
Gold nanonails for surface-enhanced infrared absorption†
Hang
Yin‡
a,
Nannan
Li‡
b,
Yubing
Si
 a,
Han
Zhang
b,
Baocheng
Yang
*a and
Jianfang
Wang
*b
a,
Han
Zhang
b,
Baocheng
Yang
*a and
Jianfang
Wang
*b
aHenan Provincial Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou, Henan 450006, China. E-mail: baochengyang@infm.hhstu.edu.cn
bDepartment of Physics, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China. E-mail: jfwang@phy.cuhk.edu.hk
First published on 15th June 2020
Abstract
Surface-enhanced infrared absorption (SEIRA) can dramatically enhance the vibrational signals of analyte molecules owing to the interaction between plasmons and molecular vibrations. It has huge potential for applications in various detection and diagnostic fields. High-aspect-ratio rod-like metal nanostructures have been the most widely studied nanomaterials for SEIRA. However, nearly all of the rod-like nanostructures reported previously are fabricated using physical methods. They suffer from damping and low areal number densities. In this work, high-aspect-ratio Au nanorods are synthesized, and Au nanonails are prepared through Au overgrowth on the as-prepared Au nanorods. The aspect ratios of the Au nanorods and nanonails can be varied in the range of ∼10 to ∼60, and their longitudinal dipolar plasmon resonance wavelengths can be correspondingly tailored from ∼1.6 to ∼8.3 μm. The Au nanonails exhibit superior SEIRA performance with 4-aminothiophenol used as the probe molecules. They are further used to detect the common biomolecule L-cysteine. Numerical simulations are further performed to understand the experimental results. They match well with the experimental observations, revealing the mechanism of the SEIRA enhancement. Our study demonstrates that colloidal high-aspect-ratio Au nanonails and nanorods can function as SEIRA nanoantennas for highly sensitive molecular detection in various situations.
New conceptsA new concept has been developed for the growth of colloidal Au nanonails for surface-enhanced infrared absorption (SEIRA) applications. The Au nanonails have two sharp tips at the ends. Their longitudinal dipolar plasmon wavelength is controlled to be located in the mid-infrared region and can be synthetically varied. The underlying growth mechanism is based on that reported widely for Au nanobipyramids, whose growth relies on the structural direction enabled by penta-twinned Au nanoparticle seeds. Similarly, the growth of the Au nanonails employs Au nanorods with five equally angularly separated twinning planes. The Au nanonails exhibiting remarkable electric field enhancement at the ends are chemically stable, uniform in length and diameter, highly flexible for dispersion in liquids and deposition on substrates, and potentially scalable in production. They offer an attractive platform for pushing forward the advancement of SEIRA, which is as important as surface-enhanced Raman scattering (SERS) in molecular detection, but has dramatically lagged behind SERS in development because of the lack of suitable plasmonic nanostructures for SEIRA. |
Introduction
Infrared (IR) spectroscopy is one of the most powerful techniques for analyzing the composition, structure and configuration of molecules. Samples in almost arbitrary states can be examined by IR spectroscopy, such as liquids, solutions, pastes, powders, films, gases and surfaces.1 IR spectroscopy is therefore widely used in health, pharmaceutical, food and forensic sciences for the analysis and identification of molecular substances.1,2 However, because of the ultrasmall molecular cross-section for IR absorption (10−20 cm2 per molecule), there are still challenging difficulties in the detection of IR absorption signals for minute amounts of molecules. Localized surface plasmon resonances (LSPRs), the collective oscillations of free charge carriers in metal or highly-doped semiconductor nanomaterials in resonance with an incident light, can provide large optical cross-sections as well as strong local field enhancements.3–5 Based on the interaction between plasmon resonances and molecular vibrations, LSPRs have made tremendous contributions in surface-enhanced spectroscopy, such as surface-enhanced Raman scattering (SERS) and surface-enhanced infrared absorption (SEIRA). SERS is a maturely developed and commonly used sensing technique and its applications cover a broad range of fields,6–8 including environmental monitoring,9 food analysis,10 chemical threat detection,11 biomedical diagnosis,12 and antique identification.13 In SEIRA, the vibrational signals of molecules can be greatly enhanced by 103–106 folds, and the detection limits of analyte molecules can be as low as the attomole or even zeptomole level correspondingly.14–18 Combining the selectivity of IR spectroscopy and the strong local field enhancement of LSPRs, SEIRA has shown huge potential for applications in various fields, such as hyperspectral infrared chemical imaging,19,20 biomolecular detection,21,22 environmental pollutant monitoring,23,24 and on-chip gas sensing.25,26SEIRA has been demonstrated with a variety of nanomaterials so far, including noble and transition metals (Ag, Au, Pt, Pd, Rh, Cu, Fe, Al, Pb),27 highly-doped semiconductors (P-doped Si,28 Si-doped InAs,29 Sn-doped indium oxide30), and graphene.31 Although doped semiconductors hold the advantages of low loss and the ability to withstand high temperatures, these materials underperform relative to noble metals in local electric field enhancement and surface plasmon propagation loss reduction.32 In terms of shape, nanostructures with various shapes have been employed for SEIRA, such as high-aspect-ratio nanorods and nanowires,33–38 nanocrosses,39,40 cruciform nanoapertures,41,42 split-ring43,44 and fan-shaped nanostructures,45 and log-periodic trapezoidal46 and dendritic nanomaterials.47 Among them, high-aspect-ratio rod-like nanostructures are probably the most widely used nanomaterials over the last decade for SEIRA. The longitudinal dipolar LSPR peak of rod-like metal nanostructures can be tailored from the visible to IR region by simply adjusting their aspect ratio.48 Such linear nanostructures feature pronounced local electric field enhancement and enhanced IR vibrational signals at the tips or in the gap regions.33–38 Metal nanostructures fabricated by physical methods possess well-designed dimensions, shapes and geometrical arrangements, while they usually suffer from large damping and inferior plasmonic properties due to their polycrystalline nature and the use of an adhesive layer.49 In addition, the SEIRA measurements on the nanoantennas fabricated by physical methods are usually carried on individual or only a small number of nanoparticles, which usually results in low signal-to-noise ratios and hampers the detection of analyte molecules. Lithographically fabricated nanostructures are therefore highly preferred for constructing optical metasurfaces. On the other hand, wet-chemistry methods provide a cheap and facile alternative for producing rod-like nanocrystals. Colloidal nanocrystals synthesized by wet-chemistry methods possess several promising features. They can be facilely prepared at large scale as well as low cost and show prominent plasmonic performances owing to their single-crystalline nature.48 In addition, colloidal metal nanocrystals can be deposited on arbitrary substrates without the use of an adhesive layer. They can be transferred into solutions and liquids easily, and show great potential in biological and medical sciences.50 However, the advancement of rod-like metal nanostructures has been slow until very recently,51 as it is hindered by the difficulties in synthesizing highly elongated nanocrystals with well-controlled dimensions, shapes and chemical compositions. Experimental realization of such elongated nanocrystals with well-defined and uniform shapes is still strongly desired.
Silver nanorods were the first demonstration of chemically synthesized metal nanocrystals with a tailorable longitudinal dipolar plasmon resonance in the near-infrared (NIR) and mid-infrared (MIR) regions in our previous work, where their application for SEIRA after coating with silica has also been demonstrated.52 Compared to Ag nanorods, Au nanocrystals used as SEIRA nanoantennas possess several advantages. Silver is well-known to be chemically unstable under many environmental conditions and is oxidized easily.53 Gold nanoparticles are much more stable than Ag ones, so their surfactant molecules can be removed completely through harsh treatment. The elimination of surfactant molecules facilitates the attachment of probe molecules, and the silica coating of nanostructures is unnecessary. Gold nanomaterials are more bio-friendly and can be transferred into tissues without harming cells.54 Moreover, Au nanorods with thin diameters and sharp tips are expected to exhibit large electric field enhancement and therefore are beneficial for SEIRA applications. Furthermore, a previous report shows that porous Au nanowires with a rough surface produce better SEIRA performance than smooth ones.38 We therefore believe that the introduction of surface protrusions on Au nanocrystals can produce more surface sites with large electric field enhancement. In terms of the metal, the dielectric function of Au follows a nearly perfect Drude-type behaviour in the IR region. Although Ag exhibits a lower intrinsic damping than Au, the difference is very small in the IR region.55 As a result, Au nanocrystals are therefore much more attractive for acting as SEIRA nanoantennas than Ag nanorods owing to their stability, biocompatibility and stronger local field enhancement.
Herein we report on the synthesis of high-aspect-ratio Au nanorods and Au nanonails, with the latter possessing sharp tips. High-aspect-ratio penta-twinned Au nanorods are synthesized and purified through a three-step seed-mediated growth method, which is modified from previous works.56,57 The lengths and diameters of the Au nanorods can be controlled by adjusting the seed concentration and the pH of the growth solution. High-aspect-ratio Au nanonails are further prepared through the overgrowth of Au on the as-prepared Au nanorods. The underlying mechanism of the sharpening of the Au nanorods is similar to that of the growth of Au nanobipyramids from penta-twinned seeds. The penta-twinned Au nanorods act as the seeds for Au overgrowth to produce the Au nanonails with two sharp tips. The diameters and lengths of the Au nanonails can be adjusted by choosing differently sized Au nanorods as the seeds. The aspect ratios of the Au nanorods and nanonails can be varied in the range of ∼10 to ∼60, and their longitudinal dipolar plasmon resonance wavelengths can be correspondingly tailored from ∼1.6 to ∼8.3 μm. By employing 4-aminothiophenol (ATP) as the probe molecule, the SEIRA performances of both Au nanorods and nanonails are investigated. The Au nanonails exhibit larger SEIRA enhancement owing to their sharp tips. The Au nanonails are further used to detect the commonly seen biomolecule L-cysteine, which marks a step forward towards the biological applications of colloidal metal nanocrystals as SEIRA nanoantennas. Finite-difference time-domain (FDTD) simulations are further performed to calculate the near-field enhancements of the Au nanocrystals and the coupling of the probe molecules with the plasmonic nanoantennas. The simulation results match well with the experimental observations, revealing the mechanism of the SEIRA enhancement. To our knowledge, the demonstration of colloidal Au nanocrystals synthesized by wet-chemistry methods as SEIRA nanoantennas has never been reported before. These colloidal Au nanocrystals are ideal candidates for SEIRA, because they are IR-responsive at both ensemble and single-particle levels, facilitating ultrasensitive molecular detection by SEIRA.
Results and discussion
High-aspect-ratio gold nanorods
The high-aspect-ratio Au nanorods were synthesized and purified through a three-step seed-mediated growth method.56,57 The directly grown products are a mixture of three types of Au nanoparticles, including spherical nanoparticles (∼50 nm), multi-faceted nanoplates (∼100–150 nm), and high-aspect-ratio nanorods. All of the nanoparticles in the solutions are stabilized by a bilayer of cetyltrimethylammonium bromide (CTAB) molecules. To separate the high-aspect-ratio Au nanorods from the other nanoparticles, the mixture solution was first left undisturbed overnight. The spherical nanoparticles have much higher dispersibility due to their smaller depletion force and remain in the supernatant indefinitely, while the Au nanorods and nanoplates slowly precipitate (Fig. S1 and S2, ESI†). The supernatant containing nearly pure nanospheres was removed, and the precipitate was redispersed in CTAB solution. Upon the oxidation of Au(III), the Au nanoplates were transformed into smooth and circular nanodisks and became dispersible in the solution. The Au nanorods, however, exhibited selective oxidation, which occurred only at their two ends, where the surface density of the capping surfactant molecules is lower due to the curvature effect.58–60 The Au nanorods hence only changed slightly in length and were still heavy enough to undergo gravitational sedimentation after being left undisturbed overnight. After several cycles of the oxidation treatment, the content of impurities became less than 1%.A previous study has shown that the pH value of the growth solution can affect the equilibrium of the redox reaction, causing a drastic difference in the size of the final Au nanorod sample.61 The length and diameter of the Au nanorods can therefore be controlled by adjusting the seed concentration and the pH of the growth solution. The scanning electron microscopy (SEM) images of the Au nanorod samples prepared under different conditions are shown in Fig. 1. The detailed growth conditions are summarized in Table S1 (ESI†). The pH values of the growth solutions are 6.83 (sample 1), 5.55 (sample 2), 4.52 (samples 3 and 4), and 2.06 (samples 5–9), respectively. The yields of the Au nanorods in these purified samples can be as high as nearly 100%. Their lengths can be widely varied from ∼200 nm to ∼1400 nm. Lowering the seed concentration leads to the production of longer and thicker Au nanorods, because each nanoparticle has more Au atoms available for growth (samples 3, 4 and 5–9). On the other hand, when the seed concentration is fixed, lowering the growth rate by reducing the pH value of the growth solution leads to the production of longer and thinner Au nanorods,62 as explained as follows. The as-prepared Au nanorods possess a penta-twinned crystal structure, and are encapsulated by five {100} side facets and ten {111} end facets.57,63 The growth rate along the {111} facets is larger than along the {100} facets owing to the selective adsorption of Br− ions on the {100} facets.62 When the seed concentration is fixed (samples 1–3), lowering the pH value of the growth solution is unfavourable for the reduction of AuBr4− to Au atoms with ascorbic acid. The Au nanorods will therefore grow much more slowly. As a result, reducing the pH value of the growth solution produces longer and thinner Au nanorods. Control of these two growth conditions allows for the production of Au nanorods with different lengths and diameters. The standard deviations relative to the average lengths and diameters of the samples are both in the range from ∼6% to ∼10%, as revealed by their length and diameter distributions, respectively (Fig. S3 and S4, ESI†).
 | ||
| Fig. 1 SEM images of the purified Au nanorod samples with different dimensions. The average length and diameter of each sample are indicated above the corresponding SEM image. | ||
