
A unique coordination-driven route for the precise nanoassembly of metal sulfides on metal–organic frameworks†
Xiang-Yu
Lin
ab,
Yue-Hua
Li
ab,
Ming-Yu
Qi
ab,
Zi-Rong
Tang
 b,
Hai-Long
Jiang
c and
Yi-Jun
Xu
*ab
b,
Hai-Long
Jiang
c and
Yi-Jun
Xu
*ab
aState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350116, P. R. China. E-mail: yjxu@fzu.edu.cn
bCollege of Chemistry, New Campus, Fuzhou University, Fuzhou, 350116, P. R. China
cHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 24th January 2020
Abstract
Incorporating different materials, such as metal sulfides, with metal–organic frameworks (MOFs) to develop MOF-based multifunctional composites with enhanced performance is an important area of research. However, the intrinsically high interfacial energy barrier significantly restricts the heterogeneous nucleation and nanoassembly of metal sulfides onto MOFs during the wet chemistry synthesis process. Herein, taking advantage of the natural tailorability of MOFs, the precise and controllable growth of metal sulfide nanoparticles (NPs) (CdS, ZnS, CuS and Ag2S) at the coordinatively unsaturated metal sites (CUSs) of MOFs to form MOF@metal sulfide composites under mild conditions is achieved via a cysteamine-assisted coordination-driven route. During the process, the molecular linker of cysteamine, possessing one amino group for chelating with the CUSs of the MOF and one thiol group as a docking site to anchor metal ions, plays a prominent role in enhancing interfacial interactions between the MOF and metal ions. The subsequent S2− anion exchange process leads to intimate surface-attached nucleation and epitaxial growth of metal sulfide NPs on the surface of the MOF. The as-formed composites exhibit enhanced charge separation and transfer capability, and thus boost photocatalytic performance. This general and simple approach provides a new avenue for the design of MOF–metal sulfide hybrids.
New conceptsAvoiding the heterophase separation originating from the intrinsically high interfacial energy barrier in two different materials such as MOFs and metal sulfides is essential for the controllable synthesis of MOF–metal sulfide hybrids at a molecular level. Previous efforts have mostly been focused on combining these two materials using external physical forces, thus inevitably leading to undesirable heterophase separation. Herein, by utilizing the inherent tailorability of the coordinatively unsaturated metal sites (CUSs) of MOFs and cysteamine as a molecular linker, we for the first time present a unique and general chemical route for the precise and controllable growth of metal sulfides on the surface of MOFs to form MOF@metal sulfide composites. The molecular linker of the cysteamine, possessing one amino group for chelating with the CUSs of the MOF and one thiol group as a docking site to anchor metal ions, acts as a lubricant in alleviating the large interfacial energy barrier between the MOF and metal sulfides. The subsequent S2− anion exchange process leads to intimate surface-attached nucleation and epitaxial growth of various metal sulfide (even Ag2S with its extremely low Ksp value) nanoparticles on the surface of MOFs. This work details a new conceptual route into the rational design of MOF–metal sulfide nanostructures based on a coordination chemistry perspective. |
Metal–organic frameworks (MOFs) are an intriguing class of porous crystalline materials comprising metal nodes bridged by multitopic organic ligands through metal–ligand coordination bonds.1–4 The microporosity and highly tailored structures of MOFs make them attractive materials to integrate with desirable materials (e.g., metal sulfides) into multifunctional systems, providing a significant route for the enhanced performance of MOF-based applications.5–15 Thus far, hydrothermal and surfactant-assisted synthesis has primarily been employed for the direct epitaxial growth of metal sulfides on MOFs. Nevertheless, the intrinsic interfacial energy barrier originating from the different anisotropic growth properties of these two functional materials still limits the precise assembly of metal sulfides on MOFs at a controllable molecular level.16,17 In principle, due to their low solubility product constant (Ksp), metal sulfide nanoparticles (NPs) tend to agglomerate during hydrothermal treatment and inevitably grow out of MOFs, leading to heterophase separation. The lack of proper sites in MOFs to dock with metal sulfides also results in inadequate surface-attached nucleation of metal sulfide NPs on the MOFs. Therefore, organic modifying agents (e.g., surfactants) are usually used to functionalize the surface of MOFs, thereby providing additional sites for the anchoring of metal precursors.18 However, the coated amount of metal sulfide NPs is restricted due to the limited capping ability of the surfactant. Besides this, excess surfactant blocks the active sites of MOFs, leading to undesirable negative effects. In this regard, alternative and efficient strategies that alleviate the large interfacial energy barrier without compromising their favorable properties are highly desirable toward the rational design of MOF–metal sulfide hybrids.
Previous studies have demonstrated that the coordinatively unsaturated metal sites (CUSs) in MOFs play key roles in their catalysis, gas separation and sorption applications.19–23 In this context, CUSs, usually generated by dislodging terminal water from a metal center, function as active Lewis acidic sites for forming strong interactions with trapped reactants. In addition, the surface chemistry of MOFs can be finely tuned by grafting different kinds of organic molecules on CUSs. However, to the best of our knowledge, there have been no examples where CUSs have been used to reduce the intrinsically high interfacial energy barrier between metal sulfides and MOFs, and to serve as docking sites for the controllable assembly of MOF@metal sulfide composites. To achieve this goal, a suitable and unique linker that bridges both the CUSs of MOFs and metal sulfides is inevitably required.
Herein, by ingeniously utilizing cysteamine as a dual-functional linker, we report a general and coordination-driven strategy for the controllable growth of metal sulfides NPs (CdS, ZnS, CuS and Ag2S) on the CUSs of the MOF to form MOF@metal sulfide composites under mild conditions (at a temperature lower than 80 °C). The molecular linker of cysteamine, possessing one amino group for chelating with the CUSs of the MOF and one thiol group as a docking site to anchor metal ions, plays a role in enhancing the interfacial interactions between the MOF and metal ions. The subsequent S2− anion exchange process leads to intimate surface-attached nucleation and epitaxial growth of metal sulfide NPs on the surface of the MOF. As a result, even for Ag2S with an extremely low Ksp value (6.3 × 10−50), excellent fine control of the hetero-epitaxial growth of Ag2S on the MOF can be realized. As a proof-of-concept application, the MIL-101@CdS composites have been used as active materials in the photoreduction of aromatic nitro compounds, which exhibit notably improved photoactivity compared to their MIL-101/CdS counterpart due to enhanced charge carrier separation and transport ability. This work is expected to make a significant contribution to the toolbox for the fabrication of MOF–semiconductor hybrid systems and inspire the development of other MOF–semiconductor composites towards various targeted functional applications.
Results and discussion
Chromium(III) terephthalate (MIL-101) was selected as an ideal model MOF for this study given its numerous potential CUSs and excellent stability,24,25 the preparation of which is illustrated in Scheme 1 (for experimental details, see the Methods section in the ESI†). MIL-101 nanoparticles, which were prepared via a hydrothermal method,19 were dehydrated at 120 °C to expose their CUSs under vacuum treatment. Subsequently, the exposed CUSs reacted with cysteamine in anhydrous ethanol, an organic molecule that strongly coordinates with different types of metal ions,26,27 to form cysteamine-grafted MIL-101 (denoted as MIL-101-S). MIL-101-S then undergone S2− anion exchange process to grow CdS NPs on MIL-101, thereby forming MIL-101@CdS composites.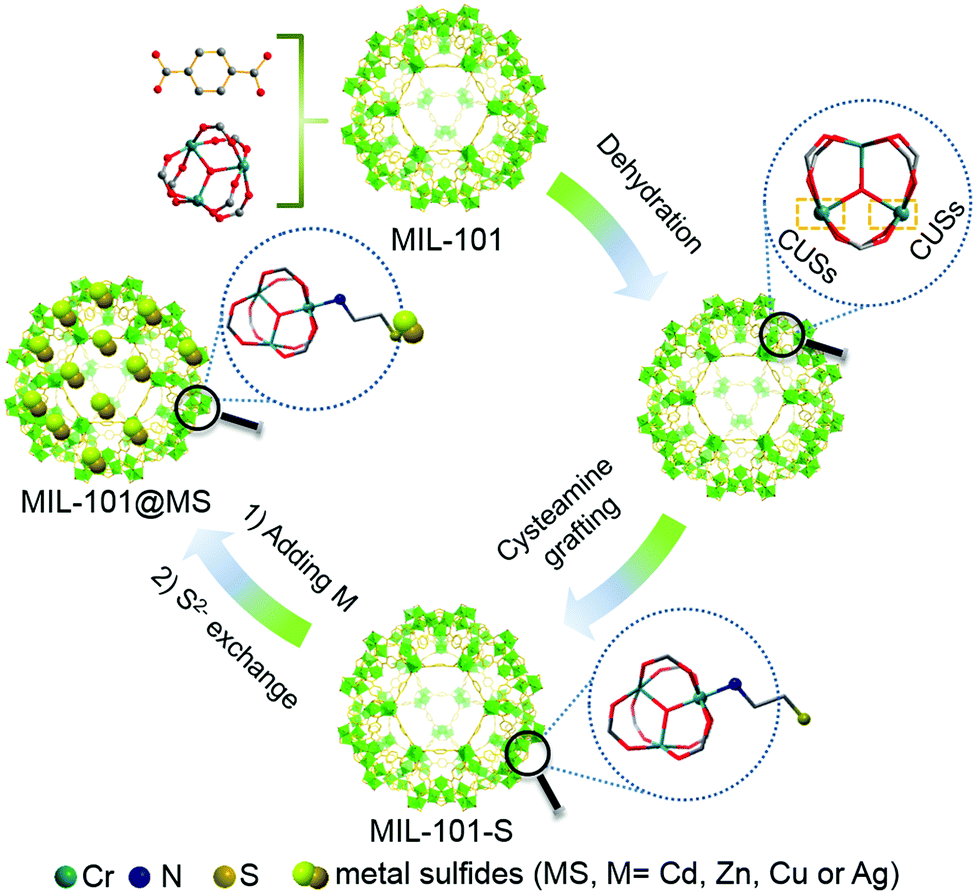 | ||
| Scheme 1 Synthesis route to MIL-101@MS composites prepared in anhydrous ethanol. The coordinatively unsaturated metal sites (CUSs) of MIL-101 are exposed by removing coordinated water in MIL-101. Color key: chromium (teal), nitrogen (purple), sulfur (dark yellow), carbon (gray), oxygen (red), hydrogen atoms are omitted for clarity. | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were employed to study the microstructures of the samples. Bare CdS particles are easily aggregated in the absence of MOF (Fig. S1, ESI†). The MIL-101 microcrystals exhibit a well-defined octahedral profile with a smooth surface (Fig. S2, ESI†). For MIL-101@CdS, small CdS NPs are dispersed evenly on the surface of MIL-101, indicating the intimate epitaxial growth of CdS NPs on MIL-101 (Fig. 1A, B and Fig. S3, ESI†). In contrast, for MIL-101/CdS prepared without cysteamine modification (Fig. 1C, D and Fig. S4, ESI†), CdS particles are severely aggregated and exhibit obvious heterophase separation from MIL-101. The different interactions between MIL-101 and CdS in these two samples were also reflected by visual observations (see the insets in Fig. 1B and C). These results indicate that cysteamine strengthens the interfacial interaction, thereby leading to surface-attached uniform nucleation of CdS NPs on MIL-101. Low-angle X-ray diffraction (XRD) analysis was used to confirm the well-retained crystallinity of the MIL-101 during the synthesis process (Fig. 1E).
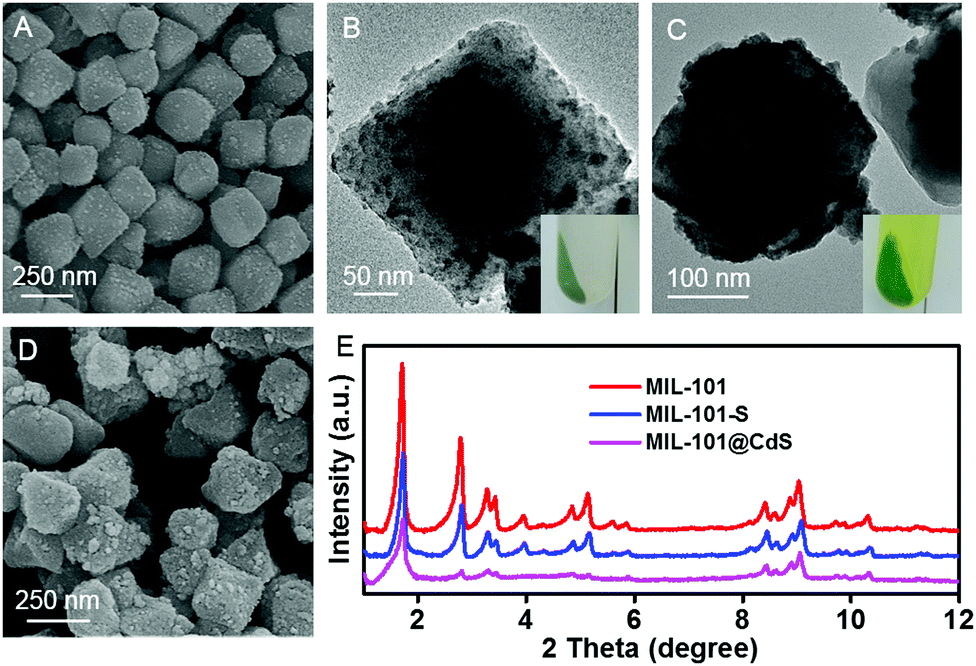 | ||
| Fig. 1 SEM and TEM images of (A and B) MIL-101@CdS, and (C and D) MIL-101/CdS. (E) Low-angle XRD patterns of MIL-101, MIL-101-S and MIL-101@CdS. The insets show the corresponding digital photos of MIL-101@CdS and MIL-101/CdS after centrifugation. | ||
Significantly, such a cysteamine enabled strategy can be well expanded by substitution of Cd2+ ions for alternative metal ions. As shown in Fig. 2, ZnS, CuS, and Ag2S NPs are uniformly distributed on the surface of MIL-101, respectively. In distinct contrast, for the composites without cysteamine grafting (Fig. 2C, F, I and Fig. S5–S7, ESI†), metal sulfide particles exhibit obvious heterophase separation from MIL-101 and an aggregated morphology, especially CuS and Ag2S, which have extremely low Ksp values. Considering that a lower Ksp value of a metal sulfide represents faster nucleation and thus harder heteroepitaxial growth on the surface of MOFs under identical conditions (Fig. 2J), it is reasonable to infer that cysteamine plays a central role in enhancing the interfacial interaction between the MOF and metal sulfides.
 | ||
| Fig. 2 SEM and TEM images of (A and B) MIL-101@ZnS, (C) MIL-101/ZnS, (D and E) MIL-101@CuS, (F) MIL-101/CuS, (G and H) MIL-101@Ag2S, and (I) MIL-101/Ag2S. (J) Table of solubility product constants of the metal sulfides. | ||
To decipher a probable formation mechanism for the MIL-101@metal sulfides, the hierarchical structure of MIL-101 containing CUSs was initially determined (Fig. S8, ESI†). It can be seen that one coordination site of the Cr3+ center is occupied by water, which is removable through thermal activation to generate CUSs. Such types of CUSs in MIL-101 can be measured by thermogravimetric analysis (Fig. S9, ESI†). The XRD analysis of the composite synthesized by replacing solvent with water (denoted as MIL-101-CdS) reveals the collapsed crystalline phase of MIL-101 (Fig. S10, ESI†), indicating that water impedes such a cysteamine-enabled assembly process and thus, both the removal of coordinated water from the CUSs of MIL-101 and the use of anhydrous ethanol as a solvent are crucial. Additionally, by swapping cysteamine for a widely-used organic modifying agent (e.g., polyvinylpyrrolidone (PVP)), the as-synthesized sample (denoted as MIL-101/CdS-PVP) exhibits obvious heterophase separation (Fig. S11, ESI†), which reflects that PVP does not have the ability to reduce the high interfacial energy barrier between MIL-101 and CdS compared to cysteamine. To further investigate the interaction between MIL-101 and cysteamine, a series of spectral measurements were carefully carried out. The UV-vis diffuse reflectance spectroscopy (DRS) of MIL-101 shows band positions at 439 and 587 nm, which can be assigned to the d–d spin-allowed transition of the Cr3+ (d5).28 After cysteamine grafting, an obvious blue shift is observed for MIL-101-S (Fig. 3A), which implies changes in the chemical environment of Cr3+ center. Such changes induced by the interaction between MIL-101 and cysteamine were further confirmed from the Raman spectra. The vibrations of MIL-101-S are shifted towards lower wavenumbers at 1606, 1436, 1132, 862 cm−1 from the MIL-101 peaks located at 1613, 1446, 1141 and 869 cm−1 (Fig. 3B), reflecting the geometry change around the metal center caused by the interplay between the cysteamine and the CUSs of the MIL-101.29,30 In the high-resolution X-ray photoelectron (XPS) Cr 2p spectra of MIL-101, two typical peaks located at approximately 577.1 and 586.8 eV can be assigned to Cr 2p3/2 and Cr 2p1/2, respectively.31 After the grafting of cysteamine, the Cr 2p peaks of MIL-101-S slightly shift toward a lower binding energy (576.8 eV for the Cr 2p3/2 and 586.6 eV for the Cr 2p1/2) (Fig. 3C), signifying an increase in the electron density of Cr3+, which can be attributed to the interaction between the N atom of cysteamine and the Cr atom of the CUSs.23,32 The grafting progress is further supported by Fourier-transform infrared spectroscopy (FT-IR) and survey XPS results (Fig. S12 and S13, ESI†). Therefore, it is reasonable to infer that cysteamine has been grafted on the CUSs of MIL-101 via a chromium–nitrogen coordinative bond during the wet chemical synthesis of MIL-101@CdS.
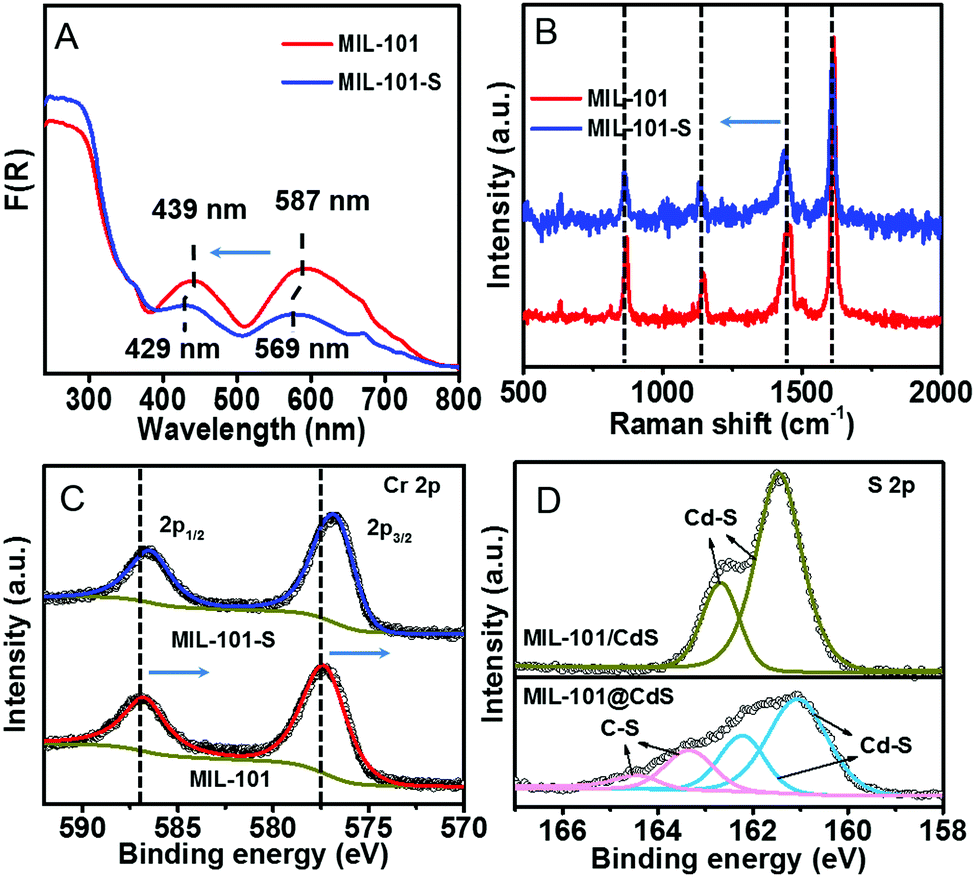 | ||
| Fig. 3 (A) DRS, (B) Raman spectra, and (C) high-resolution XPS spectra of Cr 2p of MIL-101 and MIL-101-S, respectively. (D) High-resolution XPS spectra of S 2p of MIL-101/CdS and MIL-101@CdS. | ||
The S 2p XPS spectra can further help to understand the effect of cysteamine grafting on the nucleation of CdS NPs on the surface of MIL-101-S. For MIL-101@CdS, four strong peaks with binding energies of 161.05, 162.22, 163.31 and 164.46 eV, corresponding to S 2p3/2 and 2p1/2, are assigned to the hybrid chemical bond species of Cd–S and C–S (Fig. 3D), respectively.33,34 In contrast, the high-resolution S 2p XPS spectra of MIL-101/CdS only display two peaks with binding energies of 161.40 and 162.66 eV, which can be ascribed to Cd–S. These results indicate that the thiol group of the cysteamine participates in the nucleation of CdS NPs during the synthesis process. Time-dependent monitoring of the heterogeneous nucleation of CdS on MIL-101 further indicates that cysteamine significantly reduces the nucleation rate of CdS to form MIL-101@CdS (Fig. 4A), which is significantly different from that in the absence of cysteamine (Fig. S14, ESI†). Combined with the above analyses, we thus propose a S2− anion exchange process for the heterogeneous nucleation of CdS NPs on MIL-101-S:
| Cd2+ + 2RSH → Cd(SR)2 + 2H+ | (1) |
| xS2− + mCd(SR)2 → [CdS]x[Cd(SR)2]m−x + 2xSR− | (2) |
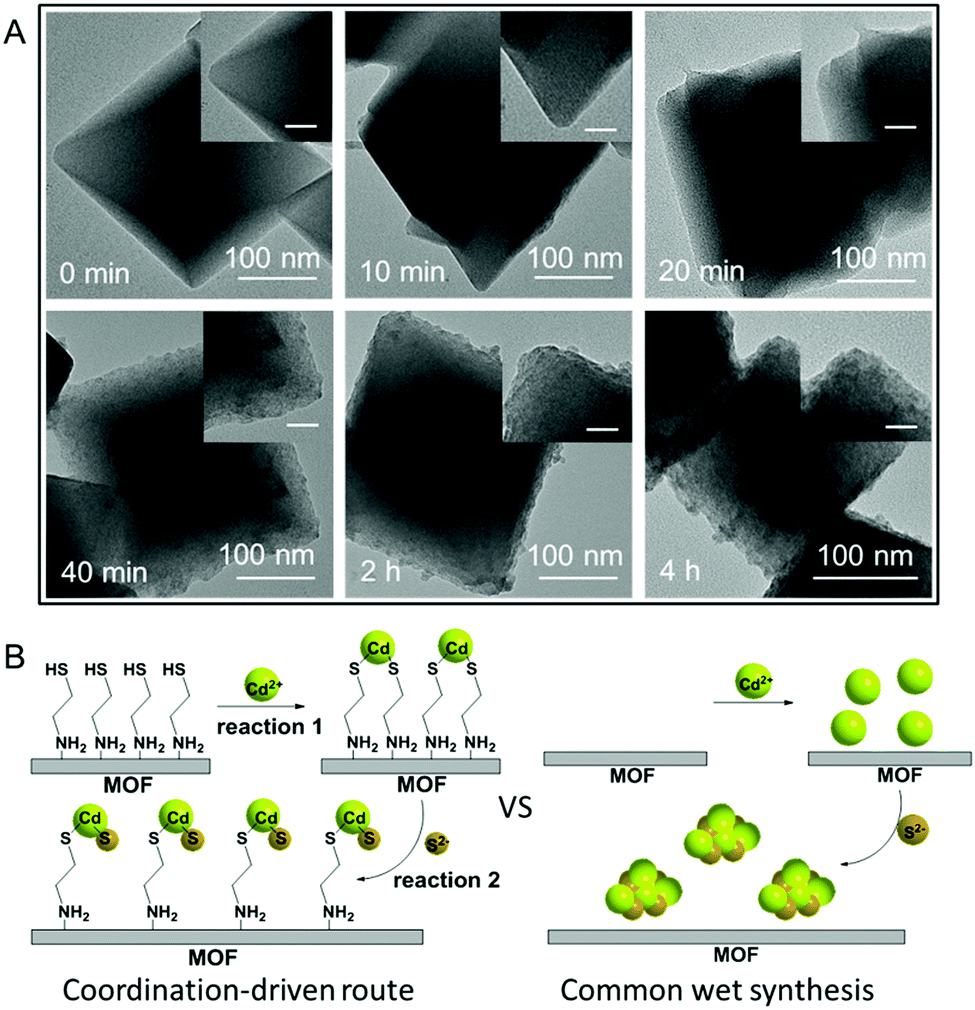 | ||
| Fig. 4 (A) Time-dependent monitoring of the heterogeneous nucleation of CdS on MIL-101-S. The scale bars in the inset images are 40 nm. (B) Schematic illustration for the formation of CdS NPs on the MOF via a coordination-driven route and common wet synthesis. The S2− anion exchange process involving reactions 1 and 2 significantly reduces the high interfacial energy barrier and thus helps the surface-attached nucleation of CdS NPs on the MOF in the coordination-driven synthesis of MIL-101@CdS. | ||
As thiols strongly bind to cadmium ions (which is reflected in the Ksp values of CdS to some extent),35,36 the exposed sulfhydryl groups on MIL-101-S firmly grab cadmium ions in solution, resulting in the formation of Cd(SR)2, where the R group is the residual part of the cysteamine anchored on MIL-101 (reaction (1)). Following the introduction of a S source, the Cd(SR)2 then undergoes S2− anion exchange to form [CdS]x[Cd(SR)2]m−x (reaction (2)), finally leading to the nucleation of CdS NPs on the surface of MIL-101 (Fig. 4B). To verify this, experiments on decreasing the ratio of S2− to cysteamine were carried out. Since an excess amount of cysteamine competes with S2− for Cd2+ to form more Cd(SR)2, the equilibrium of reaction (2) moves to the left, thereby leading to a decrease in the growth rate of CdS with the formation of smaller CdS NPs on MIL-101@CdS, which is observed in the TEM analysis (Fig. S15, ESI†). Such cysteamine-assisted synthesis progress is also reflected in the N2 adsorption/desorption measurements (Fig. S16 and S17, ESI†). Notably, the thiol group also has a high affinity for Zn2+, Cu2+, and Ag+ ions.37–39 Therefore, the probable formation mechanism of MIL-101@metal sulfides is proposed: during the process, the amino group of the cysteamine first coordinates to the CUSs of MIL-101 through chromium–nitrogen coordination bonds, resulting in the formation of MIL-101-S. The thiol group of the cysteamine that is exposed on the MIL-101-S further interacts with metal ions (M = Cd, Zn, Cu or Ag) due to their high binding affinity. Following a S2− anion exchange process, metal sulfide NPs are nucleated and epitaxially grow on MIL-101 with intimate interface contact, thereby leading to MOF@metal sulfide composites.
Next, we explored the photocatalytic performance of MIL-101@CdS toward the reduction of 4-nitroaniline (4-NA) to p-phenylenediamine (PPD) under visible light (Fig. S18, ESI†). Bare MIL-101 and MIL-101-S show no photoactivity, which can be ascribed to the low efficiency of MIL-101 in terms of exciton generation and charge separation.40 Both MIL-101@CdS and MIL-101/CdS exhibit the enhanced conversion of 4-NA compared to bare CdS. In particular, MIL-101@CdS shows higher photoactivity than that of MIL-101/CdS (Fig. 5A), which has also been evidenced by the reduction of other aromatic nitro compounds with different substituent groups (Fig. S19, ESI†). Transient photocurrent measurements and electrochemical impedance spectroscopy (EIS) were carried out to probe the origin of the enhanced photoactivity over MIL-101@CdS. MIL-101@CdS displays a higher transient photocurrent response than MIL-101/CdS (Fig. 5B), demonstrating more efficient separation of photogenerated charge carriers. The EIS results of MIL-101@CdS show a depressed semicircle at high frequency compared to MIL-101/CdS, further supporting more efficient charge carrier migration (Fig. 5C). Furthermore, the lowest unoccupied molecular orbital (LUMO) of MIL-101 was determined from Mott–Schottky plots and DRS analysis (Fig. S20, ESI†). As electron flow from the conduction band (CB) of CdS to the LUMO of MIL-101 is thermodynamically feasible,40 the intimate interfacial interaction in MIL-101@CdS contributes to more efficient separation and transfer of photogenerated charge carriers than that in MIL-101/CdS, thereby contributing to enhanced photoactivity (Fig. 5D).
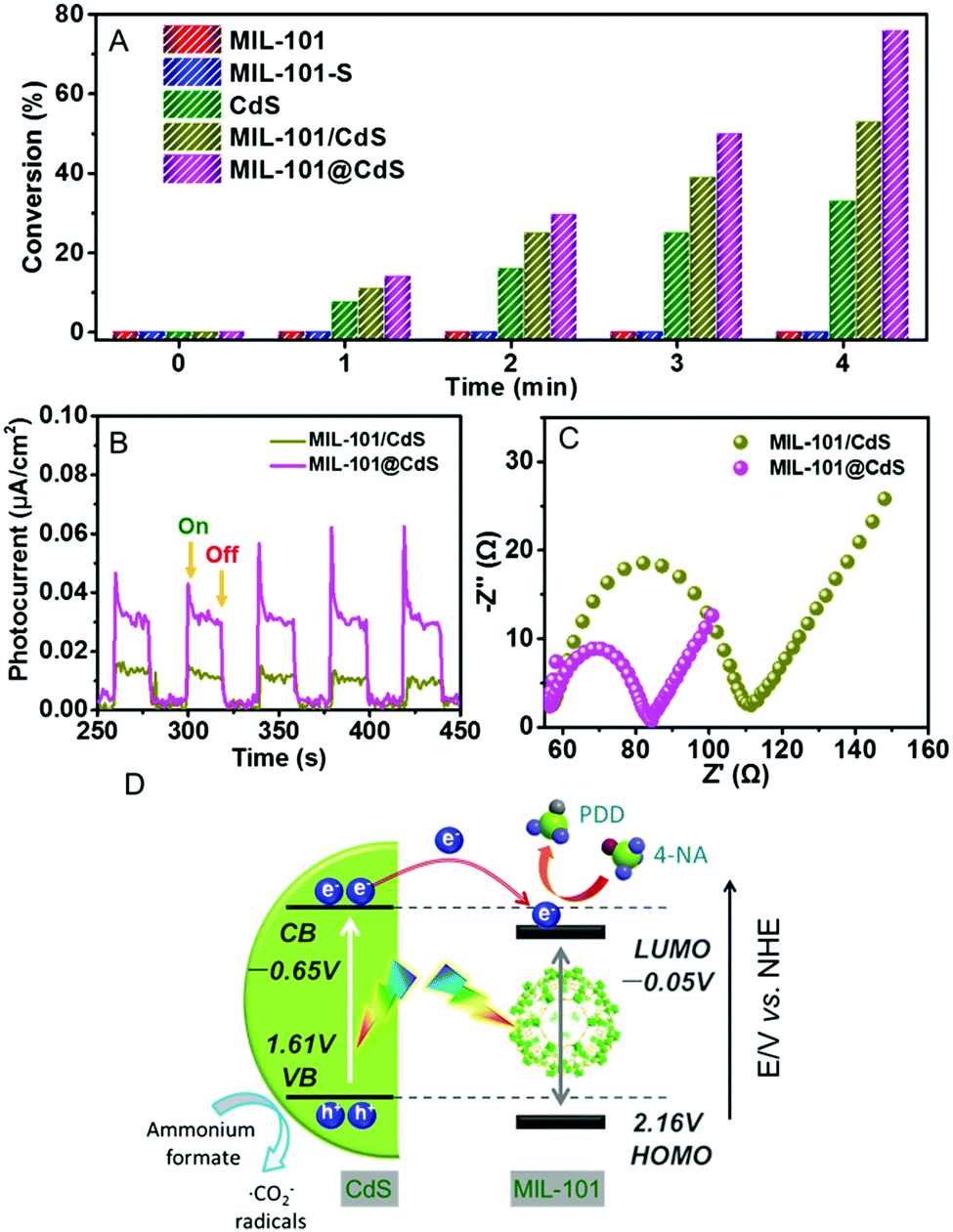 | ||
| Fig. 5 (A) Photocatalytic reduction of 4-NA under visible light irradiation within 4 min. (B) Transient photocurrent responses and (C) EIS plots of MIL-101/CdS and MIL-101@CdS. (D) Schematic illustration of the mechanism for the photocatalytic reduction of 4-NA over MIL-101@CdS. | ||
Conclusions
In summary, by combining the inherent tailorability of CUSs of MOFs with an S2− anion exchange process, we have demonstrated a cysteamine enabled strategy to strengthen the intrinsically poor interaction between a MOF (MIL-101) and metal sulfides during a wet chemical synthesis process. In this way, under mild conditions, metal sulfide NPs, even those with extremely low Ksp values, can be epitaxially and uniformly grown onto MIL-101 with intimate interfacial interactions. The as-formed composites show enhanced charge separation and transfer capability, and thus boost photocatalytic performance. This general and simple strategy opens up a new avenue for the assembly of MOF–metal sulfide hybrid systems for a broad range of applications. And, studies on more exquisite and functional structural designs toward photo- and electro-related applications are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Natural Science Foundation of China (NSFC) (21872029, U1463204, 20903023 and 21173045), the Award Program for Minjiang Scholar Professorship, the Independent Research Project of State Key Laboratory of Photocatalysis on Energy and Environment (No. 2014A05), the 1st Program of Fujian Province for Top Creative Young Talents, the NSF of Fujian Province for Distinguished Young Investigator Rolling Grant (2017J07002) and the NSF of Fujian Province (2019J0106) is gratefully acknowledged.Notes and references
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- S. Yuan, J.-S. Qin, L. Zou, Y.-P. Chen, X. Wang, Q. Zhang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6636–6642 CrossRef CAS PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- S. Jin, H.-J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 955–958 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- X. Yang, S. Yuan, L. Zou, H. Drake, Y. Zhang, J. Qin, A. Alsalme and H.-C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 3927–3932 CrossRef CAS PubMed.
- H. He, Y. Cui, B. Li, B. Wang, C. Jin, J. Yu, L. Yao, Y. Yang, B. Chen and G. Qian, Adv. Mater., 2019, 31, 1806897 Search PubMed.
- Y. Ye, W. Guo, L. Wang, Z. Li, Z. Song, J. Chen, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2017, 139, 15604–15607 CrossRef CAS PubMed.
- Q.-L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- H.-Q. Xu, S. Yang, X. Ma, J. Huang and H.-L. Jiang, ACS Catal., 2018, 8, 11615–11621 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- T. Xu, S. Wang, L. Li and X. Liu, Appl. Catal., A, 2019, 575, 132–141 CrossRef CAS.
- H. Wang, X. Liu, S. Wang and L. Li, Appl. Catal., B, 2018, 222, 209–218 CrossRef CAS.
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC.
- D. Shen, G. Wang, Z. Liu, P. Li, K. Cai, C. Cheng, Y. Shi, J.-M. Han, C.-W. Kung, X. Gong, Q.-H. Guo, H. Chen, A. C. H. Sue, Y. Y. Botros, A. Facchetti, O. K. Farha, T. J. Marks and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 11402–11407 CrossRef CAS PubMed.
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H.-L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- B. Li, Y. Zhang, D. Ma, L. Li, G. Li, G. Li, Z. Shi and S. Feng, Chem. Commun., 2012, 48, 6151–6153 RSC.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- Z. Niu, W. D. C. B. Gunatilleke, Q. Sun, P. C. Lan, J. Perman, J.-G. Ma, Y. Cheng, B. Aguila and S. Ma, Chem, 2018, 4, 2587–2599 CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- S. H. Chen and K. Kimura, Chem. Lett., 1999, 233–234 CrossRef CAS.
- L. Sheeney-Haj-Ichia, S. Pogorelova, Y. Gofer and I. Willner, Adv. Funct. Mater., 2004, 14, 416–424 CrossRef CAS.
- Y. Yang, M. A. Ratner and G. C. Schatz, J. Phys. Chem. C, 2014, 118, 29196–29208 CrossRef CAS.
- E. Elsayed, H. Wang, P. A. Anderson, R. Al-Dadah, S. Mahmoud, H. Navarro, Y. Ding and J. Bowen, Microporous Mesoporous Mater., 2017, 244, 180–191 CrossRef CAS.
- H. K. Kim, W. S. Yun, M.-B. Kim, J. Y. Kim, Y.-S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009–10015 CrossRef CAS PubMed.
- N. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2014, 8, 623–633 CrossRef CAS PubMed.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- H. B. Bao, Y. J. Gong, Z. Li and M. Y. Gao, Chem. Mater., 2004, 16, 3853–3859 CrossRef CAS.
- D. Zhao, Z. He, W. H. Chan and M. M. F. Choi, J. Phys. Chem. C, 2009, 113, 1293–1300 CrossRef CAS.
- V. Swayambunathan, D. Hayes, K. H. Schmidt, Y. X. Liao and D. Meisel, J. Am. Chem. Soc., 1990, 112, 3831–3837 CrossRef CAS.
- E. A. Forbes, A. M. Posner and J. P. Quirk, J. Soil Sci., 1976, 27, 154–166 CrossRef CAS.
- F. Karimi, H. R. Rajabi and L. Kavoshi, Ultrason. Sonochem., 2019, 57, 139–146 CrossRef CAS PubMed.
- K. Tmejova, D. Hynek, P. Kopel, S. Krizkova, I. Blazkova, L. Trnkova, V. Adam and R. Kizek, Colloids Surf., B, 2014, 117, 534–537 CrossRef CAS PubMed.
- Y. Du, B. Xu, T. Fu, M. Cai, F. Li, Y. Zhang and Q. Wang, J. Am. Chem. Soc., 2010, 132, 1470–1471 CrossRef CAS PubMed.
- Z. Jiang, J. Liu, M. Gao, X. Fan, L. Zhang and J. Zhang, Adv. Mater., 2017, 29, 1603369 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and 20 figures. See DOI: 10.1039/c9nh00769e |
| This journal is © The Royal Society of Chemistry 2020 |