
Bright, stable, and efficient red light-emitting electrochemical cells using contorted nanographenes†
Elisa
Fresta
ab,
Kevin
Baumgärtner
c,
Juan
Cabanillas-Gonzalez
 d,
Michael
Mastalerz
c and
Rubén D.
Costa
*ae
d,
Michael
Mastalerz
c and
Rubén D.
Costa
*ae
aIMDEA Materials Institute, Calle Eric Kandel 2, E-28906 Getafe, Madrid, Spain. E-mail: ruben.costa@imdea.org
bUniversidad Autónoma de Madrid, Departamento de Física Aplicada, Calle Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
cOrganisch-Chemisches Institut, Ruprecht-Karls-Universität, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
dIMDEA Nanoscience Institute, Calle Faraday 9, 28049 Madrid, Spain
eResearch Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo 165-8555, Japan. E-mail: ruben.costa@aoni.waseda.jp
First published on 6th December 2019
Abstract
This work rationalizes, for the first time, the electroluminescent behavior of a representative red-emitting contorted nanographene – i.e., hexabenzoovalene derivative – in small molecule light-emitting electrochemical cells (SM-LECs). This new emitter provides devices with irradiances of ca. 220 μW cm−2 (242 cd m−2), external quantum efficiencies (EQE) of 0.78% (<25% loss of the maximum theoretical EQE), and stabilities over 200 h. Upon optimizing the device architecture, the stability increased up to 3600 h (measured) and 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h (extrapolated) at a high brightness of ca. 30 μW cm−2 (34 cd m−2). This represents a record stability at a high brightness level compared to the state-of-the-art SM-LECs (1000 h at 0.3 μW cm−2). In addition, we rationalized one of the very rare LEC examples in which the changes of the electroluminescence band shape relates to the dependence of the relative intensity of the vibrational peaks with electric field, as corroborated by dynamic electrochemical impedance spectroscopy assays. Nevertheless, this exclusive electroluminescence behavior does not affect the device color, realizing one of the most stable, bright, and efficient red-emitting SM-LECs up to date.
000 h (extrapolated) at a high brightness of ca. 30 μW cm−2 (34 cd m−2). This represents a record stability at a high brightness level compared to the state-of-the-art SM-LECs (1000 h at 0.3 μW cm−2). In addition, we rationalized one of the very rare LEC examples in which the changes of the electroluminescence band shape relates to the dependence of the relative intensity of the vibrational peaks with electric field, as corroborated by dynamic electrochemical impedance spectroscopy assays. Nevertheless, this exclusive electroluminescence behavior does not affect the device color, realizing one of the most stable, bright, and efficient red-emitting SM-LECs up to date.
New conceptsStable, bright, and efficient red-emitting small molecule light-emitting electrochemical cells (SM-LECs) still represent a challenge (1000 h at 0.3 μW cm−2). Aggregation quenching and electrochemical formation of quenchers and trap carriers are responsible for the commonly observed poor performances. Herein, SM-LECs featuring high irradiances of ca. 220 μW cm−2, external quantum efficiencies of 0.78% (<25% loss of the theoretical maximum), and stabilities over 200 h are demonstrated using a representative red-emitting contorted nanographene – i.e., hexabenzoovalene derivative. Its distorted molecular structure avoids aggregation leading to highly emissive thin films, while its excellent electrochemical stability results in high stabilities. Upon optimization of the device architecture, outstanding stabilities of 3600 h (measured) and 13000 h (extrapolated) at a high brightness of ca. 30 μW cm−2 were achieved. Besides this new performance record compared to the SM-LEC prior-art, this works decipheres a very rare elecroluminescence behavior that is ruled by an electric-field dependence of the intensity for the radiative deactivation of the excited vibrational modes without changing the nature of the emitting excited state. This is supported by using different device architectures, driving modes as well as in operando electrochemical techniques.
|
Light-emitting electrochemical cells (LECs) consist of ionic-based active layers sandwiched between two electrodes.1 The presence of mobile ions allows for solution-based deposition techniques and low driving voltages using air-stable electrodes.2–4 Thus, LECs represent an easy-to-fabricate and low-cost alternative to the well-known organic light-emitting diodes (OLEDs). Indeed, good stabilities (320 h at 100 cd m−2) along with remarkable efficiencies (99.2 cd A−1 at 1910 cd m−2) have recently been achieved after optimizing quenching and exciton diffusion processes in a host–guest LEC.5
As a curse and a blessing, LECs hold a high versatility toward a huge variety of emitters, such as ionic transition metal complexes (iTMCs), conjugated polymers (CPs), small molecules (SMs), perovskites, and quantum dots.1,3,6–12 However, each emitter requires the optimization of the type and amount of the mobile ion source, such as ionic liquids,13 ionic polyelectrolytes,14,15 inorganic salts,16,17 as well as the device architecture with respect to thickness,18 injection layers,10,11,19etc. Along these lines, LECs based on iTMCs and CPs have extensively been optimized, reaching outstanding efficiency and brightness values.9,20 However, these emitters usually present low photoluminescence quantum yields (φ) in the low-energy emitting region, as well as low electrochemical stabilities, leading to devices with poor efficiencies and stabilities.21–23 Therefore, stable and efficient red-emitters are highly desired. Towards this end, red-emitting SMs like perylenes, benzothiadiazoles, pentacenes, porphyrins, BODIPY–porphyrin dyads, and cyanines have recently been applied to LECs.24–29 However, SM-LECs combining high irradiances along with stabilities of hundreds of hours have not been realized yet. Indeed, while the best stabilities (∼1000 hours) are accompanied by low irradiances (0.1–1 μW cm−2),26,27 the best luminances (750 cd m−2) come at the price of low stabilities (15 hours).25
Herein, we investigate the electroluminescent behavior of a highly contorted nanographene, namely a hexabenzoovalene (HBO) – i.e., 1 as shown in Fig. 1,30 highlighting its excellent prospect as an emerging family of red-emitters for SM-LECs. In general, interest in the optoelectronic application of 2D-annulated nanographenes has enormously grown in recent years.31–34 In particular, distorted nanographenes are soluble in organic solvents, as they do not present efficient π–π stacking, allowing them to be suitable candidates for solution processed devices.30,34–36 In addition, this family of SMs shows high φ, good electrochemical and thermal stabilities, and ambipolar carrier mobilities.31,37,38 Despite these excellent features, their electroluminescent behavior has barely been investigated, as highlighted by only a few interesting OLED and LEC examples.28,39
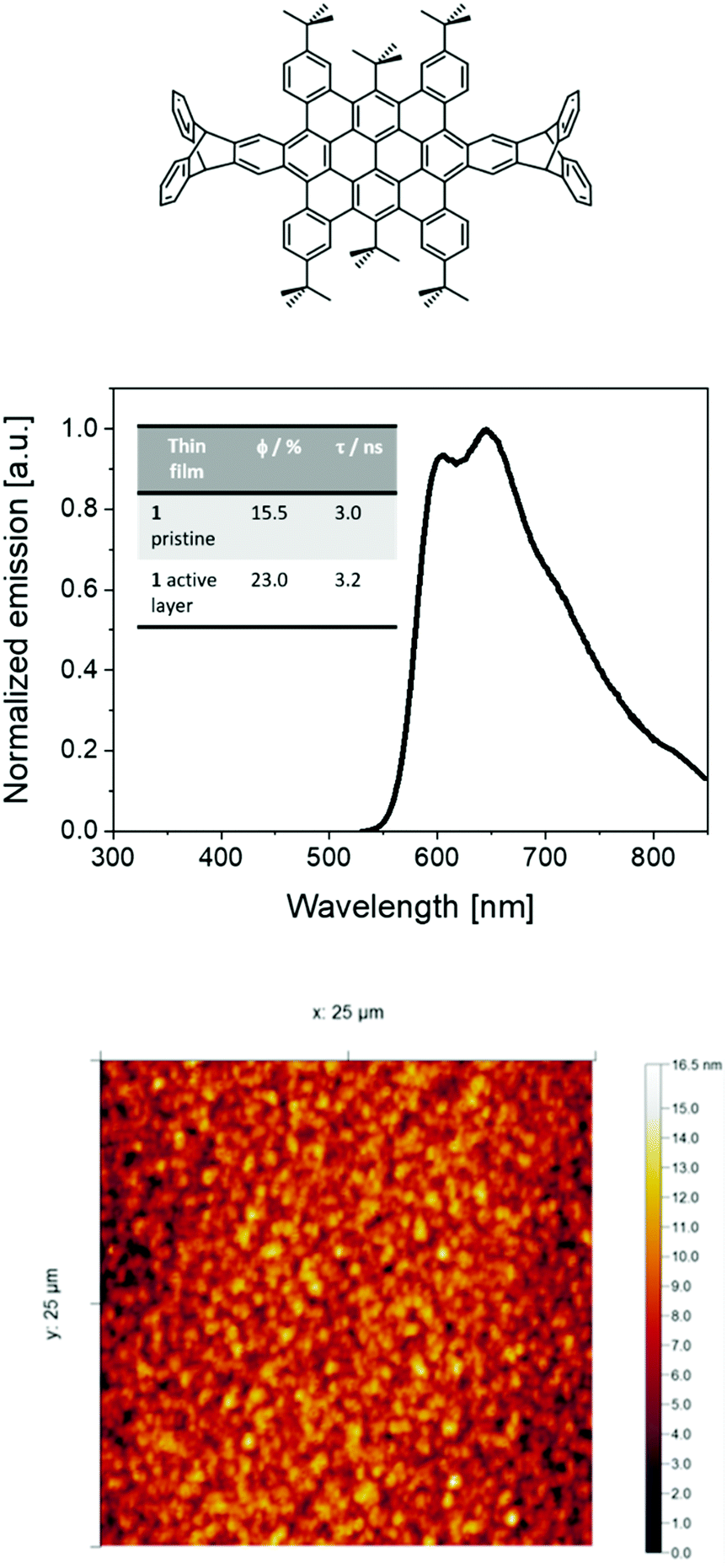 | ||
| Fig. 1 Top: Chemical structure of 1. Centre: Photoluminescence spectra (λexc = 450 nm) of thin films based on 1 with ionic matrix onto ITO substrates in air at rt. Inset: Photophysical properties (φ and 〈τ〉) of 1 thin films. Bottom: AFM picture of 1 thin films used in devices. | ||
To shed light onto the electroluminescent behavior of contorted nanographenes and its prospect in the lighting field, we decided to comprehensively investigate both photo- and electro-luminescent features of the representative HBO in thin films.30,40 The impact of this work is two-fold. On one hand, 1-LECs achieved irradiances of ca. 220 μW cm−2 (242 cd m−2) external quantum efficiencies (EQEs) of 0.78% (76% the theoretical maximum EQE), and stabilities over 200 h. Upon optimizing the active layer thickness, the devices feature stabilities of 3600 h (measured) and >13000 h (extrapolated) at a high brightness level (30 μW cm−2 or 34 cd m−2). Thus, these devices represent one the most stable and bright red-emitting SM-LECs so far reported (e.g., 1000 h at 0.3 μW cm−2).1,41 On the other hand, we encountered that, regardless of the device architecture and driving conditions, the electroluminescence behavior is highlighted by an arrangement of the vibrational intensities caused by the steady increase of the internal electric field across the active layer. This leads to minimal changes in the color quality of the devices. However, this behavior represents one of the very rare examples of electric field dependent chromaticity in single-component and single-layered lighting devices, in general, and SM-LECs, in particular.
As reported previously,30,40 HBO or 1 shows an orange-red fluorescence (maximum emission wavelength (λmax) at around 600 nm with a shoulder at 640 nm) associated to φ of 54% and excited state-lifetimes (τ) of 4 ns in solution. Thin-films of 1 mixed with TMPE (trimethylolpropane ethoxylate), PEO (polyethylene oxide), and LiOTf (lithium triflate) as ionic matrices were used as the active layers in devices – i.e., 1:TMPE:LiOTf in a mass ratio 1:0.15:0.06; see Experimental section for more details. Investigating the thin-films by atomic force microscopy (AFM) revealed a homogenous morphology with a root-mean-square roughness of 2–3 nm (Fig. 1). The photoluminescence features of these thin-films consist of a broad emission band with peaks at 598 nm and 647 nm associated to φ of 23% and τ of 3.2 ns (Fig. 1). The presence of a well-defined vibrational structure implies that thin films are not formed upon aggregation, which usually leads to broad and shapeless emission. Indeed, both the excitation spectra and τ values centered at either 598 nm or 647 nm and measured at 298 K and 77 K are similar, highlighting the lack of aggregates (Fig. S1–S4, ESI†). All-in-all, these results are in line with the well-thought molecular design that leads to a desired contorted molecular structure of 1, providing high solubilities with a low aggregation behavior even upon film forming.
After having investigated 1 and thin-films thereof in ionic matrices, the promising photophysical data encouraged us to transfer 1 to devices with the architecture ITO/PEDOT:PSS (70 nm)/1:TMPE:LiOTf 1:0.15:0.05 (70 nm)/Al (90 nm) – see Experimental section.
These devices were driven at pulsed currents of 25 mA monitoring the luminance, color, and electrical behavior over time (Fig. 2). The measurements involved the use of a pulsed driving scheme based on a block-wave at 1000 Hz and a duty cycle of 50%. In detail, the device behavior shows the typical LEC features, that is, an initial decrease of the applied voltage along with an increase of the luminance due to the formation of electrical double layers (EDLs) that assist the charge injection at the doped fronts growing from the respective electrodes. Similar to the LIV assays, the initial electroluminescence spectrum consists of a broad emission featuring the two above peaks at 600 and 650 nm. The device brightness achieved values of ca. 220 μW cm−2 in concert with stabilities over 200 h. In addition, luminous efficiency and EQE values of 0.74 lm W−1 and 0.78% were noted. Importantly, the EQE values account for 76% of the theoretical EQE (1.15%; Fig. 2 and Table 1). This is remarkable when considering the prior state-of-the-art of SM-LECs (33%),29,42 especially if taking into account that no host–guest optimization has been performed yet – e.g., EQE optimization from 33% up to 94% has recently been reported in host:guest CPs.42
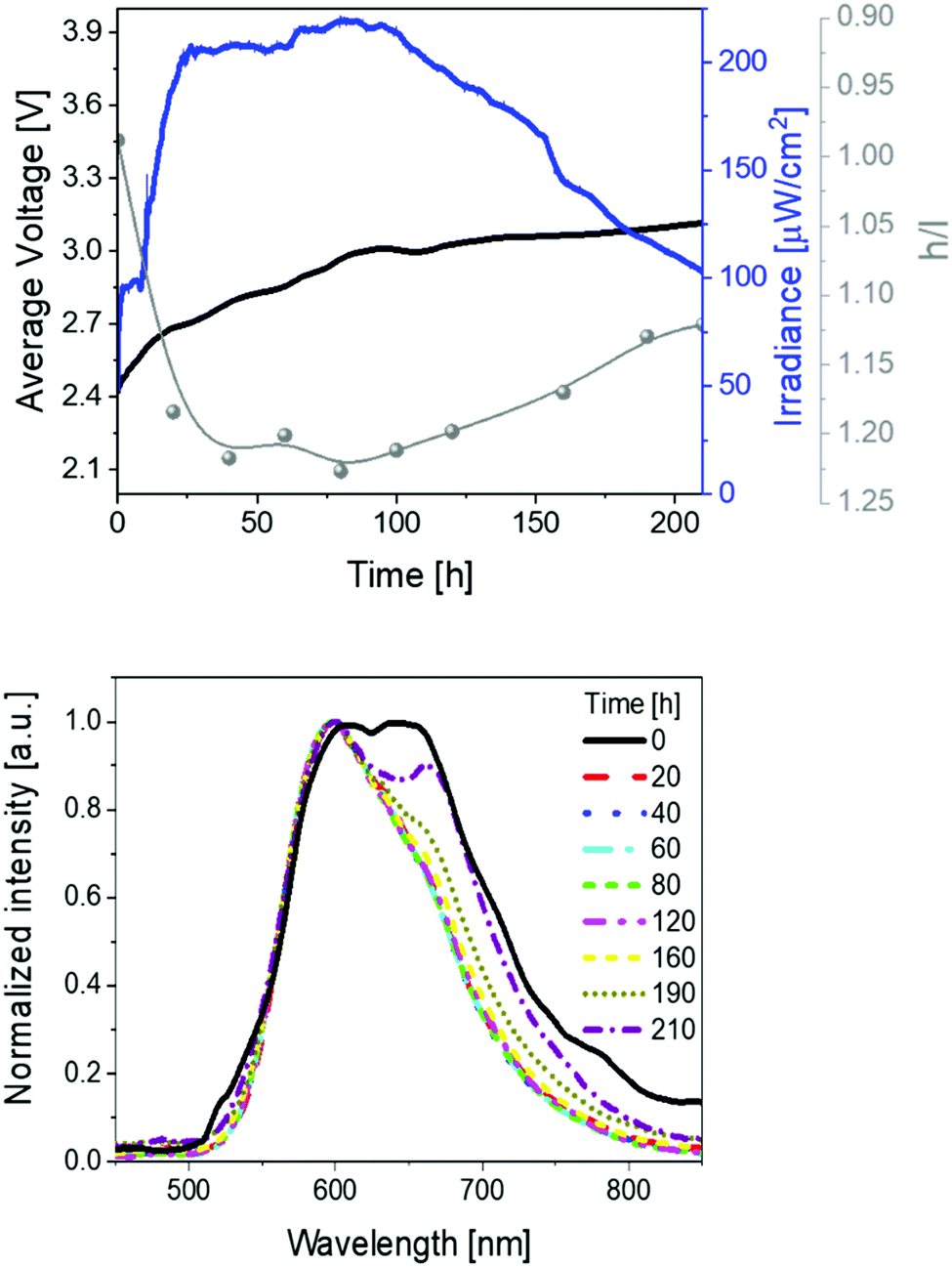 | ||
| Fig. 2 Average voltage, irradiance, and h/l (high/low energy peak) ratio (top), as well as electroluminescence spectra (bottom) over time (see legend) for 1 devices driven at a pulsed 25 mA current. | ||
| Active layer thickness [nm] | Driving current [mA] | Irradiance [μW cm−2] | t 1/2 [h] | t on [h] | Efficiency [lm W−1] | EQE [%] |
|---|---|---|---|---|---|---|
| PM = pulsed mode.a Time to reach 50% of the initial irradiance.b Time to reach the maximum irradiance. | ||||||
| 70 | 25 mA, PM | 222.8 | 204 | 27 | 0.74 | 0.78 |
| 70 | 50 mA, PM | 369.9 | 25 | 2 | 0.50 | 0.51 |
| 30 | 25 mA, PM | 31.2 | >13000 |
260 | 0.10 | 0.12 |
| 70 | 3 V | 150 | 7 | 0.40 | 0.30 | 0.43 |
Besides the excellent device performance, it is important to point out that the chromaticity changes over time (Fig. 2). This behavior has already been noticed in several LECs,1,3 showing either large λmax shifts (10–100 nm) or changes of the spectrum shape that have been attributed to a strong polarization of the emitting excited state43,44 and/or a change of nature of the emitting excited states,45–47 respectively. In contrast to these examples, the electroluminescence spectra of 1-LEC feature neither a λmax shift for both peaks nor a change of the E0–0 energy (Fig. S5, ESI†), indicating that the nature of the excited state holds upon electrical excitation over time. In short, just the intensity ratio between the high-energy vibrational peak (h-peak; 600 nm) and low-energy vibrational peak (l-peak; 650 nm), herein called h/l, changes over time, increasing from 0.99 to 1.23 during the first 40 hours, holding constant up to 100 h, and then abruptly decreasing (1.13) until the device lifetime is reached (Fig. 2). This translates to a marginal color change during the first 80 h – i.e., x/y CIE color coordinates move from 0.59/0.40 to 0.59/0.41, and then a slight change towards the red region – i.e., x/y CIE color coordinates of 0.61/0.39; Fig. S5 (ESI†). Overall, this is accompanied by a change in the correlated color temperature (CCT) of <300 K.
The reason underlying the interplay among the chromaticity changes in 1-LEC is, therefore, not trivial. Since changes in the nature of the excited state are not involved, other reasons, such as (i) unbalanced doping and movement of the emitting p–i–n regions,5 (ii) increase of the operation temperature,48,49 and (iii) microcavity and scattering effects,50–52 could explain the color evolution. They can be meaningful until the luminance reaches a steady-state regime and they are highly dependent to the thickness of the active layer. However, 1-LECs show a constant and very slow color change that extend itself throughout the entire lifetime of the device, regardless of the device architecture – vide infra. This is a hint for other reasons, such as degradation,19,53 phase separation or film morphology changes,11,54 and/or changes in the local electric field distribution and intensity.55–58
The formation of new species acting as either quenchers or emitters upon device operation may induce slow and irreversible changes over time, leading to a traceable relationship between the intensity changes of the h- and l-peaks. However, Fig. S6 (ESI†) shows that the intensity changes of the two peaks are equally affected over time. Furthermore, the E0–0 band does not shift over time, remaining constant at 2.38 eV (520 nm) (Fig. S6, ESI†). In addition, we compared the spectroscopic features of fresh and used devices (Fig. S7, ESI†), showing that both emission and UV-Vis absorption spectra, do not show any relevant change after device operation. Both findings suggest the lack of the formation of new species under electrical stimuli. As a further confirmation, we carried out static electrochemical impedance spectroscopy (EIS) of both fresh and used devices (Fig. S8–S10, ESI†). Fresh devices show a common LEC behavior, namely (i) an initial decrease of the electronic resistance at biases below the energy band-gap (ca. 2–3 V), which corresponds to the formation of the EDLs, and (ii) a further resistance decrease that holds at higher biases, which corresponds to the formation of the doped regions and the auto-sustained charge recombination (Fig. S9, ESI†). In addition, fresh and used devices showed ionic resistance values of 1 × 105 Ω and 1 × 1.9 × 104 Ω at 0 V, respectively. A decrease of the resistance in used devices suggests the lack of degradative events at the electrodes interfaces,17,59 being instead attributed to the remaining polarization upon forming the p- and n-doped regions. Upon heating the used devices at 60 °C for 30 min, the resistance is significantly recovered (82% or 0.82 × 105 Ω), indicating that electrochemical degradation processes are not taking place.17,53 Finally, we measured the figures-of merit of 1-LEC driven as described above until lifetime and heated at 60 °C for 30 min (Fig. S11 and S12, ESI†). Here, the h/l ratio of 0.85 is drastically different compared to the value at lifetime (1.15), being more similar to the one found for a fresh device (1.00). In addition, it follows the same trend over time as for fresh devices. This indicates the reversibility of the h/l change over time, as well as the absence of degradation processes.
As noticed in previous contributions, heat generation occurs under operation conditions.48 Indeed, 1-LEC experiences a temperature rise up to 65 °C during the first minutes, holding constant for the entire measurement (Fig. S13, ESI†). This contrasts with the above described behavior, in which the h/l peak intensity ratio constantly changes over much longer periods of time. Nonetheless, we also investigated the temperature dependence photoluminescence behavior of pristine films as well as device active layers (Fig. S13, ESI†). Pristine films show a broadening of the photoluminescence spectrum upon heating from 298 K to 475 K. This is expected since the thermal energy promotes excited electrons to higher vibration levels with the temperature increase, leading to new radiative transitions, which typically cause the emission band broadening.60 In contrast, the photoluminescence response of the devices does not experience significant spectral shape changes other than a slight linewidth broadening. Regardless of the surroundings of 1 in the thin-films, the increase of the temperature induces a reduction of the emission intensity. Finally, no changes in φ and layer morphology were noticed upon time when keeping the devices for several days at 353 K (Fig. S14, ESI†). Thus, we can discard temperature as a possible reason for the above-described electroluminescence response.
Finally, we investigated the relationship between electrical and chromaticity changes over time. Here, small changes in the local electric field to which the emitter is subjected may induce structural changes affecting the internal dipole moment and the reorientation of the emitter, modifying the radiative decay from vibronic states and emission quenching over time, respectively. Indeed, we have recently rationalized that the white electroluminescence in free base porphyrin LECs is related to the presence of two regioisomers, in which the inner H atoms are arranged in collinear and vicinal fashions upon increasing the electric field.53
To further confirm this hypothesis, we increased the applied electric field using four different approaches, namely (i) measuring consecutive luminance–current–voltage (LIV) assays, in which the voltage is changed over time in a ramp-manner, (ii) increasing the driving current, which is directly correlated to the average driving voltage,23,61 (iii) changing the duty cycle, as it also correlates with the duration of the applied voltage and electric field, and (iv) reducing the thickness of the active layer from 70 nm (thick) to 30 nm (thin).18,53,62
Firstly, we performed a series of LIV scans in the range of 0–6 V with a scan rate of 300 mV s−1. The devices showed a red emission response starting at low applied voltages of ∼3 V (Fig. S15, ESI†). The electroluminescence spectrum consists of a broad emission band with two peaks at 600 nm and 650 nm, indicating that the same excited state is radiative active, regardless of the type of excitation. They do not change in intensity upon consecutive scans, highlighting the sound stability of 1 upon electrical stimuli. Noteworthy, the h/l ratio changes over time assuming the same values at each step during the entire LIV measurements, indicating the reversibility of the voltage effect on the electroluminescence (Fig. S16, ESI†).
Secondly, devices driven at 50 mA pulsed current showed the same trend as above described (Fig. 3 and Table 1). In short, irradiances of 370 μW cm−2, EQEs of 0.52%, and stabilities of 24 h were noted. The electroluminescence band shows a similar trend with regard to the changes of the h/l peak intensity ratios over time, reaching a higher h/l ratio value – i.e., 1.25 vs. 1.40 at pulsed 25 and 50 mA, respectively – that hints a relationship between the electrical and chromaticity changes.
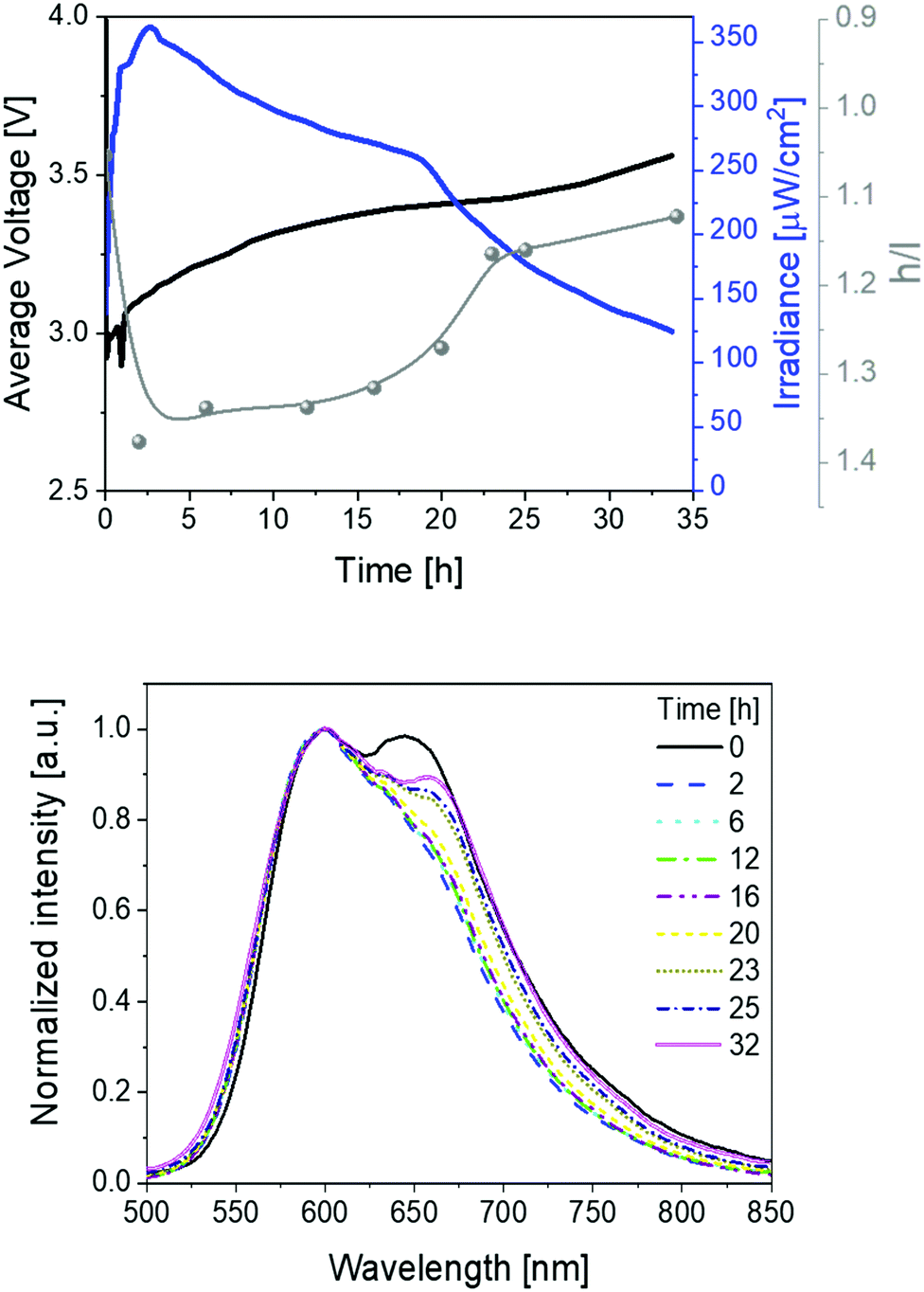 | ||
| Fig. 3 Average voltage, irradiance, and h/l (h = high energy peak, l = low energy peak) ratio (top), as well as electroluminescence spectra (bottom) over time (see legend) for 1 devices driven at a pulsed 50 mA current. | ||
Next, the duty cycle was varied in the following manner: devices were driven at 4 different duty cycle values (25, 50, 75, and 100%) for a short time (<5 minutes), in order not to generate over oxidized/reduced species that could hamper the measurement (Fig. S17, ESI†). The duty cycle scan was repeated three times. Here, two points are noteworthy, namely (i) h/l changes with the duty cycle – i.e., from 1.02 at 25% duty cycle to 0.89 at 100% duty cycle, highlighting voltage and electric field dependence, and (ii) h/l showed always similar values for the different duty cycles in each scan, indicating the reversibility of the voltage-dependency and the possibility of controlling the chromaticity with the applied electric field.
Finally, devices with thin active layers (30 nm) driven at pulsed current of 25 mA were studied (Fig. 4 and Table 1). Compared to thick devices (70 nm) (Fig. 2), the irradiance is reduced to approx. 30 μW cm−2 and the overall stability is significantly increased to >3600 h with extrapolated lifetimes of >13000 h. The device optimization with regard to the stability is also confirmed by comparing the total emitted energy of thick and thin devices – i.e., 732 J vs. 6560 J, respectively. Notice that the total emitted energy is calculated by integrating the radiant flux of the device vs. time from t = 0 (application of bias) to t = t1/5. This approach allows a clear stability comparison of devices showing different luminance levels as indicated by Bard and co-workers.63 As such, the optimized devices represent one of the most stable and bright red-emitting SM-LECs reported so far.1
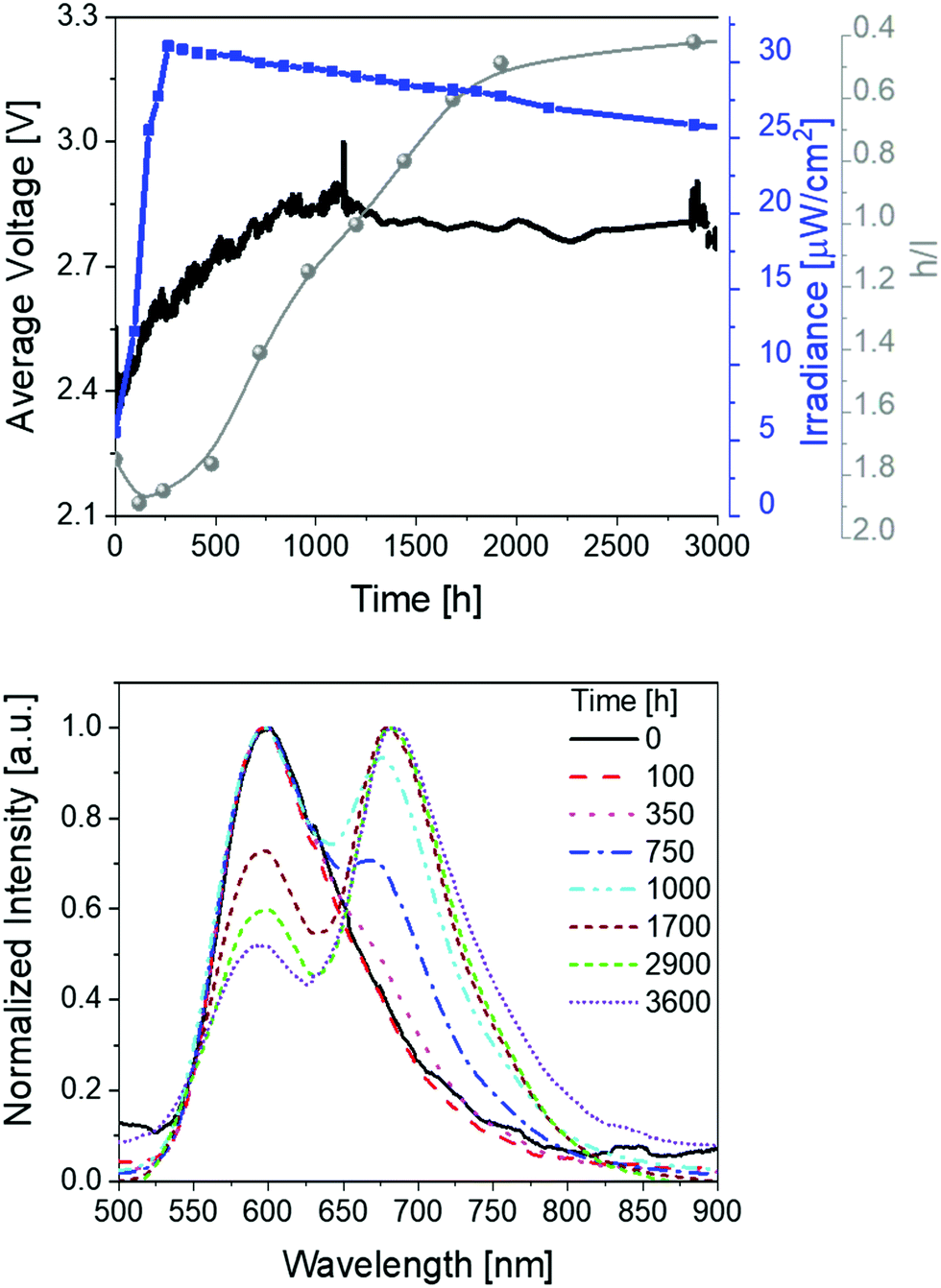 | ||
| Fig. 4 Average voltage, irradiance, and h/l (h = high energy peak, l = low energy peak) ratio (top), as well as electroluminescence spectra (bottom) over time for 1 devices driven at a pulsed 25 mA current. | ||
Besides the excellent stability performance of thin 1-LECs, the same behavior for the changes of the electroluminescence spectrum over time were observed over the whole lifetime (Fig. 4 and Table 1). In detail, the h/l ratio value increases – i.e., 1.25 vs. 1.90 for thin and thick devices, respectively. Indeed, the magnitude of the h/l peak intensity ratio is linearly correlated with respect to the externally applied electric field changing device architecture and driving conditions (Fig. S18, ESI†). Besides this aspect, the emission band shape of thin devices significantly changes showing an initial dominant emission centered at 600 nm with a low intensity shoulder at 670 nm that gets more prominent over time. After 1000 h, the l-peak becomes the major feature of the emission band. This leads to a change in the x/y CIE color coordinates from initial 0.59/0.40 to final 0.63/0.36 (Fig. S19, ESI†). After 2500 h of measurement, the h/l peak intensity ratio reaches a plateau until the end of the measurement. Noteworthy, this change accounts for a change of CCT <400 K, which is barely perceived by the human eye. As above described, the electroluminescence changes are in line with those of the applied voltage, which firstly decreases, then increases until it reaches a long plateau and then slightly decreases again, keeping its value at around 2.15 V over time (Fig. 4).
The above described relationship between the emission band shape changes and the electrical behavior over time prompted us to precisely determine the correlation with the changes of the internal electric field at the emitting neutral zone measured with dynamic EIS (see Experimental section).64 To this end, we focused on thick devices driven at constant voltage of 3 V. Their electroluminescence response shows the typical behavior of LECs driven at constant voltage, that is, a slow rise of both current and brightness followed by a slight decrease over the measuring time (Fig. 5). On top of this measurement, we carried out EIS assays at certain time intervals. The EIS data were fitted using the full electric circuit (Fig. S20, ESI†), while the figures-of-merit are gathered in Table S1 and Fig. S21 (ESI†). Details on the data calculation is provided in the ESI.† As shown in Fig. 5, the same trend is noted for the changes of both the h/l peak intensity ratios and the internal electric field experience by the emitters. In short, the internal electric field is high at the beginning, but it rapidly decreases, reaching its minimum value after 0.42 h. This is related to the growth of the doped regions as shown by both the reduction of the resistance from 86.8 Ω to 25.9 Ω and the increase of the capacitance C from 8.35 × 10−9 to 8.85 × 10−9 F (Table S1, ESI†). This nicely corresponds to a time required to reach the maximum in the h/l peak intensity ratio (Fig. 5). Subsequently, the neutral region slowly shrinks, leading to an increase of the internal electrical field that is also well-correlated to a slow decrease of the h/l peak intensity ratio over time (Fig. 5). Hence, we concluded that the electroluminescence behavior of the contorted HBO in LECs is ruled by a rare electric-field dependence of the intensity between excited vibrational modes without changing the nature of the emitting excited state.
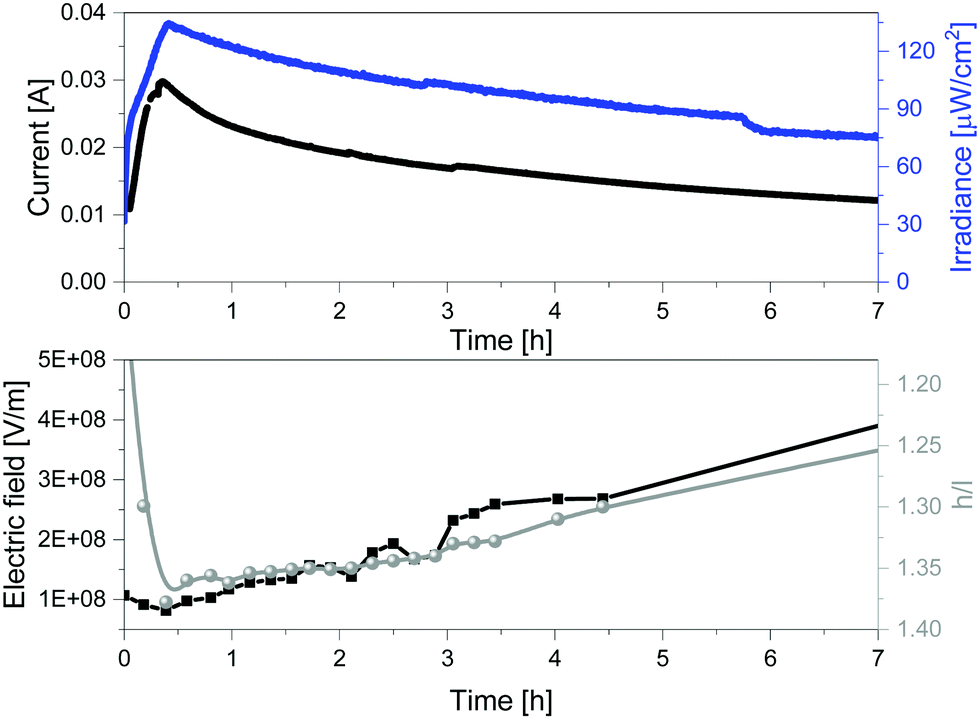 | ||
| Fig. 5 Current vs. irradiance (top) and internal electric field vs. h/l ratio (h = high energy peak, l = low energy peak) (bottom) of 1 devices driven at constant 3 V. | ||
Please notice that a possible explanation of the different chromaticity trends over time could be related to changes in the recombination dynamics moving between regions of high and low electric fields as well as changes of the average internal electric field upon changing thickness of the active layer, composition, and driving modes.
Conclusions
To sum up, the electroluminescent behavior of a representative red-emitting contorted HBO has, for the first time, been investigated using the LEC technology as a platform. Overall, this work provides two major findings. On one hand, this new family of emitters provides SM-LECs with outstanding performances, such as irradiances of ca. 220 μW cm−2 and stabilities of over 200 h in thick devices and irradiances of ca. 30 μW cm−2 and stabilities of 3600 h (measured) and 13000 h (extrapolated) in thin devices. They account for one of the most stable and bright red SM-LECs reported so far.1,26,28,29,53 On the other hand, we rationalize that the electroluminescent behavior of contorted HBO in LECs is ruled by a very rare electric-field dependence of the intensity for the radiative deactivation of the excited vibrational modes. This behavior is observed regardless of the device architecture and driving modes, while degradation, presence of aggregates, morphology changes over time, and temperature effects were excluded. It is worth mentioning that red shifted emission and broadening of the electroluminescence spectrum over time have typically been described in LECs based on iTMCs22,45 and CPs.65,66 However, they have been attributed to either a change of the nature of the excited state, or to a strong polarization as highlighted by change of the E0–0 position along with large shifts in the maximum wavelength. This contrasts to the chromaticity changes in HBO based LECs, that are ascribed to the gradual change of the internal electric field affecting the radiative rate of the excited vibrational modes without changing the nature of the excited emitting state. Finally, it is important to point out that voltage-induced changes in the electroluminescence response have also been noticed in both multi-component or multi-layered architectures.67–70 However, to the best of our knowledge, this type of electroluminescent behavior in a single-component single-layered SM-LECs has not been reported yet. Hence, this work deciphers, for the first time, the electroluminescent behavior of HBO as a new family of emitters for SM-LECs, highlighting one of the very rare examples of electric field dependent vibrational electroluminescence and one of the most efficient, stable, and bright red-emitting SM-LECs reported to date. Both aspects are of utmost relevance to design new emitters in the emerging field of SM-LECs. With this goal in mind, we illustrate the state-of-art of red-emitting LECs in Table S2 (ESI†), highlighting recent major achievements in the field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. F. and R. D. C. acknowledge the program “Ayudas para la atracción de talento investigador – Modalidad 1 of the Consejería de Educación, Juventud y Deporte—Comunidad de Madrid with the Reference No. 2016-T1/IND-1463”. R. D. C. also acknowledges the Spanish MINECO for the Ramón y Cajal program (RYC-2016-20891), the Europa Excelencia program (ERC2019-092825), and HYNANOSC (RTI2018-099504-A-C22), as well as the 2018 Leonardo Grant for Researchers and Cultural Creators from BBVA Foundation, the FOTOART-CM project funded by Madrid region under programme P2018/NMT-4367, and the Ministerio Project HYNANOSC with reference number RTI2018-099504-A-C22. K. B. and M. M. are thankful to ‘Deutsche Forschungsgemeinschaft’ supporting this project within the collaborative research center SFB 1249 “N-heteropolycyclic compounds as functional materials” (TP-A04). J. C.-G. acknowledges financial support from the Spanish Ministry of Science, Innovation and Universities (RTI2018-097508-B-I00), and from the Regional Government of Madrid (S2018/NMT4511). IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant No. SEV-2016-0686).References
- E. Fresta and R. D. Costa, J. Mater. Chem. C, 2017, 5, 5643–5675 RSC.
- J. Xu, A. Sandström, E. M. Lindh, W. Yang, S. Tang and L. Edman, ACS Appl. Mater. Interfaces, 2018, 10, 33380–33389 CrossRef CAS PubMed.
- R. D. Costa, Light-emitting electrochemical cells. Concepts, advances and challenges, Springer International Publishing, Basel, 1st edn, 2017 Search PubMed.
- S. Van Reenen, R. A. J. Janssen and M. Kemerink, Org. Electron., 2011, 12, 1746–1753 CrossRef CAS.
- S. Tang, A. Sandström, P. Lundberg, T. Lanz, C. Larsen, S. Van Reenen, M. Kemerink and L. Edman, Nat. Commun., 2017, 8, 1190 CrossRef PubMed.
- E. Fresta, G. Volpi, C. Garino, C. Barolo and R. D. Costa, Polyhedron, 2018, 140, 129–137 CrossRef.
- M. F. Aygüler, M. D. Weber, B. M. D. Puscher, D. D. Medina, P. Docampo and R. D. Costa, J. Phys. Chem. C, 2015, 119, 12047–12054 CrossRef.
- M. S. Subeesh, K. Shanmugasundaram, C. D. Sunesh, R. K. Chitumalla, J. Jang and Y. Choe, J. Phys. Chem. C, 2016, 120, 12207–12217 CrossRef CAS.
- J. Gao, Curr. Opin. Electrochem., 2018, 7, 87–94 CrossRef CAS.
- E. Fresta, G. Volpi, M. Milanesio, C. Garino, C. Barolo and R. D. Costa, Inorg. Chem., 2018, 57, 10469–10479 CrossRef CAS PubMed.
- E. Fresta, J.-M. Carbonell-Vilar, J. Yu, D. Armentano, J. Cano, M. Viciano-Chumillas and R. D. Costa, Adv. Funct. Mater., 2019, 29, 1901797 CrossRef.
- E. Fresta, M. D. Weber, J. Fernández-Cestau and R. D. Costa, Adv. Opt. Mater., 2019, 7, 1900830 CrossRef CAS.
- S. T. Parker, J. D. Slinker, M. S. Lowry, M. P. Cox, S. Bernhard and G. G. Malliaras, Chem. Mater., 2005, 17, 3187–3190 CrossRef CAS.
- S. Van Reenen, P. Matyba, A. Dzwilewski, R. A. J. Janssen, L. Edman and M. Kemerink, Adv. Funct. Mater., 2011, 21, 1795–1802 CrossRef CAS.
- J. Mindemark, S. Tang, H. Li and L. Edman, Adv. Funct. Mater., 2018, 28, 1801295 CrossRef.
- K. Y. Lin, L. D. Bastatas, K. J. Suhr, M. D. Moore, B. J. Holliday, M. Minary-Jolandan and J. D. Slinker, ACS Appl. Mater. Interfaces, 2016, 8, 16776–16782 CrossRef CAS PubMed.
- L. D. Bastatas, K. Y. Lin, M. D. Moore, K. J. Suhr, M. H. Bowler, Y. Shen, B. J. Holliday and J. D. Slinker, Langmuir, 2016, 32, 9468–9474 CrossRef CAS PubMed.
- J. K. Lee, D. S. Yoo, E. S. Handy and M. F. Rubner, Appl. Phys. Lett., 1996, 69, 1686–1688 CrossRef CAS.
- M. D. Weber, E. Fresta, M. Elie, M. E. Miehlich, J. L. Renaud, K. Meyer, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef.
- B. Pashaei, S. Karimi, H. Shahroosvand, P. Abbasi, M. Pilkington, A. Bartolotta, E. Fresta, J. Fernandez-Cestau, R. D. Costa and F. Bonaccorso, Chem. Soc. Rev., 2019, 48, 5033–5139 RSC.
- C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155–177 CrossRef CAS.
- A. F. Henwood and E. Zysman-Colman, Top. Curr. Chem., 2016, 374, 36 CrossRef PubMed.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- Z. B. Hill, D. B. Rodovsky, J. M. Leger and G. P. Bartholomew, Chem. Commun., 2008, 6594–6596 RSC.
- S. Tang, W.-Y. Tan, X.-H. Zhu and L. Edman, Chem. Commun., 2013, 49, 4926–4928 RSC.
- K. T. Weber, K. Karikis, M. D. Weber, P. B. Coto, A. Charisiadis, D. Charitaki, G. Charalambidis, P. Angaridis, A. G. Coutsolelos and R. D. Costa, Dalton Trans., 2016, 45, 13284–13288 RSC.
- M. D. Weber, V. Nikolaou, J. E. Wittmann, A. Nikolaou, P. A. Angaridis, G. Charalambidis, C. Stangel, A. Kahnt, A. G. Coutsolelos and R. D. Costa, Chem. Commun., 2016, 52, 1602–1605 RSC.
- M. D. Weber, M. Adam, R. R. Tykwinski and R. D. Costa, Adv. Funct. Mater., 2015, 25, 5066–5074 CrossRef CAS.
- A. Pertegás, D. Tordera and J. Serrano-Pérez, J. Am. Chem. Soc., 2013, 135, 18008–18011 CrossRef PubMed.
- K. Baumgärtner, A. L. Meza Chincha, A. Dreuw, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15594–15598 CrossRef PubMed.
- H. Seyler, B. Purushothaman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Pure Appl. Chem., 2012, 84, 1047–1067 CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef CAS PubMed.
- C. Nuckolls, C. Schenck, S. Xiao, M. Steigerwald, M. Ball, Y. Zhong, F. Ng and Y. Wu, Acc. Chem. Res., 2014, 48, 267–276 Search PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Chem. – Eur. J., 2015, 21, 17308–17313 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051–6056 CrossRef CAS PubMed.
- E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC.
- J. Mack, P. Vogel, D. Jones, N. Kaval and A. Sutton, Org. Biomol. Chem., 2007, 5, 2448–2452 RSC.
- J. Vollbrecht, C. Wiebeler, H. Bock, S. Schumacher and H. S. Kitzerow, J. Phys. Chem. C, 2019, 123, 4483–4492 CrossRef CAS.
- K. Baumgärtner, F. Rominger and M. Mastalerz, Chem. – Eur. J., 2018, 24, 8751–8755 CrossRef PubMed.
- A. F. Henwood and E. Zysman-Colman, Top. Curr. Chem., 2016, 374, 36 CrossRef PubMed.
- S. Tang, P. Murto, J. Wang, C. Larsen, M. R. Andersson, E. Wang and L. Edman, Adv. Opt. Mater., 2019, 1900451 CrossRef.
- Y.-M. Wang, F. Teng, Y.-B. Hou, Z. Xu, Y.-S. Wang and W.-F. Fu, Appl. Phys. Lett., 2005, 87, 233512 CrossRef.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed.
- S. B. Meier, W. Sarfert, J. M. Junquera-Hernández, M. Delgado, D. Tordera, E. Ortí, H. J. Bolink, F. Kessler, R. Scopelliti, M. Grätzel, M. K. Nazeeruddin and E. Baranoff, J. Mater. Chem. C, 2013, 1, 58–68 RSC.
- H. J. Bolink, L. Cappelli, E. Coronado, A. Parham and P. Stössel, Chem. Mater., 2006, 18, 2778–2780 CrossRef CAS.
- H. J. Bolink, L. Cappelli, S. Cheylan, E. Coronado, R. D. Costa, N. Lardiés, M. K. Nazeeruddin and E. Ortí, J. Mater. Chem., 2007, 17, 5032–5041 RSC.
- D. Asil, J. A. Foster, A. Patra, X. Dehatten, J. Delbarrio, O. A. Scherman, J. R. Nitschke and R. H. Friend, Angew. Chem., Int. Ed., 2014, 53, 8388–8391 CrossRef CAS PubMed.
- M. H. Bowler, T. Guo, L. Bastatas, M. D. Moore, A. V. Malko and J. D. Slinker, Mater. Horiz., 2017, 4, 657–664 RSC.
- H.-C. Su and C.-Y. Cheng, Isr. J. Chem., 2014, 54, 855–866 CrossRef CAS.
- G. R. Lin, H. R. Chen, H. C. Shih, J. H. Hsu, Y. Chang, C. H. Chiu, C. Y. Cheng, Y. S. Yeh, H. C. Su and K. T. Wong, Phys. Chem. Chem. Phys., 2015, 17, 6956–6962 RSC.
- N. Kaihovirta, C. Larsen and L. Edman, ACS Appl. Mater. Interfaces, 2014, 6, 2940–2947 CrossRef CAS PubMed.
- M. D. Weber, J. E. Wittmann, A. Burger, O. B. Malcioglu, J. Segarra-Martínez, A. Hirsch, P. B. Coto, M. Bockstedte and R. D. Costa, Adv. Funct. Mater., 2016, 26, 6737–6750 CrossRef CAS.
- D. R. Blasini, J. Rivnay, D. M. Smilgies, J. D. Slinker, S. Flores-Torres, H. D. Abruña and G. G. Malliaras, J. Mater. Chem., 2007, 17, 1458–1461 RSC.
- Y. J. Cho, S. Taylor and H. Aziz, ACS Appl. Mater. Interfaces, 2017, 9, 40564–40572 CrossRef CAS PubMed.
- H. Yu, Y. Zhang, Y. J. Cho and H. Aziz, ACS Appl. Mater. Interfaces, 2017, 9, 14145–14152 CrossRef CAS PubMed.
- J. Y. Li, D. Liu, C. Ma, O. Lengyel, C. S. Lee, C. H. Tung and S. Lee, Adv. Mater., 2004, 16, 1538–1541 CrossRef CAS.
- S. Yang and M. Jiang, Chem. Phys. Lett., 2009, 484, 54–58 CrossRef CAS.
- M. D. Weber, J. E. Wittmann, A. Burger, O. B. Malcıoğlu, J. Segarra-Martí, A. Hirsch, P. B. Coto, M. Bockstedte and R. D. Costa, Adv. Funct. Mater., 2016, 26, 6737–6750 CrossRef CAS.
- B. Henderson and G. F. Imbusch, Optical Spectroscopy of Inorganic Solids, Clarendon Press, Oxford, UK, 1989 Search PubMed.
- H. J. Bolink, E. Coronado, R. D. Costa, E. Ortì, M. Sessolo, S. Graber, K. Doyle, M. Neuburger, C. E. Housecroft and E. C. Constable, Adv. Mater., 2008, 20, 3910–3913 CrossRef CAS.
- H. Rudmann and M. F. Rubner, J. Appl. Phys., 2001, 90, 4338–4345 CrossRef CAS.
- G. Kalyuzhny, M. Buda, J. Mcneill, P. Barbara and A. J. Bard, J. Am. Chem. Soc., 2003, 125, 6272–6283 CrossRef CAS PubMed.
- B. M. D. Puscher, M. F. Aygueler, P. Docampo and R. D. Costa, Adv. Energy Mater., 2017, 7, 1602283 CrossRef.
- F. Habrard, T. Ouisse, O. Stéphan, L. Aubouy, P. Gerbier, L. Hirsch, N. Huby and A. Van der Lee, Synth. Met., 2006, 156, 1262–1270 CrossRef CAS.
- G. Mauthner, M. Collon, E. J. W. List, F. P. Wenzl, M. Bouguettaya and J. R. Reynolds, J. Appl. Phys., 2005, 97, 063508 CrossRef.
- A. J. Norell Bader, A. A. Ilkevich, I. V. Kosilkin and J. M. Leger, Nano Lett., 2011, 11, 461–465 CrossRef CAS PubMed.
- J. M. Leger, S. A. Carter and B. Ruhstaller, J. Appl. Phys., 2005, 98, 124907 CrossRef.
- M. L. Wu, G. Y. Chen, T. A. Shih, C. W. Lu and H. C. Su, Phys. Chem. Chem. Phys., 2018, 20, 18226–18232 RSC.
- J. Wang, S. Tang, A. Sandström and L. Edman, ACS Appl. Mater. Interfaces, 2015, 7, 2784–2789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00641a |
| This journal is © The Royal Society of Chemistry 2020 |