The longitudinal dipolar plasmon resonance peaks of the Au nanorods with such high aspect ratios are located in the IR region. The IR transmittance spectra of the Au nanorod samples were measured on a Fourier-transform infrared (FTIR) spectrometer. The FTIR measurements are within the region from 6000 cm−1 to 1200 cm−1. MgF2 substrate was chosen for the FTIR measurements because it is optically transparent in this spectral region, and its refractive index is relatively low (∼1.38). The effect of the substrate on the plasmon resonance of the samples can be ignored due to the relatively low refractive index. As shown in Fig. 2a, as the average lengths of the Au nanorod samples are increased from 229 nm to 1388 nm, the longitudinal dipolar plasmon resonance peaks redshift from ∼6000 cm−1 to ∼1200 cm−1. The extinction spectra were further obtained by converting the transmittance spectra according to the relationship E = −log(T), where E and T stand for the extinction and transmittance values, respectively. The longitudinal dipolar plasmon wavelengths of the Au nanorod samples vary from ∼1.6 μm to ∼8.3 μm (Fig. 2b), covering a part of the “chemical fingerprints” of molecular vibrations in the range14 of ∼1500–500 cm−1. The dependence of the wavelength and wavenumber of the peak position on the aspect ratio are also plotted (Fig. 2c). The relationship between the longitudinal dipolar plasmon resonance wavelength and the aspect ratio of high-aspect-ratio metal nanostructures is nonlinear, as reported in previous works.64,65 The aspect ratio can be synthetically changed from ∼12 to ∼57.
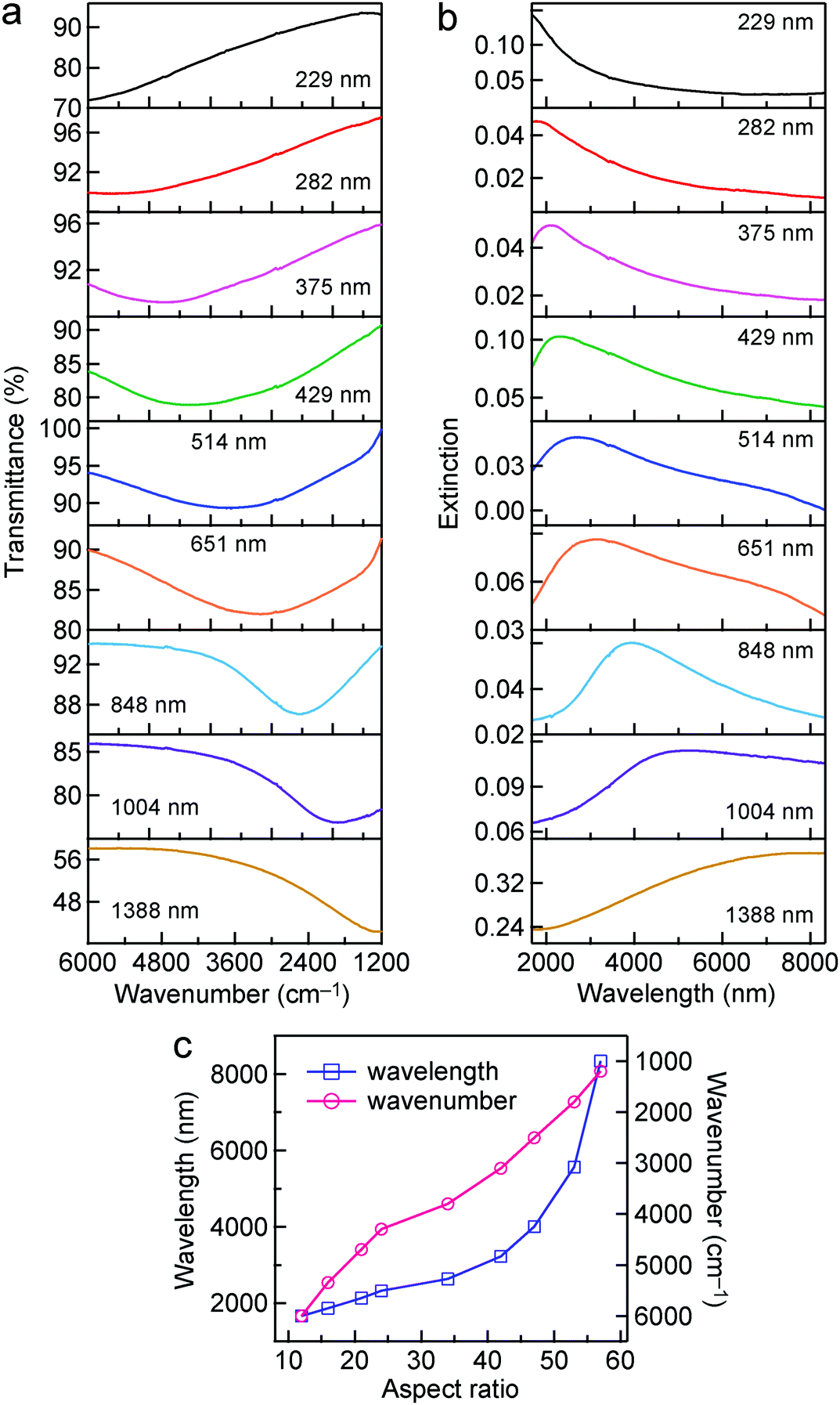 | ||
| Fig. 2 Transmittance and extinction spectra of the Au nanorod samples. (a) Transmittance spectra. (b) Extinction spectra converted from the corresponding transmittance spectra. The average length of each sample is indicated in the plot of its transmittance and extinction spectra. (c) Variations of the wavelength and wavenumber of the peak position as functions of the aspect ratio. | ||
High-aspect-ratio gold nanonails
The high-aspect-ratio Au nanonails were further grown from the as-prepared Au nanorods. Au(III) was reduced by ascorbic acid in aqueous solutions, and the Au nanorods were added into the growth solution for overgrowth. The idea of the preparation of the Au nanonails from the Au nanorods was inspired by the growth mechanism of Au nanobipyramids from penta-twinned seeds. The seed-mediated growth of Au nanocrystals has been well-known to be strongly dependent on the initial structure of the Au seeds. Previous studies have demonstrated that the single-crystalline Au seeds, which are usually stabilized by CTAB, grow into single-crystalline nanorods in the presence of Ag(I).62 In the same growth solution, however, the multiply twinned seeds stabilized by citrate grow into penta-twinned Au nanobipyramids.62 The as-prepared Au nanorods possess a penta-twinned crystal structure and are encapsulated by five {100} side facets and ten {111} end facets.57,63 The overgrowth of the penta-twinned Au nanorods can follow two possible routes, along the 〈110〉 direction that is parallel to the longitudinal axis of the nanorod and along the 〈112〉 direction that is across the side facets (transverse axis) of the nanorod.63 The overgrowth along the 〈110〉 and 〈112〉 directions produces sharp tips and wavelike rough surfaces, respectively. The concentrations of AgNO3 and CTAB in the growth solution play an important role in the final morphology of the Au nanonails. The Au nanonail samples with different aspect ratios prepared in this way are shown in Fig. 3. The average lengths and diameters of the Au nanonail samples are slightly larger than those of the original corresponding Au nanorod samples, respectively. In addition, the resultant Au nanonails possess an increased roughness on the side facets along the entire length and two sharpened needlelike tips at the ends. The dimensions and aspect ratios of the Au nanonails and nanorods are summarized in Table S2 (ESI†). The increased roughness with wavy features can be clearly observed using the transmission electron microscope (TEM) images (Fig. S5, ESI†). The Au nanonails were also characterized by using high-resolution transmission electron microscopy (HRTEM) at the tip and side facet regions (Fig. S6, ESI†). The observed spacing between the fringes at the two regions is 0.224 nm and 0.220 nm, corresponding to the spacing of the Au(111) and Au(200) lattice planes, respectively, which is consistent with previous reports.66 The detailed length and diameter distributions of the Au nanonail samples are provided in Fig. S7 and S8 (ESI†). The Au nanonails exhibit narrow distributions in both length and diameter. The IR transmittance spectra of the Au nanonail samples were acquired in the same way as the Au nanorod ones. The plasmon resonance wavelength of the Au nanonails can be tailored from ∼6000 cm−1 to 1500 cm−1 as their average aspect ratio is increased, providing another type of high-aspect-ratio Au nanocrystal with tunable plasmon resonance in the NIR and MIR regions (Fig. S9, ESI†). Their extinction spectra are also plotted for comparison. | ||
| Fig. 3 SEM images of the Au nanonail samples with different dimensions. The average length and diameter of each sample are indicated above the corresponding SEM image. | ||
SEIRA performance of the Au nanonails and nanorods
Thiol molecules are commonly used as probe molecules in SEIRA studies, because they can adsorb on the surface of Au nanostructures easily through the formation of Au–S bonds to form a self-assembled monolayer. For example, long-chain octadecanethiol molecules with their dominant –CH2– stretching mode near 2900 cm−1 have been mostly investigated.33,40,45 In our studies, ATP molecules were used as the probe molecules. They are one type of the most commonly studied probe molecules in SERS.67–69 The surface modification of the Au nanocrystals was carried out by dispersing the centrifuged Au nanorod and nanonail samples in the solution of ATP molecules to replace the adsorbed CTAB molecules. The Au nanocrystal solution was then centrifuged and concentrated before being dropped on a MgF2 substrate. After being dried under vacuum, the substrate was further washed by immersion in ethanol and then blown dry with N2 before the FTIR measurements were performed. The supported Au nanocrystals were stable throughout these treatments. Silica coating is not necessary, which is an advantage in comparison with the high-aspect-ratio Ag nanorods reported previously by us.52 As shown in Fig. S10 (ESI†), ATP molecules possess four vibrational modes, including the C–N stretching mode (1282 cm−1), C–C stretching mode (1422, 1491, 1591 cm−1), N–H bending mode (1618 cm−1), and N–H stretching mode (3349 cm−1).70 We focused mainly on the C–C stretching and N–H bending modes, which are dominant among all of the vibrational modes. The IR transmittance spectra measured from the ATP-decorated Au nanonails with different dimensions are shown in Fig. 4a. In comparison with the bare nanonails, the IR spectra of the ATP-decorated Au nanonails broaden and redshift, which can be ascribed to the higher refractive index of the probe molecules (∼1.66) surrounding the nanoantennas. The SEIRA performances of these nanoantennas were carefully investigated by varying the longitudinal dipolar plasmon resonance peak across the C–C stretching (∼1600 cm−1) region. The lineshape of the enhanced vibrational signals varies with the position of the plasmon resonance peak relative to that of the molecular vibrations. When the plasmon peak is far away from the molecular vibrations, the vibrational signals possess a weak and asymmetric lineshape. When the plasmon resonance frequency is higher than those of the molecular vibrations, the shape of the vibrational signals experiences falling and rising as the frequency is increased (Fig. 4a, the 2nd to 6th row). The shape of the vibrational signals undergoes a distinct change when the plasmon peak moves closer to those of the molecular vibrations (Fig. 4a, the 7th to 9th row). In particular, when the plasmon frequency is adjusted to match those of the molecular vibrations, the vibrational signals appear as anti-absorption peaks (transmission peak) in the final IR spectrum (Fig. 4a, the lowest row). In the resonant case, the vibrational signals are not observed as a direct increase in absorption, but instead as a modulation on the plasmon resonance. This can be understood using the model of the Fano formalism, where the enhancement of vibrational signals results from the constructive and destructive interferences between the broadband plasmon resonances of nanoantennas and the narrowband vibrations of probe molecules.34,71 For the ATP-decorated Au nanorods, the lineshape of the molecular vibrational signals varies from asymmetric dips to anti-absorption peaks with the change of the relative positions between the plasmon resonance and the molecular vibrations (Fig. 4b), which is similar to the case of the Au nanonails.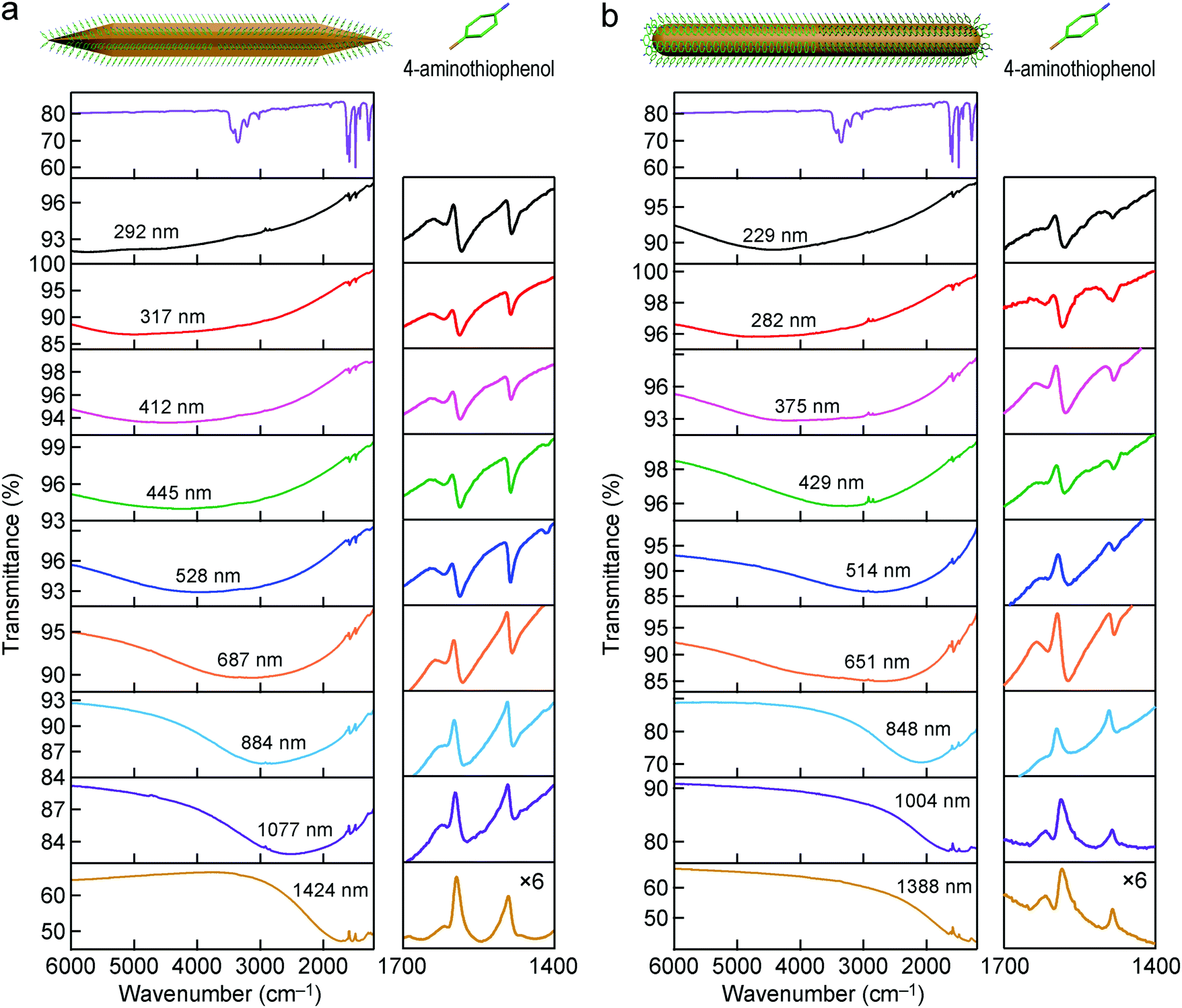 | ||
| Fig. 4 IR spectra of the Au nanoantennas decorated with ATP molecules. (a) IR transmittance spectra of the ATP molecules (top) and the nine ATP-decorated Au nanonail samples. (b) IR transmittance spectra of the ATP molecules (top) and the nine ATP-decorated Au nanorod samples. The curves in the right column for both of the nanonail and nanorod cases are zoomed-in portions around the vibrational signals (multiplied by 6 times). The average lengths of the original Au nanoantennas are indicated in the IR transmittance spectra. | ||
We focus below on the resonant cases where the plasmon resonance is tailored to spectrally match the molecular vibrations. In the resonant cases, the average lengths/diameters of the Au nanonails and nanorods are 1424 ± 104 nm/33 ± 2 nm, and 1004 ± 83 nm/18 ± 1 nm, respectively. Because only the extinction intensity is proportional to the number density of the nanoantennas, the transmittance spectra were converted into extinction ones. To reduce the experimental errors, the IR spectra of the Au nanonail and nanorod samples were each measured three times by drop-casting the ATP-adsorbed nanonail and nanorod samples on three MgF2 substrates (Fig. 5). For better comparison, the ratio of the enhanced anti-absorption signal of the probe molecules (Δh) to the extinction intensity of the nanoantennas (ΔH) was used to characterize the SEIRA performance quantitatively (Fig. S11, ESI†). The average value of the ratio (Δh/ΔH) is 0.180 ± 0.013 for the Au nanonails, which is larger by 54% than that of the Au nanorods (0.117 ± 0.008). The Au nanonails therefore exhibit stronger vibrational signals and superior SEIRA enhancement. The primary reason lies in the fact that the Au nanonails possess two sharp tips with much larger electric field enhancement, which will be further ascertained by FDTD simulations below. In addition, the nanoscale topographic features in the form of periodic ridges along the penta-facets of the Au nanonails also form extra hot-spot sites for local electric field enhancement, as demonstrated by a previous work.66 On the other hand, the needle-like sharp tips of the Au nanonails can facilitate them to penetrate into cells, which offers potential for SEIRA in biological applications.72,73
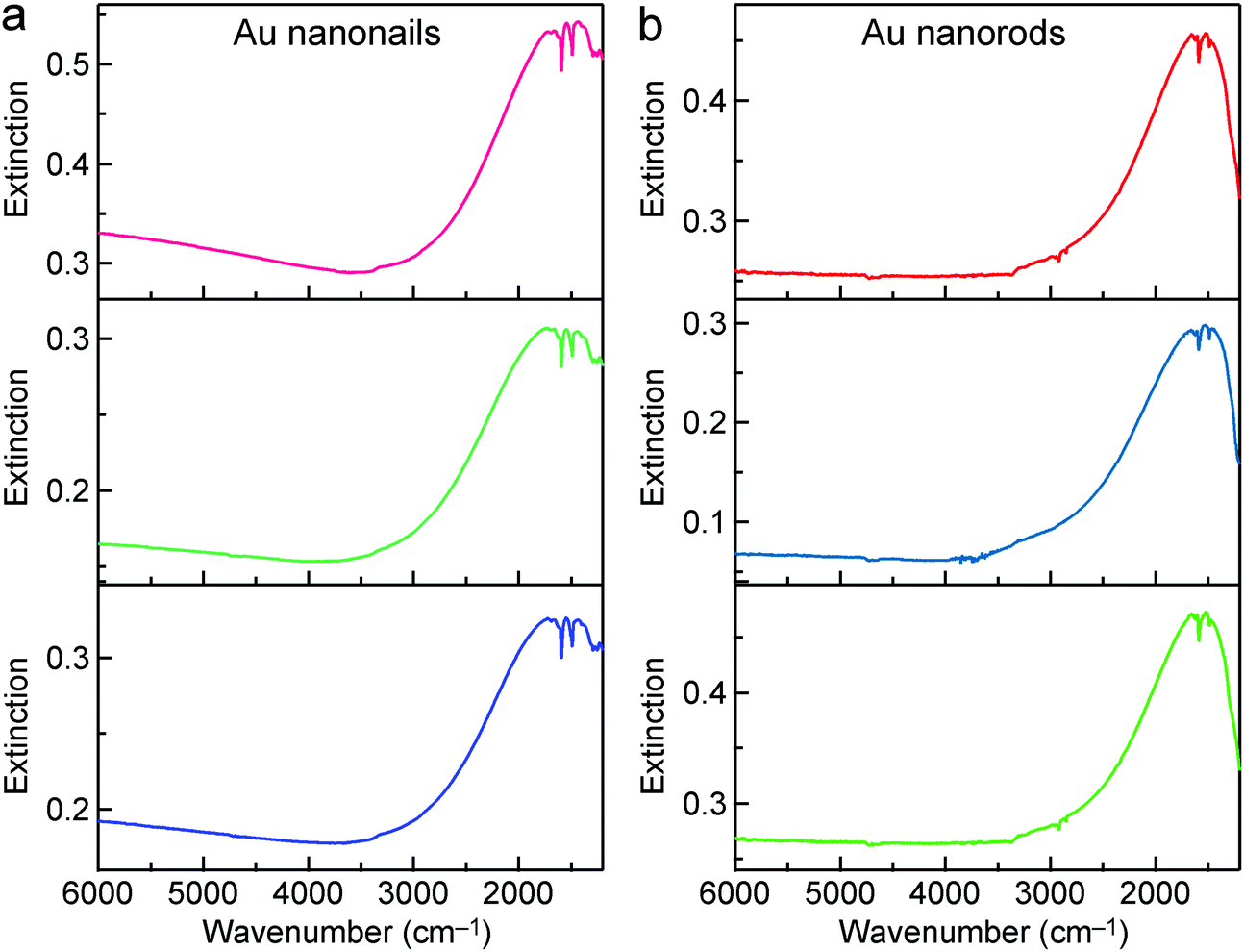 | ||
| Fig. 5 SEIRA performances of the Au nanonails and nanorods. (a) IR extinction spectra of the Au nanonail sample measured three times. (b) IR extinction spectra of the Au nanorod sample measured three times. | ||
A commonly used parameter for evaluating the SEIRA performance is the enhancement factor (EF), which relates the enhanced vibrational signal strength to that measured by the standard IR technique.33,40,52 In our study, the experimental SEIRA EF is defined as
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules) for the Au nanonail, and is 4.96 × 10−19 mol (299000 molecules) for the Au nanorod. The IR spectrum of ATP molecules at a known amount was measured and used as the reference. The EFexp values of the Au nanonails and nanorods were consequently calculated to be ∼2180 and ∼1400, respectively. The difference between the Au nanonails and nanorods is 56%. This is very close to the difference found above for Δh/ΔH.
000 molecules) for the Au nanonail, and is 4.96 × 10−19 mol (299000 molecules) for the Au nanorod. The IR spectrum of ATP molecules at a known amount was measured and used as the reference. The EFexp values of the Au nanonails and nanorods were consequently calculated to be ∼2180 and ∼1400, respectively. The difference between the Au nanonails and nanorods is 56%. This is very close to the difference found above for Δh/ΔH.
Our obtained EF values are on the same order of magnitude as the typical ones reported in previous studies,18,79 while they are smaller than the reported high EF values of 104 folds39,40 and 105 folds.33,45 One of the possible reasons for the smaller EF values in our work is that the enhanced vibrational signal mainly originates from the molecules located at the hot-spots, namely the tips of the nanonails. However, a majority of the probe molecules are located around the Au nanocrystals on the side facets, and their signals are only slightly enhanced. The averaging effect of the enhancements at the different positions results in the smaller EF values in our work. Nevertheless, the EF values are both higher than that of the Ag nanorods in our previous work (∼1020), which is ascribed to the thinner diameter of the Au nanorods and sharper tips of the Au nanonails. Thiolate polymers, such as thiol-terminated methoxy poly(ethylene glycol), can be preferentially attached to the ends of elongated Au nanocrystals.80,81 In this regard, if the probe molecules can be managed to adsorb selectively at the ends of the Au nanocrystals in the future, the EF values can be further increased. If the ends of the Au nanonails are assumed to have a penta-pyramid shape and those of the Au nanorods have a hemisphere shape, the EF values can be estimated to be ∼87000 and 31600 for the Au nanonails and nanorods, respectively, based on the surface areas of the penta-pyramids and the hemispheres. These values are actually under-estimated because maximal field enhancements are usually located in the sharpest regions for elongated plasmonic metal nanocrystals. The Au nanonails can be further used as SEIRA nanoantennas to detect other molecules, such as the commonly seen biomolecule L-cysteine. L-Cysteine is a type of amino acid containing a thiol group and has been used as a food additive. The thiol side chain in cysteine often participates in enzymatic reactions, as a nucleophile. The thiol is susceptible to be oxidized to give the disulfide derivative cystine, which plays a crucial structural role in many proteins. Analyzing the amount of L-cysteine is of great importance in biological systems. The Au nanonails with an average length/diameter of 1424 ± 104 nm/33 ± 2 nm were used to measure L-cysteine. The vibrational signal of L-cysteine at 1400 cm−1, which can be ascribed to the symmetric stretching mode of the O–C–O group,82 is enhanced and can be observed clearly in the IR spectrum (Fig. S15, ESI†). This result suggests that the Au nanonails are of huge potential for the enhancements of the vibrational signals of different molecules, which marks a step forward towards biological applications of colloidal metal nanocrystals as SEIRA nanoantennas.
FDTD-simulated SEIRA performance
The far-field optical cross-sections and near-field electric field enhancements of the Au nanorod and nanonail were further simulated using the FDTD method in order to better understand the experimental results. The refractive index of the MgF2 substrate is low (∼1.38) and the substrate was not considered in our simulations for the sake of simplicity. The lengths/diameters of the Au nanonail and nanorod were set at 1784 nm/32 nm and 1304 nm/18 nm, respectively. There was a dielectric shell with a shell thickness of 1.0 nm surrounding the Au nanonail and nanorod. The refractive index of the dielectric shell was set at 1.66, which is the same as that of ATP molecules. The diameters of the simulated Au nanonail and nanorod are the same as those of the Au nanonail and nanorod samples in the experiments, respectively. Their lengths were adjusted to make their longitudinal dipolar plasmon resonance peaks match the molecular vibrations (1555 cm−1). The lengths of the simulated nanoparticle models are slightly larger than those of the experimental samples due to the absence of the substrate in our simulations. The simulated extinction, absorption and scattering cross-sections of the Au nanonail and nanorod are plotted in Fig. S16 (ESI†). Compared to the Au nanorod, the Au nanonail possesses a larger extinction cross-section and a higher scattering-to-absorption ratio. The higher scattering-to-absorption ratio of the Au nanonail also contributes to its larger SEIRA enhancement, which has been shown in a previous report.83 The electric field is enhanced and maximized at the two ends of the Au nanocrystals and decays exponentially away from the metal surface, as shown in the electric field intensity enhancement contours (Fig. 6a). The Au nanonail exhibits larger local field enhancement than the Au nanorod due to its sharper tips. This is consistent with the knowledge that the shaper a plasmonic nanocrystal, the larger the electric field enhancement is.5 The electric field intensity enhancement (|E/E0|2) was employed to estimate the SEIRA enhancement, since the enhancement of molecular vibrational signals scales as |E|2 of the local electric field.27 The maximal field intensity enhancement factor |E/E0|2 at the two ends of the Au nanonail is 108000, which is 7 times larger than that of the Au nanorod (15000) (Fig. 6b). The Au nanonails are therefore more favourable for optical applications where large extinction cross-sections and high electric field enhancements are desired.
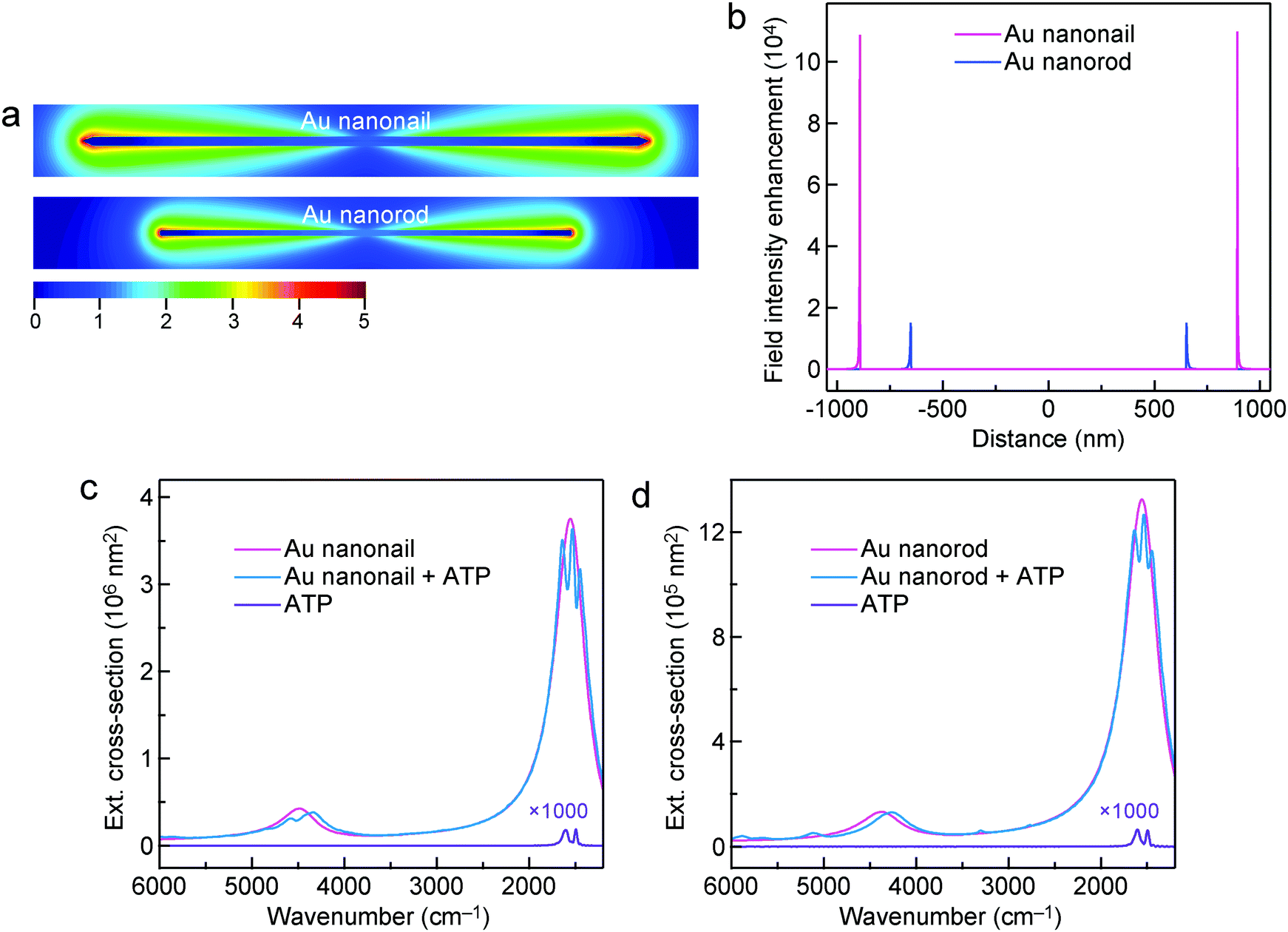 | ||
| Fig. 6 FDTD simulations. (a) Electric field enhancement (|E/E0|2) contours of the Au nanonail and nanorod under the longitudinal excitation at 1555 cm−1. The lengths/diameters of the Au nanonail and nanorod are 1784 nm/32 nm and 1304 nm/18 nm, respectively. The contours are drawn at the logarithmic scale. (b) Electric field intensity enhancement (|E/E0|2) profiles along the longitudinal direction of the Au nanonail and nanorod. The origin is at the center of the Au nanocrystals. (c) Extinction cross-sections of the Au nanonail, the Au nanonail encapsulated with ATP molecules, and the sole shell composed of ATP molecules. (d) Extinction cross-sections of the Au nanorod, the Au nanorod encapsulated with ATP molecules, and the sole shell composed of ATP molecules. | ||
The interaction between the plasmon resonance and the molecular vibration can be interpreted by introducing the probe molecules in the models of the Au nanocrystals. The dimensions of the Au nanonail and nanorod remain unchanged. The probe molecules are evenly distributed in the dielectric shell around the nanocrystals with a shell thickness of 1.0 nm. The dielectric function of the probe molecules is described by a homogeneously broadened three-oscillator Lorentzian model52,84
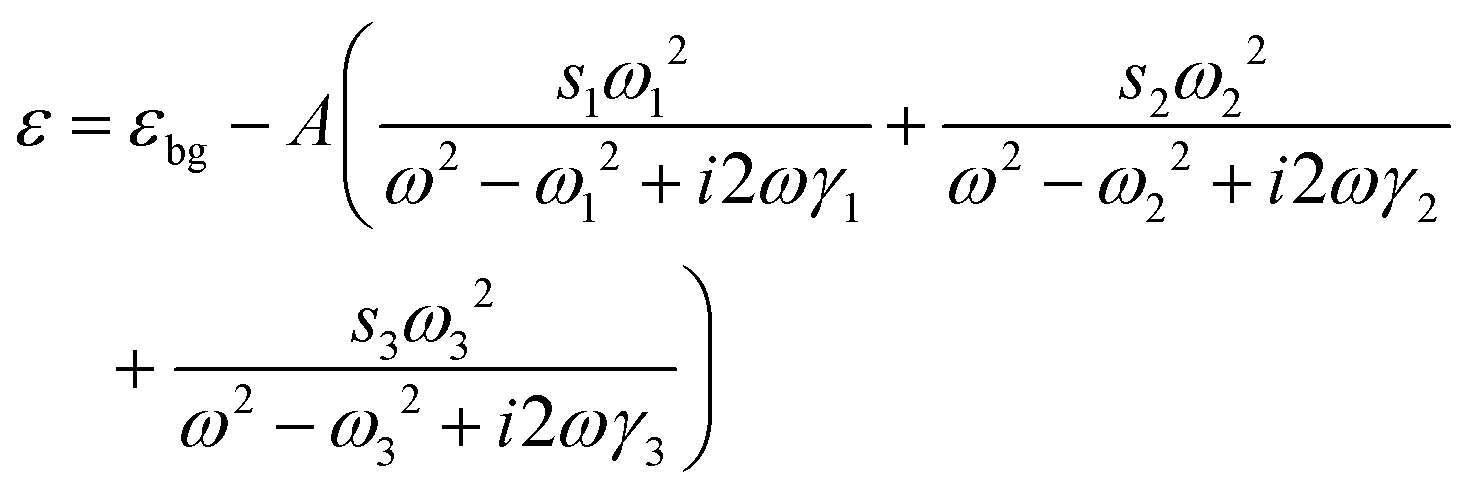 | (2) |
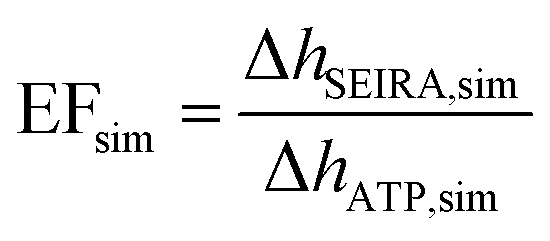 | (3) |
Conclusions
In summary, high-aspect-ratio Au nanorods with high uniformities in size and shape have been successfully prepared. Their length can be synthetically controlled from ∼200 nm to ∼1400 nm and their longitudinal plasmon resonance wavelength can be varied from the NIR to MIR region. High-aspect-ratio Au nanonails have been further prepared through Au overgrowth on the Au nanorods. Their dimensions and plasmon resonance wavelength can also be widely varied. By employing ATP as the probe molecule, SEIRA enhancements have been successfully demonstrated on the Au nanonails and nanorods. The Au nanonails exhibit better SEIRA performance owing to their sharp tips and the nearly periodic ridges along their penta-facets. The experimental SEIRA enhancement factors are 2180 and 1400 for the optimized Au nanonails and nanorods, respectively. FDTD simulations reveal that the Au nanonail possesses higher local field enhancement and superior SEIRA performance than the Au nanorod. The simulated SEIRA enhancement factors for the Au nanocrystals are higher than the experimental ones while on the same order. The Au nanonails have been further used to detect L-cysteine, which marks a step forward towards the biological applications of colloidal metal nanocrystals as SEIRA nanoantennas. Our discoveries pave the way towards the utilization of colloidal metal nanocrystals in SEIRA as well as the exploration of their applications in chemical sensing and biomolecular detection in the IR region.Experimental
Chemicals
Gold chloride trihydrate (HAuCl4·3H2O, 99%), silver nitrate (AgNO3, 99%), sodium borohydride (NaBH4, 98%), trisodium citrate (99%) and ascorbic acid (99%) were purchased from Sigma-Aldrich. CTAB (98%) was obtained from Alfa Aesar. Sodium hydroxide (NaOH, 99.9%) was purchased from Aladdin Chemical. Deionized water obtained from a Direct-Q 5 ultraviolet water purification system was used in all experiments.Preparation of the Au nanorods
The Au nanorods were synthesized using the seed-mediated growth method, as reported previously. The seed solution was prepared by adding freshly prepared, ice-cold NaBH4 solution (0.1 M, 0.25 mL) into a solution composed of HAuCl4 (0.01 M, 0.25 mL), trisodium citrate (0.01 M, 0.25 mL) and deionized water (9 mL) under vigorous stirring. The resultant seed solution was left undisturbed at room temperature for 2 h. The first two solutions (1 and 2) were made by mixing together CTAB (0.1 M, 9 mL), HAuCl4 (0.01 M, 0.25 mL), ascorbic acid (0.1 M, 0.05 mL) and NaOH (0.1 M, 0.05 mL) sequentially. The NaOH solution was replaced with HNO3 (0.1 M) at different volumes for the preparation of the Au nanorods with different aspect ratios. The third growth solution (3) was made by mixing together CTAB (0.1 M, 90 mL), HAuCl4 (0.01 M, 2.5 mL), ascorbic acid (0.1 M, 0.5 mL) and NaOH (0.1 M, 0.6 mL). The formation of the Au nanorods was initiated by adding the seed solution (0.2–1 mL) to the growth solution 1, which was followed by gentle shaking for 5 s. The resultant solution (1 mL) was then added to the growth solution 2, followed by gentle shaking for 5 s. All of the resultant solution was then added to solution 3. The obtained solution was mixed by gentle shaking and then left undisturbed at room temperature for 12 h. High-aspect-ratio Au nanorods along with a significant amount of Au nanoplates precipitated from the solution and formed a thin film at the bottom of the flask. The resultant supernatant, which contained mostly spherical nanoparticles, was carefully removed. The precipitate of the Au nanorods and nanoplates was redispersed into CTAB solution (0.05 M, 10 mL) upon sonication for 30 s. The oxidizing Au(III) solution was prepared by adding HAuCl4 (0.01 M, 0.5 mL) to CTAB (0.1 M, 10 mL). The Au(III) solution (1 mL) was then added into the suspension of the nanorods and nanoplates upon stirring and left undisturbed for 12 h. The Au nanorods along with a small amount of large nanodisks precipitated and formed a thin film on the bottom of the flask. The greenish-blue supernatant containing small nanodisks was carefully removed and the precipitate was again redispersed in CTAB (0.05 M, 10 mL) followed by the addition of another Au(III) solution (1 mL). This process of partial dissolution was repeated three times until the complete removal of the Au nanoplates. The pure Au nanorods were dispersed in deionized water (10 mL).Preparation of the high-aspect-ratio Au nanonails
The Au nanonail samples were produced by performing overgrowth on the Au nanorods of different dimensions. Each overgrowth solution was prepared by mixing CTAB (0.1 M, 1.0 mL), HAuCl4 (0.01 M, 0.01 mL), AgNO3 (0.01 M, 0.04 mL) and deionized water (6.5 mL) in sequence. Ascorbic acid (0.1 M, 0.016 mL) and the Au nanorods (2.5 mL, ∼1 × 109 particles mL−1) were subsequently added into the mixture solution. The resultant solution was kept under stirring at room temperature for 6 h to complete the overgrowth. The as-prepared Au nanonails were washed and redispersed in deionized water for further use.Decorating the Au nanorods and nanonails with the thiol molecules
The as-grown Au nanocrystal solution (1 mL) was centrifuged twice to remove the surfactant CTAB molecules and redispersed in deionized water (1 mL and 0.2 mL, respectively). The obtained Au nanocrystal solution was added into ATP solution (0.01 M, 1 mL) and then kept undisturbed for at least 6 h. The solution was centrifuged twice and redispersed in deionized water (1 mL and 0.2 mL, respectively). The resultant Au nanocrystal solution was drop-cast on a MgF2 substrate and dried under vacuum. The dried substrate was further immersed in ethanol for at least 6 h and then blown dry with N2 for the FTIR measurements. L-Cysteine molecules were adsorbed on the Au nanocrystals in a similar way.Instrumentation
The extinction spectra were collected using a Hitachi U-4100 ultraviolet/visible/NIR spectrophotometer using 0.5 cm quartz cuvettes. SEM imaging was performed using an FEI Quanta 250 FEG microscope operated at 20 kV. TEM imaging was carried out using an FEI Tecnai Spirit 12 microscope operated at 120 kV. The pH of the growth solution was measured using a FiveEasy Plus (Mettle-Toledo) pH meter. The ICP-MS measurements were performed using an iCAP 7600 ICP-OES system. Before the ICP-MS measurements, the Au nanorod and nanonail samples were centrifuged and dissolved by aqua regia (v/v, 3:1, concentrated HCl/concentrated HNO3) to dissolve the Au nanocrystals, and diluted to the original volume in deionized water. The IR spectra were measured on a Thermo Scientific Nicolet iS5 FTIR spectrometer. The spectral resolution was 0.5 cm−1, and the IR spectra were collected by averaging 36 scans. The aperture diameter of the IR beam was 8 mm and the approximate area was 50 mm2. Before the measurements, the Au nanorod and nanonail solution (0.2 mL) was dropped on the surface of a MgF2 substrate. The substrate was then placed into a vacuum chamber that was connected to a mechanical vacuum pump. After the solution was dried, the substrate was subsequently transferred into the sample chamber of the FTIR spectrometer. The IR transmittance spectra of the samples were corrected by deducting the spectrum taken on a blank MgF2 substrate to remove the background. The spectral range for the MgF2 substrates was 6000–1200 cm−1. The circular MgF2 substrate had a surface area of 113 mm2. The illuminated area was approximately half of the surface area of the substrate.
DFT calculations
The surface density of the ATP molecules on the Au nanocrystals was calculated by using DFT at the PBE0/6-31+G(d,p) level. The polarizable continuum model was implemented to consider the solvent effect, with the static dielectric constant of water (ε = 78.36) being used. The vibrational frequency analysis was performed to verify that the optimized geometry was a local minimum. The gold surface was modelled with a (5 × 5) Au(111) surface slab, which contained five Au layers and a 1.5 nm-thick vacuum layer. The periodic boundary condition (PBC) model was implemented to optimize the structure of gold, with the bottom three layers of Au atoms frozen. The HSEh1PBE functional with the LANL2DA basis set for Au atoms was employed. The Int (Grid = Ultrafine) was performed, which is generally better for PBC calculations. All of the optimizations were manipulated by the Gaussian 09 suite. In the simulations, the hydrogen atom in the thiol group of ATP was removed on account of the experimental observation that the dissociative adsorption occurs on the Au surface for the S–H bond. The structure of the adsorption system was optimized using the PBC model together with the HSEh1PBE functional.FDTD simulations
The simulations were performed using FDTD Solutions 8.7, developed by Lumerical Solutions. In the simulations, an electromagnetic plane wave in the wavelength range from 1600 nm to 8350 nm was launched into a box containing a target Au nanonail or nanorod. The mesh size was set at 2 nm in calculating the absorption, scattering and extinction spectra of the nanocrystals, and it was set at 1 nm in calculating the electric field enhancement contours. The refractive index of the surrounding medium was set at 1.0. The dielectric function of Au was fitted from Palik's data in the range of 1–8.5 μm. The dielectric function of the ATP molecule was described using a homogeneously broadened three-oscillator Lorentzian model. The shell thickness was 1.0 nm. Each Au nanorod was modelled as a cylinder with two hemispherical ends. The Au nanonail was modelled as a pentagonal cylinder with two penta-pyramid ends. The length/diameter of the Au nanonail and nanorod were set at 1784 nm/32 nm and 1304 nm/18 nm, respectively. The polarization direction of the incident light was set to be parallel to the length axis to excite the longitudinal dipolar plasmon resonance of the Au nanocrystals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51872110) and Hong Kong Research Grants Council (General Research Fund: 14306817).References
- B. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons, 2004 Search PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic, London, 3rd edn, 1990 Search PubMed.
- V. Giannini, A. I. Fernández-Domínguez, S. C. Heck and S. A. Maier, Chem. Rev., 2011, 111, 3888–3912 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W. S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- R. B. Jiang, B. X. Li, C. H. Fang and J. F. Wang, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS PubMed.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- R. Panneerselvam, G.-K. Liu, Y.-H. Wang, J.-Y. Liu, S.-Y. Ding, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, Chem. Commun., 2018, 54, 10–25 RSC.
- J. Langer, D. J. de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. G. de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. L. Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. V. Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. L. Xu, H. X. Xu, Y. K. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- H. R. Wei, S. M. Hossein Abtahi and P. J. Vikesland, Environ. Sci.: Nano, 2015, 2, 120–135 RSC.
- J. K. Zheng and L. L. He, Compr. Rev. Food Sci. Food Saf., 2014, 13, 317–328 CrossRef CAS.
- A. Hakonen, P. O. Andersson, M. S. Schmidt, T. Rindzevicius and M. Käll, Anal. Chim. Acta, 2015, 893, 1–13 CrossRef CAS PubMed.
- S. Milliken, J. Fraser, S. Poirier, J. Hulse and L.-L. Tay, Spectrochim. Acta, Part A, 2018, 196, 222–228 CrossRef CAS PubMed.
- F. Pozzi and M. Leona, J. Raman Spectrosc., 2016, 47, 67–77 CrossRef CAS.
- F. Neubrech, C. Huck, K. Weber, A. Pucci and H. Giessen, Chem. Rev., 2017, 117, 5110–5145 CrossRef CAS PubMed.
- X. X. Yang, Z. P. Sun, T. Low, H. Hu, X. D. Guo, F. J. García de Abajo, P. Avouris and Q. Dai, Adv. Mater., 2018, 30, 1704896 CrossRef PubMed.
- T. Wang, V. H. Nguyen, A. Buchenauer, U. Schnakenberg and T. Taubner, Opt. Express, 2013, 21, 9005–9010 CrossRef CAS PubMed.
- R. Adato, S. Aksu and H. Altug, Mater. Today, 2015, 18, 436–446 CrossRef CAS.
- D. Rodrigo, O. Limaj, D. Janner, D. Etezadi, F. J. G. de Abajo, V. Pruneri and H. Altug, Science, 2015, 349, 165–168 CrossRef CAS PubMed.
- C.-K. Chen, M.-H. Chang, H.-T. Wu, Y.-C. Lee and T.-J. Yen, Biosens. Bioelectron., 2014, 60, 343–350 CrossRef CAS PubMed.
- L. Kühner, M. Hentschel, U. Zschieschang, H. Klauk, J. Vogt, C. Huck, H. Giessen and F. Neubrech, ACS Sens., 2017, 2, 655–662 CrossRef PubMed.
- C. Wu, A. B. Khanikaev, R. Adato, N. Arju, A. A. Yanik, H. Altug and G. Shvets, Nat. Mater., 2012, 11, 69–75 CrossRef CAS PubMed.
- O. Limaj, D. Etezadi, N. J. Wittenberg, D. Rodrigo, D. Yoo, S.-H. Oh and H. Altug, Nano Lett., 2016, 16, 1502–1508 CrossRef CAS PubMed.
- C. V. Hoang, M. Oyama, O. Saito, M. Aono and T. Nagao, Sci. Rep., 2013, 3, 1175 CrossRef PubMed.
- T. D. Dao, H. L. T. Nghiem, K. Chen and T. Nagao, Proc. SPIE, 2016, 9929, 992904 CrossRef.
- K.-J. Kim, X. Y. Chong, P. B. Kreider, G. H. Ma, P. R. Ohodnicki, J. P. Baltrus, A. X. Wang and C.-H. Chang, J. Mater. Chem. C, 2015, 3, 2763–2767 RSC.
- X. Y. Chong, Y. J. Zhang, E. W. Li, K.-J. Kim, P. R. Ohodnicki, C.-H. Chang and A. X. Wang, ACS Sens., 2018, 3, 230–238 CrossRef CAS PubMed.
- M. Osawa, Topics in Applied Physics, Springer, Berlin, 2001 Search PubMed.
- L.-W. Chou, N. Shin, S. V. Sivaram and M. A. Filler, J. Am. Chem. Soc., 2012, 134, 16155–16158 CrossRef CAS PubMed.
- S. Law, L. Yu, A. Rosenberg and D. Wasserman, Nano Lett., 2013, 13, 4569–4574 CrossRef CAS PubMed.
- G. Garcia, R. Buonsanti, A. Llordes, E. L. Runnerstrom, A. Bergerud and D. J. Milliron, Adv. Opt. Mater., 2013, 1, 215–220 CrossRef.
- F. D’Apuzzo, A. R. Piacenti, F. Giorgianni, M. Autore, M. C. Guidi, A. Marcelli, U. Schade, Y. Ito, M. W. Chen and S. Lupi, Nat. Commun., 2017, 8, 14885 CrossRef PubMed.
- W. T. Hsieh, P. C. Wu, J. B. Khurgin, D. P. Tsai, N. Liu and G. Sun, ACS Photonics, 2018, 5, 2541–2548 CrossRef CAS.
- F. Neubrech, A. Pucci, T. W. Cornelius, S. Karim, A. García-Etxarri and J. Aizpurua, Phys. Rev. Lett., 2008, 101, 157403 CrossRef PubMed.
- V. Giannini, Y. Francescato, H. Amrania, C. C. Phillips and S. A. Maier, Nano Lett., 2011, 11, 2835–2840 CrossRef CAS PubMed.
- C. D’Andrea, J. Bochterle, A. Toma, C. Huck, F. Neubrech, E. Messina, B. Fazio, O. M. Maragò, E. D. Fabrizio, M. Lamy de La Chapelle, P. G. Gucciardi and A. Pucci, ACS Nano, 2013, 7, 3522–3531 CrossRef PubMed.
- M. Abb, Y. D. Wang, N. Papasimakis, C. H. de Groot and O. L. Muskens, Nano Lett., 2014, 14, 346–352 CrossRef CAS PubMed.
- S. Bagheri, K. Weber, T. Gissibl, T. Weiss, F. Neubrech and H. Giessen, ACS Photonics, 2015, 2, 779–786 CrossRef CAS.
- I. Schubert, C. Huck, P. Kröber, F. Neubrech, A. Pucci, M. E. Toimil-Molares, C. Trautmann and J. Vogt, Adv. Opt. Mater., 2016, 4, 1838–1845 CrossRef CAS.
- B. Cerjan, X. Yang, P. Nordlander and N. J. Halas, ACS Photonics, 2016, 3, 354–360 CrossRef CAS.
- L. V. Brown, K. Zhao, N. King, H. Sobhani, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2013, 135, 3688–3695 CrossRef CAS PubMed.
- K. Chen, R. Adato and H. Altug, ACS Nano, 2012, 6, 7998–8006 CrossRef CAS PubMed.
- Y. Q. Li, L. Su, C. Shou, C. M. Yu, J. J. Deng and Y. Fang, Sci. Rep., 2013, 3, 2865 CrossRef PubMed.
- S. Cataldo, J. Zhao, F. Neubrech, B. Frank, C. J. Zhang, P. V. Braun and H. Giessen, ACS Nano, 2012, 6, 979–985 CrossRef CAS PubMed.
- B. Lahiri, G. Holland, V. Aksyuk and A. Centrone, Nano Lett., 2013, 13, 3218–3224 CrossRef CAS PubMed.
- L. V. Brown, X. Yang, K. Zhao, B. Y. Zheng, P. Nordlander and N. J. Halas, Nano Lett., 2015, 15, 1272–1280 CrossRef CAS PubMed.
- H. Aouani, H. Šípová, M. Rahmani, M. Navarro-Cia, K. Hegnerová, J. Homola, M. H. Hong and S. A. Maier, ACS Nano, 2013, 7, 669–675 CrossRef CAS PubMed.
- G. Q. Wallace, H. C. Foy, S. M. Rosendahl and F. Lagugné-Labarthet, J. Phys. Chem. C, 2017, 121, 9497–9507 CrossRef CAS.
- D. Huo, M. J. Kim, Z. H. Lyu, Y. F. Shi, B. J. Wiley and Y. N. Xia, Chem. Rev., 2019, 119, 8972–9073 CrossRef CAS PubMed.
- L. Shao, Y. T. Tao, Q. F. Ruan, J. F. Wang and H.-Q. Lin, Phys. Chem. Chem. Phys., 2015, 17, 10861–10870 RSC.
- W. J. Parak, D. Gerion, T. Pellegrino, D. Zanchet, C. Micheel, S. C. Williams, R. Boudreau, M. A. Le Gros, C. A. Larabell and A. P. Alivisatos, Nanotechnology, 2003, 14, R15–R27 CrossRef CAS.
- R. L. S. Tan, W. H. Chong, Y. H. Feng, X. H. Song, C. L. Tham, J. Wei, M. Lin and H. Y. Chen, J. Am. Chem. Soc., 2016, 138, 10770–10773 CrossRef CAS PubMed.
- N. N. Li, H. Yin, X. L. Zhuo, B. C. Yang, X.-M. Zhu and J. F. Wang, Adv. Opt. Mater., 2018, 6, 1800436 CrossRef.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Jr, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS PubMed.
- F. Frederix, J. M. Friedt, K. H. Choi, W. Laureyn, A. Campitelli, D. Mondelaers, G. Maes and G. Borghs, Anal. Chem., 2003, 75, 6894–6900 CrossRef CAS.
- M. Tzschoppe, C. Huck, J. Vogt, F. Neubrech and A. Pucci, J. Phys. Chem. C, 2018, 122, 15678–15687 CrossRef CAS.
- B. D. Busbee, S. O. Obare and C. J. Murphy, Adv. Mater., 2003, 15, 414–416 CrossRef CAS.
- B. P. Khanal and E. R. Zubarev, J. Am. Chem. Soc., 2008, 130, 12634–12635 CrossRef CAS PubMed.
- S. Z. Zhang, X. S. Kou, Z. Yang, Q. H. Shi, G. D. Stuck, L. D. Sun, J. F. Wang and C. H. Yan, Chem. Commun., 2007, 1816–1818 RSC.
- T. Ming, X. S. Kou, H. J. Chen, T. Wang, H. L. Tam, K. W. Cheah, J. Y. Chen and J. F. Wang, Angew. Chem., Int. Ed., 2008, 47, 9685–9690 CrossRef CAS PubMed.
- X. S. Kou, Z. H. Sun, Z. Yang, H. J. Chen and J. F. Wang, Langmuir, 2009, 25, 1692–1698 CrossRef CAS PubMed.
- Y.-N. Wang, W.-T. Wei, C.-W. Yang and M. H. Huang, Langmuir, 2013, 29, 10491–10497 CrossRef CAS PubMed.
- M. Z. Liu and P. G. Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- E. R. Encina and E. A. Coronado, J. Phys. Chem. C, 2007, 111, 16796–16801 CrossRef CAS.
- X. L. Zhuo, X. Z. Zhu, Q. Li, Z. Yang and J. F. Wang, ACS Nano, 2015, 9, 7523–7535 CrossRef CAS PubMed.
- X. L. Kang, Q. F. Ruan, H. Zhang, F. Bao, J. Guo, M. H. Tang, S. Cheng and J. F. Wang, Nanoscale, 2017, 9, 5879–5886 RSC.
- X.-L. Tang, P. Jiang, G.-L. Ge, M. Tsuji, S.-S. Xie and Y.-J. Guo, Langmuir, 2008, 24, 1763–1768 CrossRef CAS PubMed.
- Y.-F. Huang, H.-P. Zhu, G.-K. Liu, D.-Y. Wu, B. Ren and Z.-Q. Tian, J. Am. Chem. Soc., 2010, 132, 9244–9246 CrossRef CAS PubMed.
- J. Ye, J. A. Hutchison, H. Uji-i, J. Hofkens, L. Lagae, G. Maes, G. Borghsa and P. V. Dorpe, Nanoscale, 2012, 4, 1606–1611 RSC.
- J. Kundu, F. Le, P. Nordlander and N. J. Halas, Chem. Phys. Lett., 2008, 452, 115–119 CrossRef CAS.
- E. J. Osley, C. G. Biris, P. G. Thompson, R. R. F. Jahromi, P. A. Warburton and N. C. Panoiu, Phys. Rev. Lett., 2013, 110, 087402 CrossRef CAS PubMed.
- S. J. Ye, G. Marston, J. R. McLaughlan, D. O. Sigle, N. Ingram, S. Freear, J. J. Baumberg, R. J. Bushby, A. F. Markham, K. Critchley, P. L. Coletta and S. D. Evans, Adv. Funct. Mater., 2015, 25, 2117–2127 CrossRef CAS.
- X. Z. Zhu, H. K. Yip, X. L. Zhuo, R. B. Jiang, J. L. Chen, X.-M. Zhu, Z. Yang and J. F. Wang, J. Am. Chem. Soc., 2017, 139, 13837–13846 CrossRef CAS PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- V. Canpean and S. Astilean, Lab Chip, 2009, 9, 3574–3579 RSC.
- Y.-T. Kim, R. L. McCarley and A. J. Bard, J. Phys. Chem., 1992, 96, 7416–7421 CrossRef CAS.
- N. Mohri, M. Inoue, Y. Arai and K. Yoshikawa, Langmuir, 1995, 11, 1612–1616 CrossRef CAS.
- M. Tachibana, K. Yoshizawa, A. Ogawa, H. Fujimoto and R. Hoffmann, J. Phys. Chem. B, 2002, 106, 12727–12736 CrossRef CAS.
- F. Le, D. W. Brandl, Y. A. Urzhumov, H. Wang, J. Kundu, N. J. Halas, J. Aizpurua and P. Nordlander, ACS Nano, 2008, 2, 707–718 CrossRef CAS PubMed.
- F. Wang, S. Cheng, Z. H. Bao and J. F. Wang, Angew. Chem., Int. Ed., 2013, 52, 10344–10348 CrossRef CAS PubMed.
- X. Z. Zhu, H. L. Jia, X.-M. Zhu, S. Cheng, X. L. Zhuo, F. Qin, Z. Yang and J. F. Wang, Adv. Funct. Mater., 2017, 27, 1700016 CrossRef.
- A. Pawlukojć, J. Leciejewicz, A. J. Ramirez-Cuesta and J. Nowicka-Scheibe, Spectrochim. Acta, Part A, 2005, 61, 2474–2481 CrossRef PubMed.
- T. Neuman, C. Huck, J. Vogt, F. Neubrech, R. Hillenbrand, J. Aizpurua and A. Pucci, J. Phys. Chem. C, 2015, 119, 26652–26662 CrossRef CAS.
- H. J. Chen, L. Shao, K. C. Woo, J. F. Wang and H.-Q. Lin, J. Phys. Chem. C, 2012, 116, 14088–14095 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Extinction spectra, size distribution histograms, TEM and HRTEM images, FTIR spectra, calculation data and definition of the relevant parameters. See DOI: 10.1039/d0nh00244e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |