
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site specific and localized structural displacements in open structured multimetallic oxides†
Thomas
Lunkenbein
 *a,
Liudmyla
Masliuk
a,
Milivoj
Plodinec
a,
Gerardo
Algara-Siller
a,
Sabrina
Jung
a,
Mateusz
Jastak
a,
Pierre
Kube
a,
Annette
Trunschke
a and
Robert
Schlögl
ab
*a,
Liudmyla
Masliuk
a,
Milivoj
Plodinec
a,
Gerardo
Algara-Siller
a,
Sabrina
Jung
a,
Mateusz
Jastak
a,
Pierre
Kube
a,
Annette
Trunschke
a and
Robert
Schlögl
ab
aDepartment of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: lunkenbein@fhi-berlin.mpg.de
bDepartment of Heterogeneous Reactions, Max Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim a. d. Ruhr, Germany
First published on 13th March 2020
Abstract
The structures of solids can locally differ from the macroscopic picture obtained by structural averaging techniques. This difference significantly influences the performance of any functional material. Measurements of these local structures are challenging. Thus, the description of defects is often disregarded. However, in order to understand the functionality, such irregularities have to be investigated. Here, we present a high resolution scanning transmission electron microscopic (STEM) study revealing local structural irregularities in open structured oxides using catalytically active orthorhombic (Mo,V,Te,Nb)Ox as a complex example. Detailed analysis of annular dark field- and annular bright field-STEM images reveal site specific local structural displacements of individual framework and channel sites in the picometer range. These experimental observables can be considered as an important structural addendum for theoretical modelling and should be implemented into the existing data in order to quantify site specific potential energies and stresses. This information can further be used to describe the impact of the structure on the catalytic performance in greater detail.
A. Introduction
Defects penetrate the structure of any crystalline solid on the nanoscale and can greatly influence their performance.1 However, detailed defect-activity correlations have not yet been established due to difficulties in capturing, localizing, and quantifying these structural deviations.2,3 Modern scanning transmission electron microscopes (STEM), equipped with annular detectors, have become powerful tools to visualize the geometric inconsistencies of surfaces and bulk in real space and on the atomic scale.2,4 In addition, these modern TEMs exhibit more robust columns enabling local structural analyses with 10 pm accuracy.5,6Among the huge variety of functional materials, high-performance catalysts, such as multimetallic molybdenum- and vanadium-based oxides, which can crystallize in an open crystal structure, are highly susceptible to geometric and compositional defects, such as different cation occupancies, polyhedral distortions or structural reconnectivities.2,4,7–9 Prominent oxides of this family have an orthorhombic unit cell (space group: Pba2) and crystallize in the so-called M1 structure. The unit cell of the M1 structure is composed of 44 metal atoms that are distributed over 13 crystallographic different metal sites (S). The main structural features of orthorhombic (Mo,V,Te,Nb)Ox are pentagonal building blocks consisting of a central bipyramidal NbO7 polyhedron that shares edges with five M1O6 octahedra (M1 = Mo) (framework). These pentagonal building blocks are connected via corner-sharing M2O6 (M2 = Mo, V) octahedra resulting in the formation of hexagonal and heptagonal structural channels.10–12 These channels can be partially occupied by cationic species of tellurium, antimony, vanadium or ammonium.7,10–13 Furthermore, complex molybdenum and vanadium mixed oxides represent non-stoichiometric compounds. Thus, in order to accomplish charge neutrality, the intrinsic oxidation states of molybdenum and vanadium within these materials depend on the metal and oxygen composition.14–16
Orthorhombic molybdenum and vanadium mixed oxides that crystallize in the M1 structure are, for instance, selective in the catalytic conversion of propane to acrylic acid or dehydrogenation of ethane.17,18 They have been the subject of numerous studies that focused on the understanding of their catalytic and structural properties also including operando/in situ investigations.7,9–13,16,19–29 A crystallographic model is shown in Fig. 1A. We adopt the numbering of individual metal sites, as recommended by Buttrey and co-workers.10,11 In this material corner-sharing octahedra, such as the S2 site, have a high Debye–Waller factor, reflecting a certain degree of disorder in the S2 sites.19 Previous in situ TEM studies of (Mo,V,Te)Ox have demonstrated that, under reaction conditions, the disordering of the S2 site may arise from a destabilizing effect induced by the formation of oxygen vacancies in the octahedral framework rather than from thermal disorder.20 In addition, single crystal analyses of different M1 oxides have shown an anisotropic displacement parameter or out-of-center shift of metal sites occupying the crystallographic S2 position. The displacement vector of shifted metal cations on this site points toward the centers of the hexagonal channels.12,13 Their spatial distribution and a detailed understanding of how these displacements affect the local chemistry and properties of the material has yet not been reported. However, this local information would be an important prerequisite to improve our current understanding of heterogeneous catalysis.30
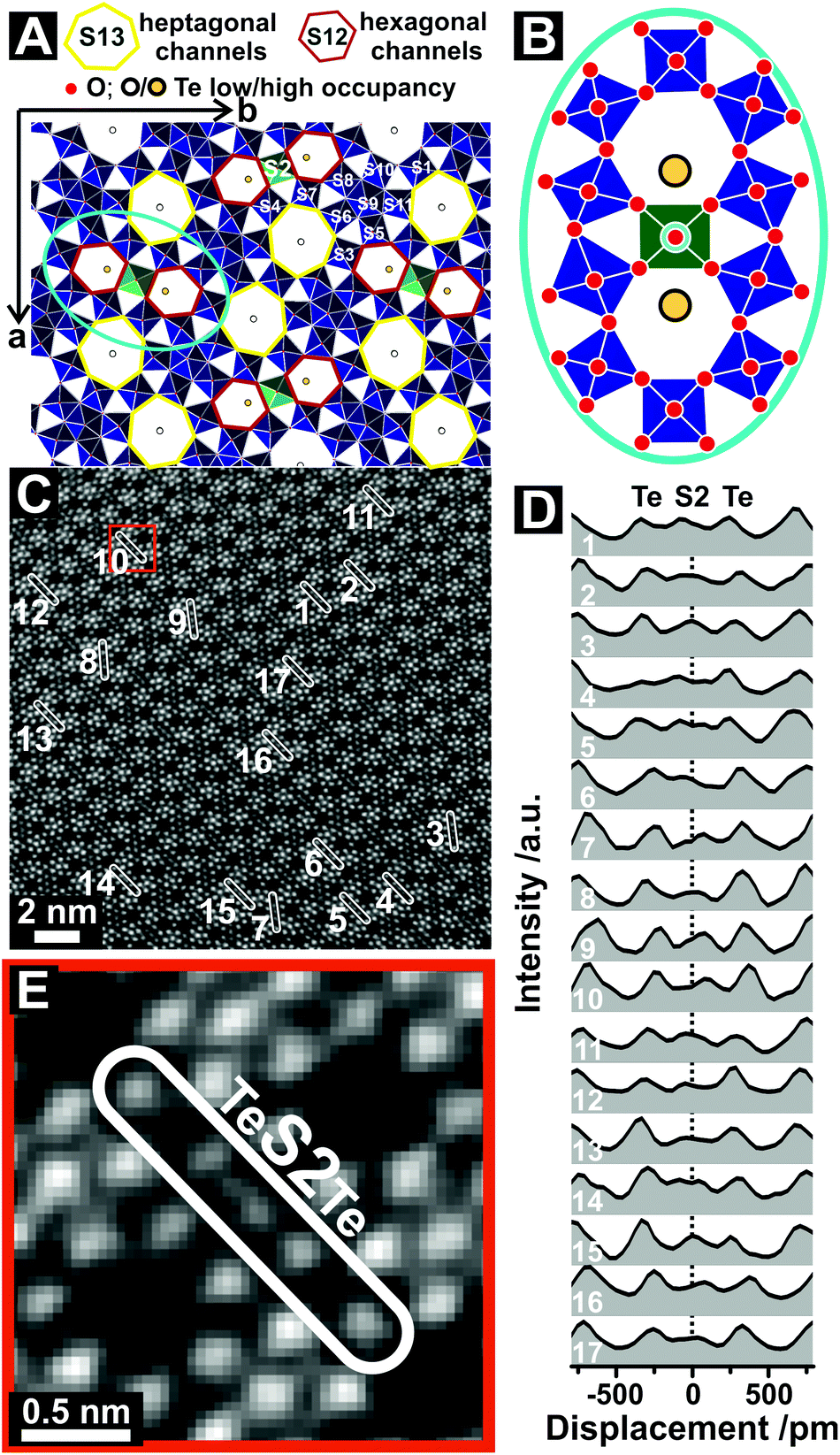 | ||
| Fig. 1 The structure of orthorhombic (Mo,V,Te,Nb)Ox. (A) Structural representation of orthorhombic (Mo,V,Nb,Te)Ox viewed along the crystallographic c-axis.11 The green polyhedra highlight the S2 sites. (B) Schematic drawing of the vicinity of the S2 site as assumed from the crystallographic model. (C) ADF-STEM image of orthorhombic (Mo,V,Te,Nb)Ox viewed along [001]. The labelled areas in (C) represent the region of interests (ROIs), from which the line profiles where taken. The oxide was synthesized via Method 1. The original ADF-STEM image is given in the ESI (Fig. S2†). (D) Corresponding line profiles taken at ROIs highlighted in (C). The dotted line (D) denotes the expected centre of the S2 site following the translational structural motif. (E) Magnified ADF-STEM image of the highlighted region in (C, red box) that displays the local environment around the S2 site. | ||
In the present study, various synthesized orthorhombic (Mo,V,Te,Nb)Ox were examined to obtain explicit real-space structure information about the shift of metal cations positioned at the S2 site. The different preparation techniques yield isostructural materials that differ in chemical composition, i.e., in the occupation of the metal sites. As we will show, the cation position in the framework is influenced by the positioning of Te in the hexagonal channels and vice versa. In addition, annular dark-field (ADF)- and annular bright-field (ABF)-STEM micrographs of (Mo,V,Te,Nb)Ox qualitatively suggest a strong Te–S2 interaction, which differs from site to site.
B. Results and discussion
The samples were prepared by hydrothermal synthesis (Method 1 and Method 2) and the slurry method (Method 3) (for a description of the synthesis see the Experimental section). The different synthesis techniques yield (Mo,V,Te,Nb)Ox with varying chemical composition and specific surface areas including MoV0.25Te0.10Nb0.42Ox (Method 1, 9.27 m2 g−1), MoV0.25Te0.23Nb0.12Ox (Method 2, 2.13 m2 g−1), and MoV0.26Te0.11Nb0.16Ox (Method 3, 7.31 m2 g−1). The samples are active in the oxidative conversion of propane and selective towards the formation of acrylic acid (Fig. S1†). The differences in the performances are mainly related to the differences in the specific surface areas and composition of the catalysts as a result of applying different synthesis protocols, which is in line with previous reports.31Detailed beam damage studies and their impact on the structure have been addressed elsewhere for similar M1 samples.2,23 Structural changes that arise due to the electron beam are expressed by an immediate amorphization that spreads from the surface to the bulk or removal of Te from the channel sites. Our studies suggest that under the applied imaging conditions (Mo,V,Te,Nb)Ox can be at least 20 minutes exposed continuously to the electron beam without noticeable morphological changes (Fig. S3†) which is sufficiently longer than the required time to collect a representative image. The presented STEM images have been post-processed after acquisition by applying a Gaussian smoothing filter.
A crystallographic model of (Mo,V,Te,Nb)Ox is presented in Fig. 1A. The model is based on scattering results and resembles the average of a multitude of scattering events, which lead to an isotropic description of the corner-sharing octahedra, such as the S2 site (Fig. 1B). Fig. 1C depicts the ADF-STEM reproduction of (Mo,V,Te,Nb)Ox, synthesized via Method 1, viewed along [001]. The observed microstructure agrees with previous reports and resembles on the first glance the crystallographic model.10,11,22
However, detailed line profile analyses taken from the framed and numbered regions of interests (ROI) in Fig. 1C over two neighboring hexagonal channels reveals insights into the unusual behavior of the Te and S2 sites (Fig. 1D). Quantitative expressions of positions of the atoms in a crystalline material can be obtained by analyzing the images with different computational techniques.32–34 Still traditional line profiling can be used to assess differences in the structure on a very localized scale.20,35 Line profiles were recorded in several directions to account for artefacts, such as sample mistilt, scanning distortions or astigmatism. In the line profile analyses the crystallographic center of the S2 site was set to 0 pm displacement (Fig. 1D, dotted line). The intensity distributions of the line profiles indicate (i) a displacement of the S2 site towards the hexagonal channels as well as (ii) an irregular Te occupancy for each hexagonal channel. None of the 17 line profiles taken from different unit cells are identical. This observation implies structural irregularities for the S2 and Te sites in an otherwise highly ordered structure. The large amount of observed displacements or elongation events corroborates the statistical relevance of this study.
To investigate the S2 and Te irregularities in greater detail we recorded high resolution ADF-STEM images of (Mo,V,Te,Nb)Ox, synthesized by Method 2. A representative image is presented in Fig. 2A. The corresponding line profiles are displayed in Fig. 2B and exemplarily highlight the magnitude of elongated S2 displacements. Respective Gaussian fits and the error of the displacements are presented in Fig. S5.† Similar to Fig. 1C, all measured profiles are different and show irregularities at the S2 and Te position. In addition, the line profiles of the S2 sites demonstrate the coexistence of anisotropic (ROI 1–5, Fig. 2B) and isotropic (ROI 6–7, Fig. 2B) S2 sites within one M1 crystal indicating the occurrence of local structural differences within this class of open structured oxide.
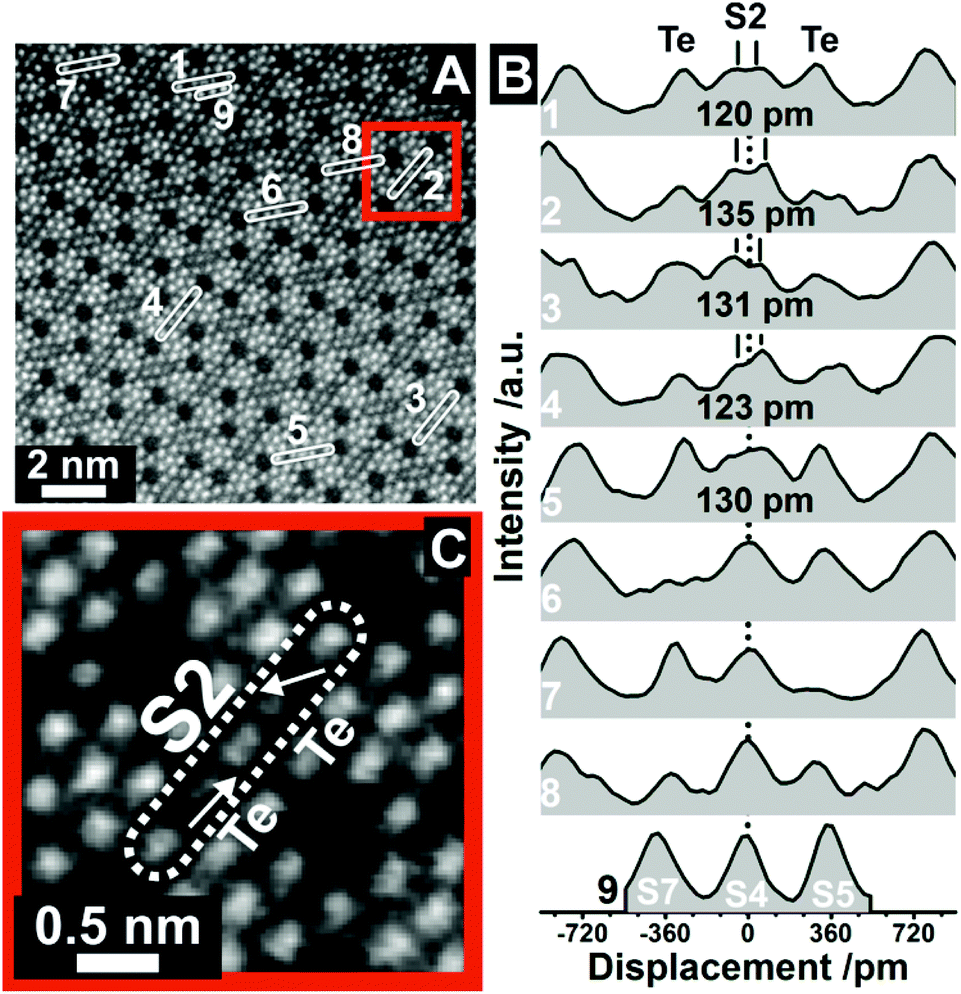 | ||
| Fig. 2 Displacement of the S2 sites at high resolution. (A) ADF-STEM image taken from orthorhombic (Mo,V,Te,Nb)Ox, which was synthesized by Method 2, viewed along the [001] direction. The numbers denote ROIs from which line profiles have been taken. The original ADF-STEM image is presented in the ESI (Fig. S4†). The line profiles in (B) correspond to the ROIs in (A). The distances in (B) denote the displacement of S2 sites as obtained by Gaussian fitting (Fig. S5†). ROI9 (S7, S4, S5 metal sites) was used for comparison to highlight the otherwise almost Gaussian shaped intensity distribution of other metal centers. (C) Magnified ADF-STEM image of the area around ROI2 in (A). The arrows in (C) denote the shift vectors of the Te centers with respect to the middle of the hexagonal channels. The dotted line (B) denotes the expected center of the S2 site following the translational structure motif. | ||
The distinctive character of the S2 sites may be interpreted as cationic columns (Mo or V), where the S2 cations diverge from their octahedral anionic coordination environment. The projected atom to atom distances range from (120 ± 13) pm to (135 ± 11) pm. Similar cation displacements have been observed in multimetallic oxides, such as ferroelectric perovskites, in which the off-centering of the cations can lead to a correlated effect that can determine the function.32 In our study, however, these displacements appear very localized.
In addition, Fig. 2B reveals that the S2 site displacement depends on the irregular occupancy of individual vicinal Te entities. If the Te centers are positioned close to the center of the axis of the hexagonal channels, which may happen for a low Te occupancy, or if only one hexagonal channel is occupied by Te, an almost isotropic character of the S2 site is obtained. On the contrary, the combination of relatively high Te occupancies with off-centered Te sites causes a shift of the S2 metal sites. For comparison, the bottom line profile (ROI 9, Fig. 2B) demonstrates the almost Gaussian peak shape of regular S7, S4 and S5 sites. The displacement of the S2 site is further highlighted in Fig. 2C, taken from ROI 2. It also demonstrates the out-of-center shift of vicinal Te centers towards the S2 site. In addition, the shift vectors of two Te entities in neighboring channels can point in different directions (Fig. 2C). While one vector indicates an out-of-center shift towards the S2 site, its counterpart in the neighboring channel reveals a shift of the Te entities towards the center of one bridging S2–O–S4/S7 bond.
We have further recorded ABF-STEM images (Fig. 3, Fig. S6†) to examine details of the interaction of Te with the lattice oxygen of the S2–O–S4/S7 bond. In the presented ABF image, which is shown in Fig. 3A, channels within the M1 microstructure appear bright, whereas individual metal and oxygen sites are dark. As opposed to ADF imaging, the O columns in the vicinity of the metal sites and electron density in the heptagonal channels can be observed in ABF-STEM images. In general, ABF-STEM images are sensitive to sample mistilt and residual aberration.36 The ABF-STEM image is in good agreement with the superimposed crystal structure and unit cell (Fig. 3A) suggesting the absence of mistilt and scan distortions. In line with the ADF-STEM images, the geometric information obtained by ABF-STEM imaging shows similar disordered trends in (Mo,V,Nb,Te)Ox, which was synthesized by Method 3 (Fig. S7†). A magnified image of the region highlighted in Fig. 3A (red box), is presented in Fig. 3B and reveals details of the S2–O–Te interaction. The combined ABF-STEM image and structural representation (Fig. 3C) demonstrate that the greyish atom columns observed in Fig. 3B are oxygen. This would allow a direct measure of the Te–O distances. In total, nine different S2 sites (Fig. S8†) have been found (Fig. 3D) from which the Te–O distances similar to Fig. 3B could be measured (Fig. 3D, Fig. S9†). The corresponding line profiles of Te–O distances have been analyzed by Gaussian peak fitting and fits are presented in the ESI (Fig. S9†). The measured Te–O distances (Fig. 3D, Fig. S9†), which were directly obtained from the high-resolution ABF-STEM images indicate the presence of varying distances between the Te and different vicinal lattice oxygens, which form the anionic configuration around the S2 site. The largest differences of vincinal Te–O distances have been observed when the measured Te–O distances vary from above 220 pm to around 180 pm, respectively. This substantial variation in the Te–O distances does not exist exclusively in orthorhombic (Mo,V,Te,Nb)Ox. It has previously been highlighted that bridging Te(IV)–O bond distances in different tellurium oxides and hydroxides are 188 pm, while apical Te–O bond distances can extent to 219 pm.37,38 The observed interaction of Te with lattice oxygen for the shorter distance (around 180 pm) may indicate the presence of covalent Te–O bonds in (Mo,V,Te,Nb)Ox, while the longer Te–O distance (>220 pm) may be characteristic of weaker van-der Waals interactions or apical Te–O bonds of the corresponding polyhedra. A covalent bond character of the hexagonal channel cation with lattice oxygen, which surrounds the S2 site, has already been discussed for (Mo,V)Ox.12
 | ||
| Fig. 3 Insights into the Te–O bond length. (A) ABF-STEM image of orthorhombic (Mo,V,Te,Nb)Ox viewed along [001] superimposed with the orthorhombic unit cell (white rectangle) and the crystal structure.11 Red spheres denote the positions of oxygen within the crystal structure. Different colours show the positions of the metal cations. The M1 oxide was prepared by Method 3. The original image is shown in the ESI (Fig. S6†). (B) Magnified region around a S2 site highlighted by the red square in (A). Pink and violet ellipsoids in (B) display Te and O distances around the S2 site that were used for measuring. (C) ABF-STEM image of the same region superimposed by the crystallographic structure showing that the greyish atom columns in the ABF-STEM image in (B) are oxygen and black atom columns correspond to metal centers. (D) Te–O distances for nine different S2 sites (Fig. S8†) after Gaussian fitting of the corresponding line profiles (Fig. S9†). The squares show the upper limit and the dots the lower limit of Te–O distance for the measured S2 sites. | ||
An unequal Te occupancy in the hexagonal channels of orthorhombic (Mo,V,Te)Ox has been previously reported.9,20 Our finding of Te disorder in the hexagonal channels of various synthesized (Mo,V,Te,Nb)Ox with different composition shows that these structural irregularities are a general feature in Te-containing molybdenum/vanadium mixed oxides. Although the origin of the observed structural displacement remains unclear, it seems reasonable that the occupancy of Te in the hexagonal channels is influenced by the nature of neighboring metal sites. There are 32 possible compositional combinations in this region (S2, S4, S7), in which all possible scenarios of the occupancy of the S2 site could be considered including the presence of partially reduced Mo5+ and/or V4+ cations.16 These partially reduced cationic species have ionic radii of 61 and 58 pm, respectively. The ionic radii of these cations are, thus, 3% (Mo5+/Mo6+) and 7% (V4+/V5+) larger than the ionic radii of their fully oxidized counterparts in the same octahedral environment.39 This rather small difference of the ionic radii compared to that of other transition metal redox couples (Fe2+/Fe3+ (17%), Ti3+/Ti4+ (9%), or Cr5+/Cr6+ (10%)) implies a higher flexibility towards redox cycling of partially reduced V and Mo species in their respective anionic configurations.39 Smaller differences in the ionic radii of redox couples may minimize the induced geometric stress and maintain structural stability. A conclusion on the nature of the S2 cations, based solely on the contrast obtained from the ADF-STEM images, is inappropriate as the individual occupancy of the S2 site is variable and suffers from displacements.
As has been discussed in a previous report12 the observed displacement of the S2 sites can be manifold. Molybdenum and vanadium in their d0 state show a large out-of-center shift in an octahedral environment.40 As opposed to V5+ species, the majority of Mo6+ cations in an octahedral environment are shifted towards the edges of the basal planes. Thus, the observed directed shift may be induced by the presence of fully oxidized molybdenum or vanadium species. Oxidation state induced distortions were also addressed in a recent ABF-STEM study on orthorhombic (Mo,V)Ox.7 In this study the projected metal–oxygen bond angles were imaged. The highly distorted polyhedra of the pentagonal building blocks were found to be occupied by fully oxidized molybdenum species, while medium distortions of the corner-sharing octahedra were assigned to a preferential V5+ occupancy. In addition, octahedra that are free of distortion were observed for the S1 and S2 sites which were expected to host partially reduced cation species. In addition, an intimate cation–cation repulsion of the S2 site with neighboring Te4+ ions (Fig. 2) could also be discussed as the origin of the repulsion of the Mo and V cations from their ideal positions. The new equilibrium positions may depend on both, the type, size, and charge of the metal cation, which occupies the S2 sites, as well as the degree of occupancy of the Te sites. Furthermore, the formation of Te–O bonds (Fig. 3) can weaken adjacent S2–O bonds on one site of the octahedron while the strengths of the opponent bonds may remain unchanged. In such an unbalanced local environment the different bond strength would, consequently, drive the shift of metal cations from their original positions. The combination of previous ABF-STEM results7 with the current results (Fig. 2 and 3) may exclude electronic effects as a cause for the observed displacement.
More importantly, it has been shown recently that such displacements can change the potential energy within individual polyhedra.41,42 In addition, this geometric distortion may induce intrinsic strain and stress43 in the bulk and surface-near channels. The stress introduced by such sub-Angstrom displacements can locally be as high as several tens of GPa as has been, for instance, demonstrated for Al/TiN and Cu/TiN interfaces by theoretical modelling.43 Such values for the local displacements significantly surmount the expected critical resolved shear stresses, i.e. the point when plastic deformation starts, of densely packed MgO.44 The strained and stressed S2 sites are randomly distributed throughout the bulk and surface of (Mo,V,Te,Nb)Ox. Furthermore, the open structure of (Mo,V,Te,Nb)Ox may contribute to their spatial separation. This spatial separation may, thus, be the origin of the high stability of these kinds of open structured oxides during function. Structural stress can then be buffered efficiently by the open microstructure allowing flexibility in the interlinking of polyhedra.
The displacement may further separate the center of mass charges. This would lead to the formation of atomic dipoles located around the S2 sites.
In addition, the out-of-position shift of the Te entities in the hexagonal channels may also result in the formation of local dipoles, whose magnitudes depend on the extent of the displacement, and may vary for different channels. For the S2 sites with unbalanced cationic displacements, and for the Te sites with inhomogeneous occupancies and displacements throughout individual hexagonal channels, the dipoles cannot offset each other and would produce a nonzero net polarization. The formation of electronic dipoles by off-positioning of Te cations in the hexagonal channels is consistent with previous reports on the formation of dipoles in molybdenum- and vanadium-based M2 crystals.45
Furthermore, as the STEM images represent a 2D projection of 3D objects, the difference in local coordination can also be interpreted as a superposition of a segregated and regular polyhedron. These factors seem to be of particular importance in the function of these oxides as neighboring Te–O chains are expected to be involved in redox processes, while the channels are considered as reservoirs of Te.46–48 It may, thus, be important that Te remains in the complex oxide to maintain its structure. It has been previously shown that this prerequisite can be met in phase-pure (Mo,V,Te,Nb)Ox catalysts.48 The observed displacements may further indicate the ability to store and retrieve free electrons. These displacements may, thus, present the geometric locations where redox equivalents can be easily parked and donated. Thereby, severe distortions of the bulk lattice can be avoided.
Scheme 1 compares the translational structural model with the results obtained within this local structural analysis. It further highlights the observed structural irregularities in the picometer regime and the huge variety of local structures that can be present in such open structured oxides.2 However, a direct correlation of the qualitatively described local displacements with the catalytic performance is not possible, due to the involved large parameter space. Prospectively, further theoretical work is needed to convert these local structural descriptors into a detailed understanding of how they impact the performance of a heterogeneous catalyst or of any functional property. In addition, this data demonstrates that integral analytical techniques are not sufficient to fully describe the structure and composition of complex solids, which can host and stabilize the active surface.
 | ||
| Scheme 1 Comparison between the averaged ideal structure (left) and the observed local real structure (right) around the S2 site of (Mo,V,Te,Nb)Ox. The arrows denote the shift direction of the S2 site. Green: Oxygen polyhedra surrounding the S2 site; yellow: tellurium; red: oxygen, blue: S2 cation, dashed: un-displaced positions. | ||
C. Experimental
Preparation of the open structured oxides
Method 1: Hydrothermal synthesis (Nb rich; internal FHI sample number: 23926). Details on the hydrothermal synthesis of MoVTeNbOx are reported elsewhere.49 In brief, MoV0.25Te0.10Nb0.42Ox was synthesized in a stirred Premex autoclave (stirring rate: 250 rpm) at 175 °C for 17 h. 11.75 g ammonium heptamolybdate were dissolved in 140 mL distilled water at 60 °C and stirred for 15 min. In a separate batch 3.80 g ammonium niobium oxalate were dissolved in 30 mL distilled water at 40 °C. Both aqueous solutions were mixed and subsequently stirred for additional 30 min and subjected for hydrothermal treatment at 175 °C. After 1.75 h at the reaction temperature, 30 mL of an aqueous solution, which contains 3.86 g VOSO4 was added. After additional 1.75 h, 30 mL of an aqueous solution of Te(OH)6 (3.52 g) was added. The hydrothermal batch was stirred for 14 h at 175 °C, washed with water and dried at 80 °C for 2 days. The solid product was annealed at 650 °C for 2 h under flowing argon (100 ml min−1) in a rotating tube furnace. Last, the activated MoVTeNbOx was washed with H2O2.Method 2: Hydrothermal synthesis (Te rich; internal FHI sample number: 11777). Details on the hydrothermal synthesis of MoVTeNbOx are reported elsewhere.49 In brief, MoV0.25Te0.23Nb0.12Ox was synthesized in a stirred Premex autoclave (stirring rate: 250 rpm) at 210 °C for 12 h and a heating rate of 5 °C min−1. 11.75 g ammonium heptamolybdate were dissolved in 200 mL of distilled water at 80 °C. Subsequently, 4.21 g of VOSO4 were added. After complete dissolution of VOSO4 3.52 g of Te(OH)6 were added. After 15 min of stirring the solution was cooled down to 40 °C. In a separate batch 3.80 g ammonium niobium oxalate dissolved in 60 mL distilled water at 40 °C. Both aqueous solutions were mixed and stirred for additional 30 min at 40 °C. After the hydrothermal treatment the solid is washed with water and subsequently dried for 16 h at 80 °C. The solid product was annealed at 650 °C for 2 h under a flow of argon (100 ml min−1) in a rotating tube furnace.
Method 3: Slurry method (internal FHI sample number: 17817). MoV0.26Te0.11Nb0.16Ox was synthesized by dissolving 57.41 g ammonium heptamolybdate in 700 mL of distilled water at 80 °C. Subsequently, 11.41 g ammonium vanadate was added to the AHM solution followed by the addition of 17.16 g of Te(OH)6 at 40 °C. In a separate batch, 18.72 g of ammonium niobium oxalate dissolved in 250 mL of distilled water at 40 °C. After dissolving the Nb-containing solution was poured into the solution of Mo/V/Te, diluted to a total volume of 1150 mL and stirred for additional 30 min. The remaining slurry was spray dried (Tinlet = 143 °C; Toutlet = 107 °C). The precursor was calcined at 275 °C (10 °C min−1) in synthetic air (21%O2, 79%Ar, total flow: 100 mL min−1) for 1 h, followed by annealing in Ar (100 mL min−1) at 600 °C (15 °C min−1) for 2 h. The resulting phase mixture was washed with a 15%H2O2 solution and annealed again under flowing argon atmosphere (100 mL min−1) at 600 °C for 2 h.
Catalytic testing
The gas-phase propane oxidation was carried out in a fixed-bed tubular reactor in continuous flow mode. The feed was composed of C3H8, O2, H2O, and N2 in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:40:51 vol%. The reaction was carried out at 400 °C with a space velocity of 10000 ml g−1 h−1. Gas analysis was performed by an online gas chromatograph (7890A, Agilent). A combination of two capillary columns (Poraplot Q and Plot Molesieve) in connection with a thermal conductivity detector is used to analyze the permanent gases. Two capillary columns (HP-FFAP and Poraplot Q) are connected to a flame ionization detector for the analysis of alkanes, olefins, and oxygenates.
:6:40:51 vol%. The reaction was carried out at 400 °C with a space velocity of 10000 ml g−1 h−1. Gas analysis was performed by an online gas chromatograph (7890A, Agilent). A combination of two capillary columns (Poraplot Q and Plot Molesieve) in connection with a thermal conductivity detector is used to analyze the permanent gases. Two capillary columns (HP-FFAP and Poraplot Q) are connected to a flame ionization detector for the analysis of alkanes, olefins, and oxygenates.
STEM characterization
Aberration-corrected scanning transmission electron microscopy (STEM) was performed on a JEM ARM-200F CF microscope equipped with CEOS CESCOR and CETCOR hexapole aberration correctors for probe and image forming lenses, respectively, operated at 200 kV. The semi-convergence angle α was set to 22 mrad. Annular bright field (ABF-STEM) and annular dark field (ADF-STEM) images were recorded with a size of 512 × 512 pixels and a maximum dose rate of 6200 e Å−2 s−1 using an acquisition time of 40 μs per pixel. The corresponding inner and outer collection semi-angles for ABF- and ADF-detectors were set from 11 to 23 mrad, and from 67 to 250 mrad, respectively. The samples for STEM investigation were prepared by dry deposition on a carbon coated Cu grids. In order to improve the signal to noise ratio STEM images were processed using the smoothing spatial filter in Digital Micrograph before the line profile was applied. Original images are presented in the ESI (Fig. S2, S4 and S6†).Nitrogen physisorption
N2 physisorption measurements were conducted on a Quantachrome Autosorb at 77 K. Prior to measurements the samples were degassed at 120 °C and 200 °C, respectively, for 2 hours. The specific surface area was derived from the method suggested by Brunauer, Emmett, and Teller (BET).D. Conclusions
In summary, we have pinpointed the displacement of the S2 sites in complex (Mo,V,Te,Nb)Ox by means of chemical electron microscopy. It has been shown that the displacement depends on the local environment and could be primarily driven by the irregular positioning of Te entrapped in the hexagonal channels, which may be again caused by irregularities in the framework. Both of these features can explain the occurrence of structural dipoles. The described structural disorder may be one way to realize materials with well separated strained sites. The study can be considered as a model case to unravel local structural displacements on different metal sites in open crystal structures which remain hidden underneath the crystallographic average. In addition, the study highlights the need to re-examine well-established structures in the light of unravelling such hidden local structural irregularities. Future work on such materials may focus on theoretical modelling of their influence on the catalytic performance and finding new synthesis protocols that allow controlled and selective variation of the displacements. Prospectively, both ways will give access to the important question whether such structural displacements are a requirement for a good heterogeneous catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Maricruz Sanchez-Sanchez for their preparative work during her postdoctoral employment at FHI. This work was conducted in the framework of the BasCat collaboration between BASF SE, TU Berlin, FHI, and the cluster of excellence “Unified Concepts in Catalysis” (UniCat http://www.unicat.tu-berlin.de). Open Access funding provided by the Max Planck Society.Notes and references
- H. S. Taylor, Proc. R. Soc. London, Ser. A, 1925, 108, 105–111 CAS.
- L. Masliuk, M. Heggen, J. Noack, F. Girgsdies, A. Trunschke, K. E. Hermann, M. G. Willinger, R. Schlögl and T. Lunkenbein, J. Phys. Chem. C, 2017, 121, 24093–24103 CrossRef CAS.
- I. Kasatkin, P. Kurr, B. Kniep, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2007, 46, 7324–7327 CrossRef CAS PubMed.
- T. Vogt, D. A. Blom, L. Jones and D. J. Buttrey, Top. Catal., 2016, 59, 1489–1495 CrossRef CAS.
- M. Saito, K. Kimoto, T. Nagai, S. Fukushima, D. Akahoshi, H. Kuwahara, Y. Matsui and K. Ishizuka, J. Electron Microsc., 2009, 58, 131–136 CrossRef CAS PubMed.
- I. MacLaren and Q. M. Ramasse, Int. Mater. Rev., 2014, 59, 115–131 CrossRef CAS.
- T. Lunkenbein, F. Girgsdies, A. Wernbacher, J. Noack, G. Auffermann, A. Yasuhara, A. Klein-Hoffmann, W. Ueda, M. Eichelbaum, A. Trunschke, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 6828–6831 CrossRef CAS PubMed.
- X. Li, D. J. Buttrey, D. A. Blom and T. Vogt, Top. Catal., 2011, 54, 614–626 CrossRef CAS.
- T. Epicier, M. Aouine, T. T. Nguyen and J. M. M. Millet, ChemCatChem, 2017, 9, 3526–3533 CrossRef CAS.
- P. DeSanto, D. J. Buttrey, R. K. Grasselli, C. G. Lugmair, A. F. Volpe, B. H. Toby and T. Vogt, Z. Kristallogr., 2004, 219, 152–165 CAS.
- P. DeSanto, D. J. Buttrey, R. K. Grasselli, C. G. Lugmair, A. F. Volpe, B. H. Toby and T. Vogt, Top. Catal., 2003, 23, 23–38 CrossRef CAS.
- A. Trunschke, J. Noack, S. Trojanov, F. Girgsdies, T. Lunkenbein, V. Pfeifer, M. Havecker, P. Kube, C. Sprung, F. Rosowski and R. Schlögl, ACS Catal., 2017, 7, 3061–3071 CrossRef CAS.
- M. Sadakane, K. Yamagata, K. Kodato, K. Endo, K. Toriumi, Y. Ozawa, T. Ozeki, T. Nagai, Y. Matsui, N. Sakaguchi, W. D. Pyrz, D. J. Buttrey, D. A. Blom, T. Vogt and W. Ueda, Angew. Chem., Int. Ed., 2009, 48, 3782–3786 CrossRef CAS PubMed.
- Y. V. Kolen'ko, W. Zhang, R. N. d'Alnoncourt, F. Girgsdies, T. W. Hansen, T. Wolfram, R. Schlogl and A. Trunschke, ChemCatChem, 2011, 3, 1597–1606 CrossRef.
- T. Ushikubo, K. Oshima, A. Kayou and M. Hatano, Spillover and Migration of Surface Species on Catalysts, 1997, vol. 112, pp. 473–480 Search PubMed.
- R. K. Grasselli, D. J. Buttrey, P. DeSanto, J. D. Burrington, C. G. Lugmair, A. F. Volpe and T. Weingand, Catal. Today, 2004, 91(2), 251–258 CrossRef.
- P. Botella, B. Solsona, A. Martinez-Arias and J. M. L. Nieto, Catal. Lett., 2001, 74, 149–154 CrossRef CAS.
- T. T. Nguyen, M. Aouine and J. M. M. Millet, Catal. Commun., 2012, 21, 22–26 CrossRef CAS.
- D. A. Blom, T. Vogt, L. F. Allard and D. J. Buttrey, Top. Catal., 2014, 57, 1138–1144 CrossRef CAS.
- M. Aouine, T. Epicier and J. M. M. Millet, ACS Catal., 2016, 6, 4775–4781 CrossRef CAS.
- E. V. Ishchenko, R. V. Gulyaev, T. Y. Kardash, A. V. Ishchenko, E. Y. Gerasimov, V. I. Sobolev and V. M. Bondareva, Appl. Catal., A, 2017, 534, 58–69 CrossRef CAS.
- W. D. Pyrz, D. A. Blom, T. Vogt and D. J. Buttrey, Angew. Chem., Int. Ed., 2008, 47, 2788–2791 CrossRef CAS PubMed.
- Y. Y. Zhu, P. V. Sushko, D. Melzer, E. Jensen, L. Kovarik, C. Ophus, M. Sanchez-Sanchez, J. A. Lercher and N. D. Browning, J. Am. Chem. Soc., 2017, 139, 12342–12345 CrossRef CAS PubMed.
- D. Melzer, P. H. Xu, D. Hartmann, Y. Y. Zhu, N. D. Browning, M. Sanchez-Sanchez and J. A. Lercher, Angew. Chem., Int. Ed., 2016, 55, 8873–8877 CrossRef CAS PubMed.
- C. Heine, M. Havecker, M. Sanchez-Sanchez, A. Trunschke, R. Schlögl and M. Eichelbaum, J. Phys. Chem. C, 2013, 117, 26988–26997 CrossRef CAS.
- G. Fu, X. Xu and P. Sautet, Angew. Chem., Int. Ed., 2012, 51, 12854–12858 CrossRef CAS PubMed.
- M. Havecker, S. Wrabetz, J. Krohnert, L. I. Csepei, R. N. d'Alnoncourt, Y. V. Kolen'ko, F. Girgsdies, R. Schlögl and A. Trunschke, J. Catal., 2012, 285, 48–60 CrossRef.
- J. S. Valente, H. Armendariz-Herrera, R. Quintana-Solorzano, P. del Angel, N. Nava, A. Masso and J. M. L. Nieto, ACS Catal., 2014, 4, 1292–1301 CrossRef CAS.
- S. Lwin, W. J. Diao, C. Baroi, A. M. Gaffney and R. R. Fushimi, Catalysts, 2017, 7, 109 CrossRef.
- S. L. Scott, ACS Catal., 2019, 9, 4706–4708 CrossRef CAS.
- P. Botella, E. Garcia-Gonzalez, J. M. L. Nieto and J. M. Gonzalez-Calbet, Solid State Sci., 2005, 7, 507–519 CrossRef CAS.
- C. L. Jia, K. W. Urban, M. Alexe, D. Hesse and I. Vrejoiu, Science, 2011, 331, 1420–1423 CrossRef CAS PubMed.
- A. De Backer, K. H. W. van den Bos, W. Van den Broek, J. Sijbers and S. Van Aert, Ultramicroscopy, 2016, 171, 104–116 CrossRef CAS PubMed.
- A. Belianinov, Q. He, M. Kravchenko, S. Jesse, A. Borisevich and S. V. Kalinin, Nat. Commun., 2015, 6, 7801 CrossRef CAS PubMed.
- Y. Jiang, Z. Chen, Y. M. Hang, P. Deb, H. Gao, S. E. Xie, P. Purohit, M. W. Tate, J. Park, S. M. Gruner, V. Elser and D. A. Muller, Nature, 2018, 559, 343–349 CrossRef CAS PubMed.
- H. G. Brown, R. Ishikawa, G. Sanchez-Santolino, N. R. Lugg, Y. Ikuhara, L. J. Allen and N. Shibata, Ultramicroscopy, 2017, 173, 76–83 CrossRef CAS PubMed.
- O. Lindqvist and J. Moret, Acta Crystallogr., Sect. B: Struct. Sci., 1973, 29, 956–963 CrossRef CAS.
- O. Lindqvist, Acta Chem. Scand., 1967, 21, 1473–1483 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- K. M. Ok, P. S. Halasyamani, D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. Mater., 2006, 18, 3176–3183 CrossRef CAS.
- P. V. Balachandran, T. Shearman, J. Theiler and T. Lookman, Acta Crystallogr., Sect. B: Struct. Sci., 2017, 73, 962–967 CrossRef CAS PubMed.
- L. E. Orgel, Discuss. Faraday Soc., 1958, 138–144 RSC.
- S. K. Yadav, R. Ramprasad, J. Wang, A. Misra and X. Y. Liu, Modell. Simul. Mater. Sci. Eng., 2014, 22, 035020 CrossRef.
- J. Amodeo, S. Merkel, C. Tromas, P. Carrez, S. Korte-Kerzel, P. Cordier and J. Chevalier, Crystals, 2018, 8, 240 CrossRef.
- Y. H. Zhu, Q. X. Wang, L. Zhao, B. Y. Teng, W. M. Lu and Y. Han, Chem. Mater., 2012, 24, 3269–3278 CrossRef CAS.
- B. Deniau, T. T. Nguyen, P. Delichere, O. Safonova and J. M. M. Millet, Top. Catal., 2013, 56, 1952–1962 CrossRef CAS.
- O. V. Safonova, B. Deniau and J. M. M. Millet, J. Phys. Chem. B, 2006, 110, 23962–23967 CrossRef CAS PubMed.
- R. N. d'Alnoncourt, L. I. Csepei, M. Havecker, F. Girgsdies, M. E. Schuster, R. Schlögl and A. Trunschke, J. Catal., 2014, 311, 369–385 CrossRef.
- M. S. Sanchez, F. Girgsdies, M. Jastak, P. Kube, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2012, 51, 7194–7197 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr09041j |
| This journal is © The Royal Society of Chemistry 2020 |