
Sound-driven dissipative self-assembly of aromatic biomolecules into functional nanoparticles†
Sukhvir Kaur
Bhangu
 a,
Gianfranco
Bocchinfuso
b,
Muthupandian
Ashokkumar
*a and
Francesca
Cavalieri
*bc
a,
Gianfranco
Bocchinfuso
b,
Muthupandian
Ashokkumar
*a and
Francesca
Cavalieri
*bc
aSchool of Chemistry, University of Melbourne, VIC 3010, Australia. E-mail: masho@unimelb.edu.au
bDipartimento di Scienze e Tecnologie Chimiche, Università di Roma “Tor Vergata”, via della ricerca scientifica 1, 00133, Rome, Italy
cDepartment of Chemical Engineering, University of Melbourne, VIC 3010, Australia. E-mail: francesca.cavalieri@unimelb.edu.au
First published on 11th December 2019
Abstract
Dissipative self-assembly processes were recently exploited to assemble synthetic materials into supramolecular structures. In most cases, chemical fuel or light driven self-assembly of synthetic molecules was reported. Herein, experimental and computational approaches were used to unveil the role of acoustic cavitation in the formation of supramolecular nanoaggregates by dissipative self-assembly. Acoustic cavitation bubbles were employed as an energy source and a transient interface to fuel and refuel the dissipative self-assembly of simple aromatic biomolecules into uniform nanoparticles. Molecular dynamics simulations were applied to predict the formation of metastable aggregates and the dynamic exchange of the interacting molecules in the nanoaggregates. The intracellular trafficking and dissipative dissolution of the nanoparticles were tracked by microscopy imaging.
New conceptsDissipative self-assembly (DSA) of “activated building blocks” leads to the formation of supramolecular structures and new functional nanomaterials. Mostly, DSA processes in man-made systems are fueled by light or chemical agents. Herein, the role of acoustic energy in the formation of supramolecular nanoaggregates by DSA is unveiled. We show that a transient energy input supplied by ultrasound (cavitation) can push the aromatic biomolecules into a high-energy state and provide transient liquid–air interface where the self-assembly of biomolecules into uniform nanoparticles can take place, on bubble collapse. Molecular dynamics simulations give an insight into the formation of metastable aggregates and the mechanism of energy dissipation. The obtained nanoparticles showed distinctive optical properties, in a wide spectral range, that were exploited to investigate the intracellular disassembly process of nanoparticles in living cells. |
Introduction
Dissipative self-assembly, DSA, processes are out-of-equilibrium ubiquitous phenomena observed in biological systems. For instance, the reversible formation of actin filaments is a case of dissipative self-assembly.1 The products of these out-of-equilibrium biochemical reactions require a continuous supply of energy to persist, and their integrity is controlled by the relative rates of formation and disassembly driven by the energy dissipation processes.2,3 Although common in biology, the DSA has only recently emerged as a strategy for designing new functional materials with programmable lifetimes.2–5Man-made precursor systems are turned into self-assembling building blocks by an activation reaction, triggered by a source of energy, typically provided by light or a fuel molecule.2–8 The assemblies can only be maintained in their out-of-equilibrium state by a continuous input of energy or fuel that is subsequently converted to thermal energy and waste products. Chemical fuel and light driven DSA processes of organic and synthetic building blocks have been investigated, such as the self-assembly of vesicular nanoreactors,9 gelation of dibenzoyl-L-cysteine,10 light-driven azobenzene self-assembly into rod-like aggregates3 and self-assembly of synthetic nucleic acid strands to design synthetic DNA-based receptors.11 Strikingly, acoustic energy has not yet been considered as a fuel in DSA processes. However, it was reported that low frequency sonication (29 and 80 kHz) can exert thermal and kinetic effects to promote the nucleation and the supramolecular reorganization of thermodynamically stable amyloid-like fibrils.12,13 In the latter study, low frequency ultrasound (80 kHz) was used to trigger temporary supramolecular reconfiguration of assembled aromatic dipeptide amphiphiles from tapes to coiled fibers and straight fibers to spherical aggregates, which revert to the initial organization state when the sound is switched off.
To the best of our knowledge, ultrasound-driven DSA has never been reported. Here, we provide an additional conceptual framework to obtain DSA of natural aromatic biomolecules. We combine experimental and computational approaches to unveil the role of the acoustic field in the formation of out-of-equilibrium nanoaggregates using, as a proof of concept, a simple amino acid, L-tryptophan. We anticipate that this approach can be used for the DSA of many other aromatic biomolecules, including phenylalanine, peptides, aromatic drugs and natural compounds.
We demonstrate that acoustic bubbles, driven by high frequency standing waves, provide a reactive surface for the dimerization of aromatic amino acids into amphiphilic molecules and an energy source to fuel and refuel their dissipative self-assembly into uniform nanoaggregates. The computational study predicts that the aggregation of tryptophan dimers occurs in less than 20 ns when a high local concentration is experienced at the cavitation bubble–solution interface. The lifetime of the nanoaggregates can be tuned by changing the pH of the media when the supply of acoustic energy is discontinued. The unique optical and bio-functional properties of nanoparticles have been employed for probing their intracellular dissipative dissolution by imaging.
Results and discussion
The ultrasonic hydroxylation and dimerization of tryptophan
To investigate the ability of ultrasound to induce tryptophan oxidation, dimerization and subsequently DSA of tryptophan dimers, an aqueous solution of tryptophan (1 mg mL−1, 4.9 mM, pH 5) was sonicated up to 5 h using an ultrasonic frequency and power of 355 kHz and 2 W cm−2, respectively (see ESI,† Experimental section). We selected 355 kHz for our experiments because this is an optimal frequency to generate the required amount of OH radicals.14 For instance, in our experimental setup, we evaluated that after 1 h of sonication 1 mM OH radicals are generated (see ESI,† Experimental section). The phenomenon occurring during sonication is depicted in Fig. 1a. We recently reported that the oscillating surface of acoustic cavitation microbubbles acts as a reactive and catalytic site for the C–C coupling of amphiphilic moieties.14 In this study, hydroxylated tryptophan species and hydroxylated tryptophan dimers, hereafter referred to as dTrp, were formed on the bubble surface (step 1 – Fig. 1a, and Fig. 1c). The dTrp ultimately self-assembled into uniformly shaped spherical nanoparticles, dTrpNPs, during bubble collapse (step 2 – Fig. 1b) with ∼40% yield. To gain insight into the chemical structure of oxidized tryptophan molecules and molecular composition of the nanoparticles, HPLC (Fig. S1, ESI†), mass spectrometry (Fig. S2, ESI†), energy dispersive X-ray spectroscopy (EDS, Fig. S3a, ESI†), FTIR (Fig. S3b, ESI†), NMR (Fig. S3c, ESI†) and fluorescence spectroscopy (Fig. S4, ESI†) analyses were performed. The HPLC analysis (Fig. S1, ESI†) shows the conversion of tryptophan (retention time, 3.2 min) into different products and the simultaneous appearance of two new peaks. The peak at a retention time of 4.4 min (Fig. S1b, ESI†) was attributed to the dimers and hydroxylated dimers of tryptophan, whereas the peak at 2.7 min (Fig. S1c, ESI†) was attributed to the hydroxylated tryptophan species. Mass spectrometry analysis performed on sonicated tryptophan solutions (Fig. S2a, ESI†) confirmed the formation of hydroxylated products such as Trp + OH, Trp + 2OH and Trp + 3OH at m/z values of 221, 237 and 253. In addition, different dimeric species with different degrees of hydroxylation were also obtained (e.g. the peaks at m/z values of 407, 423, and 439). This indicates that both tryptophan or hydroxylated trytophan can combine to form dimers. The mass spectrometry analysis of dTrpNPs (Fig. S2b, ESI†), after purification and dissolution of nanoparticles, indicated that dTrpNPs were mostly composed of monohydroxylated dTrp (m/z 445-dTrp). Correspondingly, energy dispersive X-ray spectroscopy of the dTrpNPs confirmed an increase in the percentage of oxygen which could be attributed to the presence of hydroxyl groups (Fig. S3a, ESI†). Fig. S3b (ESI†) shows the FTIR spectrum of dTrpNPs in comparison with Trp, where the peak at 1695 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, that at 1596 cm−1 is attributed to the CC aromatic stretch, and the peak at 1324 cm−1 is due to the CC indole stretch. The peak at 3400 cm−1 in Trp is due to N–H; however, a broad additional peak in dTrpNPs at 3340 cm−1 corresponds to the O–H stretch. A comparison between the NMR spectra (Fig. S3c, ESI†) of unmodified tryptophan and dissolved dTrpNPs indicate the absence of the peaks typically assigned to the H-4 aromatic proton. This can be explained by the dimerization of tryptophan via C–C coupling. The other aromatic peaks were observed in the region 6.9–7.5 ppm. The presence of ArOH was confirmed by several peaks appearing around 8 ppm.
O stretch, that at 1596 cm−1 is attributed to the CC aromatic stretch, and the peak at 1324 cm−1 is due to the CC indole stretch. The peak at 3400 cm−1 in Trp is due to N–H; however, a broad additional peak in dTrpNPs at 3340 cm−1 corresponds to the O–H stretch. A comparison between the NMR spectra (Fig. S3c, ESI†) of unmodified tryptophan and dissolved dTrpNPs indicate the absence of the peaks typically assigned to the H-4 aromatic proton. This can be explained by the dimerization of tryptophan via C–C coupling. The other aromatic peaks were observed in the region 6.9–7.5 ppm. The presence of ArOH was confirmed by several peaks appearing around 8 ppm.
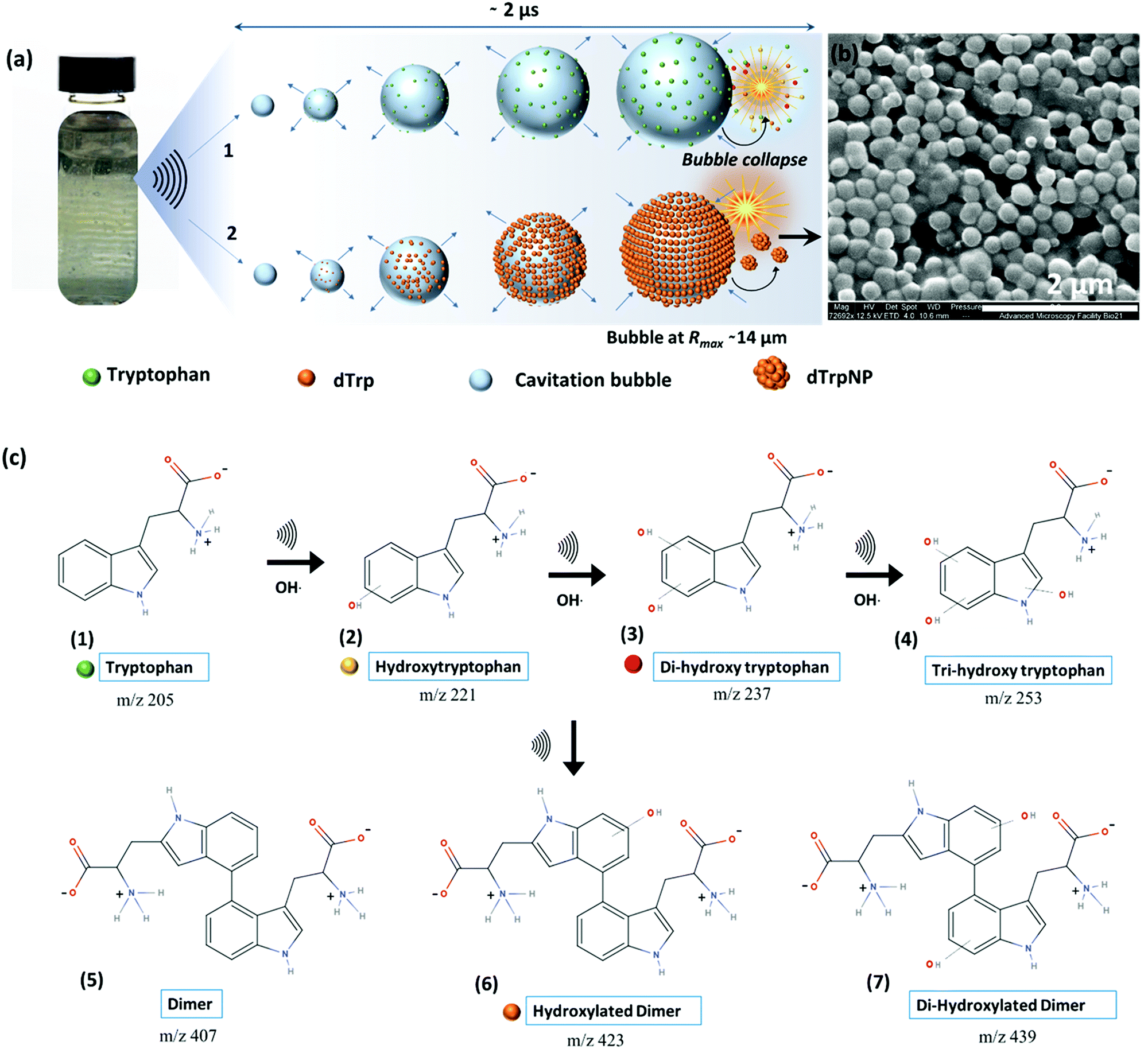 | ||
| Fig. 1 Reaction scheme and proposed mechanism for the formation dTrpNPs by dissipative self-assembly. (a) Schematic showing the formation of standing waves in the glass vial on sonication, increase in bubble size and gathering of Trp (step 1) and dTrp molecules (step 2) on the bubble–solution interface during one oscillation cycle where collapse of the bubble generates hydroxylated products and dTrp (step 1) and dTrpNPs (step 2). (b) SEM images of the dTrpNPs generated after 3 h sonication of tryptophan below the cac. (c) A reaction scheme showing the formation of the hydroxylated products due to the attack of hydroxyl radicals on the indole ring of tryptophan followed by a dimerization reaction to form dTrp species which ultimately self-assemble into nanoparticles. | ||
Fig. S4a and b (ESI†) show the fluorescence emission and excitation spectra of sonicated tryptophan solution, respectively. We observed a red shift and an increase in emission intensity of sonicated tryptophan as a function of sonication time. The emission at 370 nm was ascribed to the formation of hydroxylated tryptophan species. This was confirmed by the excitation spectra showing a consistent decrease of the tryptophan peak at 280 nm with an increase in sonication time, and the appearance of a new peak at around 330 nm (Fig. S4b, ESI†). The absorption spectra of the sonicated tryptophan solution acquired at different sonication times (Fig. S4c, ESI†) indicated the formation of species absorbing in the range of 300–600 nm. When excited in the range 360–500 nm, the sonicated solution exhibited an additional fluorescence peak at 465 nm ascribed to dTrp. The extended degree of conjugation in the dTrp dimers led to this emission peak. A shift in wavelength from 420 nm to 570 nm was detected when excitation was changed from 360 to 500 nm. This suggests the presence of multiple species which is in agreement with mass spectrometry analysis. A similar fluorescence emission band in the range 400–600 nm was observed for cyclo-ditryptophan in the self-assembled form but not in the soluble form.16 The fluorescence properties of the dTrpNP suspension were also investigated (Fig. S5a, ESI†) to confirm the chemical structure of the nanoparticle components. The fluorescence emission spectra of dTrpNPs show the characteristic peak of dTrp at 465 nm, indicating the presence of dimers. Interestingly, intense emission bands in the far-red and near IR region (650–860 nm) were observed when the dTrpNP suspension was excited at 575 nm and 640 nm (Fig. S5a and b, ESI†). As dTrps are not fluorescent in the near-red region, the emission peaks can be ascribed to the nanoaggregates. These emissive states can arise from the aggregates as a result of the π–π stacking interactions between aromatic moieties. In fact, when the fluorescence spectrum of dissolved dTrpNPs was acquired, the peaks in the red region were absent, whereas the emission of the soluble dTrp at 470 nm was preserved (Fig. S5a, green line, ESI†), confirming that the red-shifted peaks at higher wavelengths >600 nm are due to intermolecular interactions stabilizing the dTrpNPs. Taken together, this comprehensive characterization study suggests the formation of different soluble hydroxylated Trp species where the dTrpNPs are made of less hydrophilic monohydroxylated dTrp. Fig. 1c shows a schematic of the possible mechanistic pathways involved in the ultrasonic hydroxylation and dimerization of tryptophan. Previous computational and experimental studies have also shown that tryptophan bears multiple sites for the OH radical attack and hydrogen can also be abstracted from the indole rings.17 It was shown that the hydroxylation of tryptophan by Fenton's reaction and photolysis oxidation occurs in different positions of the molecule yielding many possible isomers of hydroxytryptophan and monohydroxytryptophan dimers.21,22 However, none of these studies have ever reported the formation of nanoparticles.21,22 It is well known that sonication of an aqueous solution results in the formation of H and OH radicals due to acoustic cavitation.15 In our system, OH radicals generated during the high frequency ultrasonic treatment can abstract protons from the indole moiety which then undergo OH radical addition to form hydroxylated products (structure 2, 3 and 4 in Fig. 1c). In addition, two Trp radicals can combine through C–C coupling to form dimers or hydroxylated dTrp (structure 5, 6 and 7 in Fig. 1c), where monohydroxylated dTrp self-assemble into nanoparticles.
The oscillation and collapse of transient cavitation bubbles induce DSA of mono-hydroxytryptophan dTrp.
The dynamic behavior of cavitation bubbles and the resulting physical and chemical effects are intertwined with the dissipative self-assembly of dTrp molecules as depicted in Fig. 1a. The change in bubble radius as a function of time (Fig. S6a, ESI†) was calculated using the Rayleigh–Plesset equation for the given frequency (355 kHz) and power (2 W cm−2).15,18–20 From these data the maximum radius of gas bubbles driven by the acoustic field was estimated as ∼14 μm. Fig. S6a (ESI†) shows that a single acoustic cycle lasts for approximately 2 μs under given conditions, during which the bubble size increases until Rmax is reached and the bubble collapses. Based on the diffusion coefficient of the amino acids (700–1000 μm2 s−1) and dTrp (870 ± 78 μm2 s−1), which was measured by fluorescence correlation spectroscopy (Fig. S7, ESI†),14,18 this is likely a sufficient time for both Trp and dTrp molecules to diffuse, adsorb, dimerize and further aggregate during the bubble collapse. First, the tryptophan molecules will be adsorbed at the bubble surface (step 1 – Fig. 1a) to form hydroxylated and dimeric species mediated by the bubble collapse and generated OH radicals. Next, after 3 h of sonication uniform spherical nanoparticles (dTrpNPs) were formed by the self-assembly of the dTrp species (Fig. 1a). In Fig. 1b, a representative SEM image of the uniform dTrpNP is shown. The average diameter of tryptophan nanoparticles obtained from the SEM analysis was 230 ± 50 nm. The ζ-potential of dTrpNPs measured at pH 7 was found to be −26 ± 7 mV. The dTrpNPs were negatively charged because of the deprotonation of the carboxyl and aromatic hydroxyl groups in water at pH 7. This is supported by the potentiometric titration of dTrpNPs (Fig. S8, ESI†) after dissolution indicating three pKa values at approximately 2, 6 and 10 ascribed to carboxylic acid, hydroxyl and ammonium group deprotonation respectively. Interestingly, we noticed that under these experimental conditions, the dTrps have a strong tendency to aggregate into nanoparticles only when ultrasound is applied. Conversely, after the rapid dissolution of dTrpNPs under alkaline conditions at the same concentration as they were formed during sonication (50 μg mL−1, 1.6 mM), they were unable to self-aggregate, once the pH was brought back to neutral or acidic conditions. This indicates that the self-assembly of dTrp is thermodynamically disfavoured in the bulk at that concentration and it is likely to be energetically fuelled by high frequency ultrasound. To confirm this, the surface and aggregation properties of dTrp were evaluated. The surface tension of an aqueous solution of dTrp dissolved at the same concentration (1.6 mM) was 67.3 mN m−1 indicating surface activity and, therefore, ability to adsorb on the cavitation bubbles. The critical aggregation concentration, cac, and the surface excess concentration (determined using Gibbs adsorption equation) of the dTrp were 100 μg mL−1 and 1.1 × 10−6 mol m−2, respectively. These results confirm that the ultrasound assisted self-assembly of dTrp occurs well below the cac (50 μg mL−1). Hence, the association of dTrp building blocks to give aggregates is not thermodynamically favoured because the molecules reside in a global minimum in the energy landscape as depicted in Fig. 2a.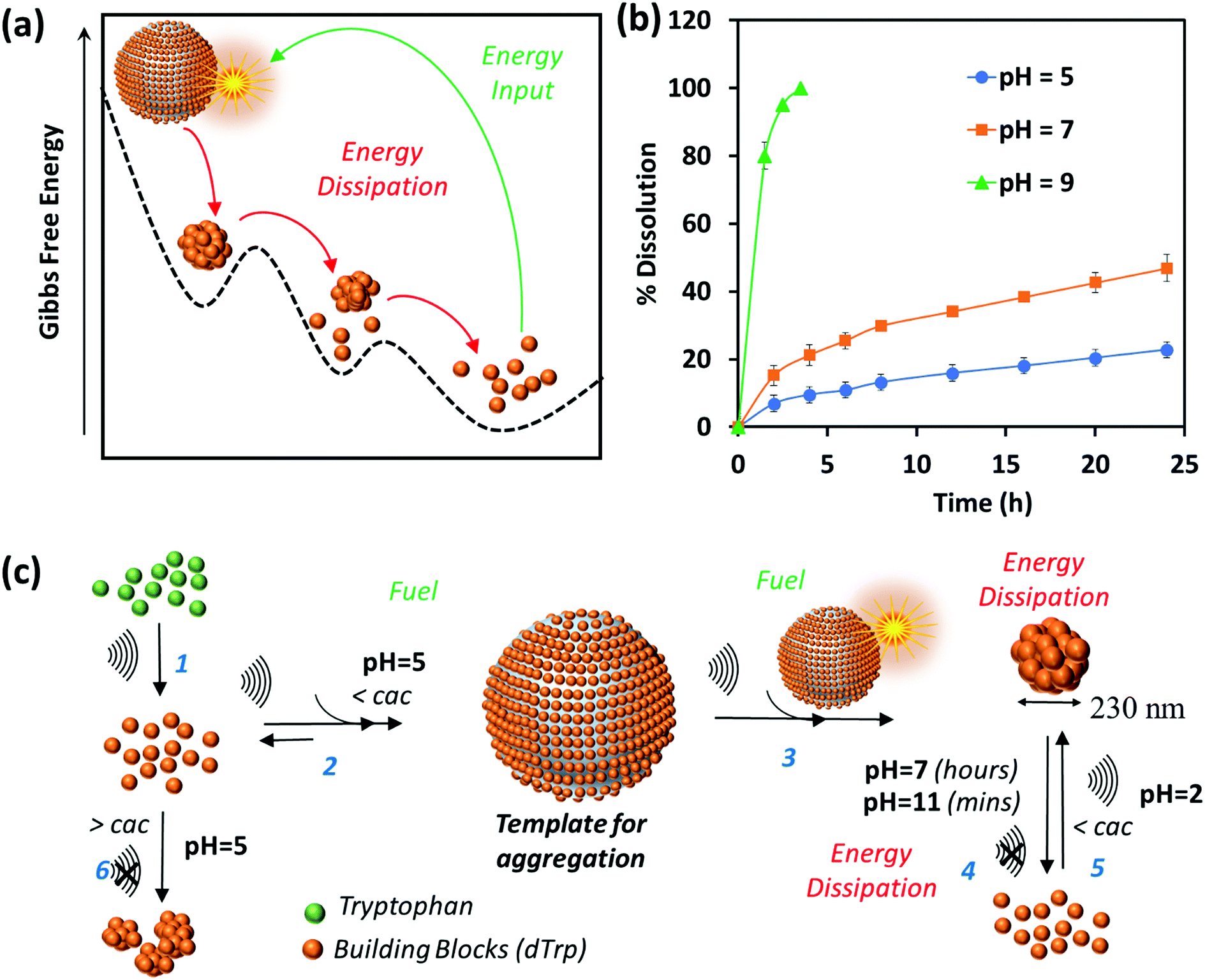 | ||
| Fig. 2 (a) The Gibbs free energy landscape of dissipative self-assembly of dTrp to form dTrpNPs where dTrp building blocks reside in global minimum and transient energy input due to acoustic cavitation leads to out of equilibrium self-assembly of dTrp into dTrpNPs. Furthermore, dTrpNPs undergo subsequent energy dissipation to recover the original non-activated building block state over a prolonged period of time. (b) Dissolution kinetics of dTrpNPs at 37 °C at different pH values as a function of time suggesting slow dissolution of dTrpNP at pH 5 and pH 7 and prompt dissolution at a higher pH (pH 9) around the pKa of the aromatic hydroxyl and amine groups. (c) General scheme for the ultrasound driven dissipative self-assembly of dTrp to dTrpNPs. Step 1 shows the ultrasonic conversion of Trp to building blocks dTrp; steps 2 and 3 show how the cavitation bubble acts as a fuel providing a template for adsorption of dTrp and high concentration conditions to facilitate the aggregation upon collapse, leading to the formation dTrpNPs. Step 4 shows the energy dissipation step upon increasing pH to form the pristine building blocks; step 5 shows how the acoustic energy can refuel the dissipative self-assembly of dTrp below the cac at pH 2. | ||
However, a transient energy input provided by ultrasound (cavitation) can push the building blocks into a high-energy state where the self-assembly can take place (Fig. 2a). The thermodynamically favoured dTrp nanoaggregates can be formed only at concentrations higher than the cac and showed a different morphology with micrometer sized clusters which lack structural organization (Fig. S6b, ESI†). The lifetimes of out of equilibrium dTrpNPs can be tuned by altering the kinetics of the dissipative step, i.e., by changing the pH (Fig. 2b). The dTrpNPs disassemble at pH 5 over one week, dissolve within 48 h at pH 7 whereas dissolve within 5 hours upon increasing the pH above the pKa of the aromatic hydroxyl and amine groups (>pH 10) (Fig. 2b). This indicates that the repulsive electrostatic interactions between negatively charged molecules can affect the kinetics of nanoparticle dissolution into water soluble building blocks.
We speculate on the possible role of cavitation bubbles in driving DSA processes. As shown in Fig. 2c, the oscillating bubbles, driven by the high frequency acoustic field, provide a transient liquid–air interface where dTrp molecules are collected and preorganized (step 2 in Fig. 2c). The experimental value of area occupied by each molecule on the gas–liquid interface was calculated using surface excess measurements and it was found to be approximately 1.52 nm2. Assuming the molecules as spherical, the number of dTrp molecules covering the surface of oscillating bubbles at the maximum expansion (step 2 – Fig. 1c) can be approximately estimated as 2.4 × 108. Under these transient conditions, the dTrp molecules experience an activated state and high local concentration. Upon bubble collapse, the formation of uniform spherical nanoparticles far below the cac occurs (step 3 in Fig. 2c).
This is also energetically allowed because higher local concentration and strong intermolecular interactions are simultaneously experienced by the building blocks. Overall the dissipative self-assembly of dTrp molecules is coupled to a chemical reaction network that includes (i) the irreversible chemical conversion of precursors (Trp) into building blocks (dTrp) (step 1 – Fig. 2c), (ii) the reversible activation of building blocks on the surface of cavitation bubbles which results in high local concentration of dTrp (step 2 – Fig. 2c) and (iii) a reversible reaction where the activated building blocks self-assemble to form nanoaggregates (step 3 – Fig. 2c). The nanoaggregates dissipate the free energy acquired during steps (ii) and (iii) to recover the original non-activated building block state over a prolonged time, depending upon the pH conditions (step 4 – Fig. 2c). The overall process is described schematically in Fig. 2c. The nanoaggregates dissipate the energy and relax to the equilibrium state when the acoustic energy supply stops. This is because the nanoaggrates are out of equilibrium structures that return to the lower free energy, non-assembled state, mainly favored by the entropic terms. At any given pH the nanoaggregates relax spontaneously to the thermodynamic stable state over time (step 4 – Fig. 2c). However, in the presence of OH– the kinetics of dissolution of the nanoaggregates is faster.
Two assembly–disassembly cycles were performed to demonstrate that the system can be re-fuelled by the acoustic energy. A solution of dissolved dTrpNPs was sonicated under acidic conditions at an ultrasonic frequency and power of 355 kHz and 2 W cm−2, respectively, for 1 h to obtain the nanoparticles. In the second cycle the protonation of carboxyl groups was required to assist the self-assembly of dTrp (step 5 – Fig. 2c). Fig. S9a and b (ESI†) show the size distribution and SEM image of the nanoparticles obtained in the second cycle. The Z-average size of nanoparticles was ∼188 ± 50 nm. This may indicate that the amphiphilic properties of the building block changed upon sonication by further hydroxylation of dTrp molecules and the surface properties must be tuned by the protonation of carboxyl groups to maximize the surface activity and diffusion at the gas–liquid interface of dTrp. To support this hypothesis, the surface activity of dTrp in aqueous solution was determined using surface tension measurements at different pH values. The surface tensions at pH 2, pH 7 and pH 11 were 58.9 mN m−1, 70.5 mN m−1 and 80.7 mN m−1, respectively, suggesting higher surface activity of dTrp molecules at low pH.
One can reasonably deduce that the sound driven self-assembly of dTrp is favoured when the carboxyl groups are protonated. It is worth mentioning that the cavitation bubble causes the dissociation of N2 and O2 present in air. The dissociated species and N2 and O2 can form NOx, HNO3, HNO2 and HNO which decrease the pH during the ultrasonic treatment to pH 2–3.15 We noticed a shift in pH during the sonication from 5 to 2 in the first few hours of sonication when the precursors (Trp) convert into the building block (dTrp). This could explain why in the second cycle a pH adjustment of the solution is necessary to recover the original structure of the building block. Overall, our study indicates that acoustic cavitation is a fuel that provides both energy input and protons for self-assembly to take place.
Molecular dynamics (MD) simulations predict the formation of metastable aggregates at high local concentration
To gain insight into the driving forces behind the formation of dTrpNPs and confirm the vital role of acoustic cavitation bubbles, Molecular Dynamics (MD) simulations of tryptophan dimers were performed. To this end, six replicas of dTrp were randomly inserted in a 64 nm3 cubic box in the presence of water (Fig. 3a). This condition can mimic the high local concentration experienced by the amphiphilic dTrp molecules in proximity to the bubble collapse region (see Fig. 1a). MD simulations are usually unable to reproduce the spatial and temporal scales required for analysis of larger aggregates; however, the early stages of aggregation can be simulated, and the data can be used to characterize the structural features of the final aggregates.23 Representative snapshots from MD simulations up to 865 ns are shown in Fig. 3a. In Fig. 3b-1, the Solvent Accessible Surface Area (SAS) of the six dTrp molecules during the first 50 ns of simulation is reported. The global SAS value of the solutes typically decreases when aggregation takes place, and this parameter is sensitive to both the number of aggregates present in the simulation box and their compactness.23,24 At the simulation start point, dTrp molecules are far from each other and the global SAS value is slightly below 33 nm2. Fig. 3b-1 shows that during the first 20 ns the SAS value of the six dTrp molecules decreases from 33 to roughly 20 nm2. The observed decrease in the global SAS is likely due to the occurrence of contacts between molecules upon aggregation. To confirm this, the SAS of each single dTrp molecule, during the whole simulation, was evaluated (Fig. 3b-2). In this case, the surface of each single dTrp molecule is considered as totally exposed to the solvent and its value could change only because of intra-molecular conformational transition. Fig. 3b-2 shows that the starting SAS value of each single dTrp molecule is retained during the whole simulation, irrespective of the presence or absence of aggregates. A value of 5.5 ± 0.2 nm2 was measured for all the molecules, which is exactly a sixth of the starting value of the global SAS. This suggests that the intramolecular conformational changes only slightly affect the SAS of the single dTrp molecule and the global SAS variation effectively probes the aggregation process. In addition, data reported in Fig. 3b-1 indicate that, under the simulated conditions, stable aggregates form in less than 25 ns. This lag time has been excluded from the further simulation analysis run up to 1 μs. Fig. 3c shows the behavior of the global SAS from 25 ns to the end of the simulation (1 μs). From the minimum distances between all the possible pairs of monomers, we have evaluated at each time, the number of units present in the aggregates, i.e. aggregation number. Fig. 3c shows that a single aggregate involving all the six molecules is present for almost all the simulation time (continuous red line). In detail, aggregates of 6, 5, 4 and 3 dTrp molecules are present for 87.6, 8.1, 4.2 and 0.1% of the simulation time (dashed red lines) respectively. Fig. 3c shows that when only an aggregate of six dTrp molecules is present, the global SAS remains roughly below 22 nm2; on the other hand, when single monomers or dimers come off from the aggregate the SAS suddenly increases to higher value.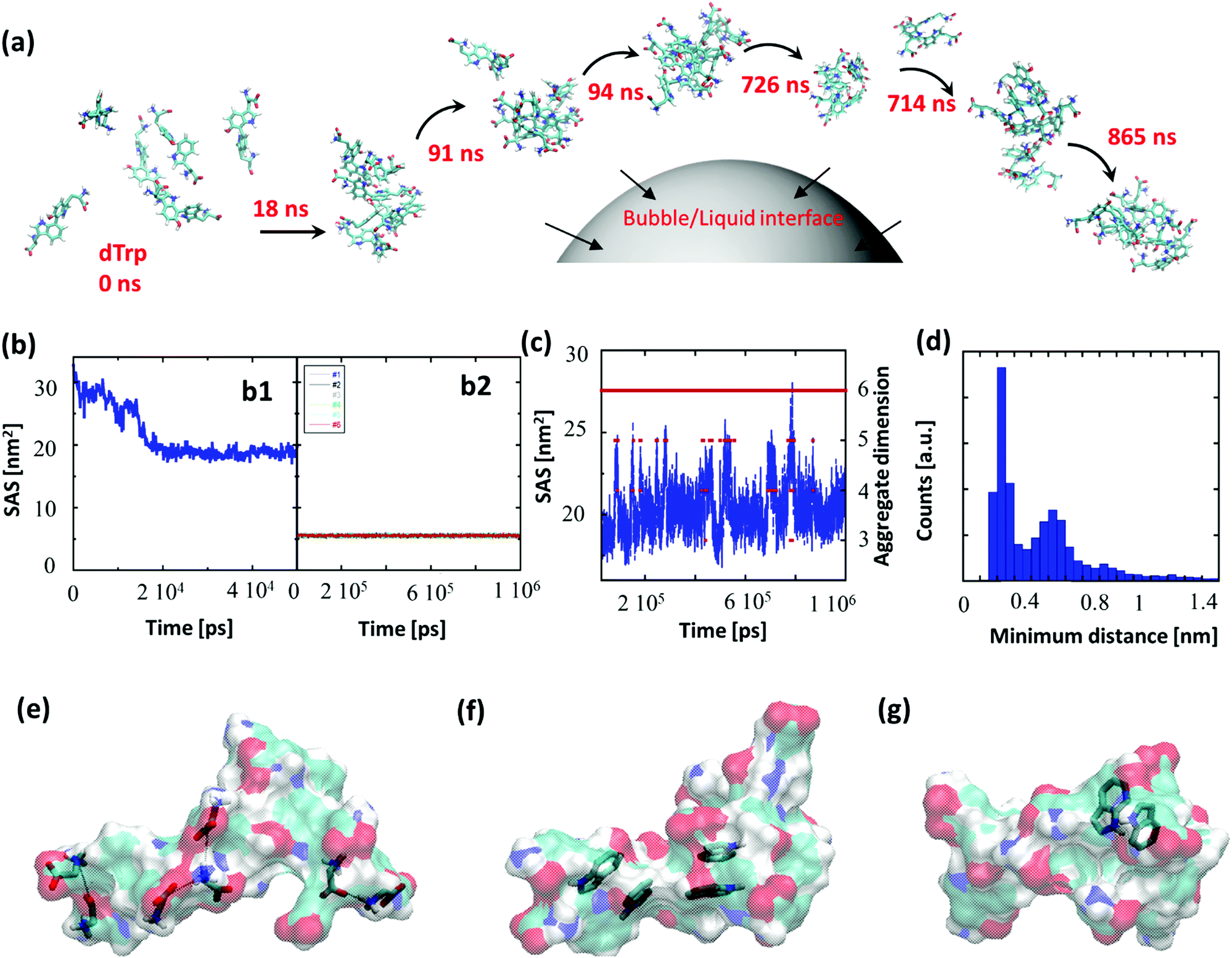 | ||
| Fig. 3 (a) Molecular dynamics simulation of six replicas of dTrp molecules, snapshots at 0 ns, 18 ns, 91 ns, 94 ns, 714 ns, 726 ns, and 868 ns and their schematic adsorption at the bubble–liquid interface. The first aggregate was formed just after 18 μs. (b1) Global SAS of the six dTrp molecules during the first 50 ns of the simulation. (b2) SAS calculated for all the six dTrp molecules singularly considered during the whole trajectory (1 μs). (c) SAS value calculated on all the six dTrp molecules (blue line, y axis on the left) and number of monomers in the aggregates (red points, y axis on the right) during the simulation time. (d) Distribution of the minimum distance of a pair of dTrps during the simulation. The distance has been calculated between 25 ns and 1 μs. (e–g) snapshots from MD simulations. The dTrp molecules are shown as semitransparent surfaces. Models of (e) ionic interactions, (f) π–π interactions and (g) H-bonds involving aromatic rings as acceptors. The involved atoms are represented as solid sticks. The C, H, N and O atoms are colored in cyan, white, blue and red, respectively. | ||
To discriminate between the different contributions to the stabilization of the formed aggregates, the different interactions observed during the simulation were screened. The average number of hydrogen bonds detected during the simulations was 5 ± 2; however, other interactions can play a role in the aggregate stabilization. Fig. 3e–g depicts the different interactions observed during the simulation. Ionic interactions between the charged carboxyl and amine groups have been first identified (Fig. 3e). The presence of aromatic moieties in the aqueous environment favors the stabilization of π–π interactions (Fig. 3f) between the indole moieties present in the dTrp molecules, which may result in peculiar optical features later investigated. Finally, in Fig. 3g an example of unusual hydrogen bonds in which the aromatic ring acts as an acceptor is also reported. This kind of interaction, which is about half as strong as a normal hydrogen bond, is known to play a significant role in molecular associations.25 All these types of interactions have been systematically observed throughout the simulation process and we can assume they all contribute to the formation and stabilization of the nanoaggregates.
To investigate the persistence of the interactions between couples of molecules in the aggregate, we have calculated the distribution of the minimum distances for all the possible 15 pairs of dTrp molecules during the simulations. All distributions are similar; Fig. 3d reports an example of these distribution profiles. The distribution shows that the pairs populate both low and high minimum distance values during the simulation. This suggests that, although the six-molecule aggregate persists for almost the entire simulation, the molecules assembled into the aggregates dynamically interact with each other. Overall the computational results are consistent with the rapid formation and stability of dTrp nanoparticles via salts bridges and π–π and H-bonding interactions. In addition, the dynamic exchange of the interacting molecules in the aggregates may contribute to the dissipation of the energy process resulting in the dissolution of the nanoaggregates.
Finally, from the MD data we have evaluated the number of dTrp molecules present in a single 230 nm sized nanoparticle. From the six dTrp molecule aggregate's SAS value (20 ± 1 nm2) and assuming a spherical shape for the formed aggregates, we can estimate a volume of 8.4 nm3. As a result, the estimated number of dTrp molecules present in a single 230 nm sized nanoparticle is approximately 5 × 106. From the SAS data of isolated dTrp molecules (surface and planar area of 5.5 ± 2 nm2 and 1.5 nm2 respectively), we can also estimate the maximum number of dTrp molecules that can be placed into a monolayer covering the whole surface of a microbubble which is approximately 245 × 106. This is in good agreement with the experimentally measured surface excess concentration (2.4 × 108). These results suggest that the collapse of a single bubble can in principle produce a single or more NPs. As the number of dTrp molecules covering the bubble surface exceeds the estimated number of molecules present in a single dTrpNP we can speculate that each collapsing bubble can give rise to the formation of a few nanoparticles as shown in Fig. 1a. In conclusion, the experimental and computational results are consistent with the hypothesis that the dTrpNPs arise from the rapid collapse of the microbubble and that they are formed by DSA of the molecules adsorbed on the surface of the same microbubble during the acoustic cavitation. The reported out-of-equilibrium system can be classified as a DSA system because (i) it is fuelled and sustained by the input of acoustic energy and associated pH change, (ii) the nanoaggregates have a finite lifetime and relax spontaneously to the thermodynamic stable state over time, when the fuel is removed and (iii) the system can be refuelled by the acoustic energy and protons to promptly regenerate the nanoparticles.
Probing the intracellular disassembly processes of dTrpNPs in different cellular compartments by imaging
A key challenge, however, is understanding the interactions of dTrpNPs with biological systems at the cellular and subcellular levels. The kinetics and mechanism of cellular uptake, intracellular trafficking, dissolution and ultimately bioactivity of dTrpNPs strongly depend on whether the kinetics of disassembly of dTrpNPs investigated in a test tube can take place in the crowded and complex intracellular milieu. We found that the optical properties of dTrpNPs offer a tool to gain insight into their intracellular behavior. The dTrpNPs showed good photostability under irradiation and at room temperature (Fig. S10a and b, ESI†) and good quantum yield (QY575 = 0.39). On the other hand, fluorescence probes like Rhodamine 6G, FITC, Cy3, etc. undergo bleaching in a few seconds and require storage at −20 °C.26–28 These spectral features can be advantageous to track their intracellular trafficking and disassembling. The colloidal stability and integrity of dTrpNPs in the presence of serum proteins (100% FBS) was investigated using nanoparticle tracking analysis. A slight increase in particle size from 230 ± 50 nm to 280 ± 65 nm was likely induced by adsorption of protein corona on the surface of nanoparticles, whereas no aggregation or disassembly of dTrpNPs was observed. Interstingly, the adsorption of protein corona on the dTrpNP surface produces a 3-fold increase in green emission (Fig. S10c, ESI†). This increase in emission was also observed when dTrpNPs were suspended under alkaline conditions (Fig. S10d, ESI†), therefore it was ascribed to the deprotonation of aromatic hydroxyl groups upon protein adsorption.Next, we verified the possible cellular cytotoxicity effects exerted by dTrpNPs. For that purpose, MDA-MB-231 cells were incubated with dTrpNPs at different concentrations from 3 to 100 μg mL−1 for 24 h and 48 h. Fig. S11a (ESI†) shows that the particles exhibit negligible cytotoxicity even after 24 h and 48 h at all the tested concentrations. We evaluated the association of dTrpNPs with MDA-MB-231 cells as a function of time using flow cytometry under different fluorescent channels (Fig. S11b, ESI†). The association relies on both membrane binding and intracellular uptake processes. We observed rapid and complete association of dTrpNPs with cells in the first 6.5 h of incubation (Fig. S11b, ESI†). Furthermore, to study the uptake of dTrpNPs, cells were incubated with a 10 μg mL−1 nanoparticle suspension for 5 h and the medium was replaced followed by further incubation for up to 8 h, 24 h and 48 h at 37 °C in fresh medium.
The imaging of live cells incubated with dTrpNPs was carried out using confocal microscopy to investigate the kinetics of disassembly of dTrpNPs inside the cells. Fig. 4c shows blue, green and red fluorescence under different fluorescence channels. After 8 h and 24 h of incubation, the cells show mainly the punctuate fluorescence pattern with limited fluorescence diffusely spread throughout the cytosol in the blue, green and red channels. After 48 h of incubation, the cells exhibit blue and green fluorescence signals spread in the cytosol whereas the red fluorescence disappears. The punctuate pattern indicates the partial confinement of dTrpNPs into acidic endo-lysosomes (pH 5–6). As the red emission is indicative of the dTrpNPs aggregate state, these results suggest that a time dependent dissolution of dTrpNPs occurs in the cytosol (pH 7) after escaping from endo-lysosomes.
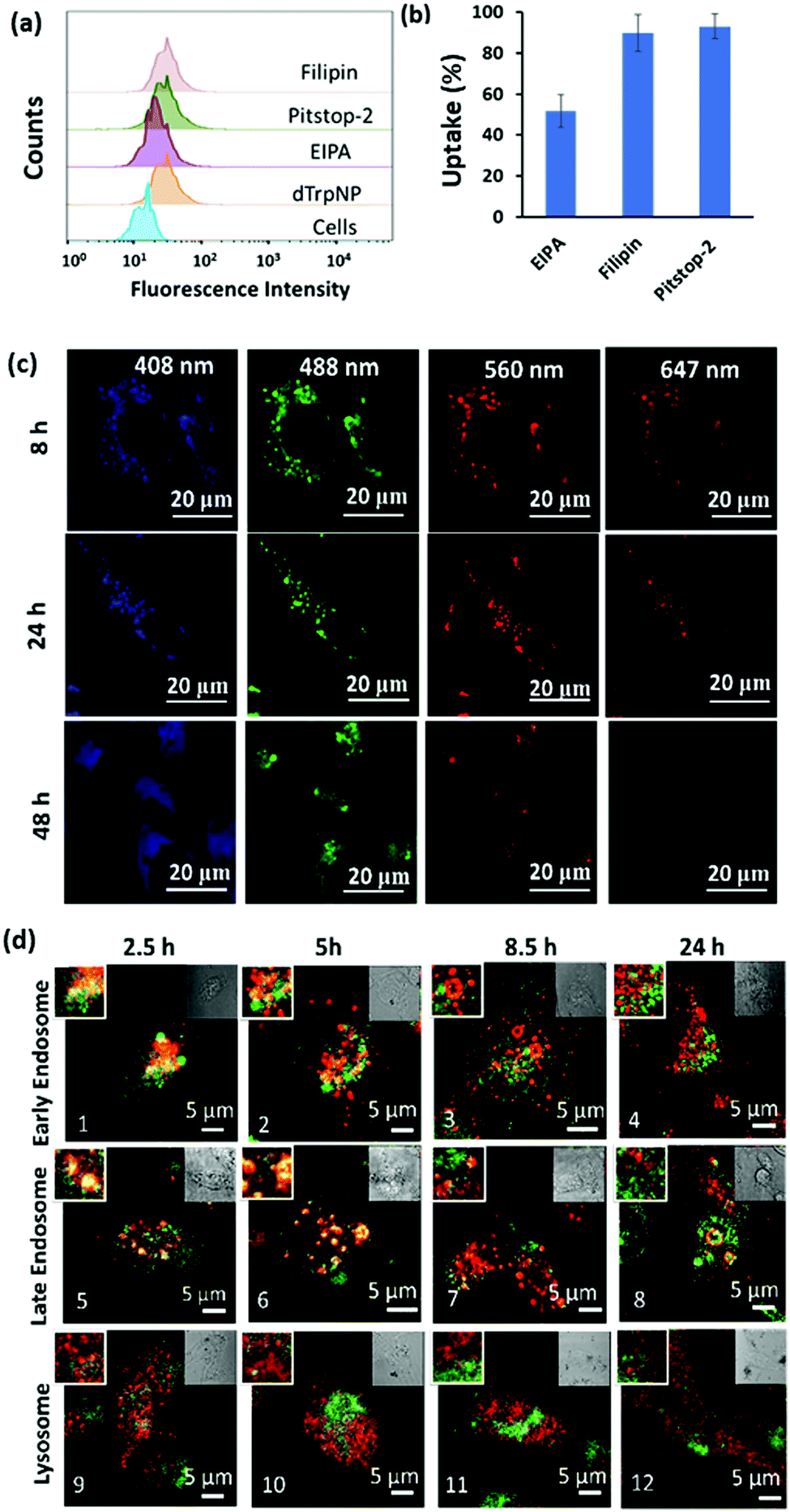 | ||
| Fig. 4 (a) Fluorescence intensity histogram of MDA-MB-231 cells before and after treatment with endocytic inhibitors and dTrpNPs measured using flow cytometry. (b) The effect of different endocytic inhibitors on the uptake of dTrpNPs. (c) Confocal microscopic images of the MDA-MB-231 cells incubated with the dTrpNPs at 37 °C for 8 h, 24 h and 48 h. (d) Colocalization studies of dTrpNPs (green) with different constructs (red) after 2.5 h washout and further 5 h, 8 h and 24 h incubation with confocal microscopy. Early endosomes, late endosomes and lysosomes were stained with rabbit anti-EEA1 monoclonal antibody, rabbit anti-Rab7 monoclonal antibody and rabbit anti-LAMP-1, respectively, followed by staining with the anti-rabbit secondary Alexa Fluor-647 conjugated antibody. The left inset is the magnified image and the right inset is the bright field image of the principal image. | ||
To gain further insight into the cell internalization mechanism, the MDA-MB-231 cells were incubated with filipin, pitstop-2, and ethylisopropyl amiloride (EIPA) to inhibit caveolae-dependent endocytosis, clathrin-dependent endocytosis and macropinocytosis, respectively, and then incubated with dTrpNPs (10 μg mL−1). Fig. 4a and b suggest that almost no uptake inhibition was observed with filipin and pitstop-2; however, EIPA led to 50% inhibition of uptake of dTrpNPs. Therefore, the uptake of dTrpNPs can be mediated through macropinocytosis. To advance the understanding of the intracellular route of dTrpNPs, we examined their endocytic trafficking as a function of time in fixed cells by using the immunostaining of organelles. The MDA-MB-231 cells were incubated with dTrpNPs for 2.5 h, washed with fresh medium to remove the extracellular dTrpNPs, and cultured for further 2.5 h, 5 h and 21.5 h at 37 °C corresponding to observations after 2.5 h, 5 h, 8.5 h and 24 h. Immunostaining of early endosomes, late endosomes and lysosomes was performed using EEA1 antibody, Rab7 antibody, LAMP 1 markers and AF 647 secondary antibody. Fig. 4d shows the representative confocal microscopy images of cell vesicles (red signal) and dTrpNPs (green signal) acquired using 640 nm and 560 nm lasers, respectively, after 2.5 h, 5 h, 8.5 h and 24 h observation times. Confocal parameters were adjusted to minimize the intrinsic fluorescence of dTrpNPs under 640 laser scanning. It was observed that for all three organelles, i.e., late endosomes, early endosomes, and lysosomes, the colocalization (yellow signal) was maximum until the first 5 h and almost negligible after that. The PCC (Pearson correlation coefficient) values were used to analyze the images and quantify the extent of colocalization (yellow signal) of organelles with dTrpNPs (Table S1, ESI†). The PCC values estimated at different incubation times indicate the amount of colocalization. Fig. 4d-1–2 and d-5–6 and the corresponding PCC values (0.5–0.6) suggest that dTrpNPs partially colocalize with both early and late endosome compartments after 2.5 h and 5 h observation times. Conversely after 8 h and 24 h, the confocal microscopic images (Fig. 4d-3–4 and d-7–8) and the corresponding PCC values (0.2–0.3) indicate a weak colocalization of dTrpNPs with both early and late endosomes. In addition, negligible colocalization with lysosomes at all times was found (Fig. 4d-9–12) indicating that lysosomes are not involved in the dTrpNP trafficking. Taken together, these results clearly suggest that the endosomal escape of dTrpNPs toward the cytosol progressively occurs after 2.5 h of incubation. Subsequently, the shift in pH from 5.5 to 7 can trigger the dissipative dissolution of dTrpNPs in the cytosol. This is in agreement with our study in the test tube showing that dTrpNPs dissolve very slowly at the endosomal acid pH 5–6. A possible mechanism for dTrpNP endosomal escape could be the “proton sponge effect” depicted in Fig. S12 (ESI†). This is typically mediated by species with high buffering capacity in the endosomal “pH change window”, such as amine-rich molecules with a pKa of around 6,29,30 which can produce osmotic imbalance inside the endosome and ultimately lead to the disruption of the endosomal membrane.30 Indeed, the potentiometric titration curve of dTrp (Fig. S8, ESI†) shows its buffering capacity in the biologically relevant region, between pH 5 and 7 because of protonation/deprotonation of aromatic hydroxyl groups. Hence, the endosomal escape of the dTrpNPs could be potentially mediated by the “proton sponge effect” triggered by dTrpNPs.
In conclusion, our results show that the dTrpNPs provide a multispectral bioimaging tool suitable for tracking their intracellular fate. The dTrpNPs are promptly internalized in MDA-MB-231 cells, trafficked from early to late endosomes, released into the cytosol by escaping the endosomes and dissolved in the neutral cytosolic environment. It is worth noting that apart from pH, in the intracellular milieu dTrpNPs may also experience alternative experimental conditions which may induce the disassembly of the aggregates. Competitive interactions with lipids or associating proteins with dTrp could also potentially promote the dissolution of dTrpNPs in the cytosol.
Therapeutic properties of dTrpNPs
We finally sought to investigate the possible applications of dTrpNPs in the biomedical field. For instance, Fig. S13 (ESI†) shows that dTrpNPs possess high antioxidant and radical scavenging properties. Therefore, dTrpNPs can potentially be used to protect cells against any oxidative damage. Furthermore, the applicability of dTrpNP as a platform for sustained anticancer drug delivery was also considered. The sustained release of drugs from nanoparticles fuelled by the cytosolic pH can be advantageous to attain intracellular therapeutic concentrations with higher selectivity and specificity.Doxorubicin, DOX, was incubated with dTrpNPs at pH 5–6 and a high loading efficiency (70% with 0.38 mg DOX/1 mg of dTrp) was observed. The high loading capacity can be attributed to the electrostatic and hydrophobic interactions between dTrpNPs and DOX. The release of DOX was studied in PBS (pH 7.4) at 37 °C by monitoring the intensity of fluorescence emission spectra at λex = 480 nm. Fig. 5a shows the percentage release of doxorubicin as a function of time. The nanoparticles show slow and bimodal release profile of the drug where the initial release could be due to some adsorbed drug onto the surface of dTrpNPs. The surge in the release of the drug after 10 h could be due to the erosion or dissolution of dTrpNPs. Fig. 5b illustrates the viability of MDA-MB-231 cells when treated with DOX and DOX loaded dTrpNP (dTrpNP + DOX) as a function of different concentrations of DOX ranging from 0.19 to 12 μg mL−1 after 24 h and 48 h. These data show that the cell viability decreases with an increase in the concentration of DOX. The toxicity of dTrpNP + DOX was less than that of free DOX after 24 h due to the slower release of DOX from the nanoparticles. However, the cytotoxicity of dTrpNP + DOX was comparable with that of free DOX after 48 h. Fig. 5c shows the confocal microscopy images of the cells incubated with dTrpNP + DOX particles at different incubation times of 2 h and 24 h. The intrinsic green fluorescence signal arising from dTrpNPs remains confined in the cytosol whereas an increase in red fluorescence as a function of time is observed in the nucleus. This clearly indicates the slow accumulation of doxorubicin into the nucleus upon release from dTrpNPs. The accumulation of doxorubicin into the nucleus is clear evidence of the release of the drug from dTrpNPs. The drug can be released as a result of the disassembly of dTrpNPs or because of diffusion across the nanoparticles. On the other hand, free DOX at the same concentration can completely accumulate into the nucleus just after 1–2 h of incubation as shown in Fig. S14 (ESI†). Overall this study suggests that the dTrpNPs can be successfully used to attain the controlled release of the drug so that the required concentration of the drug can be maintained within therapeutic levels over an extended period of time.
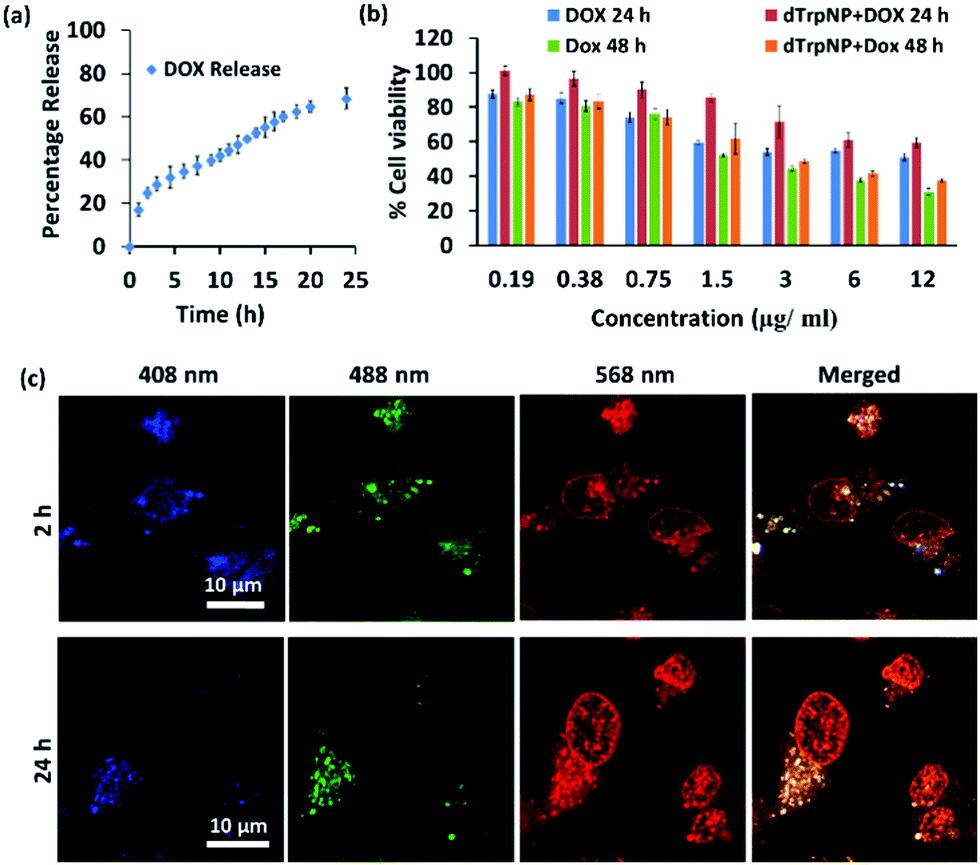 | ||
| Fig. 5 Intracellular release of DOX by dTrpNPs. (a) The percentage release of DOX from the dTrpNP + DOX (blue) dissolution in PBS (pH = 7.4) at 37 °C as a function of time. (b) Cell viability evaluation of DOX and dTrpNP + DOX at different concentrations of DOX after 24 and 48 hours towards MDA-MB-231 cells. (c) Confocal microscopic images of the MDA-MB231 cells incubated with dTrpNP + DOX for 2 and 24 h showing the release of DOX from nanoparticles into the nucleus. | ||
Conclusion
In conclusion we have presented computational and experimental approaches to demonstrate the role of the acoustic field in the formation of out-of-equilibrium nanoaggregates using a simple amino acid, L-tryptophan, as a building block. The collapse of acoustic cavitation bubbles first triggered the formation of tryptophan dimers and then ultimately acted as a fuel for dissipative self-assembly of tryptophan dimers to form uniform nanostructures with multifarious properties. The characteristic optical and bio-functional properties of synthesized nanoparticles made them a powerful tool for sustained release of anti-cancer drugs and for probing their intracellular trafficking. In addition, the insight provided in this work paved the way for ultrasound driven dissipative self-assembly applicable on sets of biomolecules which are currently under investigation. An alternative strategy to pursue is the rational design of ad-hoc building blocks combining experiments, simulation and acoustic theory to predict ultrasound-driven self-assembly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (ARC) under a Future Fellowship (F. Cavalieri, FT140100873) and the University of Melbourne Establishment Grant (F. Cavalieri). This work received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 690901 (NANOSUPREMI). This work was performed in part at the Materials Characterization and Fabrication Platform (MCFP) at The University of Melbourne. We acknowledge the University of Melbourne for their support through an MRS scholarship and a Norma Hilda Schuster Scholarship (S. K. Bhangu). We also thank the CINECA consortium for providing computational resources. Special thanks to Mr Yang Shen for his contribution to bubble dynamics studies.Notes and references
- E. Reisler and E. H. Egelman, J. Biol. Chem., 2007, 282, 36133–36137 CrossRef CAS PubMed.
- S. A. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- S. De and R. Klajn, Adv. Mater., 2018, 30, 1706750 CrossRef PubMed.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70 CrossRef CAS PubMed.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111 CrossRef CAS PubMed.
- F. Rakotondradany, M. Whitehead, A. M. Lebuis and H. F. Sleiman, Chem. – Eur. J., 2003, 9, 4771–4780 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. Kowlgi, G. J. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., 2010, 122, 4935–4938 CrossRef.
- J. Boekhoven, A. M. Brizard, K. N. Kowlgi, G. J. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- E. Del Grosso, A. Amodio, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2018, 57, 10489–10493 CrossRef CAS PubMed.
- K. Nakajima, H. Ogi, K. Adachi, K. Noi, M. Hirao, H. Yagi and Y. Goto, Sci. Rep., 2016, 6, 22015 CrossRef CAS PubMed.
- C. G. Pappas, T. Mutasa, P. W. J. M. Frederix, S. Fleming, S. Bai, S. Debnath, S. M. Kelly, A. Gachagan and R. V. Ulijn, Mater. Horiz., 2015, 2, 198–202 RSC.
- F. Cavalieri, E. Colombo, E. Nicolai, N. Rosato and M. Ashokkumar, Mater. Horiz., 2016, 3, 563–567 RSC.
- S. K. Bhangu and M. Ashokkumar, Sonochemistry, Springer, 2017, pp. 1–28 Search PubMed.
- K. Tao, Z. Fan, L. Sun, P. Makam, Z. Tian, M. Ruegsegger, S. Shaham-Niv, D. Hansford, R. Aizen, Z. Pan and S. Galster, Nat. Commun., 2018, 9, 3217 CrossRef PubMed.
- J. I. Mujika, J. Uranga and J. M. Matxain, Chem. – Eur. J., 2013, 19, 6862–6873 CrossRef CAS PubMed.
- D. Sunartio, M. Ashokkumar and F. Grieser, J. Am. Chem. Soc., 2007, 129, 6031–6036 CrossRef CAS PubMed.
- K. Yasui, Acoustic Cavitation and Bubble Dynamics, Springer, 2018, pp. 37–97 Search PubMed.
- Y. Shen, K. Yasui, T. Zhu and M. Ashokkumar, Phys. Chem. Chem. Phys., 2017, 19, 20635–20640 RSC.
- L. Carroll, D. I. Pattison, J. B. Davies, R. F. Anderson, C. Lopez-Alarcon and M. J. Davies, Free Radical Biol. Med., 2017, 113, 132–142 CrossRef CAS PubMed.
- V. Paviani, G. T. Galdino, J. N. D. Prazeres, R. F. Queiroz and O. Augusto, J. Braz. Chem. Soc., 2018, 29, 925–933 CAS.
- A. Farrotti, P. Conflitti, S. Srivastava, J. K. Ghosh, A. Palleschi, L. Stella and G. Bocchinfuso, Molecules, 2017, 22, 1235 CrossRef PubMed.
- G. Bocchinfuso, C. Mazzuca, P. Conflitti, D. Cori, T. Coviello and A. Palleschi, Z. Phys. Chem., 2016, 230, 1395–1410 CAS.
- M. Levitt and M. F. Perutz, J. Mol. Biol., 1988, 201, 751–754 CrossRef CAS PubMed.
- C. Eggeling, J. Widengren, R. Rigler and C. Seidel, Anal. Chem., 1998, 70, 2651–2659 CrossRef CAS PubMed.
- H. NamáChan, Chem. Commun., 2014, 50, 13578–13580 RSC.
- Y. Q. Sun, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Angew. Chem., Int. Ed., 2012, 51, 7634–7636 CrossRef CAS PubMed.
- D. W. Pack, D. Putnam and R. Langer, Biotechnol. Bioeng., 2000, 67, 217–223 CrossRef CAS PubMed.
- C. Lin and J. F. Engbersen, J. Controlled Release, 2008, 132, 267–272 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, characterization of dTrpNP-HPLC, EDS, mass spectrometry, fluorescence spectroscopy, and therapeutic properties of dTrpNPs. See DOI: 10.1039/c9nh00611g |
| This journal is © The Royal Society of Chemistry 2020 |