|---|---|
|
a The solution of Py-C14/FI-C14 at the concentration noted in the table was mixed with a saturated COR solution at 1:1 ratio.
|
|||
| Py-C14/FI-C14 | 3.16 ± 0.07 | 3.73 ± 0.07 | 70 ± 3 |
| Py-C15/FI-C15 | 6.93 ± 0.09 | 6.93 ± 0.09 | 60 ± 1 |
| Py-C14/FI-C14/COR | |||
| 25 mM | 6.69 ± 0.03 | 6.69 ± 0.03 | 61 ± 2 |
| 17 mM | 6.67 ± 0.06 | 6.67 ± 0.06 | 60 ± 1 |
Considering the I⋯N halogen bonding and 1:1 ratio of the components on the HOPG surface, a molecular model is proposed, as shown in Fig. 2C. Note that the molecular arrangements of the head groups for Py-C14 and FI-C14 were determined by DFT calculations (Fig. 2D). Py-C14 and FI-C14 formed a complex via I⋯N halogen bonding and became a pair. The halogen-bonded complexes arranged with opposite orientations in relation to each other and their positions shifted slightly due to Ar–F⋯H–Py interactions. Consequently, 2D chiral helical assemblies were found at the vertices of the lozenge lattice (Fig. 2B and S2 (ESI)†), as reported by many others.35,43–48 The total interaction energy of the 4 head groups (Fig. 2D) was calculated to be −20.2 kcal mol−1 (Fig. S3, ESI†), indicating that the complex formation even at the head groups contributes significantly to the stabilization of the 2D structure. In addition, alkyl chain moieties assisted the 2D arrangements in the lozenge structure: one of the two alkyl chains derived from FI-C14 in the complex was aligned along the HOPG lattice, accompanied by two alkyl chains of Py-C14.
Even when the Py-C14:FI-C14 blend ratio was changed between 1:3 and 3:1, the 2D structure remained unchanged and showed the same lozenge-shaped lattice (data not shown), suggesting that the lozenge structure is thermodynamically favored in the binary blend of Py-C14/FI-C14.29 Note that the lozenge structure was observed within the concentration range of 10–25 mM, and no change in the 2D structure was found even after 15 h of storage at room temperature (25 °C).
Effect of the alkyl chain length on the binary blend
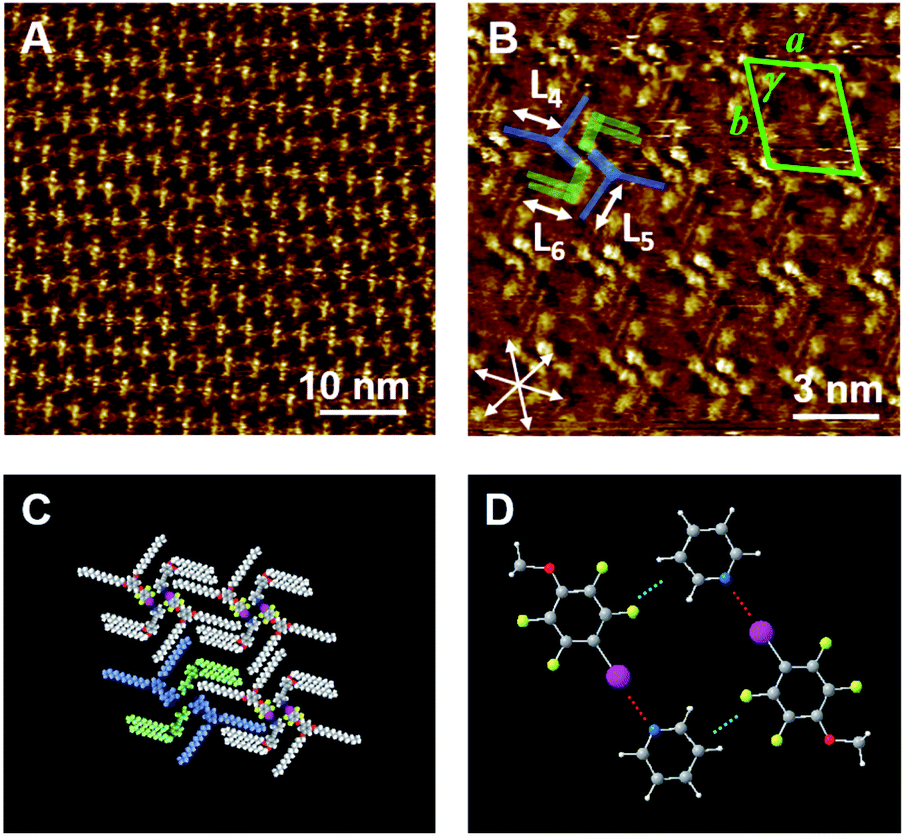
In our previous study,35 we observed the formation of halogen-bonded hexagonal networks in a binary blend of Py-C18/FI-C18. In this context, we examined the effect of the alkyl chain length on the 2D structure formation in the bi-component blend. Fig. 3A and B show the STM images of Py-C15/FI-C15 at the HOPG/1-phenyloctane interface. Within the concentration range of 10–25 mM, Py-C15/FI-C15 dominantly formed hexagonal arrays throughout the physisorbed monolayer, although the individual components (Py-C15 and FI-C15) displayed linear structures (Fig. S4, ESI†). The triangular helical assembly due to the I⋯N halogen bonding and Ar–F⋯H–Py interactions (Fig. 3C) allowed the formation of a honeycomb structure with hexagonal pores. The lattice constants of the honeycomb structure in Py-C15/FI-C15 are listed in Table 2. The results in Fig. 2 and 3 suggest that there is a specific alkyl chain length at which the blend forms either a honeycomb or lozenge structure: hexagonal arrays are constructed by the building blocks of Py-Cn/FI-Cn (n = 15 and 18), whereas a lozenge structure is formed by those of Py-C14/FI-C14.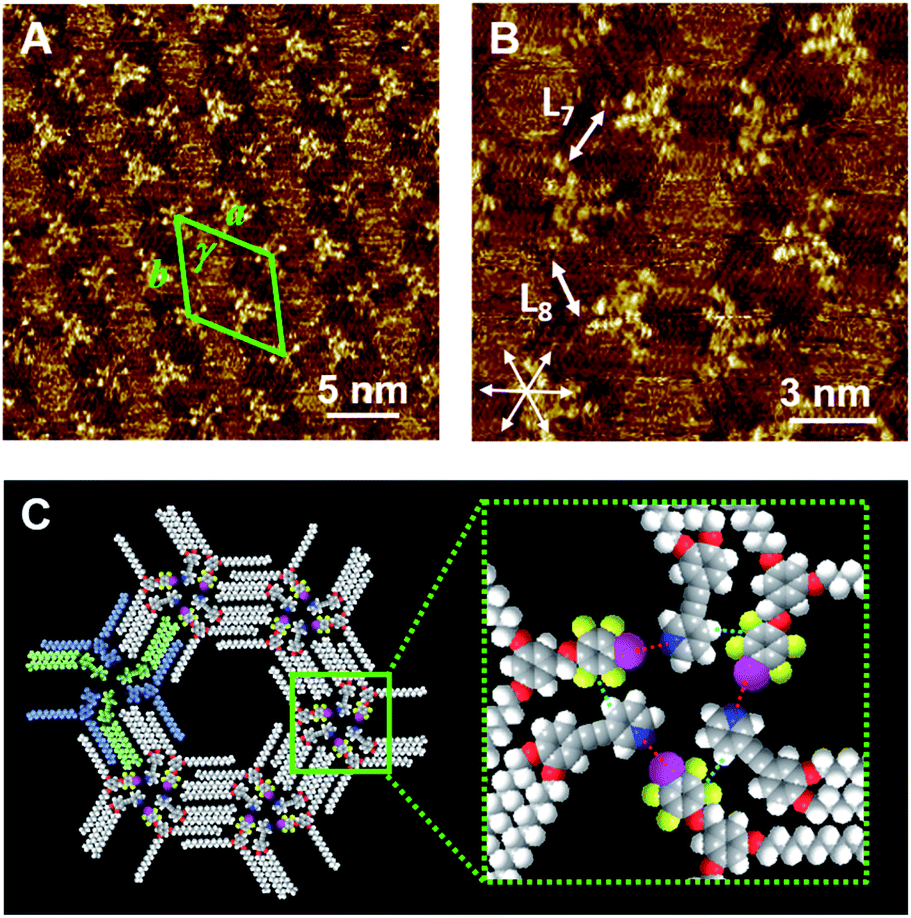 | ||
| Fig. 3 STM images of the bi-component blends of Py-C15/FI-C15 (A, B: 5 mM) physisorbed at the HOPG/1-phenyloctane interface. A set of arrows indicates the HOPG lattice directions. The lengths of L7 and L8 were measured to be 1.92 ± 0.09 nm, and they correspond to that of the pentadecyloxy chain (1.90 nm). The proposed molecular model of the honeycomb structure (C) is depicted based on the STM image in (B). The Py-C15 and FI-C15 units in one of the triangular assemblies are highlighted in green and blue, respectively in (C). Inset in (C): I⋯N halogen bonding and Ar–F⋯H–Py interactions are represented as red and cyan dotted lines, respectively. Tunneling conditions: (A) I = 25 pA, V = −909 mV; (B) I = 25 pA, V = −909 mV. | ||
We propose that the alkyl chain length-dependence of the 2D structure formation can be explained in terms of both solvent co-adsorption and packing density. The pores of the hexagonal arrays cannot be completely vacant, and they can be dynamically filled with solvent molecules that aid the stabilization of the pore structure.21,49 The longer the alkyl chain length is, the larger the pore size is. This implies that building blocks with long alkyl chains are more advantageous compared to those with short chains in terms of the number of solvent molecules that can be co-adsorbed. When the alkyl chain is short, a sufficient number of solvent molecules cannot be introduced into the pore to maintain the honeycomb structure.
In general, building blocks physisorbed on the surface tend to cover the entire surface and increase the packing density to a large extent, in order to maximize the intermolecular interactions. The surface coverage calculated from the number of building blocks and lattice constant indicates that the lozenge structure (0.356 molecule per nm2) has ca. 16% larger packing density than that of the hexagonal structure (0.307 molecule per nm2), if we assume that Py-C14/FI-C14 forms a hexagonal structure.50 Taking the solvent co-adsorption and packing density into account, it can be concluded that the lozenge structure is energetically favored in comparison to the porous hexagonal structure in the case of Py-C14/FI-C14.
Host–guest co-adsorption
Although Py-C14/FI-C14 formed a lozenge structure without pores, we studied the host–guest behavior of the 2D networks. COR was selected as a flat guest molecule. Fig. 4A–C and S5 (ESI)† show the STM images of the Py-C14/FI-C14/COR mixture physisorbed at the HOPG/1-phenyloctane interface. Interestingly, hexagonal arrays were formed throughout the monolayer, where the lengths of L9 and L10 were measured to be 1.77 ± 0.07 nm. The lattice constants of the hexagonal structure are listed in Table 2. In some pores, close-packed COR with seven individual molecules was found with clear contrast, as indicated by the arrows in Fig. 4A, whereas the other pores showed fuzzy disk-like spots, suggesting the dynamic adsorption and mobility of COR in the pore.24–26 Note that STM allowed the capture of snapshots indicating the dynamic behavior of guest inclusion. Therefore, variable numbers of COR were observed in the pore, that changed during the STM imaging (Fig. S6 and S7, ESI†). According to the STM image in Fig. 4B, a molecular model with the maximum number of adsorbed COR molecules is proposed, as shown in Fig. 4D. The I⋯N halogen bonding and Ar–F⋯H–Py interactions resulted in triangular assemblies (see Fig. 3C), which organized into hexagonal arrays through the interdigitation of alkyl chains. Remarkably, the I⋯N halogen bonding was found in both 2D structures before (Fig. 2) and after guest inclusion (Fig. 4), that is, the halogen bonding remained substantially unchanged. Therefore, the arrangements of the halogen-bonded supramolecular complex and the alkyl chains in Py-C14/FI-C14 adaptively changed to form hexagonal arrays to accommodate the guest molecules in the 2D structure. Although there are numerous reports on 2D structural change accompanying guest inclusion in molecular assemblies composed of C3-symmetric rigid molecules,24–26 the present study demonstrates the dynamic host–guest chemistry in halogen-bonded 2D molecular networks formed with linear molecular building blocks. | ||
| Fig. 4 STM images of the Py-C14/FI-C14/COR assembly physisorbed at the HOPG/1-phenyloctane interface. A 25 mM solution of Py-C14/FI-C14 was mixed with a saturated solution of COR at a blend ratio of 1:1. A set of arrows indicates the HOPG lattice directions. The proposed molecular model in (D) was drawn based on the STM image in (C). Tunneling conditions: (A–C) I = 25 pA, V = −752 mV. | ||
We further studied the effect of the blend ratio on the guest inclusion. Fig. 5 shows the STM images of Py-C14/FI-C14/COR obtained by tuning the relative ratio of Py-C14/FI-C14 and COR. When the concentration of the 25 mM solution of Py-C14/FI-C14 was decreased to 17 mM and then mixed with the saturated COR solution, the hexagonal structure and its lattice constants remained unchanged (Table 2). However, a larger number of the hexagonal pores were filled with close-packed COR molecules (seven in total) (Fig. 5A–C) than that observed in Fig. 4. This result suggests that increasing the guest concentration relative to that of the host network allows more COR molecules to reside in the pores without the alteration of the hexagonal structure.
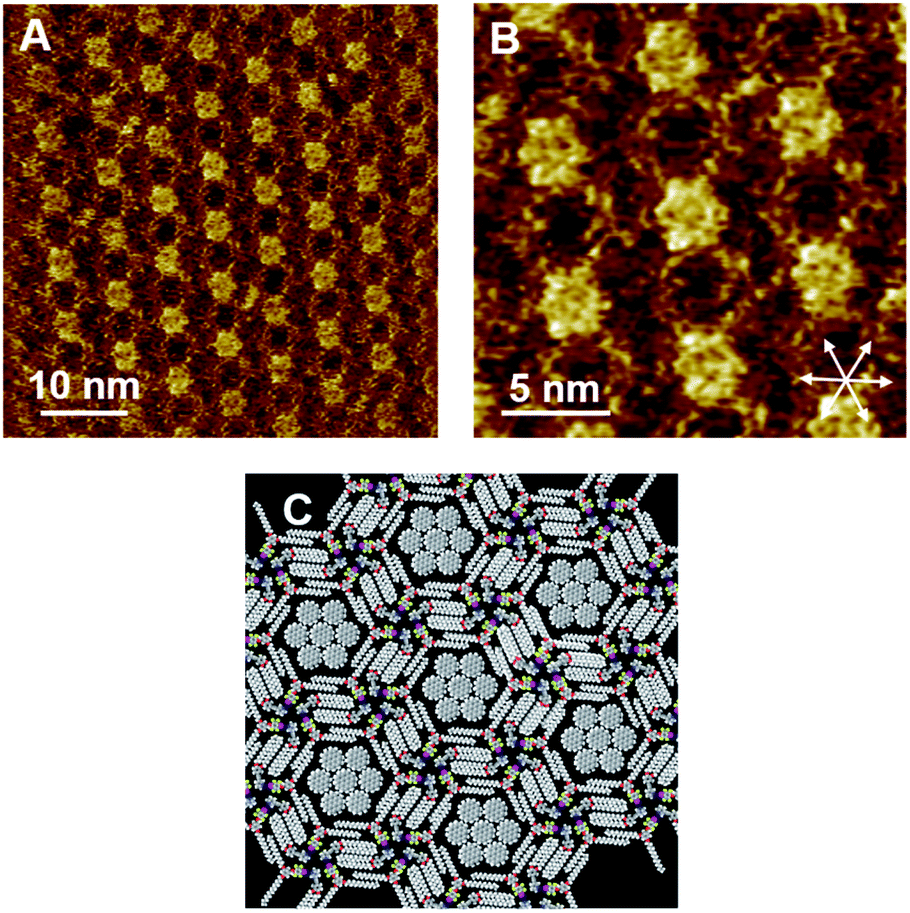 | ||
| Fig. 5 STM images of the Py-C14/FI-C14/COR assembly physisorbed at the HOPG/1-phenyloctane interface. A 17 mM solution of Py-C14/FI-C14 was mixed with a saturated solution of COR at a blend ratio of 1:1. A set of arrows indicates the HOPG lattice directions. The proposed molecular model in (C) is drawn based on the STM image in (B). Tunneling conditions: (A) I = 25 pA, V = −1209 mV; (B) I = 50 pA, V = −948 mV. | ||
Considering that the pore is surrounded by 1.77 nm long tetradecyloxy chains, the diagonal length of the hexagonal pore (pore size) could be 1.77 × 2 = 3.44 nm. The reported lattice constants of the close-packed structure of COR on HOPG are a = b = 1.11 nm and γ = 60°,51 suggesting that three molecules of COR with a size of 1.11 × 3 = 3.33 nm can be incorporated along each diagonal direction in the pore. Furthermore, as the size of the pore matches with that of the close-packed COR consisting of seven units, a higher packing density (0.485 nm2 per molecule) could be achieved than in the lozenge structure (0.356 nm2 per molecule). Therefore, the halogen-bonded Py-C14/FI-C14 assembly spontaneously transformed its 2D arrangement from a lozenge to the hexagonal structure so as to capture COR, resulting in a stable molecular assembly. Thus, we have successfully demonstrated the dynamic host–guest behavior in halogen-bonded 2D molecular networks.
Conclusions
Halogen-bonded 2D assemblies for host–guest chemistry were fabricated at the solid/liquid interface, and the 2D structures were visualized by STM. The building blocks of Py-C14 and FI-C14 formed double-columnar and single-columnar structures, respectively. The bi-component blend of Py-C14/FI-C14 displayed a lozenge-shaped structure, whereas that of Py-C15/FI-C15 showed hexagonal arrays with pores. Although both structures included the I⋯N halogen bonding, the arrangements of the halogen-bonded complex and alkyl chains were different, possibly because of the solvent co-adsorption and the molecular packing density. This result suggests that there is a specific alkyl chain length that leads to either a lozenge or hexagonal structure. Once the guest molecule, COR, is introduced into the system, the 2D structure of Py-C14/FI-C14 changed to a hexagonal structure to accommodate the guest into the lattice; some pores were filled with close-packed COR molecules while the others were dynamically accommodated with COR. A relative increase in the guest concentration allowed an increase in the number of close-packed COR molecules. Thus, the halogen-bonded molecular networks are suitable for efficiently assembling guest molecules.In this work, we have shown that guest molecules can be captured and immobilized in halogen-bonded molecular networks composed of simple, linear building blocks. This work could inspire the controlled organization of functional molecules for constructing multi-component 2D nanoarchitectures via supramolecular chemistry at surfaces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI (Grant No. 17K05851 and 18KK0156).Notes and references
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS.
- E. Gomar-Nadal, J. Puigmartí-Luis and D. B. Amabilino, Chem. Soc. Rev., 2008, 37, 490–504 RSC.
- C. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 322, 429–432 CrossRef CAS.
- D. B. Amabilino, Supramolecular Chemistry at Surfaces, Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- M. Gobbi, S. Bonacchi, J. X. Lian, Y. Liu, X. Y. Wang, M. A. Stoeckel, M. A. Squillaci, G. D'Avino, A. Narita, K. Müllen, X. Feng, Y. Olivier, D. Beljonne, P. Samorì and E. Orgiu, Nat. Commun., 2017, 8, 1–8 CrossRef.
- K. Tahara, T. Ishikawa, B. E. Hirsch, Y. Kubo, A. Brown, S. Eyley, L. Daukiya, W. Thielemans, Z. Li, P. Walke, S. Hirose, S. Hashimoto, S. De Feyter and Y. Tobe, ACS Nano, 2018, 12, 11520–11528 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS.
- S. Tsuzuki, A. Wakisaka, T. Ono and T. Sonoda, Chem.–Eur. J., 2012, 18, 951–960 CrossRef CAS.
- S. Tsuzuki, T. Uchimaru, A. Wakisaka, T. Ono and T. Sonoda, Phys. Chem. Chem. Phys., 2013, 15, 6088–6096 RSC.
- E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS.
- J. Poznański, M. Winiewska, H. Czapinska, A. Poznańska and D. Shugar, Acta Biochim. Pol., 2016, 63, 203–214 CrossRef.
- B. Li, S. Q. Zang, L. Y. Wang and T. C. W. Mak, Coord. Chem. Rev., 2016, 308, 1–21 CrossRef CAS.
- J. Teyssandier, K. S. Mali and S. De Feyter, ChemistryOpen, 2020, 9, 225–241 CrossRef CAS.
- K. E. Plass, A. L. Grzesiak and A. J. Matzger, Acc. Chem. Res., 2007, 40, 287–293 CrossRef CAS.
- J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7333 CrossRef CAS.
- Y. Tobe, K. Tahara and S. De Feyter, Bull. Chem. Soc. Jpn., 2016, 89, 1277–1306 CrossRef CAS.
- J. Otsuki, Coord. Chem. Rev., 2010, 254, 2311–2341 CrossRef CAS.
- S. Uemura, R. Tanoue, N. Yilmaz, A. Ohira and M. Kunitake, Materials, 2010, 3, 4252–4276 CrossRef CAS.
- Y. Yang and C. Wang, Curr. Opin. Colloid Interface Sci., 2009, 14, 135–147 CrossRef CAS.
- J. A. A. W. Elemans, Adv. Funct. Mater., 2016, 26, 8932–8951 CrossRef CAS.
- X. Peng, F. Zhao, Y. Peng, J. Li and Q. Zeng, Soft Matter, 2019, 16, 54–63 RSC.
- J. Teyssandier, S. De Feyter and K. S. Mali, Chem. Commun., 2016, 52, 11465–11487 RSC.
- K. Iritani, K. Tahara, S. De Feyter and Y. Tobe, Langmuir, 2017, 33, 4601–4618 CrossRef CAS.
- J. Li, Y. Qian, W. Duan and Q. Zeng, Chin. Chem. Lett., 2019, 30, 292–298 CrossRef CAS.
- S. Furukawa, K. Tahara, F. C. De Schryver, M. Van Der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2007, 46, 2831–2834 CrossRef CAS.
- Y. Shen, L. Zeng, D. Lei, X. Zhang, K. Deng, Y. Feng, W. Feng, S. Lei, S. Li, L. Gan, Q. Zeng and C. Wang, J. Mater. Chem., 2011, 21, 8787–8791 RSC.
- M. Li, P. Xie, K. Deng, Y. L. Yang, S. Bin Lei, Z. Q. Wei, Q. D. Zeng and C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 8778–8782 RSC.
- S. Chang, R. Liu, L. Wang, M. Li, K. Deng, Q. Zheng and Q. Zeng, ACS Nano, 2016, 10, 342–348 CrossRef CAS.
- M. Shen, Z. Luo, S. Zhang, S. Wang, L. Cao, Y. Geng, K. Deng, D. Zhao, W. Duan and Q. Zeng, Nanoscale, 2016, 8, 11962–11968 RSC.
- X. Peng, Y. Geng, M. Zhang, F. Cheng, L. Cheng, K. Deng and Q. Zeng, Nano Res., 2019, 12, 537–542 CrossRef CAS.
- J. Li, B. Tu, X. Li, C. Ma, C. Chen, W. Duan, X. Xiao and Q. Zeng, Chem. Commun., 2019, 55, 11599–11602 RSC.
- A. Mukherjee, J. Teyssandier, G. Hennrich, S. De Feyter and K. S. Mali, Chem. Sci., 2017, 8, 3759–3769 RSC.
- Y. Kikkawa, M. Nagasaki, E. Koyama, S. Tsuzuki and K. Hiratani, Chem. Commun., 2019, 55, 3955–3958 RSC.
- C. P. Rosenau, B. J. Jelier, A. D. Gossert and A. Togni, Angew. Chem., Int. Ed., 2018, 57, 9528–9533 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- B. J. Ransil, J. Chem. Phys., 1961, 34, 2109–2118 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. De Feyter and F. C. De Schryver, in Scanning Probe Microscopies Beyond Imaging, ed. P. Samorì, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, ch. 1, pp. 3–35 Search PubMed.
- N. Katsonis, H. Xu, R. M. Haak, T. Kudernac, Ž. Tomović, S. George, M. Van Der Auweraer, A. P. H. J. Schenning, E. W. Meijer, B. L. Feringa and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 4997–5001 CrossRef CAS.
- S. Lei, M. Surin, K. Tahara, J. Adisoejoso, R. Lazzaroni, Y. Tobe and S. De Feyter, Nano Lett., 2008, 8, 2541–2546 CrossRef CAS.
- J. A. A. W. Elemans, I. De Cat, H. Xu and S. De Feyter, Chem. Soc. Rev., 2009, 38, 722–736 RSC.
- T. Chen, W. H. Yang, D. Wang and L. J. Wan, Nat. Commun., 2013, 4, 1388–1389 CrossRef.
- Y. Fang, K. Tahara, O. Ivasenko, Y. Tobe and S. De Feyter, J. Phys. Chem. C, 2018, 122, 8228–8235 CrossRef CAS.
- O. Ochs, O. Ochs, M. Hocke, M. Hocke, S. Spitzer, S. Spitzer, W. M. Heckl, W. M. Heckl, N. Martsinovich, M. Lackinger and M. Lackinger, Chem. Mater., 2020, 32, 5057–5065 CrossRef CAS.
- A lattice of the lozenge structure contains 2 molecules for each component (4 molecules in total), whereas that of a hexagonal structure consists of 6 molecules (12 molecules in total). The lattice of the hexagonal structure with a vacant pore is assumed from the lattice constants of the hexagonal structure containing COR, which is shown in Fig. 3.
- K. Walzer, M. Sternberg and M. Hietschold, Surf. Sci., 1998, 415, 376–384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional STM images and interaction energies of the complex. See DOI: 10.1039/d0na00616e |
| This journal is © The Royal Society of Chemistry 2020 |