
Chiral gold–PPA nanocomposites with tunable helical sense and morphology†
Julián
Bergueiro‡
 ,
Manuel
Núñez-Martínez‡
,
Sandra
Arias
,
Emilio
Quiñoá
,
Ricardo
Riguera
and
Félix
Freire
*
,
Manuel
Núñez-Martínez‡
,
Sandra
Arias
,
Emilio
Quiñoá
,
Ricardo
Riguera
and
Félix
Freire
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS) and Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: felix.freire@usc.es
First published on 10th December 2019
Abstract
A novel type of stimuli-responsive dynamic helical polymer–metal nanoparticle nanocomposite formed by a helical poly(phenylacetylene) (PPA) combined with gold nanoparticles (AuNPs) is described. Thus, several PPA copolymers containing the ethynyl-4-benzamide of (S)-phenylglycine methyl ester (M1) to dictate the helical structure/sense of the copolymer, and the ethynyl-4-benzamide of the 11-((2-(2-(2-aminoethoxy)ethoxy)ethyl)amino)undecane-1-thiol (M2) to link the copolymer to the AuNPs are prepared. Different morphologies of these nanocomposites were obtained by considering the thiol ratio and the self-assembly properties of the PPA, which generates from dispersed AuNPs to fibre-like structures. All these nanocomposites show a dynamic chiral behaviour, it being possible to manipulate their helical sense by the action of external stimuli. Moreover, it is possible to control the aggregation of these nanocomposites into macroscopically chiral nanospheres with low polydispersity by using Ba2+ as a crosslinking agent.
New conceptsIn this work, we describe for the first time, how it is possible to prepare chiral dynamic plasmonic nanocomposites—composed by gold nanoparticles (AuNPs) and a thiol containing co-poly(phenylacetylene) (PPA)—where the macroscopic chiral content of the nanocomposite can be modulated resorting to external stimuli, which can affect the helical sense of the organic counterpart of the nanocomposite. We report in this manuscript the control over the morphology of the chiral nanocomposites by tuning the thiol content in the copolymer chain, which in addition to the self-assembly properties of PPAs can generate from dispersed AuNPs to fiber-like structures. The aggregation of these nanocomposites was also controlled by using metal ions as crosslinking agents. It was found that for those nanocomposites where the thiol content is low—dispersed AuNPs—it is possible to generate macroscopically chiral nanospheres, whose size can be modulated by tuning the nanocomposite/Ba2+ ratio. Moreover, these novel and unprecedented results on the dynamic behavior of chiral nanocomposites in addition to their self-assembling properties will allow novel materials to be generated with potential applications in fields such as theragnostics, diagnosis, sensing or chiral recognition among others. |
Introduction
Hybrid nanomaterials1,2 have become a very important area in nanoscience thanks to the synergistic combination of the properties of their building blocks. In this way, new nanoengineered materials with morphological diversity and potential applications in photonics,3 optics,4 asymmetric catalysis,5 selective separation,6 chiral sensing and recognition7 and drug delivery,8 have been described.Gold nanoparticles (AuNPs) have been widely employed as inorganic building blocks to form these nanocomposites. This is due to their plasmon related unique optical properties that facilitate applications like fluorescence, surface enhance Raman spectroscopy (SERS),9–11 phototransducers in photothermal therapy (PTT)12,13 and ultrasound among others. Moreover, the easy surface modification of the AuNPs with thiolated organic molecules allows their functionalization and the production of a variety of nanomaterials.14–16 As organic building blocks to form these kinds of nanocomposites, different chiral small molecules, supramolecular polymers17–20 and natural biomacromolecules such as proteins, RNA or DNA were used in combination with AuNPs.21,22 From these studies, only a very limited number of reports employed chiral synthetic polymers for this purpose.23 Moreover, in most of the AuNP based nanocomposites reported, the chiral coating of the AuNPs, is static. This means that the helix of the biomolecule (protein, DNA, polymer) constituting the coating, is not altered – inverted or enhanced – by the formation of the nanocomposite. An important breakthrough in this aspect has been recently presented by Prof. Zhao et al. They showed that the chirality of a hybrid material composed by an organogelator and AuNPs can be tuned by manipulation of the coordination of the organogelator to the Au(III) atoms, before the formation of the gold nanoparticles.24
Herein, we describe how the combination of AuNPs with dynamic helical polymers (DHP)25–28 produces new nanocomposites with tunable helical sense, and nanoaggregates with controlled size and morphology (Fig. 1a). We reasoned that the use of preformed helical thiolated copolymers in the synthesis of hybrid materials would allow us to achieve a better control over the morphology of the nanocomposites obtained. Thus, by tuning the amount of thiolated monomer within the copolymer it is possible to control the number of connecting points towards the AuNP surface. Moreover, considering the helical structure defined by the helical copolymer, it is expected that the different thiol groups point to different directions from the helix (Fig. 1b).
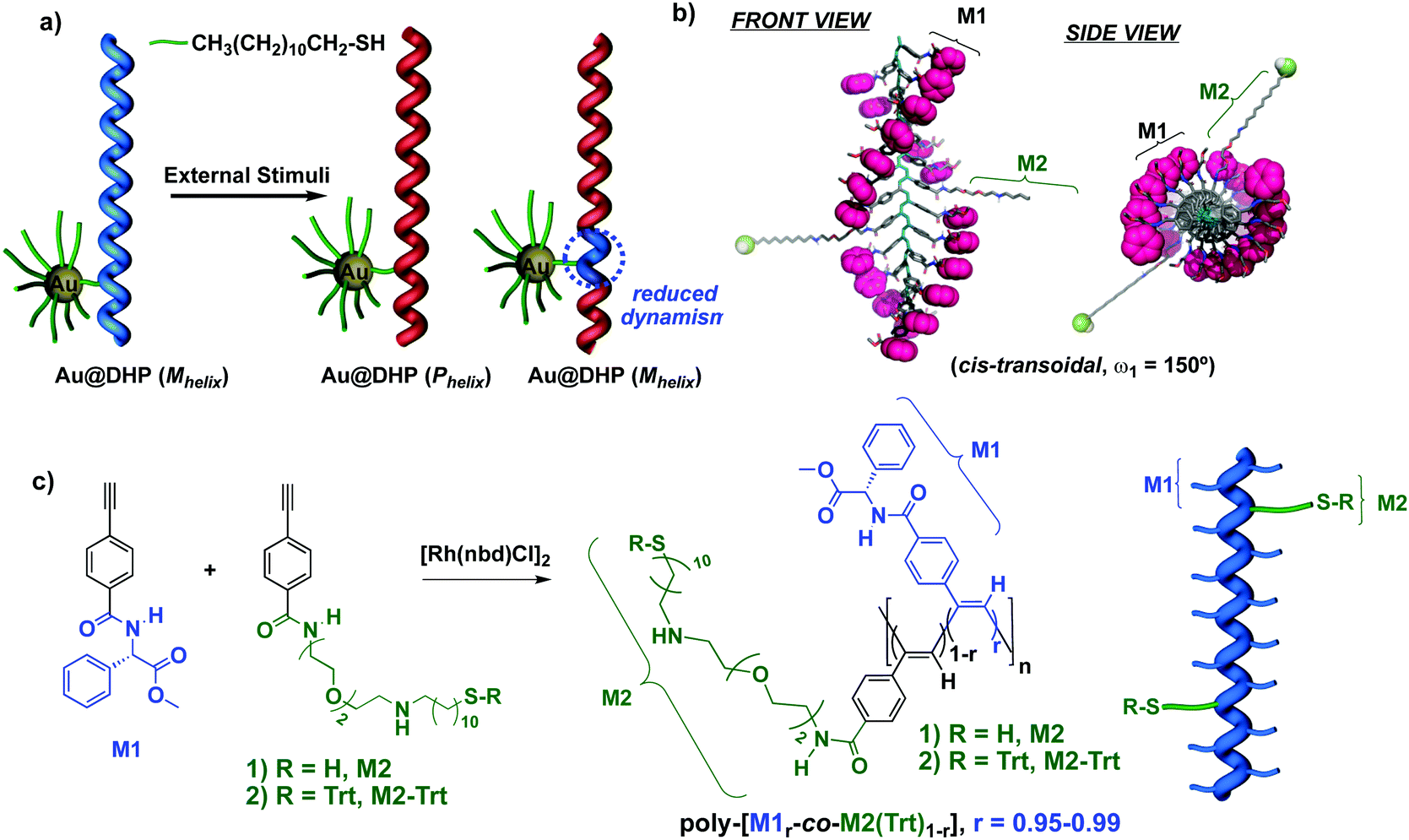 | ||
| Fig. 1 (a) Conceptual representation of Au–DHP nanocomposites with a dynamic chiral soft component. (b) Graphic illustration of the potential helical structure adopted by poly-[M1r-co-M21−r] highlighting the possible distribution of monomer 2 within the helix scaffold. (c) Synthesis and structure for the poly-(M1r-co-M21−r) copolymers series. | ||
Furthermore, the metal nanoparticles can be used to crosslink different polymer chains, a fact that is favored by the different spatial orientation of the thiol groups within the helical structure. As a result, nanocomposites with different morphologies should be obtained.
Results and discussion
As the organic component of the composite, we chose poly(phenylacetylene)s29–37 (PPA)s bearing amino acid residues as substituents. This class polymers adopts helical structures where the P/M helix is stabilized by hydrogen bonds between neighboring pendants—nth and (n + 2)th or nth and (n + 3)th—which are arranged into a β-sheet like orientation.34 Nevertheless, external stimuli that modify the conformation of the pendants or their hydrogen bonding are known to produce relevant structural changes—helix inversion and/or elongation.38–45 The preparation of the Au–PPA nanocomposite requires the presence in the PPA polymer of thiol groups necessary to link the PPA to the metal nanoparticle. Thus, we decided to use a copolymer composed by a major component designed to keep the helical structure and its response to the stimuli and another co-monomer that bears a thiol group to link the PPA to the metal nanoparticle (minor component).In this way, we selected the 4-benzamide of the phenylglycine methyl ester (M1) as the comonomer that controls the helical structure and the 4-benzamide of the 11-((2-(2-(2-aminoethoxy)ethoxy)ethyl)amino)undecane-1-thiol (M2) as the component designed to link to the gold (Fig. 1c).
The polymerization of the mixture M1–M2 in the final copolymer poly-(M1r-co-M21−r) was determined by NMR. All copolymers were fully characterized by NMR, UV, CD, IR, Raman, TGA, DSC and GPC (see the ESI†).
The concentration of thiolated groups in the copolymers was found to have a strong effect on the solubility of the material and in fact, only insoluble gels are obtained when the molar fraction of M2 is higher than 0.05. In the literature, it is found that addition of large amounts of thiolated molecules to a helical polymer presenting a polyene skeleton could result in the addition of the thiolated molecule to the polymer main chain.50 In our case, this addition could explain the crosslinking ability of the thiolated monomer and the formation of a gel when the amount of M2 is higher than 0.05.
Therefore, we selected for our study copolymers with a thiol molar fraction in the range of 0.01 to 0.05 and explored measures to avoid thiol oxidation during the synthesis and purification. The following two synthetic approaches were examined and compared:
(1) Copolymerization of M1 with the unprotected thiolated monomer M2 in the presence of TIPS-SH to avoid thiol oxidation (see the ESI† for details) (route 1).
(2) Copolymerization of M1 with the trityl protected monomer M2, followed by deprotection of the resulting copolymers with TFA/TIPS-SH (see the ESI† for details) (route 2).
Thus, copolymers poly-(M10.99-co-M20.01); poly-(M10.98-co-M20.02); poly-(M10.97-co-M20.03); poly-(M10.96-co-M20.04), and poly-(M10.95-co-M20.05) were prepared allowing us to study by CD the influence of the two different approaches and comonomers over the net PPA helicity. These studies revealed that the trityl protected copolymers (method 2) present practically the same CD as the parent homopolymer poly-M1. This fact indicates that the introduction in the main chain of the (Trt)-M2 protected monomer at low comonomer ratios does not affect the helical structure determined by the chiral phenylglycine monomer M1. In contrast, the copolymers obtained using the unprotected M2 (method 1) present with respect to poly-M1, an important helix decay of around 40–50%. This fact indicates that the introduction of the thiolated comonomer in the main chain, produces some destabilization of the helical arrangement. This helical disturbance should occur nearby the thiolated group within the copolymer chain, and therefore should depend on thiol group ratio. Nevertheless, the depletion of the helical folding is practically the same within 0.01 to 0.05 molar fraction, suggesting that perhaps the thiolated groups are not randomly distributed but somehow accumulated likewise in a block copolymer.
Alternatively, when the (Trt)-M2 protected copolymers were deprotected with TFA (1% v/v), an 80% reduction of the CD trace was observed (Fig. 2b). NEt3 and acid resin scavengers were added to the polymer solution to remove the acidic media but no changes in the CD trace were obtained (see the ESI†).
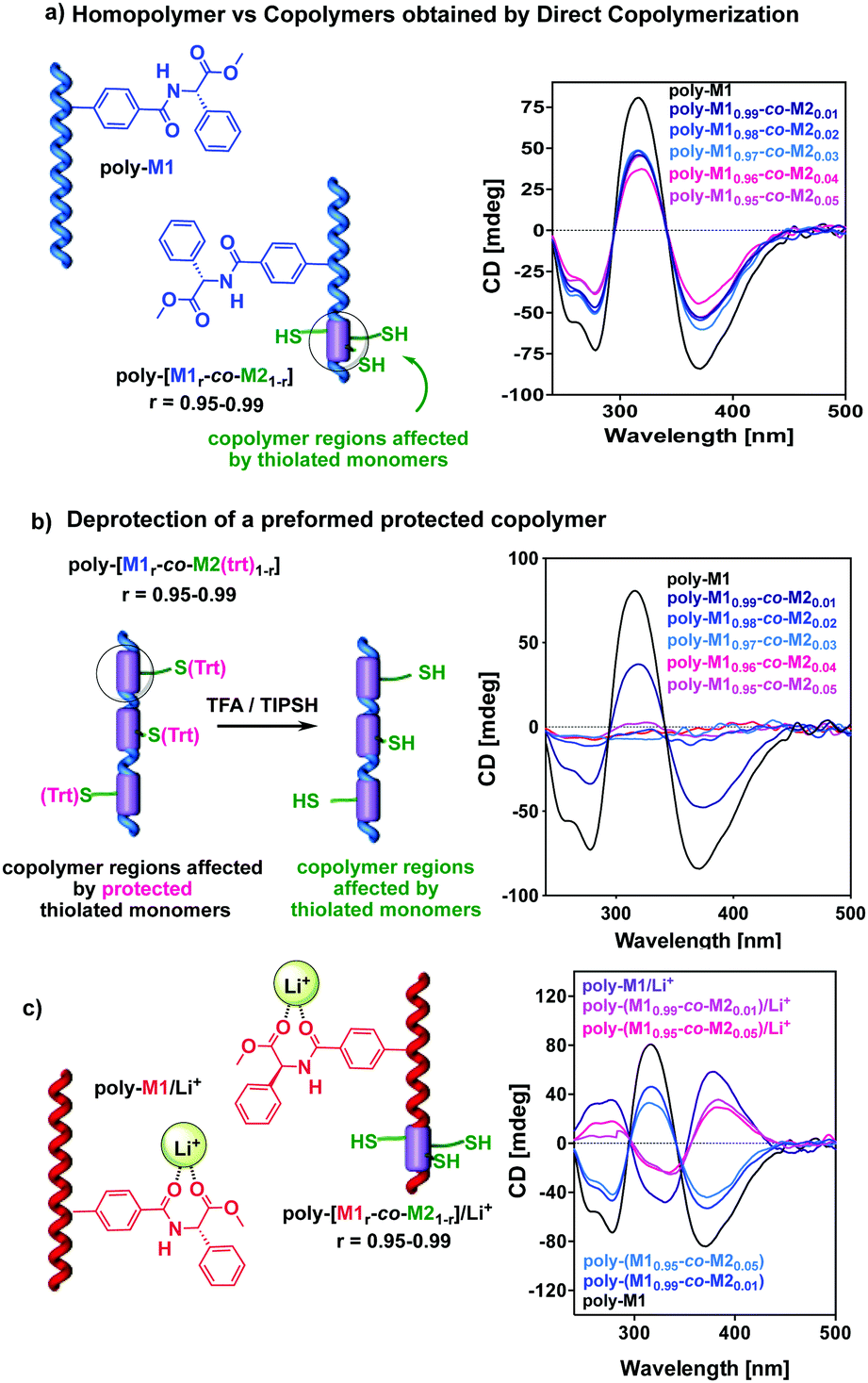 | ||
| Fig. 2 Comparative CD values for poly-M1 and poly-(M1r-co-M21−r) copolymers (r = 0.99–0.95) prepared by (a) direct synthesis or (b) by a protection/deprotection approach. (c) Dynamic behavior of poly-M1 and poly-(M1r-co-M21−r) (r = 0.99–0.95) obtained by subsequent direct copolymerization. | ||
In the literature, it is found that TFA can disrupt the H bond pattern between pendant groups; thus, we added TFA to a chloroform solution of poly-M1. Interestingly, the presence of TFA does not affect the secondary structure of poly-M1 (see the ESI†). Therefore, we assume that the factor that triggers this important loss of helical preference in the copolymer is the presence of the thiol group. In any case, this unfolding renders useless for our purpose the copolymers prepared by the protection/deprotection approach. Therefore, we centered our studies on the copolymer series obtained by direct polymerization of comonomers M1 and M2, which conserve a significant helical preference.
Structural studies—DSC, CD, UV (see the ESI†)—on the poly-(M1r-co-M21−r) (r = 0.99–0.95) series indicated that copolymers containing thiolated units conserve the helical structure commanded by monomer M1, in poly-M1 (CD (−), Mhelix), and that the folded part of the chain adopts the same cis–transoidal helical scaffold.
In addition, these copolymers respond to stimuli such as solvent polarity and Li+ ions47,48 as the parent homopolymer M1. Thus, the series poly-(M1r-co-M21−r) (r = 0.99–0.95), were shown to invert their helical sense by polarity changes of the solvent or by addition of LiClO4 (Fig. 2c and ESI†).
During these studies we found that the presence of external stimuli affects the folded fragment of the polymer chain only, producing a helix inversion or amplification proportional to the folded content of the copolymer but never reaching the levels of the parent homopolymer.
For instance, the CD spectrum of poly-(M10.99-co-M20.01) shows with respect to poly-M1, a 40% decay and conversely, when compared to the poly-M1/Li+ complex, the CD of the copolymer metal complex poly-(M10.99-co-M20.01)/Li+ suffers a 40% decay too.
Once copolymers with dynamic response to stimuli were obtained, thanks to comonomer M1, we tried to link the thiolated monomers M2 to the AuNPs. To that end, poly-(M10.99-0.95-co-M20.01-0.05) were combined with AuNPs using a variation of the Brust–Schiffrin method (BS).46
Dodecanethiol was added during the nanocomposite formation to improve the stabilization of the AuNPs (see the ESI† for experimental details). The resulting Au–poly(M1r-co-M21−r) hybrid materials were purified by precipitation, re-dispersion and centrifugation. Finally, the different products were further purified by gel permeation chromatography (GPC) (Fig. 3 and see the ESI†). These nanocomposites are very stable, and can be stored in the solid state for months, it being possible to redisperse them in solution without altering their properties.
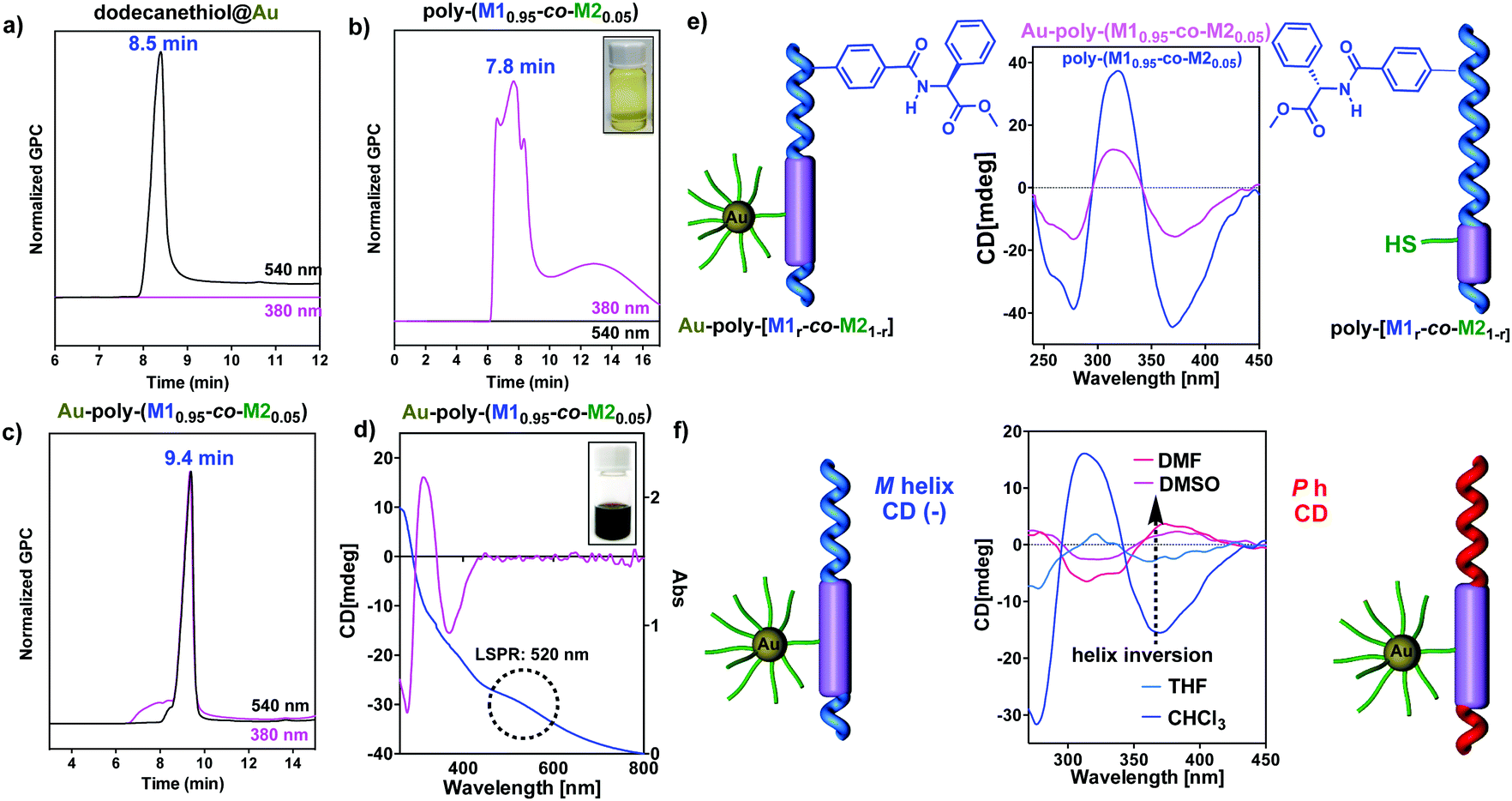 | ||
| Fig. 3 (a) GPC chromatograms of (a) AuNPs@dodecanethiol, (b) poly-(M10.95-co-M20.05), and (c) Au–poly-(M10.95-co-M20.05) at 380 and 540 nm. (d) CD and UV spectrum of Au–poly-(M10.95-co-M20.05) in CHCl3. (e) Comparison of the CD spectra for Au–poly-(M10.95-co-M20.05) and poly-(M10.95-co-M20.05). (f) Dynamic helical behaviour of Au–poly-(M10.95-co-M20.05) in polar and low-polar solvents (see the ESI† for experimental details). | ||
Fig. 3 shows the GPC chromatograms recorded at 380 (helical polymer absorption) and 540 nm (nanoparticle plasmon band) for three different THF solutions containing the copolymer (solution 1), dodecanethiol@AuNPs (solution 2) and the hybrid material—Au–poly-(M10.95-co-M20.05)—which clearly demonstrate the formation of the nanocomposites (see the ESI† for the GPC data). In accordance with that, the UV spectra of the nanocomposites (0.7 mg mL−1, CHCl3) showed the plasmon band at 520 nm indicating the presence of AuNPs (Fig. 3d), while the CD spectra show the trace typical for a helical PPA. Therefore, these results indicate that nanocomposites are chiral, showing an excess of helical sense in the polymer attached to the metal nanoparticle (Fig. 3e, f and ESI†). Interestingly, the formation of the composite is accompanied by a CD signal decay when compared to the corresponding parent copolymers (Fig. 3e and ESI†). This reduction of the helical content is not related to the synthetic procedure used to prepare the nanocomposites, which does not act on the helicity of the polymer (see the ESI†). Therefore, the helix depletion is produced by the presence of AuNPs in the material. In our view, this helical decay could be interpreted as the result of the close proximity of the AuNPs to the helical scaffold, which results in a partially distorted scaffold due to its dynamic behavior.
Stimuli responsive studies on these nanocomposites reveal that these hybrid materials are dynamic and respond to the presence of external stimuli such as polarity changes (Fig. 3f). Interestingly, in spite of the well-known response of the helix of poly-M1 and copolymers poly-(M1r-co-M21−r) to complexation with Li+ ions, the helicity of the nanocomposites is not affected by the addition of Li+ salts. The same effect was observed when Li+ was added to a mixture of poly-M1 and AuNPs@dodecanethiol, leading us to conclude that, for some reason, in the presence of AuNP the lithium ion is not able to complex with the polymer.
