
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle molecule distribution of RhD binding epitopes on ultraflat erythrocyte ghosts†
Sarah
Stainer‡
a,
Sara
Reisetbauer‡
a,
Joan E. A.
Ahiable
a,
Leon
Ebner
a,
Rong
Zhu
a,
Dagmar
Reindl
a,
Günther F.
Körmöczi
b and
Andreas
Ebner
 *a
*a
aMolecular Biosensing group, Institute of Biophysics, Johannes Kepler University Linz, Austria. E-mail: andreas.ebner@jku.at
bDepartment of Blood Group Serology and Transfusion Medicine, Medical University of Vienna, Austria
First published on 27th October 2020
Abstract
The Rh blood group system plays a key role in transfusion and organ transplant medicine. The complex transmembrane Rh polypeptides RhD and RhCE carry numerous antigens, including the extremely immunogenic D antigen. The Rh polypeptides form multimolecular Rh complexes with certain transmembrane and skeletal proteins, with so far only incompletely understood physiological functions. Determination of the energy landscape of individual Rh binding epitopes towards their specific interaction partners as well as their localization across the red blood cell (RBC) membrane requires single molecule approaches including large area high resolution recognition imaging. Atomic force microscopy based molecular recognition force spectroscopy in combination with single molecule recognition imaging fulfills these requirements. For unbiased single molecule results, nano-mechanical influences due to cell elasticity have to be eliminated. This is realized by generation of ultra flat erythrocyte ghosts on a solid support. We developed a protocol for the preparation of complete ultraflat erythrocyte ghosts and determined the molecular binding behaviour of different anti-D antibodies towards their binding epitopes on RhD positive and negative erythrocytes. Performing optimized topography and recognition imaging at 16 Mpixel resolution allowed localisation of individual RhD molecules at the single molecule level across an entire RBC. A map of Rh antigens across integer ultraflat RBC ghosts was generated with nanometer resolution. Here we show a homogeneous distribution on rim and dimple regions with comparable receptor densities. Furthermore, differences in the energy landscape between specific monoclonal antibodies were determined at the single molecule level.
1. Introduction
Among the 39 blood group systems, the highly polymorphic Rh system is the most important after ABO. Of the 55 defined Rh antigens residing on RhD and RhCE polypeptides on the erythrocyte surface (according to the actual status of the International Society of Blood Transfusion), the D antigen is by far the most immunogenic. Anti-D alloimmunization may lead to potentially fatal hemolytic transfusion reactions and hemolytic disease of the fetus and newborn.1,2 The Rh polypeptides are in close relation to Rh-associated glycoprotein, CD47, LW, glycophorin B and Band 3, with strong mutual interdependence regarding their expression.3 These multi-molecular Rh complexes on the red cell surface exert functions that are only incompletely understood,4–8 and their skeletal anchors may be relevant for cell shape, deformability and integrity.9 Exact Rh complex configuration and cell surface distribution are not fully characterized. In the late 80s, Paccaud et al.10 reported a homogeneous D antigen distribution without any large clusters appreciating 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 antigenic sites/erythrocyte in immunofluorescence assays. Due to technical and physical limitations, neither individual antigens nor small clusters were detectable. Following studies mainly investigated the average density on the bulk level using immunoradiometry,11 enzyme-linked immunosorbent assay,12 and flow cytometry.9,13 They allowed determination of the total number of different D epitopes, often with a focus on genetically variant RhD phenotypes.14 To provide new insights into structure, assembly and physiological function of red cell molecules, methods providing insights into the cellular distribution at the molecular level appear advantageous. Atomic force microscopy has been developed into a valuable tool in nanoscience with promising potential in nanomedicine15–17 allowing single molecule research. Since its invention,18 red blood cells (RBCs) have been in the focus of AFM research.19 With increasing performance and technical innovations, more and more studies were able to reveal molecular details. Besides research on (often disease related) morphological details20–22 using imaging methods, single molecule interactions got into focus.23–27 The combination of both approaches – morphological investigations based on AFM imaging and functional studies using single molecule bio-functionalized tips – allows the observation of structure–function relationships at single molecule level.
000 antigenic sites/erythrocyte in immunofluorescence assays. Due to technical and physical limitations, neither individual antigens nor small clusters were detectable. Following studies mainly investigated the average density on the bulk level using immunoradiometry,11 enzyme-linked immunosorbent assay,12 and flow cytometry.9,13 They allowed determination of the total number of different D epitopes, often with a focus on genetically variant RhD phenotypes.14 To provide new insights into structure, assembly and physiological function of red cell molecules, methods providing insights into the cellular distribution at the molecular level appear advantageous. Atomic force microscopy has been developed into a valuable tool in nanoscience with promising potential in nanomedicine15–17 allowing single molecule research. Since its invention,18 red blood cells (RBCs) have been in the focus of AFM research.19 With increasing performance and technical innovations, more and more studies were able to reveal molecular details. Besides research on (often disease related) morphological details20–22 using imaging methods, single molecule interactions got into focus.23–27 The combination of both approaches – morphological investigations based on AFM imaging and functional studies using single molecule bio-functionalized tips – allows the observation of structure–function relationships at single molecule level.
In one of the first combined AFM topography and recognition imaging (TREC) studies,28 the molecular distribution and local density of cystic fibrosis transmembrane conductance regulators on the inner membrane of RBCs was determined. Anti-Band III studies29 and investigations of Na+–K+ ATPases at the same system allowed location of those molecules with nanometre resolution at areas of the inner cell membrane. Although these studies showed promising results in biosensing based AFM on the inner RBC membrane, the molecular distribution of RhD binding epitopes on the entire outer membrane was not possible yet. The realization including an optimized protocol for preparation of complete intact and ultraflat (i.e. perfectly emptied and completely flattened) outer erythrocyte ghosts membranes, as well as – for the first time ever – large area high-resolution 16 Mpixel recognition imaging is presented.
2. Results and discussion
2.1 Ultraflat erythrocyte ghosts
Erythrocytes are known as round donut shaped nucleus free blood cells with a diameter of 5–10 μm.30 Because of their shape, properties like cell elasticity31,32 or membrane orientations33 differ significantly, depending on the position of investigation. This behaviour may play a major role in the application of techniques at the nanoscale. Accurate rupture lengths and sharp distributions of the effective spring constant enhance the accuracy and reproducibility of dynamic force spectroscopy experiments at the single molecule level. More importantly, the atomic force microscopy technique of topography and recognition imaging34,35 is based on accurate signal processing of the oscillation amplitudes.36 Especially the damping of the upper part of oscillation is influenced by the spring constant of the pulled system, which varies with local differences in elasticity. Additionally, the local resolution of AFM images increases with a decrease in softness and energy dissipation of the support.22 Thus, a surface with uniform high stiffness is optimal to gain highly reproducible structural and functional data at a maximum of resolution. In earlier studies, the preparation of inside-out28,29,37,38 and only partially integer outside-out RBC membranes38 was demonstrated. Within this study, we developed and optimized a new protocol to prepare complete integer ultraflat RBC ghosts in a perfectly flattened outside-out configuration: after venepuncture, RBCs should immediately be separated from freshly drawn blood to avoid any denaturation processes. RBC isolation was facilitated by a series of centrifugation steps as described previously.37 AFM, besides other single molecule techniques, requires a tight attachment of the cells to a substrate. Therefore, immobilization was realized through electrostatic adhesion using a thin positively charged layer of poly-L-lysine – a common approach in cell adherence.In addition to immobilizing RBCs to mica and glass slides – used for AFM experiments – we tested the same coupling protocol on gold coated sensors like quartz crystal microbalance (QCM) crystals and surface plasmon resonance (SPR) chips (Fig. 1A and B). The process of RBC adhesion works rather simple by incubation of a diluted and purified RBC suspension on the chosen substrate (Fig. 1C).
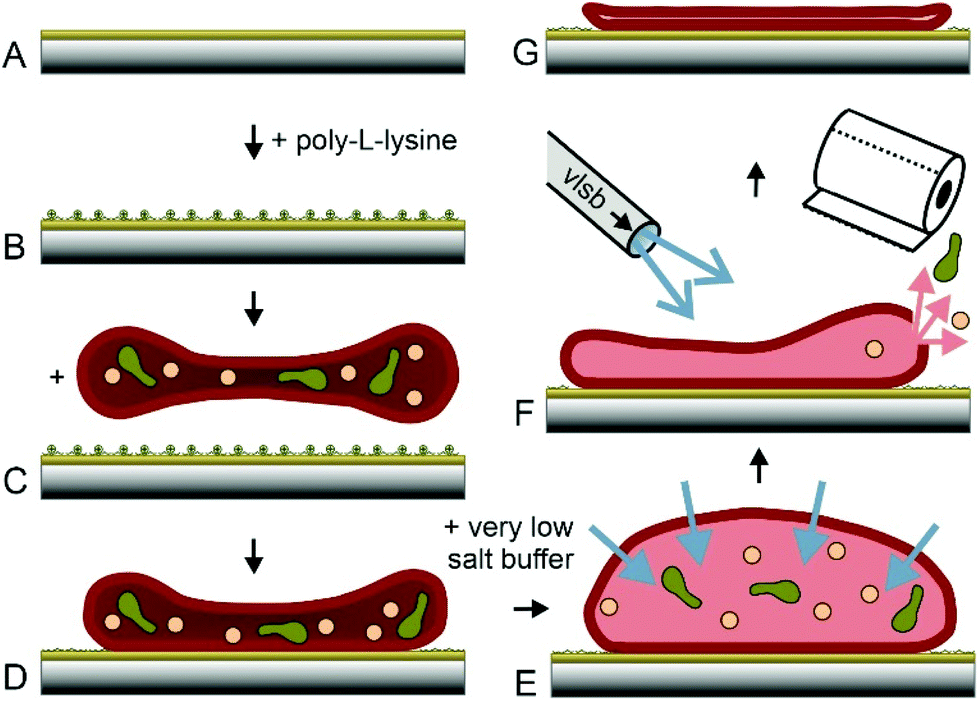 | ||
| Fig. 1 Preparation of ultraflat erythrocyte ghosts. Glass slides or gold coated surfaces (A) are incubated with poly-L-lysine (B). Purified erythrocytes are incubated on PLL and (C) attached to PLL via electrostatic adhesion (D). PBS is exchanged with VLSB (E) resulting in osmotic influx. Rinsing with 4 °C VLSB at one side and sucking on the other side (F) resulting a shear force applied to the RBCs, ending up in empty and ultraflat RBC ghosts (G). | ||
Gentle sample treatment during the previously described steps is crucial for successful ultraflat RBC generation. Mechanical stress exerted to the RBCs should be avoided. The isotonic salt concentration has to be kept accurate. At these conditions, cells stay intact and adhered RBCs (Fig. 1D) will still show their typical height of ∼2 μm. The most important step in this protocol is the quasi simultaneous opening, emptying and flattening of the immobilized RBCs. Lowering the salt concentration of the buffer results in an osmotic pressure and consequently in a significant increase of the inner cell volume (Fig. 1E). The appearance of the cells changes from donut to more or less round shape. For generation of ultraflat ghosts, the salt concentration has to be lowered sufficiently to force an osmotic induced burst of the cell. At the same time, a complete depletion of the cytosol is required. Furthermore, the upper membrane has to adhere on the lower cell membrane with a minimum of wrinkles and protrusions. All these three steps have to be performed quasi simultaneously, intertwining, and finely tuned. Lowering the temperature slows down the burst and reclosing step of the cellular membrane. To profit from this extended time scale it is necessary to cool down both, the very low salt buffer (VLSB) as well as the cells itself. Working at the temperature of ∼4 °C turned out to be the best approach. Gently rinsing with VLSB using a Pasteur pipette starting on the upper side of the slide and simultaneous withdrawal of the liquid on the opposite side (Fig. 1F, more details can be found in the ESI†) gave the highest success rate in generating completely integer ultraflat outside-out oriented RBC ghosts (Fig. 1G). To lower the risk of abrogation of the native membrane composition and to avoid any denaturation process, all steps have to be done briskly and at low temperature, followed by gently chemical fixation. Minor deviations from this protocol may result in a failure within the formation process. In the following, cause and effect of the most prominent errors are described. The first steps (Fig. 1A–D) are only critical with respect to RBC denaturation. However, the opening/closing and flattening process (Fig. 1E+F) is much more error prone. The intensity of the applied flow of VLSB is critical.
Weak streams (i.e. 0.6 mL s−1 or less) do not allow for a simultaneous opening and flatting process and yield partially filled erythrocyte ghosts. As shown in Fig. 2A (prepared with a flow of 0.6 mL s−1), the height of the membrane protuberances reaches the limit of one micrometre and they are highly moveable. This explains the loss in lateral resolution of these areas. Too weak streams result in incomplete emptying including liquid bubbles within the cell (Fig. 2A), whereas too high shear forces (i.e. flow rates above 1.1 mL s−1) prohibit correct closure of the ghosts. As a result, large areas of the prepared RBC ghosts are ruptured, exhibiting regions with heights typical for single membrane leaflets. The desired double membrane staples partially exist, but often appear convoluted (Fig. 2B, prepared with a flow rate of 1.15 mL s−1). Thus, for optimal results, the flow rate should be in the range between 0.75 and 0.85 mL s−1. If osmotic burst, depletion and membrane collapse are not perfectly aligned and performed simultaneously, the preparation results again in incompletely emptied erythrocyte ghosts. Fig. 2C shows a preparation where the osmotic burst was delayed and incomplete as result of incorrect PBS/VLSB exchange. RBC membranes may be of interest in both, dry state as well as in buffer. Drying up ultraflat erythrocyte ghosts is possible, but should be done carefully. Besides the complete exchange of buffer or serum to distilled water, the drying process itself is critical. This process has to be done slowly (e.g. overnight) at a low temperature (4–8 °C) to avoid membrane ruptures caused by spontaneous changes in surface tension. Membrane ruptures caused by incorrect drying are shown in Fig. 2D. Usually circular holes with diameters typically ranging from 50–2000 nm in the outer membrane are observable. Following our newly developed preparation protocol results in correctly oriented and fully flattened outside-out facing RBC ghosts (Fig. 2E and F). The surface-occupancy rate of cells can be easily adjusted by varying the incubation time. 15 min incubation of the centrifuged erythrocyte suspension results in a surface coverage of 31.96 ± 3.58% (i.e. area covered by ultraflat red blood cell ghosts divided by the total area), 25 min in 47.22 ± 4.72%, and 30 min in 52.19 ± 8.00%.
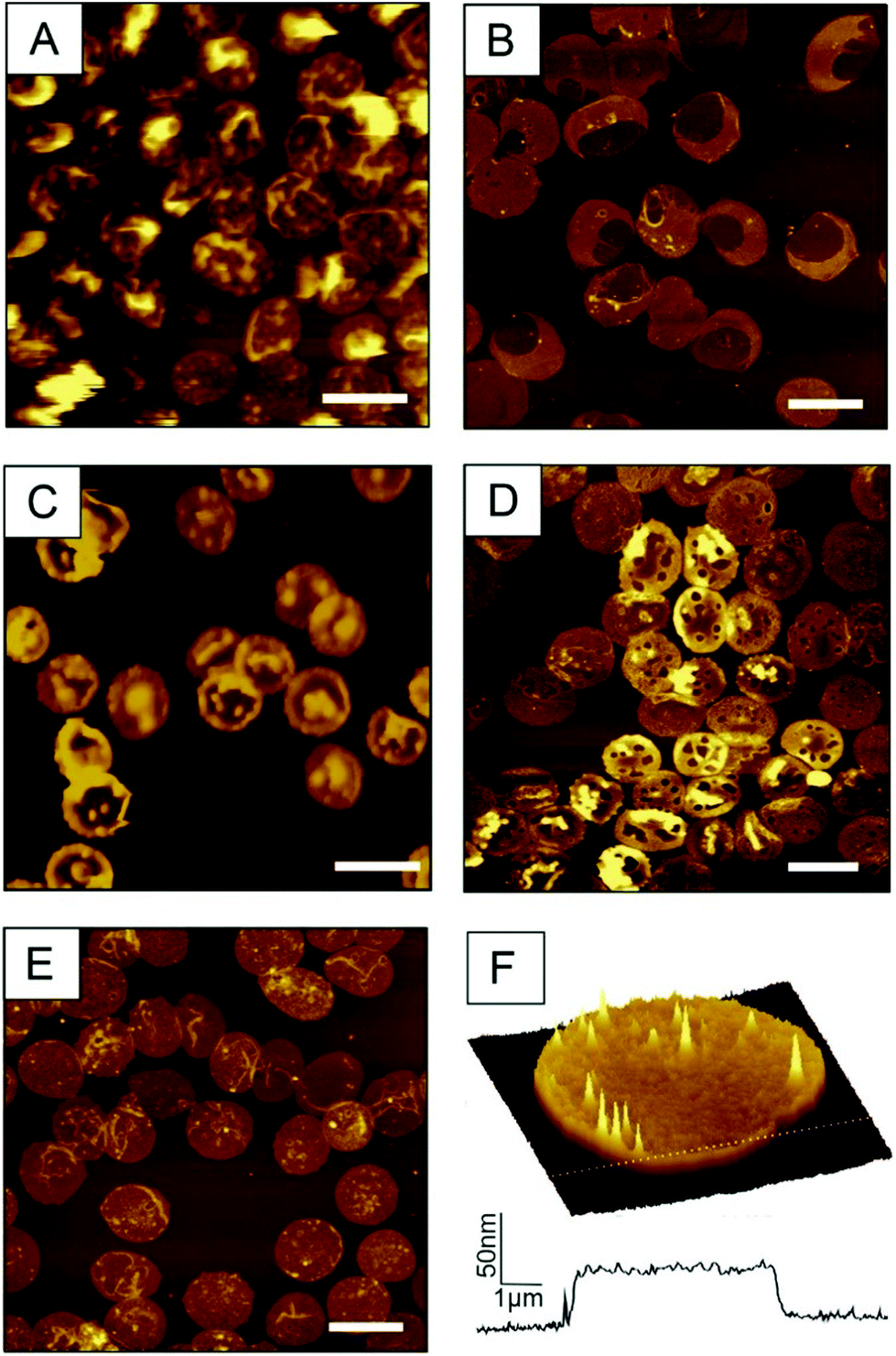 | ||
| Fig. 2 AFM images of artefacts in ultraflat RBC preparations. (A) Insufficient intensity of the VLSB shear flow causes incomplete depletion. z-Scale is 1.5 μm (B) In contrast too high shear force hinder the closing process result in ruptures erythrocyte ghosts. z-Scale is 300 nm. (C) Too slow addition of VLSB shows again partly filled erythrocytes. z-Scale is 600 nm (D) fast drying yield round shaped holes. z-Scale is 300 nm (E) correct preparation yield ultraflat outside-out RBC ghosts. z-Scale is 300 nm. (F) 100 μm2 3D image of a typical isolated ultraflat erythrocyte ghost including a cross-section. Scale bar for A–E: 10 μm. | ||
2.2 Molecular investigation of BRAD5 and ESD-1 interactions with RhD
Monitoring biorecognition on the molecular scale gives additional insights into the dynamics and nature between the binding epitopes of the D antigen and directed antibodies against these epitopes like (hidden) energy barriers, molecular dissociation rates, and the width of the energy barrier in pulling direction. There exists an enormous diversity of monoclonal anti-D antibodies that were developed for serologic RhD typing and the study of the complex D antigen consisting of at least 30 different D epitopes.3 The two representative monoclonal antibodies BRAD5 and ESD-1 used in this study were selected based on their differential fine specificity recognizing two distinct D epitopes, 6.8 and 4.1, respectively.39We used Molecular Recognition Force Spectroscopy and Molecular Recognition Imaging28,34,35,40 for the investigation of the interaction between BRAD5/ESD-1 mAbs with the 6.8 and 4.1 epitopes on RhD embedded in the erythrocyte membrane. Requirement to reach the single molecule level are ultrasharp AFM tips with a radius <10 nm and a suited tethering of the antibodies. This was realized via gas phase deposition of APTES at controlled environmental conditions,41 resulting in ∼1 nm thick amino-functionalized coating. To allow separation between unspecific adhesion and specific recognition a distensible hetero-bifunctional PEG linker was used. The latter was coupled covalently to both, the amino group on the tip apex, as well as to the antibody following established protocols.42
Force distance curves (FDCs) were recorded with these antibody functionalized tips. In case of RhD-BRAD5 complex formation, the rupture force (i.e. force acting on the cantilever directly before the forced dissociation of the complex) under a given force load can be measured directly between these two molecules. As shown in Fig. 3A, the trace (red line) shows only a sharp kink followed by a linear upwards bending of the cantilever. In contrast, in the retracting period (black line) an additional – but downwards – bending of the cantilever can be observed. The maximum in this bending reflects the rupture force. Because of the preparation of ultraflat erythrocyte ghosts, this rupture is not biased by the viscoelastic properties of the erythrocyte. Adding free (unbound) BRAD5 antibodies into the measurement chamber results in disappearance of such rupture events (Fig. 3, inset) and successfully proves the specificity of the interaction. The measured rupture forces give information on the complex strength, when a given force is applied along the axis of separation. To gain more valuable information on bond characteristics, pulling velocities can be varied. This results in a different force load acting on the complex and thus in different rupture forces. According to the Bell–Evans theory,43,44 number of energy barriers, bond lifetime (τ = 1/koff), and width of the barrier (xβ) along the pulling direction can be calculated through determination of the mean rupture force as a function of the force loading rate. The model of Evans neglects the possibility of rebinding during the retraction process. In case of low loading rates, reversible binding may occur and the model has to be adapted to take this into consideration. Friddle et al.45 introduced two regimes during the unbinding process, an equilibrium and a kinetic regime. This results, beside koff and xβ, in an additional parameter namely the equilibrium force Feq. At Feq association and dissociation are in equilibrium in a given system. The energy landscapes of both, BRAD5 and ESD-1 monoclonal antibodies were investigated towards erythrocyte embedded RhD polypetides. For this, we varied the pulling velocity from 50–10000 nm s−1. At each given velocity, at least 1000 FDCs were measured and evaluated. We compared both antibodies (BRAD5 and ESD-1) on ultraflat erythrocyte ghosts of RhD positive and RhD negative probants. As shown in Fig. 3B, the binding probability (BP, i.e. the number of FDCs exhibiting a molecular rupture event divided by the total number of recorded FDCs) differ significantly. In contrast to BPs on RhD positive cells 16.64 ± 3.79% (n = 6) for BRAD5 and 21.28% (n = 8) for ESD-1, erythrocytes of RhD negative persons showed only 1.43 ± 0.31% (n = 3) BP for BRAD5 and 3.93 ± 0.93% (n = 3) for ESD-1. Here n represents the number of datasets for each setting, always consisting of at least 1000 FDCs. The results clearly proved the specificity of interaction since Rh negative erythrocytes lack of the binding epitopes 6.8 and 4.1.
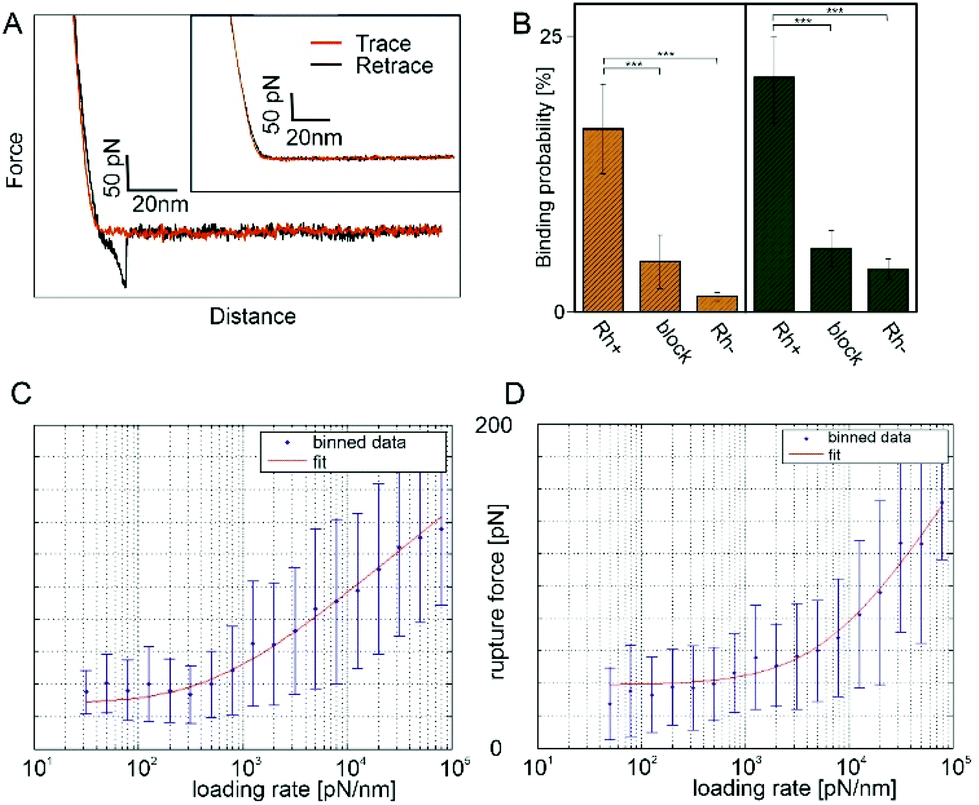 | ||
| Fig. 3 Molecular recognition force spectroscopy on ultraflat erythrocyte ghosts. (A) Typical FDC using a BRAD5 functionalized tip showing a single ligand receptor bond. Inset: Specificity proof shows no interaction. (B) Binding probabilities of BRAD5 and ESD-1 of RhD positive and negative samples successfully proving the specificity of interaction. Adding of free antibody to the system also caused a significant decrease in the binding probability. This proves that recognition events are only caused by the tip tethered antibody. ** p ≤ 0.01, *** p ≤ 0.001; (C) loading rate dependence of the rupture force using BRAD5 and (D) ESD-1 functionalized AFM tips. | ||
To prove that no other molecule than the tip tethered antibody causes rupture events, experiments on RhD positive red blood cell membranes have been repeated in the presence of the same antibody as tethered to the AFM tip. This caused a blocking of the RhD epitopes on the membranes and vanished the rupture events in FDCs (4.60 ± 2.43%, n = 3 for BRAD5 and 5.83 ± 1.63%, n = 3 for ESD-1). The higher number of bindings at blocked RhDs compared to RhD negative cells may origin from a partially incomplete block of all receptors, which is typical for a surface block in force spectroscopy experiments. Comparison of all BPs (including Student T-tests) at comparable force loading rates are shown in Fig. 3B.
Using the approach of loading rate binned data, we could show independence of the rupture force values from the loading rate in the region of low loading rates. Thus, we applied the Friddle approach45 as fitting model. The loading rate dependence including the Friddle fit is shown in Fig. 3C for BRAD5 and Fig. 3D for ESD-1. For the BRAD5 interaction with RhD, the equilibrium force is 28.16 ± 7.06 pN, the kinetic off rate is 4.73 ± 3.49 s−1, and the width of the energy barrier in pulling direction is 1.73 Å. In contrast, the equilibrium force between ESD-1 and RhD is 38.86 ± 4.53 pN with a ∼8-fold faster off rate of 36.98 ± 33.02 s−1. The width of the energy barrier is xβ is 0.92 Å.
2.3 Lateral D antigen distribution using high resolution TREC
MRFS gives insights into the nature of anti-D antibody interactions with the corresponding binding epitope at the single molecule level. Since this technique is based on movements in z-direction no lateral information is obtained. Topography and recognition imaging (TREC) allows simultaneous acquisition of sample topography and mapping of recognition sites without loss of lateral resolution. The working amplitude has to be adjusted to allow receptor ligand complexes to stay alive over a multitude of oscillations. This is a key requirement to ensure that the correct number of binding sites is detected since in this case the kinetic rate of association kon plays a less critical role.As shown in Fig. 4 left, the cantilever functionalized with one single anti D (BRAD5 or ESD-1) antibody at the apex of the tip is oscillated over the ultraflat erythrocyte ghost surface at frequencies close to the cantilevers resonance frequency (typically 7–12 kHz). Changes of the lower part of the oscillation (blue), caused by sample morphology, result in the topographical image of the membrane (Fig. 4, lower left image). In contrast, damping of the upper part of the oscillation (red) can be directly related to molecular RhD–anti-D interactions, resulting in the recognition image (Fig. 4, upper left image). To deconvolute upper amplitude reductions from noise, accurate processing is required. This results in a landscape of recognition sites (Fig. 4 upper right image). It has to be mentioned, that, in contrast to force spectroscopy including force volume mode, no qualitative data of the interaction intensities are given. Superposition of the sample topography and recognition sites allows for cross-correlation (Fig. 4 lower right image).
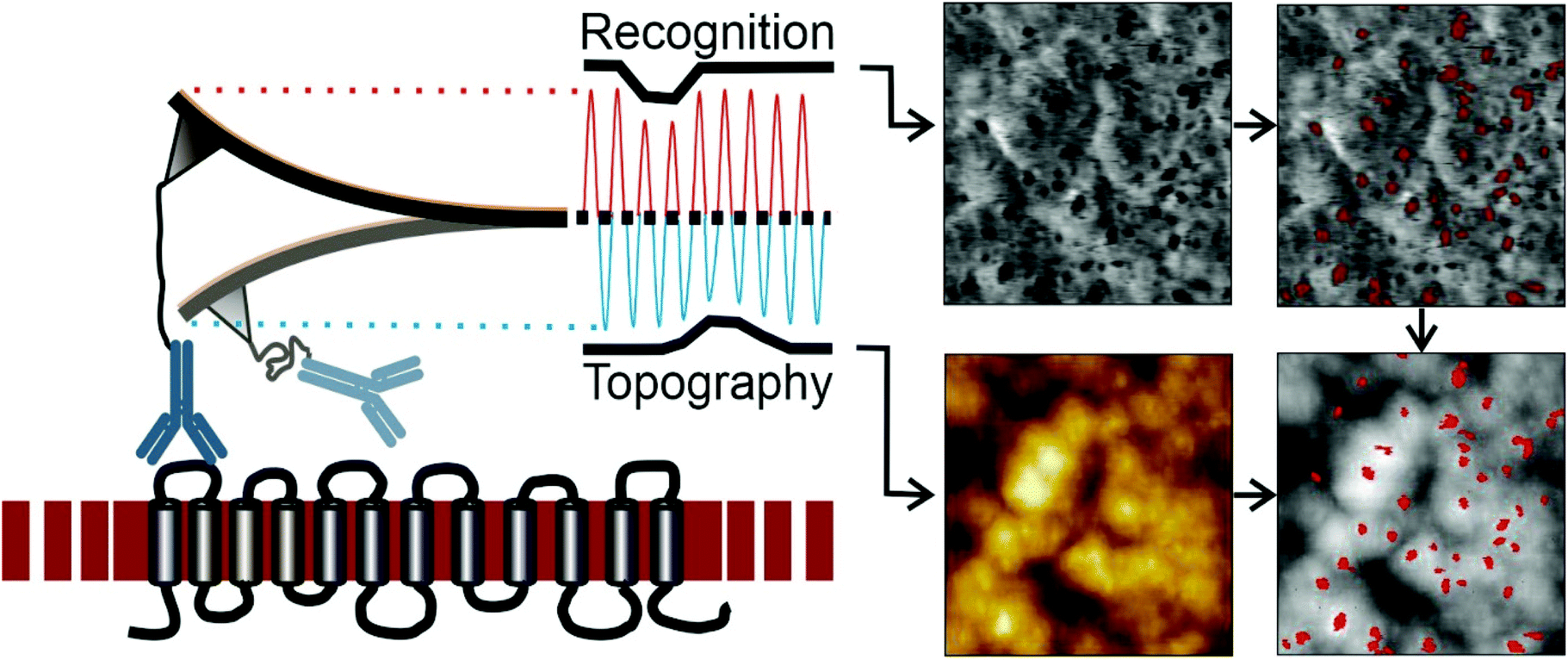 | ||
| Fig. 4 Topography and recognition imaging on utraflat RBC ghosts. Cantilever oscillation is split into upper and lower part. Changes of the lower part are caused by height changes of the membrane surface (i.e. morphology) and yield the topographical image (lower left). Reductions of the upper part of the oscillation are caused by BRAD5 IgG – RhD recognition (upper left image). Recognition spots showing ≤5-fold of the RMS of the noise are shown in the upper right image. Superposition of the erythrocyte membrane topography with the recognition spots is shown in the upper right image. Image size 1 μm2. | ||
Typically, TREC images are recorded with 1–2 lines per s for an image size of 1–2.5 μm2. Slower imaging speeds result in the same number of binding sites, whereas higher velocities cause a reduction of traced recognition sites since kon causes limitations. These limitations result in a scanning velocity of maximal 1–8 μm s−1 depending on the ligand receptor pair. For TREC imaging of complete ultraflat RBCs, which are typically 7.5–8.7 μm in diameter,30 adjustments regarding the imaging parameters had to be made to allow large area TREC images (80–100 μm2 instead of 1–10 μm2). A scanning speed of 1 line per s would result in velocities of 20 μm s−1 (since 1 Hz means that the cantilever has to move a complete line forwards and backwards). For an image comprising 100 μm2, the x movement of the cantilever had to be set to maximal 0.2 lines per s to ensure a scanning velocity of maximal 4 μm s−1. These conditions ensure a close to 100% probability to detect all surface receptors.46 Furthermore, the data acquisition frequency was set to at least 4000 data points (i.e. pixel) per line to be able to detect single RhD polypeptides and to ensure a sufficiently high TREC image resolution (i.e. 16 megapixel per image instead of typically 0.06–0.26 megapixel). Summing up, using scan speeds in the range of 2–4 μm s−1 and at least 4000 pixel per line (in each scanning direction) allows for high resolution TREC imaging with the ability to detect single RhD polypeptides and their position dependent distribution on ultraflat erythrocyte ghosts. In such a 16 megapixel image, the size of one pixel equates to 2.44 × 2.44 nm. Using either a BRAD5 or an ESD-1 functionalized tip, we were able to perform TREC imaging on single complete outside-out oriented ultraflat erythrocyte ghosts with single molecule resolution for the first time ever. A typical 16 Mpixel topographical image (Fig. 5 upper left) and a simultaneously acquired 16 Mpixel recognition image (Fig. 5 right) after data processing of a single ultraflat erythrocyte ghost are shown. It is obvious that a high number of recognition sites (i.e. recognized RhD polypeptides) is rather homogeneously scattered all over the whole cell membrane. The vast majority of D epitopes is conformation dependent. Any denaturation eliminates antibody binding to these receptors. The preservation of antibody binding to D epitopes on ultraflat ghosts underscores careful cell preparation and argues for retention of structural integrity.
 | ||
| Fig. 5 Whole cell 16 Mpixel topography (upper left) and recognition image (right) of an RhD positive RBC investigated with a BRAD5 mAb functionalized AFM tip. Scale bar is 1 μm. Surface occupancy rate does not vary significant between rim and dimple regions of the very same cell surface. | ||
In detail analysis also taking into account the grain size of a single interaction (as described in the experimental part) resulted in 177.16 ± 32.33 (n = 6) RhD polypeptides per μm2 on the inner half of the erythrocyte cell membrane (dimple region), whereas the outer part (rim region) showed 169.57 ± 23.51 RhD polypeptides per μm2 (Fig. 5 lower left).
This proves that there is no significant difference between inner (dimple) and outer part (rim) of the erythrocyte membrane, which is in agreement with fluorescence imaging data (see ESI†). Both BRAD5 and ESD-1 antibodies recognized similar numbers of D antigens per erythrocyte (BRAD5 13597 ± 2747, n = 18; ESD-1 15098± 5113, n = 9), which is in the same order as previous studies.47,48
In the following, we compared the density of recognized RhD polypeptides on RhD positive, negative and weak D RBC membranes using ESD-1 and BRAD5 antibodies by performing high resolution TREC. Performing recognition imaging using the same antibody functionalized tip on the surface of RhD negative patients resulted, as expected, in a significant loss of binding events, i.e. 544 ± 335, n = 10 for BRAD5 and 550 ± 32, n = 2 for ESD-1. In addition, weak D red blood cells showed 1078, n = 1 RhDs for BRAD5 and 2234 ± 655 for ESD-1 mAbs. Here n defines the number of TREC imaging for each setting. These data are in good agreement to previous studies. In a pioneering publication of Paccaud et al.10 a number of 16400 RhD/erythrocyte was reported using immune-gold electron microscopy and 10000 sites/cell by performing a radioimmunoassay. In a more recent study by Chapman et al.49 using a 1:2 blend of BRAD3:BRAD5 mABs 13600 RhD sites per red blood cell were determined on the same blood group phenotype as in his study (CcDEe).
Our study shows that nanoscale TREC analysis of RhD on the red cell surface is feasible at the real single molecule level. RhD has a pivotal role in transfusion medicine owing to its tremendous immunogenicity for RhD-negative individuals when coming into contact with it via transfusion, transplantation or pregnancy. The consequences of anti-D alloimmunization may be life-threatening, including hemolytic transfusion reactions and hemolytic disease of the fetus and newborn. In sharp contrast to these well-defined implications for clinical medicine, the function of the RhD polypeptide on the red cell surface remains obscure. The overall structure of RhD exhibiting six transmembrane loops resembles a channel protein.50 Rh polypeptides may, however, have only a stabilizing function for the assembly of the Rh/Band3 macrocomplex that involve also RhAG, CD47, LW and some others.51 A promising strategy to elucidate the functions of these tightly connected molecule complexes could be a TREC-based analysis of red cells that lack some of the Rh complex constituents, as in Rh or Band3 deficiency, known to be associated with characteristic pathological signs.50,52,53
Taken together, this nanoscale approach shows a homogenous distribution of 8.6 and 4.1 epitopes at the single molecule level. The realization using the homogeneity of RhD epitopes may aid in investigating the distribution and quantity of other red cell surface receptors. Furthermore single molecule force spectroscopy experiments have the potential to explore binding differences of various RhD phenotypes and genotypes including rare events and variations in the interaction forces at the single molecule level.
3. Experimental
3.1 Materials
If not stated otherwise, all chemicals were used in their highest available grade. APTES, ethylene glycol-bis(β-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), glutaraldehyde (25%), sodiumcyanoborohydride (NaCNBH3), triethylamine (TEA), ethanolamine hydrochloride, and PLL were purchased from Sigma–Aldrich (Austria), NaCl, 2-propanol, chloroform and citric acid form VWR (Austria), and Na2HPO4 from Fluka (Austria). Monoclonal human IgG anti-D antibodies were obtained from IBGRL Research Products (Bristol, UK; BRAD5) and from Bio-Rad (Vienna, Austria; ESD-1), recognizing the D epitope 6.8 and 4.1, respectively.39 All antibodies were purified to get rid of azide and other substances disturbing the tip functionalization and the interaction process.| Isotonic PBS buffer | 5 mM Na2HPO4, 150 mM NaCl, 0.2 mM EGTA, pH adjusted to 7.4 (with HCl) |
| PBS buffer | 5 mM Na2HPO4, 150 mM NaCl, pH adjusted to 7.4 (with HCl) |
| Very low salt buffer (VLSB) | 0.3 mM Na2HPO4, 0.2 mM EGTA, pH adjusted to 7.4 (with HCl/NaOH) |
| Centrifuge | Eppendorf mini spin |
3.2 Ultraflat erythrocyte ghost sample preparation
Glass cover slips (Fischer, Austria) were mounted into a Teflon™ slide holder, covered with isopropanol, sonicated for 5 minutes, followed by replacement of isopropanol. After repeating this step 3 times, the cleaned glass slides were dried in a gentle stream of nitrogen gas and placed on a Petri dish. A drop of 300 μl 0.01% PLL in ultrapure water (resistance >18 mΩ) was placed on the glass slide and incubated for 30–90 minutes.Routine serologic Rh blood group typing of RBCs was performed as described.54 For purification of erythrocytes four drops of freshly drawn venous blood were diluted in 1000 μl isotonic PBS and centrifuged at 3800 rpm for four minutes. The supernatant was discarded and the pellet was re-suspended in 1000 μl isotonic PBS. This procedure was repeated three times. ∼3 μl of the pellet were dissolved in 1800 μl of PBS (ERY suspension). After completion of the PLL incubation time, the PLL covered glass slides were washed twice with ultrapure water and dried with a N2 or argon gas stream. The following steps were executed on ice. 400 μl of the ERY suspension were transferred onto the glass slide. The liquid should form a droplet on the same location where previously PLL coating occurred. The suspension was incubated for 15 minutes on ice. When the incubation time ended, the VLSB (4 °C, in a spray bottle) was sprayed on the glass slide. The flow rate should be adjusted to 0.75–0.85 mL s−1 (when using conventional 200–500 mL spray bottles). To do so, the glass slide was held almost perpendicular to the ground. A facial tissue was placed right under the slide on the Petri dish to remove excess liquid. After removing the ERY incubation solution the VLSB was sprayed in a zig-zag motion from the top of the slide to the bottom with low pressure applied to the bottle. Immediately afterwards, 200 μl of 1% vol/vol glutaraldehyde in PBS was added to the erythrocyte covered side of the glass slide and incubated for 30–45 minutes at room temperature. The sample was washed three times with 2000 μl PBS with 5 minutes incubation time between the PBS exchanges. The samples were used immediately or stored for a short time at 4 °C. For preparation of dried samples, PBS was exchanged by ultrapure water (10 washing steps, 5 min incubation time in between). Drying was performed at room temperature or overnight at lowered temperature (4–8 °C).
3.3 Tip functionalization
Sharp clean AFM Si3N4 cantilevers MSCT AUHAW (Bruker, France) were amino-functionalized using gas phase deposition of APTES.41 For this, a vial containing 60 μL monomeric APTES and a second vial filled with 20 μL trimethylamine (TEA) were placed in a desiccator, which was previously filled with argon gas. After 120 min reaction time, both vials were removed and the desiccator was extensively rinsed with Ar gas. For increasing the amount of formed siloxane bonds, curing was done for 48 h. Amino-functionalized tips were used either immediately or stored under Ar to avoid oxidation of the amine. As a next step the heterobifunctional PEG crosslinker NHS-PEG-aldehyde55 (synthesized by Prof. Gruber, JKU Linz, Austria) was coupled. 1 mg of the linker was dissolved in 0.5 mL chloroform. APTES functionalized cantilevers were added into this solution using a small Teflon chamber. 50 μL of TEA as a catalyst were added and the cantilevers were allowed to react for 120 min. After cleaning with chloroform (3 × 5 min), tips were dried in a gentle stream of N2 or argon gas. To deprotect the acetal residue, tips were immersed in an aqueous solution containing 1% citric acid. The tips were washed with water (3 × 5 min) and dried again. Immediately afterwards, the tips were placed in a Petri dish on a clean Parafilm™. 30 μL of a PBS buffer containing the antibody (0.1 mg ml−1) were incubated. It needs to be mentioned that these antibody solution has to be free of any other protein like BSA and other molecules showing reactivity towards benzaldehyde. 2 μL of an aqueous solution containing 1 M sodium cyanoborohydride and 20 mM NaOH were gently added to the antibody solution, carefully mixed, and allowed to react for one hour. To inactivate any unreacted aldehyde, 5 μL of an ethanolamine solution (1 M, pH 8.0) were added and allowed to react for 10 min. Finally antibody functionalized tips were washed with PBS (3 × 5 min) and used immediately or stored at 4 °C for not more than a few days. Functionalisation was done either with purified BRAD5 or with purified ESD-1 antibodies.3.4 Molecular recognition force spectroscopy
All measurements were performed on a PicoPlus 5500 AFM (Keysight, USA). Each erythrocyte ghost sample was investigated with AFM imaging in contact or tapping mode prior to MRFS experiments. Only optimal preparations with high level of surface occupancy and ultraflat intact preparations were used. The tips of AFM cantilever were functionalized as described previously. The soft cantilever B, C, and D with nominal spring constants of 20, 10, and 30 pN nm−1 were used for pulling experiments. The correct spring constants were determined using Keysight Thermal K program. Pulling velocity was varied from 0.087 to 9.794 μm s−1. At each rate at least 1000 FDC were recorded. The position was varied all 200 FDCs to avoid surface specific artefacts. Each dataset was evaluated using the homebuilt Matlab™ based software kspec. For each FDC, the sensitivity, unbinding length, rupture force and effective spring constant was determined. Furthermore, the binding probability (number of FDCs showing a ligand receptor interaction divided by the total number of FDCs collected at these conditions) was determined. The complete datasets of BRAD5 or ESD-1 functionalized tips with RhD polypeptides embedded in the erythrocyte membrane at the same conditions were split with respect to the loading rate. In this loading rate binning,56 the data were divided into segments with equal length in the semi-logarithmic plot. A MATLAB™-based framework was developed, that allows to define the number of bins per decade, sorting all data in a loading rate range stretching over one decade into N segments with apparently equidistant boundaries. Bins containing less than 40 data points were excluded from further evaluation. After binning the data, weighted mean values and standard deviations were calculated for the rupture force data within each bin. For the collected data points within each bin, a probability density function was calculated. Fitting of the loading rate binned data was performed using the model of Friddle, Noy and De Yoreo.45 For specificity proof experiments, positively tested cantilevers (i.e. with mAb functionalized tips showing BP > 10%) were used. After performing a dataset of at least 1000 FDC at a given loading rate, 200 μL PBS solution of containing the same type of antibody were incubated to gain a final concentration of 0.16 mg mL−1. After 30–60 min a second dataset is collected at the very same conditions.3.5 Fluorescence imaging
Fluorescence measurements were performed on a ZEISS Axio Observer·D1 Inverses Microscope equipped with an Oligochrome (1012-022-0-01) light source (TILL Photonics, Germany). A 63× objective (NA = 1.46, ZEISS, Germany) with immersion oil and a Cy5-4040A Filter Set (excitation/bandwidth: 628/40 nm; emission/bandwith: 692/40 nm) (AHF Analysentechnik, Germany) were used for the measurement. Fluorescence images were recorded by a HAMAMATSU digital camera C11440 ORCA flash 2.8. The image shown in the supplementary section has a pixel size of 3.63 μm. The measurement was performed with the software Micro-Manager-1.3. The exposure time was 10 s. Monoclonal BRAD5 antibodies were labelled by Cy5-NHS ester as described previously57 resulting in 3.8 dyes per antibody. Erythrocytes were incubated for 15 minutes with a 2.38 μmol L−1 mAb solution in PBS, followed by careful washing (15 times with PBS buffer).3.6 Recognition imaging
All TREC measurements were performed on a PICOPlus 5000 AFM (Keysigth, USA) equipped with a TREC™ box. Bio-functionalized cantilevers (as described previously) with a nominal spring constant of 100 pN nm−1 were used. Scan areas less than 3 × 3 μm were imaged with 1 line per s and 256 or 512 lines per image (conventional resolution TREC). Large area scans were imaged with 0.2 line per s and 4096 lines per image (high resolution TREC). The excitation frequency was set 0.5–1 kHz below the resonance frequency of the cantilever. For each functionalized tip, the optimal working oscillation amplitude was determined by observing the recognition signal as function of the amplitude. Too high as well as too low amplitudes result in a loss of recognition signal.36 (details are given in the ESI section†). Specificity proof experiments with a blocked tip were performed after collecting recognition images exhibiting recognition events. For this, the cantilever was withdrawn from the surface. 200 μL of measurement buffer were removed and the same amount of the blocking antibody (i.e. BRAD5 or ESD-1) dissolved in measurement buffer was added resulting a final concentration of 0.16 mg mL−1. After an incubation time of 30–60 min, recognition imaging was performed at the very same conditions. Gwyddion (version 2.55) and SPIP (version 7.4) were used for data processing. Statistics on recognition events were extracted from the recognition image. A reduction of the upper oscillation amplitude by at least a fivefold of the root mean square of the noise (RMS) was defined as recognition noise threshold criterion. The RMS was determined at min. 5 different positions devoid of recognition events and crosstalk. To remove scanning artefacts, a minimal grain size threshold of recognition sites was set to 5 × 5 pixel (i.e. 12 × 12 nm). This size results from the total contact area of an antibody tethered to the tip via a PEG27 crosslinker. To calculate the number of single RhD binding events the total area meeting the recognition noise threshold criterion and the minimal grain size threshold of recognition sites was divided by the expected area of a single binding event (5 × 5 pixel). For calculating the differences on rim and dimple regions, TREC images of ultraflat erythrocyte ghosts were analysed for D antigen distribution patterns: (i) binary images of recognition sites were generated using the previously mentioned noise and recognition spot grain size threshold criteria; (ii) the erythrocyte surface was split into dimple (central) and rim (outer) regions. The outer rim region was defined as the outer 2 μm of the round shaped membrane; (iii) receptor densities (grains per μm2) and grain areas in % (area of grains divided by imaged erythrocyte area) were calculated.4. Conclusions
Biosensing single molecule AFM investigation on erythrocyte membranes requires an optimal preparation of the cells. To the best of our knowledge, we present for the first time, a protocol for stable and reproducible formation of completely intact ultraflat erythrocyte ghosts. This allows for gaining large area maps of recognition sites superimposed with topography at nanometre resolution realized by performing 16 Mpixel TREC images. With this approach, we could prove a homogeneous distribution of functional RhD polypeptides on erythrocyte surfaces on both rim and dimple region. Single Molecule Force Spectroscopy experiments using monoclonal anti-D antibodies gave insights into the molecular binding behaviour towards the D antigen. Differences in the width of the energy barrier as well as in the kinetic off-rate were observable. In addition to these technical achievements, this RhD-centred study represents a new platform for further investigating the complexity of the red cell surface. Such nanometre resolution and label-free research may give novel insights into RBC biology as well as blood group phenotype analysis relevant for transfusion medicine with a focus on localizing local receptors and their distribution over the outer red blood cell membrane.Conflicts of interest
The authors declare no conflict in interest.Acknowledgements
We are thankful to the Ordensklinikum Linz, Barmherzige Schwestern Elisabethinen for their support in blood drawing, Tom Netzmann, Alexandra Seyer, Verena Arminger (beside some of the authors) for blood donating, and the Blood Center Linz of the Upper Austrian Red Cross for Rh typing of investigated blood samples. This study was supported by the Linz Institute of Technology (LIT), University Linz and the Government of Upper Austria, Project Nanoblood (LIT-2019-7-SEE-113).Notes and references
- R. M. Fasano, E. K. Meyer, J. Branscomb, M. S. White, R. W. Gibson and J. R. Eckman, Transfus. Med. Rev., 2019, 33, 12–23 CrossRef.
- M. Franchini, G. L. Forni, G. Marano, M. Cruciani, C. Mengoli, V. Pinto, L. De Franceschi, D. Venturelli, M. Casale and M. Amerini, Blood Transfus., 2019, 17, 4 Search PubMed.
- N. D. Avent and M. E. Reid, Blood, 2000, 95, 375–387 CrossRef CAS.
- L. J. Bruce, R. Beckmann, M. L. Ribeiro, L. L. Peters, J. A. Chasis, J. Delaunay, N. Mohandas, D. J. Anstee and M. J. Tanner, Blood, 2003, 101, 4180–4188 CrossRef CAS.
- M. E. Reid and N. Mohandas, Semin. Hematol., 2004, 41, 93–117 CrossRef CAS.
- F. Gruswitz, S. Chaudhary, J. D. Ho, A. Schlessinger, B. Pezeshki, C.-M. Ho, A. Sali, C. M. Westhoff and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9638–9643 CrossRef CAS.
- S. Mu, Y. Cui, W. Wang, L. Wang, H. Xu, O. Zhu and D. Zhu, Transfus. Med., 2019, 29, 121–127 CrossRef CAS.
- N. Burton and G. Daniels, Vox Sang., 2011, 100, 129–139 CrossRef CAS.
- M. Salomao, X. Zhang, Y. Yang, S. Lee, J. H. Hartwig, J. A. Chasis, N. Mohandas and X. An, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8026–8031 CrossRef CAS.
- J.-P. Paccaud, J.-L. Carpentier and J. A. Schifferli, J. Immunol., 1988, 141, 3889–3894 CAS.
- E. Rochna and N. Hughes-Jones, Vox Sang., 1965, 10, 675–686 CAS.
- E. Austin, L. Smith and R. Walker, Vox Sang., 2001, 80, 179–183 CrossRef CAS.
- G. F. Körmöczi, E.-M. Dauber, O. A. Haas, T. J. Legler, F. B. Clausen, G. Fritsch, M. Raderer, C. Buchta, A. L. Petzer and D. Schönitzer, Blood, 2007, 110, 2148–2157 CrossRef.
- S. G. Sandler, L. N. Chen and W. A. Flegel, Br. J. Haematol., 2017, 179, 10–19 CrossRef CAS.
- J. J. O. Toro and F. C. Gracia, Biomed. J. Sci. Technical Res., 2019, 22, 17034–17037 Search PubMed.
- S. Logothetidis, Nanomedicine and nanobiotechnology, Springer Science & Business Media, 2012 Search PubMed.
- D. Nikova, T. Lange, H. Oberleithner, H. Schillers, A. Ebner and P. Hinterdorfer, in Applied Scanning Probe Methods III, Springer, 2006, pp. 1–26 Search PubMed.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930 CrossRef.
- S. Gould, B. Drake, C. Prater, A. Weisenhorn, S. Manne, H. Hansma, P. Hansma, J. Massie, M. Longmire and V. Elings, J. Vac. Sci. Technol., A, 1990, 8, 369–373 CrossRef CAS.
- A. Kamruzzahan, F. Kienberger, C. M. Stroh, J. Berg, R. Huss, A. Ebner, R. Zhu, C. Rankl, H. J. Gruber and P. Hinterdorfer, Biol. Chem., 2004, 385, 955–960 CAS.
- R. Nowakowski, P. Luckham and P. Winlove, Biochim. Biophys. Acta, Biomembr., 2001, 1514, 170–176 CrossRef CAS.
- A. Ebner, H. Schillers and P. Hinterdorfer, in Atomic Force Microscopy in Biomedical Research, Springer, 2011, pp. 223–241 Search PubMed.
- J. L. Maciaszek, K. Partola, J. Zhang, B. Andemariam and G. Lykotrafitis, J. Biomech., 2014, 47, 3855–3861 CrossRef.
- J. J. Valle-Delgado, P. Urbán and X. Fernandez-Busquets, Nanoscale, 2013, 5, 3673–3680 RSC.
- Y. Pan, F. Wang, Y. Liu, J. Jiang, Y.-G. Yang and H. Wang, Nanoscale, 2014, 6, 9951–9954 RSC.
- F. A. Carvalho, S. Connell, G. Miltenberger-Miltenyi, S. V. Pereira, A. Tavares, R. A. Ariëns and N. C. Santos, ACS Nano, 2010, 4, 4609–4620 CrossRef CAS.
- A. F. Guedes, C. Moreira, J. B. Nogueira, N. C. Santos and F. A. Carvalho, Nanoscale, 2019, 11, 2757–2766 RSC.
- A. Ebner, D. Nikova, T. Lange, J. Häberle, S. Falk, A. Dübbers, R. Bruns, P. Hinterdorfer, H. Oberleithner and H. Schillers, Nanotechnology, 2008, 19, 384017 CrossRef.
- Y. Shan, Z. Wang, X. Hao, X. Shang, M. Cai, J. Jiang, X. Fang, H. Wang and Z. Tang, Anal. Methods, 2010, 2, 805–808 RSC.
- M. Diez-Silva, M. Dao, J. Han, C.-T. Lim and S. Suresh, MRS Bull., 2010, 35, 382–388 CrossRef CAS.
- Y. Park, C. A. Best, T. Kuriabova, M. L. Henle, M. S. Feld, A. J. Levine and G. Popescu, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 051925 CrossRef.
- K. E. Bremmell, A. Evans and C. A. Prestidge, Colloids Surf., B, 2006, 50, 43–48 CrossRef CAS.
- H. Deuling and W. Helfrich, Biophys. J., 1976, 16, 861–868 CrossRef CAS.
- C. Stroh, H. Wang, R. Bash, B. Ashcroft, J. Nelson, H. Gruber, D. Lohr, S. Lindsay and P. Hinterdorfer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12503–12507 CrossRef CAS.
- C. M. Stroh, A. Ebner, M. Geretschläger, G. Freudenthaler, F. Kienberger, A. Kamruzzahan, S. J. Smith-Gill, H. J. Gruber and P. Hinterdorfer, Biophys. J., 2004, 87, 1981–1990 CrossRef CAS.
- J. Preiner, A. Ebner, L. Chtcheglova, R. Zhu and P. Hinterdorfer, Nanotechnology, 2009, 20, 215103 CrossRef.
- A. Swihart, J. Mikrut, J. B. Ketterson and R. Macdonald, J. Microsc., 2001, 204, 212–225 CrossRef CAS.
- H. Wang, X. Hao, Y. Shan, J. Jiang, M. Cai and X. Shang, Ultramicroscopy, 2010, 110, 305–312 CrossRef CAS.
- M. Scott, Transfus. Clin. Biol., 2002, 9, 23–29 CrossRef CAS.
- F. Kienberger, A. Ebner, H. J. Gruber and P. Hinterdorfer, Acc. Chem. Res., 2006, 39, 29–36 CrossRef CAS.
- A. Ebner, P. Hinterdorfer and H. J. Gruber, Ultramicroscopy, 2007, 107, 922–927 CrossRef CAS.
- A. Ebner, L. Wildling and H. Gruber, in Atomic Force Microscopy, Springer, 2019, pp. 117–151 Search PubMed.
- G. I. Bell, Science, 1978, 200, 618–627 CrossRef CAS.
- R. Merkel, P. Nassoy, A. Leung, K. Ritchie and E. Evans, Nature, 1999, 397, 50–53 CrossRef CAS.
- R. W. Friddle, A. Noy and J. J. De Yoreo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13573–13578 CrossRef CAS.
- M. Leitner, N. Mitchell, M. Kastner, R. Schlapak, H. J. Gruber, P. Hinterdorfer, S. Howorka and A. Ebner, ACS Nano, 2011, 5, 7048–7054 CrossRef CAS.
- J.-P. Cartron, Blood Rev., 1994, 8, 199–212 CrossRef CAS.
- J. Jones, P. Lloyd-Evans and B. Kumpel, Vox Sang., 1996, 71, 176–183 CrossRef CAS.
- G. Chapman, J. Ballinger, M. Norton, D. Parry-Jones, N. Beharry, C. Cousins, C. Dash and A. Peters, Clin. Exp. Immunol., 2007, 150, 30–41 CrossRef CAS.
- C. H. Huang and M. Ye, Cell. Mol. Life Sci., 2010, 67, 1203–1218 CrossRef CAS.
- N. M. Burton and D. J. Anstee, Curr. Opin. Hematol., 2008, 15, 625–630 CrossRef CAS.
- R. R. Gammon, A. Delk, P. Houtz, H. Alvarez and N. Benitez, Lab. Med., 2020 DOI:10.1093/labmed/lmaa009.
- L. Kager, L. J. Bruce, P. Zeitlhofer, J. F. Flatt, T. M. Maia, M. L. Ribeiro, B. Fahrner, G. Fritsch, K. Boztug and O. A. Haas, Pediatr. Blood Cancer, 2017, 64, e26227 CrossRef.
- G. F. Körmöczi, T. J. Legler, G. L. Daniels, C. A. Green, R. Struckmann, C. Jungbauer, S. Moser, M. Flexer, D. Schönitzer and S. Panzer, Transfusion, 2004, 44, 575–580 CrossRef.
- L. Wildling, B. Unterauer, R. Zhu, A. Rupprecht, T. Haselgrübler, C. Rankl, A. Ebner, D. Vater, P. Pollheimer and E. E. Pohl, Bioconjugate Chem., 2011, 22, 1239–1248 CrossRef CAS.
- F. Rico and V. T. Moy, J. Mol. Recognit., 2007, 20, 495–501 CrossRef CAS.
- M. Cooper, A. Ebner, M. Briggs, M. Burrows, N. Gardner, R. Richardson and R. West, J. Fluoresc., 2004, 14, 145–150 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr04393a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |