
Decomposition of amyloid fibrils by NIR-active upconversion nanoparticles†
Takunori
Harada
 *a,
Hiraku
Matsuzaki
a,
Ryohei
Oyama
a,
Takuma
Takeuchi
a,
Tomoaki
Takei
a,
Taisuke
Ninomiya
a,
Kouta
Takami
a,
Takanori
Inoue
a,
Hiroyasu
Nishiguchi
b,
Emi
Hifumi
b,
Hiroyuki
Shinto
c,
Hiromi
Takahashi
d and
Kazuo
Umemura
e
*a,
Hiraku
Matsuzaki
a,
Ryohei
Oyama
a,
Takuma
Takeuchi
a,
Tomoaki
Takei
a,
Taisuke
Ninomiya
a,
Kouta
Takami
a,
Takanori
Inoue
a,
Hiroyasu
Nishiguchi
b,
Emi
Hifumi
b,
Hiroyuki
Shinto
c,
Hiromi
Takahashi
d and
Kazuo
Umemura
e
aDepartment of Integrated Science and Technology, Faculty of Science and Technology, Oita University, Dannoharu, 700, Oita city 870-1192, Japan. E-mail: tharada@oita-u.ac.jp; Tel: +81-(0)97-554-7622
bResearch Promotion Institute, Oita University, Dannoharu, 700, Oita city 870-1192, Japan
cDepartment of Chemical Engineering, Fukuoka University, 8-19-1 Nanakuma, Jonan-ku, Fukuoka 814-0180, Japan
dSystem instruments Co. Ltd, 776-2, Komiya, Hachioji, Tokyo 192-0031, Japan
eTokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan
First published on 14th January 2020
Abstract
We demonstrate amyloid fibril (AF) decomposition induced by NIR-active upconversion nanoparticles complexed with photosensitisers. The process is triggered by upconversion, which initiates a photochemical reaction cascade that culminates in the generation of the highly reactive singlet-oxygen product 1O2 close to the amyloid superstructures, resulting in AF decomposition.
Introduction
Almost all protein exists in two structurally distinct forms: a globular soluble form and an aggregated form that is often produced at extreme temperature or pH, at incompatible ionic strength, at very high protein concentration, or in the presence of certain metal ions.1 The structures of protein aggregates vary considerably, ranging from amorphous forms with no defined intermolecular interactions to highly ordered structures, so-called amyloid fibrils (AFs).2 The formation of extracellular amyloid plaques is a characteristic of protein-folding diseases. In these diseases, native proteins undergo conformational change and are deposited in the extracellular space of the brain or in the walls of cerebral blood vessels as amyloid plaques, ultimately resulting in pathological changes in cells.3 The conversion of natively folded proteins, which are usually found in bodily fluids in a soluble form (partially α-helical), to linear β-structure-rich aggregates, is an intrinsic problem in the aetiology of many protein-folding diseases, including Alzheimer's and Parkinson's diseases.4 Although the underlying pathophysiology of the transformation of the originally released monomeric peptide into the abnormal form is still not well understood, a significant body of evidence suggests that AF deposition is in some way central to the pathogeneses of folding diseases.5 Recent results indicate that the toxicity of amyloidogenic proteins lies not in the insoluble fibrils but rather in prefibrillar oligomeric intermediates that exist in the early stages of fibril formation.6–8 The toxicity of the early aggregates has been attributed to their large exposed hydrophobic surfaces, which may inappropriately interact with a wide range of cellular components.9Much effort has been directed towards the development of therapeutic approaches or agents that can prevent or suppress the aggregation of the native protein. Among these, photodynamic therapy (PDT)10 and photothermal therapy11 have attracted considerable attention recently. The techniques are driven by the absorption of near IR (NIR) light radiation wavelengths corresponding to an absorbance band of a photosensitive compound. Compared to visible light, NIR light can penetrate deep within tissues in the human body; the optical absorption coefficients of most tissue chromophores and biological fluids reach their minimum in the NIR wavelength region.12 Moreover, NIR light has reduced phototoxicity. For the generation of photons of sufficient energy for PDT, compared with the use of NIR absorbing photosensitiser (PS) species, upconversion nanoparticles (UCNPs) have several advantages: in clinical PDT use, low toxicities towards biological tissues have been measured; in particular, low phototoxicity is seen, because UC excitation does not occur in the presence of low-power (∼mW cm−2) sources such as natural light. The first reported use of UCNPs in PDT was of Ln3+ nanoparticles complexed with PS molecules. The UCNPs converted NIR light into visible-wavelength radiation via a two- or multi-photon mechanism and were functionalised with a tumour-targeting antibody.13 As another example of the use of UCNPs, Kuk et al. recently published results on NIR-light-induced suppression of amyloid-beta (Aβ) aggregation with reduced toxicity, via photosensitisation of UCNPs using NIR light.14
In this paper, our focus is on AF decomposition induced by NIR-active UCNPs, a subject that is important for the development of therapeutic approaches for protein folding diseases. We present the first example of AF decomposition induced by NIR-active UCNPs, via a process termed an AF decomposition cascade and consisting of four essential components: NIR light, UCNPs, PS, and singlet oxygen. This AF cascade resembles the reaction mechanism for producing active oxygen via visible-light-activated photosensitisation of a PDT agent in cancer treatment, and in both cases, cytotoxicity toward tissues other than the target tissue could potentially prove problematic. However, the AF decomposition cascade that we present is characterised by the generation of a highly reactive product, 1O2, only in the vicinity of amyloid superstructures, facilitating the selective attack of these proteins.
Experimental
Polyethylenimine (PEI) coated UCNPs (UCNPs@PEI) were synthesised via a solvothermal procedure.15–18 X-ray powder diffraction (XRD) revealed that the UCNPs had highly crystalline structures (Fig. S1†). Amyloid fibril (AF) formation without seeding was performed by a stepwise heating process.19 Further details of syntheses are given in the ESI experimental section† along with the results of nanoparticle characterisation, spectroscopy, and fluorescence measurement.Results and discussion
Surface modifications of the UCNPs were required to electrostatically couple the PS to the UCNPs. PEI is positively charged under physiological conditions and hence it is capable of electrostatically coupling with negatively charged PS species. The UCNPs@PEI synthesis produced α-NaYF4 particles co-doped with Yb3+ (18.7%) and Er3+ (2.1%), as characterised using EDS (data not shown). The XRD pattern shows pure cubic α-NaYF4 nanoparticles, with peaks that match those of the α-NaYF4 standard XRD spectrum (Fig. S1† and JCPDS standard card no. 77-2042). The synthesised UCNPs@PEI particles are spherical with a narrow particle size distribution (ca. 50 nm), in good agreement with the size determined from the XRD analysis, and the thin PEI surface layer can be seen to be in the range of 2 to 8 nm (TEM images, Fig. S2†). To make the UCNPs@PEI suitable for conjugation with the cationic PS, a dextran sodium sulfate (DSS) layer was coated onto their surface via a layer-by-layer method.20,21 The layers consist ∼10 nm-thick DSS (Fig. S2C†). The morphology of the core–shell nanoparticles was maintained after the DSS was coated onto the PEI layer (Fig. S2C†), which suggests that the amino-modification process does not modify the uniformity of the NaYF4:Yb/Er nanoparticles. Measurements before and after the DSS-modification process revealed a change in the surface charge of the core–shell nanoparticles from positive (ζ = +15 mV) to negative (ζ = −21 mV), confirming the successful grafting of DSS onto the nanoparticles (Fig. S3†).The emission spectra of UCNPs@PEI (Fig. 1) overlaps extensively with the absorption spectra of PS candidates such as eosin Y (EY), rose bengal (RB), orange G (OG), etc., thus making energy transfer from the UCNPs to the PS possible through Förster resonance energy transfer (FRET) and/or electronic transition. We determined the internal upconversion quantum yield of NaYF4:Yb/Er (18/2 mol%) as 0.016%, using an irradiance of 1.0 W cm−2 at 980 nm; this result is of a similar order of magnitude as a published result, 100–10−3%.22 If the UCNPs were excited with a diode laser, using 1.0 W cm−2 at 980 nm, the power of the light emitted from the excited states is estimated to be 0.12 mW cm−2, which is equivalent to the power of a 1 nm wavelength band of the excitation light of a general-purpose fluorometer.
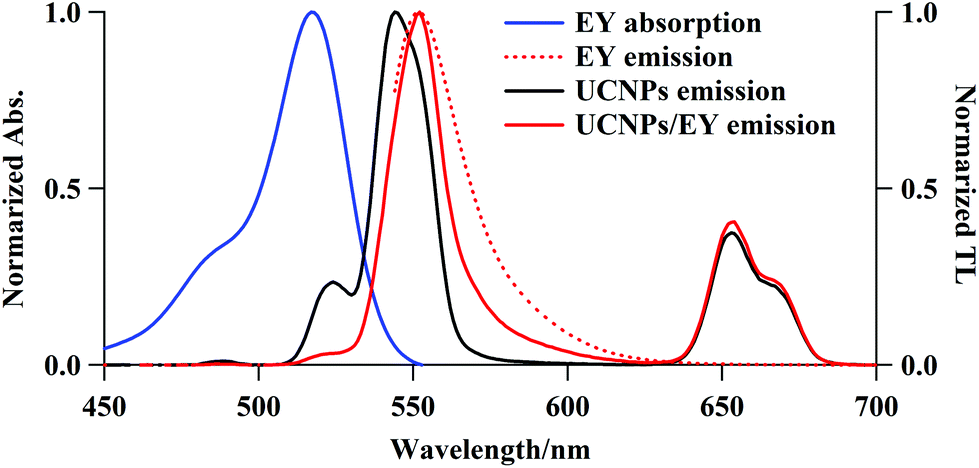 | ||
| Fig. 1 Free EY electronic absorption (solid blue line), and total luminescence (TL) spectra for EY (dotted red line), UCNPs@PEI (solid black line), and UCNPs@PEI/EY (solid red line). Excitation wavelengths: 980 and 524 nm for UCNPs or UCNPs/EY and EY, respectively. [UCNPs] = 1.8 mg mL−1, [free EY] = 5.0 μM, and [EY adsorbed UCNPs] = 3.7 μM. | ||
High encapsulation efficiencies were determined for the different anionic PS species adsorbed on the surface of the UCNPs@PEI after the electrostatic coupling process: approximately 3.9% (EY), 5.9% (RB), and 1.8% (OG). Using the anionic UCNPs@DSS, the encapsulation efficiency of the cationic PS (methylene blue (MB)) was measured as 4.5%. We observed in these experiments that the PS loaded onto the UCNPs@PEI or UCNPs@DSS leaked very little over 6 h, although a small fraction (EY: <1%, RB: 2%, other PS ≈5%) was released after 2 h (Fig. S4†). EY is therefore the most stable among the anionic PS. The amount of adsorbed PS on the UCNP surfaces was estimated using UV–Vis spectroscopy as 2–9 μM in 0.5 mg mL−1 of UCNPs@PEI or UCNPs@DSS. We did not attempt to optimise the experiment to increase adsorption because we observed that the originally selected amount of PS was sufficient to facilitate the NIR-induced sequential-cascade decomposition of AF (detailed later).
In future, for higher 1O2 conversion efficiency, we expect that there will be demand for increased amounts of PS adsorbed on the surface of UCNPs and surface modification to prevent self-quenching due to self-aggregation of PS molecules.23–25 Here, the amount of 1O2 generation by the UCNPs@PEI/PS upon excitation with a 980 nm NIR diode laser was determined through the photobleaching of pseudoisocyanine (PIC) and/or anthracene-9,10-dipropionic acid disodium salt (ADPA) as 1O2-trapping agents. Because these molecular proofs are destroyed by the 1O2 species, the concentrations of 1O2-trapping agents, determined by absorbances at 523 and 374 nm, were acquired as a measure inversely proportional to the effectiveness of the energy transfer to molecular oxygen from the PS in its triplet excited state. The capacity of the UCNPs@PEI/PS to destroy the 1O2-trapping agents increased with NIR irradiation time (Fig. S5†). Phenalenone (PN), an efficient 1O2 sensitiser with a quantum yield for 1O2 production close to unity,26 was used as a criterion for quantitative evaluation of 1O2 generation. PIC and ADPA are bleached to the photooxidation products N-ethyl-2-quinolone and nonfluorescent endoperoxide, respectively, via photooxidative reactions of singlet O2. By measuring the decrease in the absorption bands of the 1O2-trapping agents, 1O2 production can be monitored.17 The 1O2 quantum yields (Φ1O2) of some PS species are shown in Fig. 2. The most efficient PS is RB (Φ1O2 = 0.84, computed relative to that of PN using PIC). For UCNPs/MB, in comparison to free MB, a low Φ1O2 is seen.27
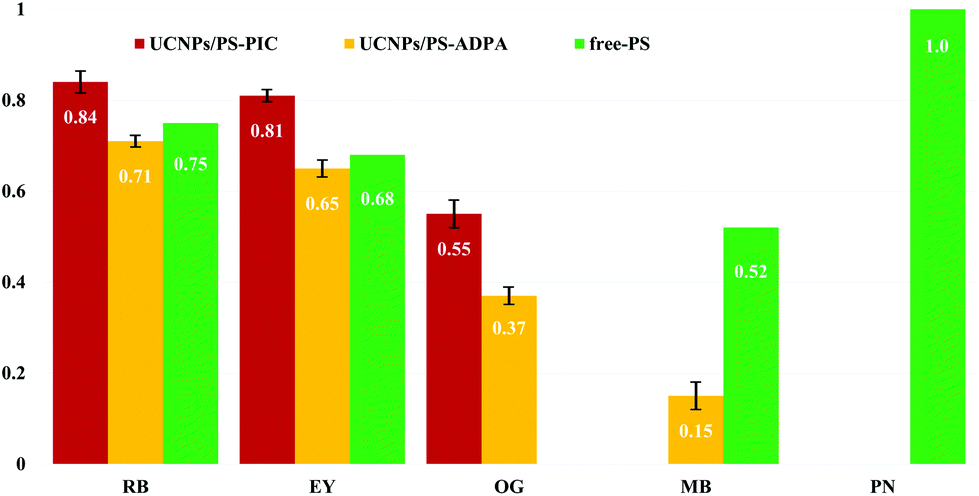 | ||
| Fig. 2 Singlet O2 quantum yields for photosensitisers in aqueous buffer solution irradiated by 980 nm NIR laser light. Red bars: UCNPs/PS (1O2 trapping agent—PIC). Yellow bars: UCNPs/PS (1O2 trapping agent—ADPA). Green bars: free PS.26 | ||
It is believed that the thickness of the capping DSS layer (ca. 10 nm) causes a drop in the efficiency of the energy transfer from the UCNP cores to the PS. Considering their high encapsulation efficiency, low capacity for PS release, and high Φ1O2, EY–UCNP and RB–UCNP composites were used in our subsequent AF decomposition experiments. Fig. 1 shows the electronic absorption spectrum of free EY and the total luminescence (TL) spectra of free EY, UCNPs@PEI, and UCNPs@PEI/EY. The absorption spectrum of free EY has an excitation maximum at about 540 nm that has considerable overlap with the red 4S3/2, 2H11/2 → 4I15/2 UCNP emission peak at approximately 530 nm. The NIR-excitation fluorescence spectrum of UCNPs@PEI/EY strongly resembles the fluorescence spectrum of free EY, and the emission spectrum of pure UCNPs drops to almost zero in this range. Judging from the renewed emission by the PS adsorbed on the UCNPs@PEI surface (solid red line), which is red-shifted with respect to the original UCNPs@PEI emission (solid black line), the coupling of the UCNPs@PEI and the PS was successful. A similar result was obtained for RB. Moreover, upon hybridisation, the surface charge, ζ, of the core–shell nanoparticles shifted from +15 to +10.1 mV, confirming that the PS had been adsorbed onto the UCNPs (Fig. S3†). When cationic MB species were loaded onto UCNPs@PEI, the positive ζ potential was disrupted such that the MB molecules were not able to approach the UCNP surfaces. Hence, the UC emission remained almost unchanged, and fluorescence emission originating from MB was not detected in this case (data not shown). It may be deduced, therefore, that the UCNP-to-PS energy transfer was dominated by FRET rather than electromagnetic radiation.
In this experiment, we used insulin AFs, a commonly used artificial AF model. The formation of AFs from native insulin was confirmed by atomic force microscopy (AFM), circular dichroism (CD), and thioflavin T (ThT) fluorescence assays. Regular rod-shaped structures are observed in the AFM images of the AF (Fig. S6†); these are rectangular in cross section, with a height of ∼5–9 nm, breadth of ∼20–40 nm, and length of the order of hundreds of nm—nearly identical to previously reported rod-like AF structures (height: 3–15 nm; width: 40–50 nm; length: 0.5–1.5 μm).28,29 These observations are consistent with the change from α-helix-enriched structures to β-sheet dominance during the process of AF formation, as observed by CD analysis. Finally, the UCNP–AF complex was electrostatically bound; a negatively charged AF (ζ = −31.1 mV, as shown in Fig. S3†) was added to the positively charged PEI-capped UCNPs in the UCNPs@PEI/PS buffer solution (pH = 8.6). The stoichiometric ratio of PS to AF was adjusted in the range of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19–46, with a fixed UCNP concentration of 0.5 mg mL−1. However, when this ratio was greater than 1:19, the AF/PS/UCNP complex was less stable over a long period of time. A reference solution of the UCNP–AF complex (UCNPs@PEI/AF) without PS was prepared from the UCNP stock solution and AF buffer solution. During the UCNP–AF hybridisation process, the AF zeta potential increased from −31.1 to −0.9 mV, confirming that the AFs had been successfully grafted onto UCNPs@PEI/EY. For UCNPs@PEI/RB, a similar change in the zeta potential was obtained.
:19–46, with a fixed UCNP concentration of 0.5 mg mL−1. However, when this ratio was greater than 1:19, the AF/PS/UCNP complex was less stable over a long period of time. A reference solution of the UCNP–AF complex (UCNPs@PEI/AF) without PS was prepared from the UCNP stock solution and AF buffer solution. During the UCNP–AF hybridisation process, the AF zeta potential increased from −31.1 to −0.9 mV, confirming that the AFs had been successfully grafted onto UCNPs@PEI/EY. For UCNPs@PEI/RB, a similar change in the zeta potential was obtained.
As a final step, the decomposition of AF by the NIR-induced sequential cascade was confirmed via a ThT fluorescence assay; this method uses the direct proportionality between the amount of AF and ThT fluorescence intensity to estimate AF quantities. Fig. 3 shows the fluorescence spectra of ThT specifically bound to the AF-complexing UCNPs/PS. The ThT fluorescence intensities acquired after NIR irradiation (for 30 min) for AF/UCNPs/EY and AF/UCNPs/RB are markedly lower than the florescence intensities for the AF/UCNPs/PS that had not undergone NIR irradiation (control, dotted line). Further, this reduction in fluorescence is greater for EY than it is in the case of RB. This suggests that the decomposition of AF occurred via the NIR-induced sequential cascade, with EY having a higher efficiency than RB for this process.
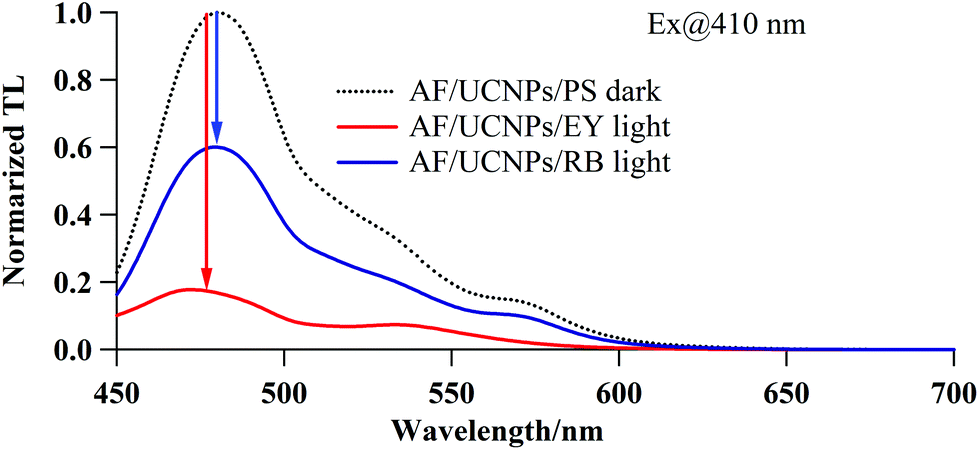 | ||
| Fig. 3 Characterisation of AF decomposition by NIR-induced sequential cascade using spectroscopic total luminescence. Dotted line: fluorescence emission spectra of ThT bound to AF-complexing UCNPs/PS, acquired without NIR irradiation. Solid lines: AF/UCNPs/EY (red) and AF/UCNPs/RB (blue) luminescence, acquired after NIR irradiation (980 nm, 1 W cm−2, 30 min). The arrows indicate the direction of the ThT fluorescence intensity changes induced by the NIR irradiation. [AF] = 0.01 mg mL−1, [UCNPs] = 0.5 mg mL−1, [EY] = 3.7 μM, [RB] = 9.0 μM, and [ThT] = 5 μM. | ||
In order to obtain direct evidence of the AF decomposition, the dispersed AF size distribution was examined using SDS-PAGE (Fig. S7A and B†). From this experiment, we found evidence of AF amounts decreasing during the irradiation process, via NIR-excited, UCNPs@PEI/PS-mediated decomposition into smaller-sized fibrils and selective singlet-O2 attack. There is significant correspondence between this result and those of the standard ThT fluorescence assay (Fig. 3), proving that the NIR-active nanoparticles decomposed the model insulin AF in vitro.
Conclusions
We demonstrated the first successful decomposition of AFs into fragmented fibrils via a cascade of four consecutive energy-transfer steps. (1) NIR light is used to excite the higher energy states, e.g.2H11/2, 4S3/2, and 4F9/2 ← 4I15/2, corresponding to the visible wavelength ranges, in UCNPs. (2) This energy is effectively transferred to the PS on the UCNP surface by FRET. (3) 3O2 is excited via intersystem crossing from a PS excited state. (4) Finally, active oxygen, 1O2, generated close to an AF decomposes it into fragmented fibrils. An important characteristic of the AF decomposition cascade is that both NIR light and NIR-light-sensitised UCNPs have low toxicities towards biological tissues. Furthermore, UC activity is not lost in the presence of oxygen; by contrast, triplet–triplet annihilation UC, for example, is inactive under the same condition. In addition, the lifetime of 1O2 is very short (approximately 10–320 ns), limiting its diffusion to only 10–55 nm in vivo,30 and hence photodynamic damage will occur only very close to the PS. As a result, this AF decomposition cascade mechanism can provide reduced cytotoxicity towards other healthy tissues surrounding the target because the generation of 1O2 occurs close to the AF; thus, AF decomposition is selective. This approach has potential as a future therapeutic treatment for folding diseases as well as in chemotherapy. To minimise the side effects on other native peptides/proteins and tissues, we plan to introduce high-amyloid-selectivity to AF decomposition cascades for states in which native peptides/proteins and denatured AF coexist. A new approach, using covalent attachment to UCNPs via AF-binding linkers such as ThT derivatives, is also planned. Under such conditions, the AF decomposition reaction will occur only at the site of 1O2 generation when a compound possessing the capacity to selectively combine with AF is introduced into an AF decomposition cascade as a PS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Y. Matsumoto of the National Institute of Technology, Oita College, Prof. T. Tsumura of Oita University, and Ms S. Suzuki of JASCO Engineering, for technical assistance. Mr K. Tanaka and Mr T. Saito (Otsuka Electronics Co., Ltd) are acknowledged for their help with zeta-potential measurements. This research was supported by Grants-in-Aid for Scientific Research, Grant No. 18K05062, from the Japan Society for the Promotion of Science, an A-STEP fund (D218H01014) from the Japan Science and Technology Agency (JST), and System Instruments Co., Ltd, Tokyo, Japan. We would like to thank Editage for English language editing.References
- S.-T. Liu, G. Howlett and C. J. Barrow, Biochemistry, 1999, 38, 9373 CrossRef CAS PubMed; J. Ryu, K. Girigoswami, C. Ha, S. H. Ku and C. B. Park, Biochemistry, 2008, 47, 5328 CrossRef PubMed.
- I. Sirangelo, C. Malmo, C. Iannuzzi, A. Mezzogirno, R. M. Bianco, M. Papa and G. Irace, J. Biol. Chem., 2004, 279, 13183 CrossRef CAS.
- P. T. Lansbury Jr., Acc. Chem. Res., 1996, 29, 317 CrossRef.
- C. M. Dobson, Nature, 2003, 426, 884 CrossRef CAS PubMed.
- J. E. Maggie and P. W. Mantyh, Brain Pathol., 1996, 6, 147 CrossRef PubMed.
- B. M. Taylor, R. W. Sarver, G. Fici, R. A. Poorman, S. B. Lutzke, A. Molinari, T. Kawabe, K. Kapppenman, A. E. Buhl and D. E. Epps, J. Protein Chem., 2003, 22, 31 CrossRef CAS PubMed.
- D. L. Brody, H. Jiang, N. Wildburger and T. J. Esparza, Alzheimers Res. Ther., 2017, 9, 62 CrossRef PubMed.
- B. Penke, F. Bogár, G. Paragi, J. Gera and L. Fülöp, Curr. Protein Pept. Sci., 2019, 20, 577 CrossRef CAS PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507 CrossRef CAS PubMed.
- P. Agostini, K. Berg, K. A. Cengel, T. H. Foste, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250 CrossRef PubMed.
- G. Shan, R. Weissleder and S. A. Hilderbrand, Theranostics, 2013, 3, 267 CrossRef CAS PubMed.
- J. Shen, G. Y. Chen, A.-M. Vu, W. Fan, O. S. Bilsel, C.-C. Chang and G. Han, Adv. Opt. Mater., 2013, 1, 644 CrossRef.
- M. Buchner, P. G. Calavia, V. Muhr, A. Kröninger, A. J. Baeumner, T. Hirsch, D. A. Russell and M. J. Marin, Photochem. Photobiol. Sci., 2019, 18, 98 RSC.
- S. Kuk, B. I. Lee, J. S. Lee and C. B. Park, Small, 2017, 13, 1603139 CrossRef PubMed.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS PubMed.
- R. S. Niedbala, H. Feindt, K. Kardos, T. Vail, J. Burton, B. Bielska, S. Li, D. Milunic, P. Bourdelle and R. Vallejo, Anal. Biochem., 2001, 293, 22 CrossRef CAS PubMed.
- P. Zhang, W. Steelant, M. Kumar and M. Scholfield, J. Am. Chem. Soc., 2007, 129, 4526 CrossRef CAS PubMed.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526 RSC.
- E. Chatani, H. Imamura, N. Yamamoto and M. Kato, J. Biol. Chem., 2014, 289, 10399 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693 CrossRef CAS.
- C. F. Gainer and M. Romanowski, J. Innovative Opt. Health Sci., 2014, 7, 1330007 CrossRef.
- H. S. Qian, H. C. Guo, P. C.-L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285 CrossRef CAS PubMed.
- S. Gai, P. Yang, C. Li, W. Wang, Y. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166 CrossRef CAS.
- S. L. Datao, T. P. Hu, J. X. R. Li, M. Wang, Z. Chen, M. Huang and X. Chen, Angew. Chem., Int. Ed., 2015, 54, 7915 CrossRef.
- E. Oliveros, S. H. Bossmann, S. Nonell, C. Martí, G. Heit, G. Tröscher, A. Neuner, C. Martínez and A. M. Braun, New J. Chem., 1999, 23, 85 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS.
- J. Brange, L. Andersen, E. D. Laursen, G. Meyn and E. Rasmussen, J. Pharm. Sci., 1997, 86, 517 CrossRef CAS PubMed.
- R. Khurana, C. Ionescu-Zanetti, M. Pope, J. Li, L. Nielson, M. Ramírez-Alvarado, L. Regan, A. L. Fink and S. A. Carter, Biophys. J., 2003, 85, 1135 CrossRef CAS.
- J. Moan, K. Berg, E. Kvam, A. Western, Z. Malik, A. Rück and H. Schneckenburger, Ciba Found. Symp., 1989, 146, 107 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experimental and results and discussion, including seven figures. See DOI: 10.1039/c9pp00356h |
| This journal is © The Royal Society of Chemistry and Owner Societies 2020 |