
A soft X-ray activated lanthanide scintillator for controllable NO release and gas-sensitized cancer therapy†
Mingyang
Jiang
a,
Zhenluan
Xue
a,
Youbin
Li
a,
Hongrong
Liu
a,
Songjun
Zeng
 *a and
Jianhua
Hao
*b
*a and
Jianhua
Hao
*b
aSynergetic Innovation Center for Quantum Effects and Application, Key Laboratory of Low-Dimensional Quantum Structures and Quantum Control of Ministry of Education, School of Physics and Electronics, Hunan Normal University, Changsha, 410081, P. R. China. E-mail: songjunz@hunnu.edu.cn
bDepartment of Applied Physics, The Hong Kong Polytechnic University, Hong Kong, China. E-mail: jh.hao@polyu.edu.hk
First published on 9th September 2019
Abstract
Multifunctional light stimuli-responsive NO-based gaseous theranostic systems are highly desirable due to them directly killing cancer cells at high concentration (>1 μM) with minimized adverse effects. However, on-demand NO-releasing nano-platforms for deep-tissue gas-based cancer therapy have not yet been explored. Herein, we develop a new type of soft X-ray-activated NaYF4:Gd/Tb scintillator as a light transducer for depth-independent NO release and on-demand gas-sensitized cancer therapy. Benefiting from the excellent radioluminescent property of NaYF4:Gd/Tb, deep-tissue (up to 3 cm) NO release can be achieved by ultralow dosage soft X-ray (45 kVp, 0.18–0.85 mGy) irradiation. More importantly, the designed scintillator-based NO gasotransmitter presents significant inhibition of tumor growth under ultralow dosage soft X-ray irradiation. Therefore, such a soft X-ray activated NO-releasing nanoplatform provides a promising depth-independent gaseous therapy mode for defeating deep-seated cancer in the future.
New conceptsMultifunctional light stimuli-responsive NO-based gaseous theranostic systems are highly desirable due to them directly killing cancer cells at high concentration (>1 μM) with minimized adverse effects. However, on-demand NO-releasing nanoplatforms for deep-tissue gas-based cancer therapy have not yet been explored. Here, we develop a new type of soft X-ray-activated NaYF4:Gd/Tb scintillator as a light transducer for depth-independent NO release and on-demand gas-sensitized cancer therapy. On the basis of energy transfer from NaYF4:Gd/Tb nanorods to the NO donor (RBS), ultralow dosage soft X-ray (∼0.85 mGy) triggered NO release could be achieved in deep tissues even up to 3 cm depth, which broke the depth limitation suffered by the traditional UV/vis and near infrared light. Moreover, the soft X-ray-activated NaYF4:Gd/Tb–RBS agent exhibits efficient inhibition effect for tumor growth. These findings open up the opportunity for developing a new type of soft X-ray activatable gaseous transducer for deep tissue gas-sensitized tumor therapy in vivo. |
Introduction
The development of theranostic systems based on endogenous gas has emerged as the new green treatment paradigm and opens up new frontiers for carcinoma therapy due to its non-invasive nature and great benefits to metabolism.1 As a new gaseous therapeutic molecule, nitric oxide (NO) plays a key role in various biological processes including neuronal communication regulation, blood vessel modulation, and other physiological/pathophysiological activities.2–4 Recent reports5–8 revealed that NO presented a concentration-dependent dual effect for promotion or inhibition of tumors. At low concentration (1 pM–1 nM), NO has a promoting effect on tumor growth. In contrast, a high concentration of NO (>1.0 μM) may remarkably inhibit tumor growth.5–8 Therefore, developing stimuli-responsive NO releasing systems with precisely controlled NO generating concentration for on-demand cancer therapy with little systemic toxicity is urgently demanded.In response to this issue, some NO releasing systems for controllable generation of NO by certain stimuli, including heat,9 pH,10–12 or light13–15 have been well developed. Among these systems, photo-triggered NO releasing platforms have emerged as promising NO-releasing transducers due to the precise control of timing, location, and dosage.16,17 Most of the photoactive NO-releasing molecules, such as metal-NO compounds (Roussin's black salt, RBS), nitroimines and bis-N-nitroso complexes, only respond to ultraviolet (UV) and visible light.18–20 However, the low penetration depth in tissue of UV/visible light greatly impedes their further application for controllable NO release in vivo. In contrast to UV/visible light, near-infrared (NIR) light located in the “optical transparency window” (700–900 nm) has been considered as an alternative light source stimulus for in vivo NO release owing to the improved tissue penetration depth.21,22 Recently, upconversion (UC) luminescent nanomaterials23–27 capable of converting low energy photons (NIR light) to high energy UV/visible light through a frequency UC process have emerged as promising NIR light-sensitive sensors14,28–30 for NO release. However, the low UC luminescent quantum yield and relatively low penetration depth of NIR light still hinder their deep-tissue gas-based therapy of tumors. In comparison to visible and NIR light, X-ray photons with greatly increased penetration depth in tissues have been widely applied in diagnosis and radiotherapy for clinical applications, especially for X-ray activated radioluminescence.31–35
Therefore, developing an X-ray activated NO-releasing system is highly desirable for deep-tissue on-demand NO release. Recently, Shi and his co-workers demonstrated a pioneering study for X-ray-induced NO-release by using high energy X-rays to break down S–N bonds.36 Nevertheless, high-dosage X-rays (>5.0 Gy) were required in this system, which inevitably caused side effects to normal tissues.37,38 In this regard, we propose a scintillating nanomaterial as the energy transducer to convert low dosage X-ray photons to visible light, and further activate the surrounding photoactive NO donors for generating NO gas.
Herein, a lanthanide-based scintillating nanomaterial (NaYF4:Gd/Tb nanorod) as the energy transducer (Scheme 1) for controllable NO release was explored. When absorbing RBS molecules, the designed scintillator presented soft X-ray triggered on-demand NO release under ultralow dosage (0.18–0.85 mGy) of X-ray irradiation via energy transfer (ET) from the scintillator to RBS owing to the efficient conversion of X-ray photons to visible light. Additionally, deep tissue NO release up to 3 cm was also achieved, which broke the major challenge of the depth barrier suffered by the traditional UV/visible and NIR light. More importantly, in vivo gas-sensitized tumor therapy was successfully performed, demonstrating direct inhibition of tumor growth.
 | ||
| Scheme 1 Schematic illustration of designing NaYF4:Gd/Tb–RBS nanocomposites for ultralow dosage soft X-ray triggered NO release. | ||
Results and discussion
For exploring the soft X-ray activated NO-releasing system, silica-coated NaYF4:40%Gd/15%Tb rare-earth (RE) nanorods were used for absorbing the [Fe4S3(NO)7]− (RBS) NO-donor via electrostatic interaction (Fig. 1a). Firstly, pure hexagonal phase NaYF4:Gd/Tb nanorods with uniform rod-like morphology (Fig. 1b and c) were prepared via a hydrothermal method by doping Gd3+ (Fig. S1, ESI†). The interplanar distance was measured to be 2.95 Å via the high resolution transmission electron microscopy (HR-TEM) image (Fig. 1d), corresponding to the (110) crystal plane of the hexagonal-phase NaYF4. The X-ray diffraction (XRD) results indicated that all the diffraction peaks were well indexed to the hexagonal phase structure (JCPDS No. 16-0334) and no impurities were detected (Fig. 1h). Then, a silica layer with thickness of about 10 nm was coated on NaYF4:Gd/Tb nanorods, which not only transferred the hydrophobic nanorods to the water phase, but also provided the ability to absorb the RBS molecules. As shown in Fig. 1e and f, the TEM results provided evidence of the formation of NaYF4:Gd/Tb@SiO2 core–shell structure. Then, NaYF4:Gd/Tb@SiO2 nanocomposites were modified with 3-aminopropyltriethoxysilane (APTES) to form –NH3+ groups on the surface, and finally reacted with RBS to form NaYF4:Gd/Tb@SiO2-RBS (hereafter referred to as NaYF4:Gd/Tb–RBS) nanocomposites via electrostatic interaction. And the stability of the NaYF4:Gd/Tb–RBS nanocomposites was evaluated in a physiological solution for 24 hours. As demonstrated, only less than 7% of the RBS was detached from the NaYF4:Gd/Tb (Fig. S2, ESI†), indicating the high stability of NaYF4:Gd/Tb–RBS for biomedical applications. Previous reports14 have demonstrated that RBS can release NO due to the photolysis effect under UV/visible light irradiation. To reveal the soft X-ray triggered NO release, the emission spectrum (Fig. 1i) of NaYF4:Gd/Tb nanorods under soft X-ray excitation was first tested, which was similar to the one excited by UV light (Fig. S3, ESI†). And, the emitting peaks were nicely overlapped with the broad-band absorption region of the RBS, leading to the occurrence of ET from the lanthanide nanorods to RBS. Moreover, after grafting RBS molecules, the soft X-ray triggered emitting intensity of the NaYF4:Gd/Tb–RBS nanohybrids was significantly decreased, further verifying the efficient ET from the lanthanide scintillator to the RBS.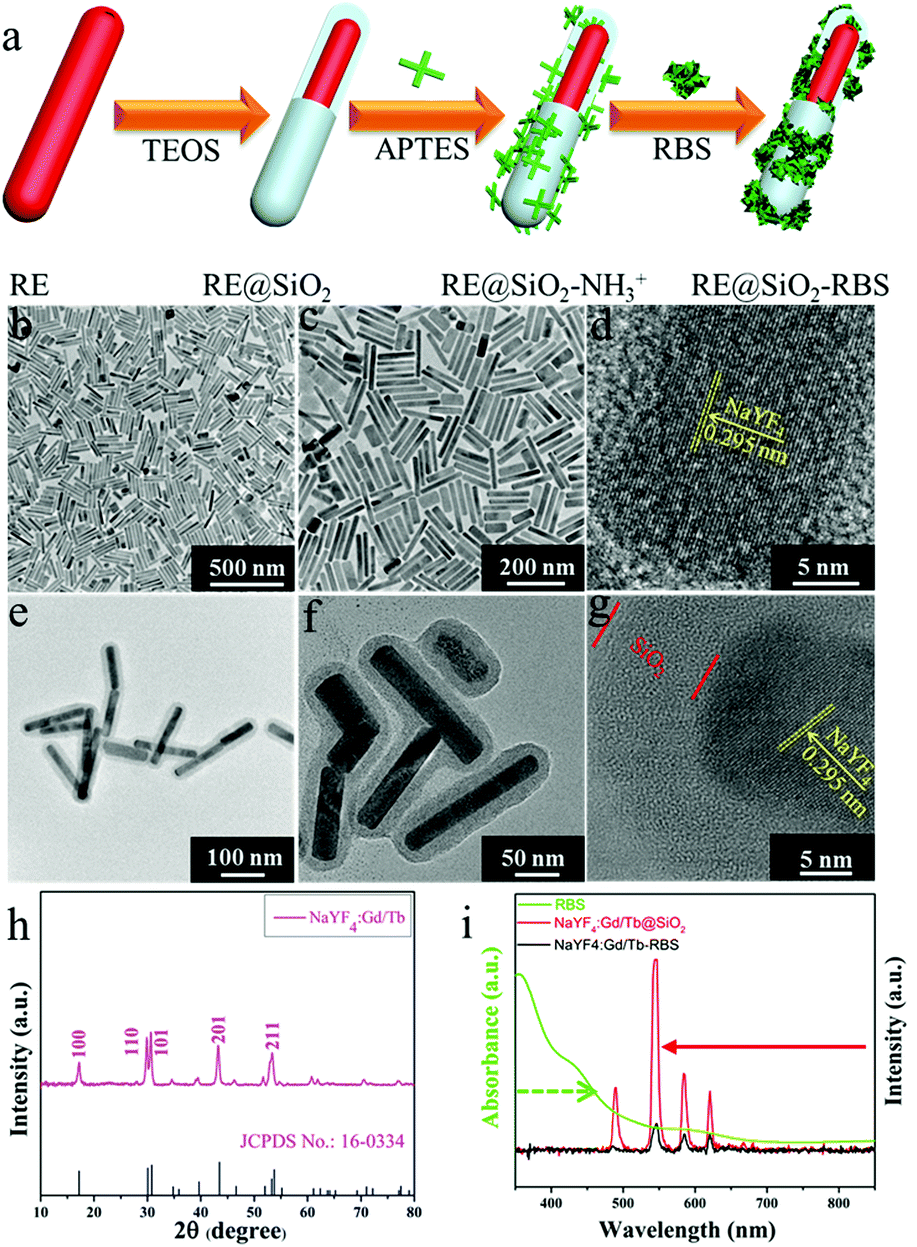 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of the NaYF4:Gd/Tb–RBS nanocomposites. (b and c) TEM images of NaYF4:Gd/Tb nanorods. (d) HR-TEM image of a single NaYF4:Gd/Tb nanorod. (e and f) TEM images of NaYF4:Gd/Tb@SiO2 nanorods. (g) HR-TEM image of a single NaYF4:Gd/Tb@SiO2 nanorod. (h) XRD pattern of NaYF4:Gd/Tb nanorods. (i) UV/Vis absorption spectrum of RBS aqueous solution and the emission spectra of NaYF4:Gd/Tb@SiO2 and NaYF4:Gd/Tb–RBS nanocomposites under the excitation of soft X-rays. | ||
To demonstrate the ability of NaYF4:Gd/Tb@SiO2 nanorods for soft X-ray stimuli-responsive NO release, in vitro soft X-ray induced visible light emitting experiments were first carried out. A series of 96-well tubes containing different concentrations of NaYF4:Gd/Tb@SiO2 were used for green radioluminescence tests under soft X-ray irradiation with low dosage (Table S1, ESI†). Fig. 2a showed that the green emission signals were significantly increased with increasing the concentrations of NaYF4:Gd/Tb@SiO2. In addition, the radioluminescent signals of the same samples were also greatly enhanced by improving the X-ray tube voltage. As illustrated in Fig. 2b, the average radioluminescent intensity visually confirmed that high tube voltage promoted the X-ray-induced green emission. To further reveal the irradiation time-dependent radioluminescent properties, NaYF4:Gd/Tb@SiO2 samples were irradiated with different irradiation times from 1 to 4 min under the same X-ray tube voltage of 45 kVp. As demonstrated in Fig. 2c, long irradiation time was highly desirable for achieving the intense radioluminescence, which was also vividly presented in the average intensity distribution diagram (Fig. 2d). Additionally, the photo-stability of the nanocomposite was also studied in biological environments. The photo-stability curve and in vitro phantom bioimaging (Fig. S4, ESI†) revealed that the designed nanocomposite presented high photo-stability both in water and PBS. Moreover, covered with different thicknesses of pork slabs (0–3 cm), the radioluminescent signals were still observed, even up to 3 cm depth, indicating the depth-independent behaviors of the X-ray light source (Fig. S5, ESI†). To further examine the capability of NaYF4:Gd/Tb@SiO2 for in vivo X-ray induced optical imaging, NaYF4:Gd/Tb@SiO2 solution (150 μL, 2 mg mL−1) was subcutaneously injected into a mouse. As shown in Fig. S6a and b (ESI†), significant signals were observed in the treated mouse. And the signal intensity was increased with improving the X-ray tube voltage, indicating the feasibility of the designed scintillating nanomaterials for in vivo X-ray induced optical imaging. Therefore, these findings demonstrate that NaYF4:Gd/Tb@SiO2 presents highly efficient radioluminescence under low dosage soft X-ray activation, which can be used as a promising light-responsive transducer for NO release under low dosage soft X-ray irradiation.
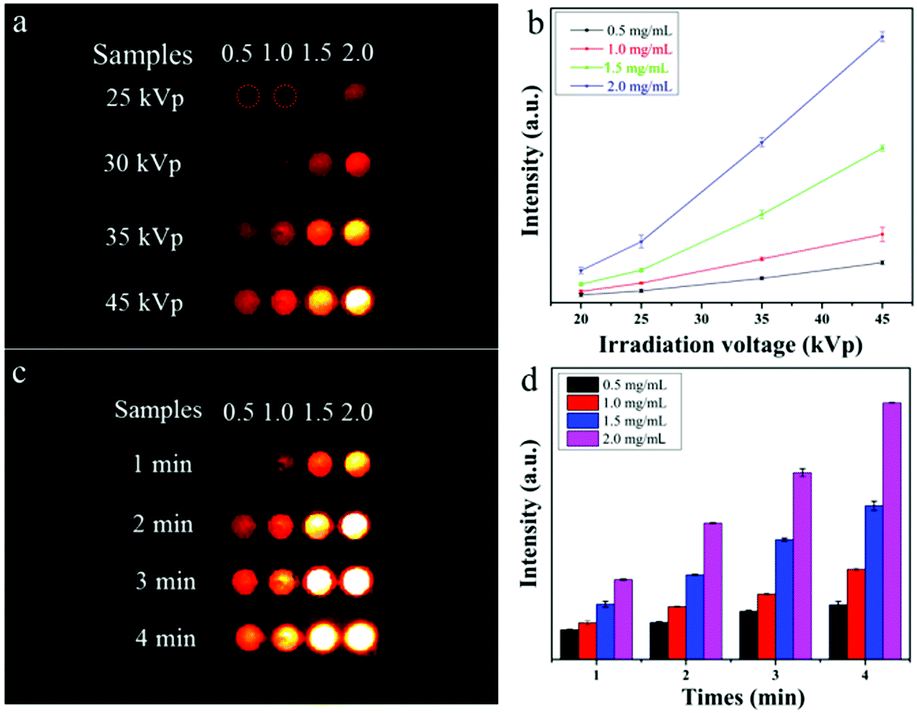 | ||
| Fig. 2 (a) Soft X-ray induced optical imaging with different concentrations of NaYF4:Gd/Tb@SiO2 solutions under different X-ray tube voltages and the same irradiation time of 2 min. (b) The corresponding average signal intensity under different tube voltages. (c) X-ray induced optical imaging under different irradiation times and the same tube voltage of 45 kVp. (d) The corresponding average signal intensity. | ||
Owing to the significant X-ray induced green radioluminescence and spectral overlap between NaYF4:Gd/Tb and RBS, soft X-ray triggered NO release can be readily achieved via ET from NaYF4:Gd/Tb nanorods to RBS (Fig. 3a). In order to verify the soft X-ray triggered NO release, the NO releasing properties of the different solutions containing deionized water, NaYF4:Gd/Tb@SiO2, and NaYF4:Gd/Tb–RBS were studied by using a typical Griess assay.39 As shown in Fig. 3b and c, there is nearly no NO generation from deionized water and NaYF4:Gd/Tb@SiO2 solution when irradiated under different X-ray tube voltages from 0 to 45 kVp with an irradiation time of 4.5 min. Meanwhile, the designed NaYF4:Gd/Tb–RBS (Fig. 3d) presents obvious soft X-ray triggered NO release and the release rate increases by improving the X-ray tube voltage. In addition, the quantitative NO release concentrations from RBS and NaYF4:Gd/Tb–RBS were further studied under soft X-ray irradiation with different tube voltages and irradiation times. As demonstrated in Fig. 3e, the X-ray triggered NO release content from 3.7 to 6.4 μM could be achieved by adjusting the X-ray tube voltage from 25 kVp to 45 kVp, which was mainly ascribed to the efficient ET from NaYF4:Gd/Tb nanorods to RBS molecules. The absorption spectra (Fig. 3f) of the NaYF4:Gd/Tb–RBS + Griess kit were further tested and presented the enhancement of the absorption value with increasing soft X-ray irradiation times, further verifying the soft X-ray activated NO release. More importantly, controllable gas release plays an important role in realizing on-demand gas-sensitized therapy. Then, the precisely controllable NO release triggered by soft X-rays was further investigated. As demonstrated in Fig. 3g, with continuous irradiation for 30 s, a bursting NO release was initiated at this stage. Then after stopping X-ray irradiation for 1 min, the NO release rate is greatly decreased, indicating the on-demand NO release by utilizing intermittent X-ray irradiation.
 | ||
| Fig. 3 (a) Schematic diagram of the NO releasing mechanism induced by soft X-rays. (b–d) Quantitative soft X-ray induced NO release from deionized water, NaYF4:Gd/Tb@SiO2, and NaYF4:Gd/Tb-RBS. (e) NO-releasing curves of NaYF4:Gd/Tb–RBS and RBS triggered by different tube voltages of X-rays and irradiation times. (f) UV/Visible absorption spectra of NaYF4:Gd/Tb–RBS + Griess kit solution under 45 kVp X-ray irradiation with different irradiation times. (g) On-demand NO-releasing behavior induced by X-ray irradiation. | ||
In addition, deep tissue NO release is another key factor for gas-sensitive tumor therapy in biomedicine application. To reveal the deep tissue NO releasing nature, X-ray triggered NO releasing properties of the designed NaYF4:Gd/Tb–RBS system covered with various thicknesses of pork slices from 0 to 3 cm were further studied (Fig. S7, ESI†). With soft X-ray irradiation for 30 s at 45 kVp, the designed NaYF4:Gd/Tb–RBS sample covered with/without pork slap can sustainably release NO gas under X-ray irradiation, and the release content increases with increasing irradiation time. It is worth noting that the release amount of NO can also reach upto 1.5 μM (Fig. S7b, ESI†) at 3 cm deep tissue after 270 s irradiation, which is higher than the concentration (>1 μM) for directly killing cancer cells. Therefore, the designed scintillator-based X-ray activated NO releasing system can break through the limitation of penetration depth, promoting deep-tissue gas-sensitized cancer therapy in vivo.
It also should be pointed out that low dosage of X-rays is a crucial factor for X-ray induced nanomedicine in clinical applications. In our designed scintillator-based NO-releasing gasotransmitter, only ultralow dosage of X-rays (∼0.85 mGy, Table S1, ESI†) is required for soft X-ray activatable NO release, which was far lower than the clinically used X-ray dosage for cancer treatment40 and previously reported high energy X-ray (>5.0 Gy) induced NO release.36
Prior to in vivo therapy, the long-term biotoxicity of NaYF4:Gd/Tb–RBS nanocomposites was first studied. The major organs including heart, liver, spleen, lung, and kidneys separated from the mice treated with NaYF4:Gd/Tb–RBS nanocomposites were analyzed by hematoxylin and eosin (H&E) staining analysis. As shown in Fig. S8 (ESI†), compared with the control group, no obvious histopathological abnormalities or lesions in organs from the NaYF4:Gd/Tb–RBS treated group (even up to 30 days) were observed, suggesting the negligible biotoxicity and high biocompatibility. And the in vitro therapy efficiency of the designed NaYF4:Gd/Tb–RBS nanocomposites was first evaluated before in vivo therapy. The fluorescent imaging results (Fig. S9a, ESI†) of A549 cells after calcein AM/propidium iodide (CA–PI) staining revealed that significant cell apoptosis was achieved in the NaYF4:Gd/Tb–RBS + X-ray group. In addition, we have further evaluated the cell viability. As demonstrated, compared with the control groups, the cell survival ratio (Fig. S9b, ESI†) of NaYF4:Gd/Tb–RBS + X-ray treatment was remarkably reduced to 27%, verifying the ultralow dosage soft X-ray triggered NO gas-based anti-cancer effect.
Then, to reveal in vivo NO gas-sensitized tumor therapy triggered by ultralow dosage soft X-rays, the tumor-bearing mice were randomly divided into four groups: control (group 1), PBS + soft X-ray irradiation (group 2), NaYF4:Gd/Tb–RBS (group 3), and NaYF4:Gd/Tb–RBS + soft X-ray irradiation (group 4). After injection of PBS and NaYF4:Gd/Tb–RBS solution (2 mg mL−1, 150 μL), the tumors were irradiated by soft X-rays with 45 kVp for 2 min. As shown in Fig. 4, the tumors in group 1, group 2 and group 3 grow fast, indicating that only low dosage X-ray irradiation or the NaYF4:Gd/Tb–RBS nanocomposite shows low efficiency tumor inhibition. In contrast, the combination of NaYF4:Gd/Tb–RBS solution and X-ray irradiation in group 4 presents the obvious inhibition of tumor growth (Fig. 4b), which is mainly attributed to the soft X-ray induced NO release for directly killing tumors. Moreover, the death state of cancer cells (Fig. 4e) in tumor tissue was further evaluated by TdT-mediated dUTP nick end labeling (TUNEL). And obvious cancer cell damage was observed in group 4 treated with NaYF4:Gd/Tb–RBS and soft X-rays, matching well with the in vivo therapy results. This finding further confirms the significant anti-cancer efficacy of NO-gas sensitized cancer therapy triggered by ultralow dosage soft X-rays in vivo.
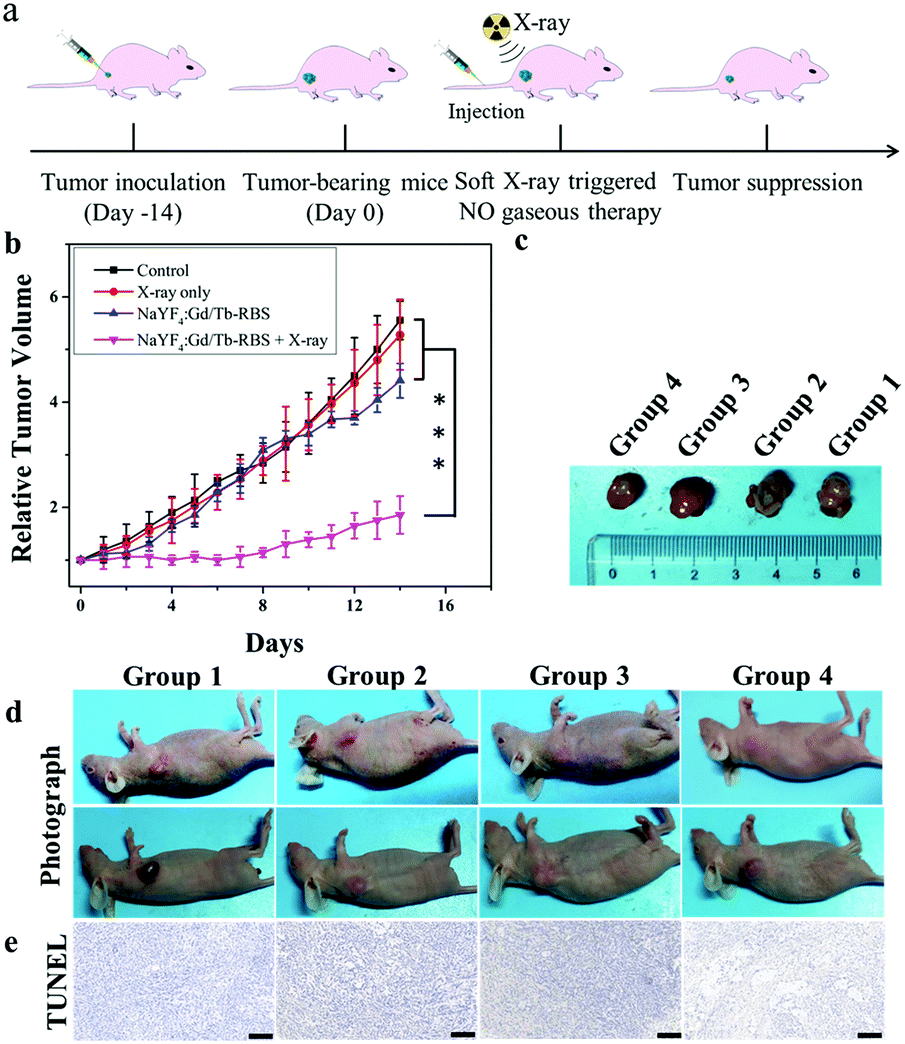 | ||
| Fig. 4 (a) Schematic diagram of soft X-ray induced NO-based gaseous therapy of cancer in vivo. (b) The relative volume changes of tumors from the control, PBS + X-ray, NaYF4:Gd/Tb–RBS and NaYF4:Gd/Tb–RBS + X-ray groups. ***P < 0.001. (c and d) Photographs of tumor-bearing mice before and after treatment. (e) TUNEL staining of mouse tumor sections after treatment. (Scale bar = 200 μm). | ||
Conclusions
In summary, a soft X-ray inducible scintillator-based NO generating nanoplatform by integrating NaYF4:Gd/Tb nanorods with NO donor (RBS) was developed for the first time. On the basis of the ET process between the NaYF4:Gd/Tb nanorods and NO donor (RBS), ultralow dosage soft X-ray (0.85 mGy) triggered NO release could be achieved in deep tissues even up to 3 cm depth, breaking the depth limitation suffered by the traditional UV/visible and NIR light. By adjusting the tube voltage of X-rays, on-demand NO release was also realized, which was important for concentration-dependent NO gas therapy. Moreover, the X-ray activated NaYF4:Gd/Tb–RBS agent exhibits an efficient inhibition effect for tumor growth. These findings open up the opportunity for developing a new type of soft X-ray activatable gasotransmitter for deep tissue gas-sensitized tumor therapy in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21671064), Science and Technology Planning Project of Hunan Province (No. 2017RS3031) and Natural Science Foundation of Hunan Province, China (No. 2019JJ10002).Notes and references
- Y. Qian and J. B. Matson, Adv. Drug Delivery Rev., 2017, 110, 137–156 CrossRef.
- K. R. Vega-Villa, J. K. Takemoto, J. A. Yanez, C. M. Remsberg, M. L. Forrest and N. M. Davies, Adv. Drug Delivery Rev., 2008, 60, 929–938 CrossRef CAS.
- B. Fadeel and A. E. Garcia-Bennett, Adv. Drug Delivery Rev., 2010, 62, 362–374 CrossRef CAS.
- W. P. Fan, B. C. Yung and X. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 8383–8394 CrossRef CAS.
- W. M. Xu, L. Z. Liu, M. Loizidou, M. Ahmed and I. G. Charles, Cell Res., 2002, 12, 311–320 CrossRef.
- L. A. Ridnour, D. D. Thomas, S. Donzelli, M. G. Espey, D. D. Roberts, D. A. Wink and J. S. Isenberg, Antioxid. Redox Signaling, 2006, 8, 1329–1337 CrossRef CAS.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- L. J. Chen, Q. J. He, M. Y. Lei, L. W. Xiong, K. Shi, L. W. Tan, Z. K. Jin, T. F. Wang and Z. Y. Qian, ACS Appl. Mater. Interfaces, 2017, 9, 36473–36477 CrossRef CAS.
- S. M. Yu, G. W. Li, R. Liu, D. Ma and W. Xue, Adv. Funct. Mater., 2018, 28, 1707440 CrossRef.
- L. A. Tai, Y. C. Wang and C. S. Yang, Nitric oxide, 2010, 23, 60–64 CrossRef CAS.
- V. Kumar, S. Y. Hong, A. E. Maciag, J. E. Saavedra, D. H. Adamson, R. K. Prud’homme, L. K. Keefer and H. Chakrapani, Mol. Pharmaceutics, 2010, 7, 291–298 CrossRef CAS.
- W. P. Fan, N. Lu, P. Huang, Y. Liu, Z. Yang, S. Wang, G. C. Yu, Y. J. Liu, J. K. Hu, Q. J. He, J. L. Qu, T. F. Wang and X. Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 1229–1233 CrossRef CAS.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- S. P. Nichols, W. L. Storm, A. Koh and M. H. Schoenfisch, Adv. Drug Delivery Rev., 2012, 64, 1177–1188 CrossRef CAS.
- J. S. Xu, F. Zeng, H. Wu, C. P. Hu, C. M. Yu and S. Z. Wu, Small, 2014, 10, 3750–3760 CrossRef CAS.
- M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS.
- P. C. Ford, Nitric oxide, 2013, 34, 56–64 CrossRef CAS.
- J. Fan, Q. J. He, Y. Liu, F. W. Zhang, X. Y. Yang, Z. Wang, N. Lu, W. P. Fan, L. Lin, G. Niu, N. Y. He, J. B. Song and X. Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13804–13811 CrossRef CAS.
- P. G. Wang, M. Xian, X. P. Tang, X. J. Wu, Z. Wen, T. W. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS.
- J. Hu, Y. Tang, A. H. Elmenoufy, H. B. Xu, Z. Cheng and X. L. Yang, Small, 2015, 11, 5860–5887 CrossRef CAS.
- Z. G. Yi, X. L. Li, Z. L. Xue, X. Xiao, W. Lu, H. Peng, H. R. Liu, S. J. Zeng and J. H. Hao, Adv. Funct. Mater., 2015, 25, 7119–7129 CrossRef CAS.
- S. J. Zeng, Z. G. Yi, W. Lu, C. Qian, H. B. Wang, L. Rao, T. M. Zeng, H. R. Liu, B. Fei and J. H. Hao, Adv. Funct. Mater., 2014, 24, 4051–4059 CrossRef CAS.
- X. M. Li, Z. Z. Guo, T. C. Zhao, Y. Lu, L. Zhou, D. Y. Zhao and F. Zhang, Angew. Chem., Int. Ed., 2016, 55, 2464–2469 CrossRef CAS.
- Q. S. Chen, X. J. Xie, B. Huang, L. L. Liang, S. Y. Han, Z. G. Yi, Y. Wang, Y. Li, D. Y. Fan, L. Huang and X. G. Liu, Angew. Chem., Int. Ed., 2017, 56, 7713–7717 CrossRef.
- Y. S. Liu, S. Y. Zhou, Z. Zhuo, R. F. Li, Z. Chen, M. C. Hong and X. Y. Chen, Chem. Sci., 2016, 7, 5013–5019 RSC.
- X. Zhang, G. Tian, W. Y. Yin, L. M. Wang, X. P. Zheng, L. Yan, J. X. Li, H. R. Su, C. Y. Chen, Z. J. Gu and Y. L. Zhao, Adv. Funct. Mater., 2015, 25, 3049–3056 CrossRef CAS.
- L. J. Tan, R. Huang, X. Q. Li, S. P. Liu and Y. M. Shen, Acta Biomater., 2017, 57, 498–510 CrossRef CAS.
- C. X. Li, J. W. Shen, J. F. Yang, J. Yan, H. J. Yu and J. Liu, Part. Part. Syst. Charact., 2018, 35, 1700281 CrossRef.
- A. Kamkaew, F. Chen, Y. H. Zhan, R. L. Majewski and W. B. Cai, ACS Nano, 2016, 10, 3918–3935 CrossRef CAS.
- L. Song, X. H. Lin, X. R. Song, S. Chen, X. F. Chen, J. Li and H. H. Yang, Nanoscale, 2017, 9, 2718–2722 RSC.
- D. J. Naczynski, C. Sun, S. Türkcan, C. Jenkins, A. L. Koh, D. Ikeda, G. Pratx and L. Xing, Nano Lett., 2015, 15, 96–102 CrossRef CAS.
- H. Wang, B. Lv, Z. M. Tang, M. Zhang, W. Q. Ge, Y. Y. Liu, X. H. He, K. L. Zhao, X. P. Zheng, M. Y. He and W. B. Bu, Nano Lett., 2018, 18, 5768–5774 CrossRef CAS.
- W. J. Sun, T. H. Shi, L. Luo, X. M. Chen, P. Lv, Y. X. Zhuang, J. J. Zhu, G. Liu, X. Y. Chen and H. M. Chen, Adv. Mater., 2019, 31, 1808024 CrossRef.
- W. P. Fan, W. B. Bu, Z. Zhang, B. Shen, H. Zhang, Q. J. He, D. L. Ni, Z. W. Cui, K. L. Zhao, J. W. Bu, J. L. Du, J. N. Liu and J. L. Shi, Angew. Chem., Int. Ed., 2015, 54, 14026–14030 CrossRef CAS.
- H. G. Chen, G. D. Wang, Y. J. Chuang, Z. P. Zhen, X. Y. Chen, P. Biddinger, Z. L. Hao, F. Liu, B. Z. Shen, Z. W. Pan and J. Xie, Nano Lett., 2015, 15, 2249–2256 CrossRef CAS.
- K. Rothkamm and M. Lobrich, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5057–5062 CrossRef CAS.
- J. Sun, X. J. Zhang, M. Broderick and H. Fein, Sensors, 2003, 3, 276–284 CrossRef CAS.
- S. B. Oh, H. R. Park, Y. J. Jang, S. Y. Choi, T. G. Son and J. Lee, Br. J. Pharmacol., 2013, 168, 421–431 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00564a |
| This journal is © The Royal Society of Chemistry 2020 |