Finally, electron microscopy studies were carried out to study the morphology of the hybrid materials. SEM/TEM images showed that the morphology of the composite is directly related to the amount of thiolated monomer in the copolymer. In this way, while composite Au–poly-(M10.99-co-M20.01) is formed mainly by isolated nanoparticles that should be decorated with a single polymer chain (Fig. 4), other copolymers containing larger amounts of the thiolated monomer, e.g. Au–poly-(M10.97-co-M20.03), form fibers due to the presence of more than one nanoparticle per copolymer chain (Fig. 4).
 | ||
| Fig. 4 Electronic microscopy studies and graphic illustration of the nanoassemblies adopted by poly-(M1r-co-M21−r) highlighting the crosslinking ability of the AuNPs and the self-assembling properties of the PPA. | ||
Thus, the Au nanoparticles are acting as crosslinking agents, bringing together several copolymer chains, which can further self-assemble by hydrogen bond interactions between pendants (Fig. 4). This fact also explains the formation of a microgel in Au–poly-(M10.95-co-M20.05).
In a complementary assay, we tested the action of Ba2+ ions —known to induce aggregation on poly-M1—on the composites and it was found that—as before with Li+—the results differed depending on the thiolated content of the copolymer. In this way, upon addition of Ba(ClO4)2 solution, nanocomposites with low thiol content produced new materials—Au–poly-(M10.99-co-M20.01)/Ba2+ and Au–poly-(M10.98-co-M20.02)/Ba2+—easily visualized by the formation of new bigger aggregates (Fig. 4),49 shown to be nanospheres with a size that depends on the amount of thiolated group in the copolymer and barium added.
Thus, particles with an average hydrodynamic diameter size of 482 and 534 nm are obtained when Au–poly-(M10.99-co-M20.01) is treated with 0.2 and 0.4 equiv. of Ba2+ respectively, while particles with 366 and 420 nm result when 0.2 and 0.4 equiv. of Ba2+ are added to Au–poly-(M10.98-co-M20.02).
For its part, in the case of the composites with larger thiolated content, Au–poly-(M10.97-co-M20.03), Au–poly-(M10.96-co-M20.04) and Au–poly-(M10.95-co-M20.05), the addition of Ba2+ solution had no impact on the original fibers nor in the gel probably due to the already compact structure of the starting materials (see the ESI†).
Conclusions
In conclusion, we have studied the role of a macromolecular helical structure in the formation of a helical polymer–Au nanocomposite. To perform these studies a poly(phenylacetylene) bearing the benzamide of a phenylglyicine methylester (M1) as a pendant group was chosen.This selection was carried out based on our knowledge of the structure and dynamic behavior of the polymer. To form the nanocomposite, a thiolated monomer was introduced into the polymer chain. This thiolated monomer has a structural effect on the copolymer secondary structure and its dynamic behavior before and after the formation of the hybrid material.
Thus, the introduction of the thiolated comonomer in the copolymer chain destabilizes the helical arrangement of the M1 monomers placed close to this residue. Interestingly, we found that the thiol groups should be located in nearby positions, because the depletion of the helix folding it is not further affected by increasing the amount of the thiol group in a molar fraction range of 0.01–0.05.
Additionally, hybridization of these thiolated copolymers with AuNPs produces another perturbation in the polymeric secondary structure, although its dynamic behavior is not affected, it being possible to modulate the nanocomposite chirality by the action of external stimuli such as solvent polarity.
Next, we found that the morphology of the nanocomposites can be modulated through the thiolated monomer ratio within the copolymer chain. Thus, at the lowest thiolated molar fraction (0.01), the nanocomposite appears as dispersed AuNPs in electron microscopy. Interestingly, at higher molar ratios of the thiolated monomer, the controlled formation of fiber like structures is produced by combining the crosslinking ability of the AuNPs with the self-assembly properties of the copolymer.
Finally, a further controlled self-assembly of a nanocomposite was achieved by using metal ions (Ba2+) as crosslinking agents. Thus, in the case of Au–poly-(M10.98-co-M20.02) and Au–poly-(M10.99-co-M20.01) it was possible to generate macroscopically chiral nanospheres, whose size could be controlled by tuning the nanocomposite/Ba2+ ratio.
Therefore, a first insight into the control over the chiral dynamicity of plasmonic nanocomposites was demonstrated. Notably, a first controlled assembly into different nanomorphologies was achieved by considering the thiol comonomer content and the helix-to-helix supramolecular interactions. Besides, by using metal ions as crosslinking agents, some of these morphologies are further nanostructured into macroscopically chiral nanospheres with low polydispersity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Servicio de Microscopía Electrónica (RIAIDT, USC). Financial support from MINECO (CTQ2015-70519-P), Xunta de Galicia (ED431C 2018/30, Centro singular de investigación de Galicia accreditation 2016-2019, ED431G/09) and the European Regional Development Fund (ERDF) is gratefully acknowledged. M. N.-M. thanks MICINN for an FPI contract (BES-2016-078107).Notes and references
- S. Zhang, R. Geryak, J. Geldmeier, S. Kim and S. V. Tsukruk, Chem. Rev., 2017, 117, 12942 CrossRef CAS PubMed.
- M. Molina, M. Asadian-Birjand, J. Balach, J. Bergueiro, E. Miceli and M. Calderon, Chem. Soc. Rev., 2015, 17, 6161 RSC.
- G. Zheng, Z. Bao, J. Pérez-Juste, D. Ruolan, W. Liu, J. Dai, W. Zhang, L. Lee and K.-Y. Wong, Angew. Chem., Int. Ed., 2018, 57, 16452 CrossRef CAS PubMed.
- X. Lu, D.-P. Song, A. Ribbe and J. Watkings, Macromolecules, 2017, 50, 5293 CrossRef CAS.
- Y. Zhou, H. Sun, S. Matysiak, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 16791 CrossRef CAS PubMed.
- Y.-L. Lin, J. L. Chu, H. J. Lu, N. Liu and Z. Q. Wu, Macromol. Rapid Commun., 2018, 39, 1700685 CrossRef PubMed.
- D. Liu, H. Chen, J. Deng and W. Yang, J. Mater. Chem. C, 2013, 1, 8066 RSC.
- E. Dreaden, A. Alkilany, X. Huang, C. Murphy and M. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740 RSC.
- M. Fleischmann, P. Hendra and A. McQuillan, Chem. Phys. Lett., 1974, 26, 163 CrossRef CAS.
- L. Kvítek, R. Prucek, A. Panácek, A. Novotny, J. Hrbác and R. Zoboril, J. Mater. Chem., 2005, 15, 1099 RSC.
- K. Faulds, W. Ewen Smith and D. Graham, Anal. Chem., 2004, 76, 412 CrossRef CAS PubMed.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. Plaza, E. Martín-Rodriguez and G. Solé, Nanoscale, 2014, 6, 9494 RSC.
- J. Chen, C. Glaus, R. Laforest, Q. Zhang, M. Yang, M. Gidding, M. Welch and Y. Xia, Small, 2010, 6, 811 CrossRef CAS PubMed.
- A. Lee, G. Andrade, A. Ahmed, M. Souza, N. Coombs, E. Turmarkin, K. Liu, R. Gordon, A. Brolo and E. Kumacheva, J. Am. Chem. Soc., 2011, 133, 7563 CrossRef CAS PubMed.
- P. Pramod and K. Thomas, Adv. Mater., 2008, 20, 4300 CrossRef CAS.
- J. M. Abad, S. F. Mertens, M. Pita, V. M. Fernández and D. J. Schiffrin, J. Am. Chem. Soc., 2005, 127, 5689 CrossRef CAS PubMed.
- D. Tao, C. Feng, Y. Cui, I. Manners, M. Winnik and X. Huang, J. Am. Chem. Soc., 2017, 139, 7136 CrossRef CAS PubMed.
- D. Tao, C. Feng, Y. Lu, Y. Cui, X. Yang, I. Manners, M. Winnik and X. Huang, Macromolecules, 2018, 51, 2065 CrossRef CAS.
- X. Yang, J. Ruan, C. Ma, B. Hao, X. Huang, G. Lu and C. Feng, Polym. Chem., 2019, 10, 4718 RSC.
- C. Feng and X. Huang, Acc. Chem. Res., 2018, 51, 2314 CrossRef CAS PubMed.
- P. Taladriz-Blanco, N. Buurma, L. Rodríguez-Lorenzo, J. Pérez-Juste, L. Liz-Marzán and P. Hervés, Mater. Chem., 2011, 21, 16880 RSC.
- J. Ou, H. Tan, X. Chen and Z. Chen, Nanomaterials, 2018, 8, 994 CrossRef PubMed.
- C. Zhang, C. Song, W. Yang and J. Deng, Macromol. Rapid Commun., 2013, 34, 1319 CrossRef CAS PubMed.
- L. Zhu, X. Li, S. Wu, K. T. Nguyen, H. Yan, H. Ågren and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 9174 CrossRef CAS PubMed.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166 CrossRef CAS PubMed.
- E. Yashima, K. Maeda, H. Lida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed.
- J. Liu, J. W. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS PubMed.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275 CrossRef CAS PubMed.
- E. Schwartz, M. Koepf, H. J. Kito, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33 RSC.
- F. Freire, E. Quiñoá and R. Riguera, Chem. Commun., 2017, 53, 481 RSC.
- T. Van Leeuwen, H. G. Heideman, D. Zhao, S. J. Wezenberg and B. L. Feringa, Chem. Commun., 2017, 53, 6393 RSC.
- S. Wang, J. Chen, X. Feng, G. Shi, J. Zhang and X. Wan, Macromolecules, 2017, 50, 4610 CrossRef CAS.
- F. Freire, E. Quiñoá and R. Riguera, Chem. Rev., 2016, 116, 1242 CrossRef CAS PubMed.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, Adv. Polym. Sci., 2013, 262, 123 CrossRef CAS.
- E. Yashima, Polym. J., 2010, 42, 3 CrossRef CAS.
- E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3 CrossRef CAS.
- V. Percec, J. G. Rudick, M. Peterca and P. A. Heiney, J. Am. Chem. Soc., 2008, 130, 7503 CrossRef CAS PubMed.
- J. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745 CrossRef CAS PubMed.
- S. Arias, F. Freire, E. Quiñoá and R. Riguera, Polym. Chem., 2015, 6, 4725 RSC.
- S. Arias, M. Núñez-Martinez, E. Quiñoá, R. Riguera and F. Freire, Polym. Chem., 2017, 8, 3740 RSC.
- R. Rodríguez, S. Arias, E. Quiñoá, R. Riguera and F. Freire, Nanoscale, 2017, 8, 17752 RSC.
- E. Suárez-Picado, E. Quiñoá, R. Riguera and F. Freire, Chem. Mater., 2018, 30, 6908 CrossRef.
- R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, Chem. Mater., 2018, 30, 2493 CrossRef.
- S. Arias, F. Freire, M. Calderón and J. Bergueiro, Angew. Chem., Int. Ed., 2017, 56, 11420 CrossRef CAS PubMed.
- R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, J. Am. Chem. Soc., 2016, 138, 9260 CrossRef PubMed.
- M. Brust, V. Walker, D. Bethell, D. J. Schiffrin and R. J. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- M. Alzubi, S. Arias, I. louzao, E. Quiñoá, R. Riguera and F. Freire, Chem. Commun., 2017, 53, 8573 RSC.
- B. Fernandez, R. Rodríguez, A. Rizzo, E. Quiñoá, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2018, 57, 3666 CrossRef CAS PubMed.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, J. Am. Chem. Soc., 2012, 134, 19374 CrossRef CAS PubMed.
- Y. Gao, X. Wang, L. Tong, A. Qin, J. Zhi Sun and B. Zhong Tang, Polym. Chem., 2014, 5, 2309 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, names and codes, synthesis of monomers, synthesis of homo- and copolymers, thermal studies for copolymers, CD and UV-vis studies for copolymers, synthesis of gold nanoparticles, thermal studies for nanoparticles, CD and UV-vis studies for nanocomposites, microscopy studies for nanocomposites, and supporting references. See DOI: 10.1039/c9nh00659a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |