
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe robustness of the terminal emitter site in major LHCII complexes controls xanthophyll function during photoprotection†
Francesco
Saccon
 a,
Milan
Durchan
b,
Tomáš
Polívka
b and
Alexander V.
Ruban
*a
a,
Milan
Durchan
b,
Tomáš
Polívka
b and
Alexander V.
Ruban
*a
aQueen Mary University of London, School of Biological and Chemical Sciences, Mile End Road E1 4NS, London, UK. E-mail: a.ruban@qmul.ac.uk; Fax: +44(0) 8983 0973; Tel: +44(0)20 7882 6314
bUniversity of South Bohemia, Institute of Physics, Faculty of Science, České Budějovice, Czech Republic
First published on 14th August 2020
Abstract
Xanthophylls in light harvesting complexes perform a number of functions ranging from structural support to light-harvesting and photoprotection. In the major light harvesting complex of photosystem II in plants (LHCII), the innermost xanthophyll binding pockets are occupied by lutein molecules. The conservation of these sites within the LHC protein family suggests their importance in LHCII functionality. In the present work, we induced the photoprotective switch in LHCII isolated from the Arabidopsis mutant npq1lut2, where the lutein molecules are exchanged with violaxanthin. Despite the differences in the energetics of the pigments and the impairment of chlorophyll fluorescence quenching in vivo, we show that isolated complexes containing violaxanthin are still able to induce the quenching switch to a similar extent to wild type LHCII monomers. Moreover, the same spectroscopic changes take place, which suggest the involvement of the terminal emitter site (L1) in energy dissipation in both complexes. These results indicate the robust nature of the L1 xanthophyll binding domain in LHCII, where protein structural cues are the major determinant of the function of the bound carotenoid.
1. Introduction
Xanthophylls are oxygenated carotenoids widely found in biological systems. Besides their relevance in behavioural ecology, from being responsible for the flamboyant plumage of birds to being precursors of many molecules responsible for the scent of flowers, they perform several “unseen” tasks as anti-oxidants and scavengers of reactive oxygen species.1–4 Among their natural hosts, photosynthetic organisms are an exceptional example of their protective activities. In plants, they are mainly found in the thylakoid membranes, associated with light harvesting complexes within the photosystems.5–7 Here, they perform a well-known role as light harvesters, whereby they collect photons in the blue-green region of the visible spectrum and transfer their energy to chlorophylls.8–10 However, their quintessential role is the photoprotection of the light harvesting apparatus during high light exposure. Xanthophylls can quench chlorophyll triplet states and prevent the accumulation of harmful reactive oxygen species.11When this barrier fails or is insufficient, they can act as singlet oxygen (1O2) scavengers.12,13 Additionally, an increasing body of evidence suggests that xanthophylls participate directly in the de-excitation of chlorophylls during non-photochemical quenching processes (NPQ).14–16 NPQ is the physiological response of photosynthetic organisms to quickly de-activate the light harvesting machinery during stressful environmental conditions. In particular during high-light exposure, when the thylakoids are overflown with high energy photons, the excess chlorophyll excitation that is not photochemically utilised is quickly quenched through the formation of dissipative pigment interactions in plant's major light harvesting complexes (LHCII).17 Isolated LHCII display an intrinsic plasticity and can switch reversibly to low-fluorescence conformations.18,19 The conformational change of LHCII is controlled by components, such as pH, detergent concentration and zeaxanthin binding, that mimic the thylakoid environment under low or high light conditions in leaves.20,21 Therefore, this inherent switch has been proposed to underlie the formation of NPQ in vivo, together with the contribution of other membrane-linked factors such as protein–protein aggregation and PsbS activation.22The LHCII monomer presents 4 xanthophyll binding sites, namely N1, L1, L2 and V1.23 A 9-cis neoxanthin binds strongly in the N1 position, in a chlorophyll b-enriched pocket.24 The L1 and L2 sites are occupied by two spectrally distinct lutein molecules, both having light harvesting and photoprotective roles.25,26 L1 is in close contact with the chlorophylls a of the terminal emitter site.27 Additionally, a V1 binding pocket is found in a peripheral location of the LHCII monomer and is of central importance for the xanthophyll cycle operation, involving the de-epoxidation of violaxanthin to form zeaxanthin during NPQ.28,29 These carotenoid binding sites are somewhat flexible and can accommodate different pigments upon reconstitution.30–32 The site of quencher formation during NPQ within the LHCII complex and the nature of the xanthophyll involved are still a matter of debate and both lutein and zeaxanthin have been proposed as candidates.14,15,33,34
Here, we investigated the nature of the quenching switch in an Arabidopsis double mutant impaired in xanthophyll biosynthetic pathways, unable to accumulate lutein and zeaxanthin during light exposure and instead enriched in violaxanthin. The npq1lut2 mutant has a drastically impaired capacity to form NPQ, highlighting the importance of both lutein and zeaxanthin in energy dissipation.35,36 Yet, its isolated LHCII complexes can still undergo the dissipative switch, despite to a different extent than in the wild type (WT).37 Our previous analysis showed that the behaviour of carotenoid triplet states is independent of the substitution of lutein with violaxanthin in LHCII, revealing the robustness of the L1 xanthophyll pocket in the triplet photoprotective mechanism.38 In this study, we produced a careful analysis of the spectroscopic signatures of the quenching induction in monomers of major LHCII complexes, with WT and npq1lut2 xanthophyll composition. Our data show that, despite the xanthophyll exchange causes subtle changes in pigment site energies and excitonic chlorophyll clusters, the position and extent of the alterations induced by the protein conformational switch are extremely well conserved. The L1 binding site is prominently affected by the switch, suggesting it is the primary site of quenching formation. The properties of LHCII are thus such that xanthophyll structure is not the key factor for the photoprotective function.
2. Materials and methods
2.1. LHCII isolation and monomerisation
Major LHCII complexes were purified from adult WT and npq1lut2 Arabidopsis plants via iso-electric focussing and resuspended in 25 mM HEPES, 0.01% β-dodecyl-D-maltosyde (βDM), pH 7.6 (resuspension buffer, RB).38 Due to the absence of luteins, crucial for trimerisation, npq1lut2 complexes were exclusively isolated in the monomeric state.39 WT LHCII were instead collected as trimers and underwent chemical monomerisation by incubation in RB with 20 mM CaCl2 and phospholipase A2 (Sigma-Aldrich) (500 μg ml−1 total chlorophyll).40 After 21 hours, the LHCII suspension was separated on a sucrose density gradient, the monomeric fraction was collected and washed through PD-10 columns (GE Healthcare) in RB.2.2. Immobilisation in polyacrylamide gels and quenching induction
Embedding of LHCII monomers in polyacrylamide gels was performed as previously described.19,38 Quenching induction was achieved via overnight incubation of LHCII gels in RB without βDM and their absorption and fluorescence properties were monitored at 1, 2, 3, 4, 20 hours from the beginning of induction. The total chlorophyll concentration of the samples was 10 μg ml−1 for the low-temperature fluorescence experiments and the data presented in ESI,† while 100 μg ml−1 was used in all other experiments.2.3. Pigment analysis
Pigment analysis was performed via reverse-phase HPLC, using a LiChrospher 100 RP-18 column and Dionex Summit chromatography system.38 The xanthophyll absorption spectra were analysed with the Chromeleon software (Dionex).2.4. Steady-state absorption and fluorescence spectroscopy
Quenching formation was monitored measuring the steady-state chlorophyll a fluorescence signal at different points during the induction using a Dual PAM fluorometer (Walz, Germany), applying a weak measuring blue light beam (<12 μmol photons m−2 s−1). Room-temperature steady-state absorption spectra were measured on an Aminco DW-2000 UV/Vis spectrophotometer (Olis Inc., USA). Second derivatives were calculated using the GRAMS software (Thermo-Fisher), applying a Savitzky–Golay filter with a 5th order polynomial function. Fluorescence emission spectra were recorded at 77 K using a Jobin Yvon FluoroMax-3 spectrophotometer equipped with a liquid nitrogen cooled cryostat. Excitation was performed at 435 nm with 5 nm slit width and the fluorescence spectral resolution was 0.5 nm. Integration time was set to 0.1 s. Every spectrum is the average of 5 scans.2.5. Ultrafast transient absorption spectroscopy
Transient absorption spectra were measured as described before.38 Briefly, a modular laser system was used, consisting of an ultrafast Ti:sapphire regenerative amplifier (Spitfire Ace-100F, Spectra-Physics, USA) seeded with a Ti:sapphire oscillator (MaiTai SP, Spectra-Physics, USA), and pumped by Nd:YLF laser (Empower 30, Spectra-Physics, USA). The generated ∼100 fs pulses (800 nm) at 1 kHz repetition rate were divided into excitation and probe beams by a beam splitter. Tunable excitation pulses were generated by an optical parametric amplifier (TOPAS-C, Light Conversion, Lithuania). A 2 mm sapphire plate was used to generate a broadband (450–750 nm) white light pulse. The probe beam was focused to the sample, overlapping with the excitation beam. Probe and reference beams were then focused to the entrance slit of a spectrograph where the beams were dispersed onto a double CCD detection system (Pascher Instruments, Sweden). The time delay between the excitation and probe pulses was introduced by a computer-controlled delay line. The mutual polarisation of the excitation and probe beams was set to the magic angle (54.7°). Excitation intensity in all experiments was kept at ∼4.0 × 1013 photons per pulse per cm2. The LHCII gel sample of a 10 × 10 mm size and of 1 mm thickness was squeezed between two quartz windows with a 1 mm Teflon spacer. The cuvette was placed into a holder attached to a Lissajous 2D scanner that moves the sample during the measurement to prevent degradation. Decay kinetics of chlorophyll a QY absorption band were described using a triple exponential function, with formulawhere Ai and τi are the fractional amplitude and the lifetime of the i-th component. The average lifetimes were calculated as
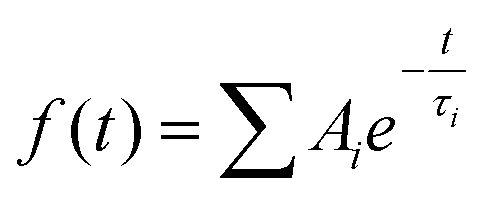
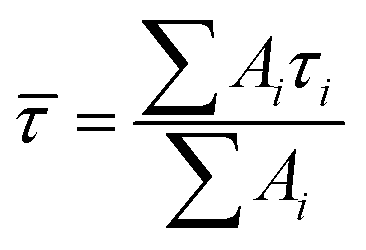
3. Results and discussion
Violaxanthin differs from lutein for the presence of two epoxy groups in the end rings (Fig. 1A). This subtle change causes a pronounced effect on the polarity of the xanthophyll41 and an increase in the conjugation length of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C backbone (N = 10, lutein; N = 9, violaxanthin). This in turn results in a visible blue shift of the ground state absorption profile (85 cm−1, Fig. 1B). Lutein is structurally related to zeaxanthin, a component of the xanthophyll cycle that promotes NPQ.2 Both carotenoids display similar optical and chemical properties and were both shown to be crucial for the correct formation of NPQ in vivo.35,36 Violaxanthin, on the contrary, has been demonstrated to act antagonistically during NPQ formation.42,43 The npq1lut2 mutant, impaired in both lutein and zeaxanthin accumulation, lost completely its capacity to form the fast qE-type quenching,36 similar only to mutants lacking the protein PsbS.44 A closer inspection to the quenching inductions of the double mutant revealed that the cause of the abolished qE phenotype, might reside on its shifted ΔpH dependency when the xanthophyll composition is changed.45 Backing up this suggestion, later work showed that the chemically-induced exacerbation of the thylakoid ΔpH can restore WT amplitudes and kinetics of qE, the main component of NPQ, in npq1lut2.46 Thus, neither lutein nor zeaxanthin are strictly essential in qE development. On the contrary, it is suggested that a correct xanthophyll asset guarantees the stability of pigment-protein complexes in the thylakoids and in turn allows a fine tuning of the pK of qE, in response to physiological needs.41,45 Complexes isolated from different xanthophyll biosynthesis mutants, retain their ability to form low-fluorescent aggregates and their sensitivity to factors such as detergent and pH that mimic the changes in the membrane occurring upon light exposure.37,47 In the analysis described hereafter, we induced chemical monomerisation of WT LHCII, first isolated as trimers, to consistently study the quenched state of single lutein or violaxanthin-enriched protein complexes. The lut2 mutation, indeed, is known to induce monomerisation of the LHCII antenna, due to the lack of lutein in the L2 position, essential for trimer stability.39 The incorporation in polyacrylamide gels provided a platform to study functional changes in LHCII without affecting the monomeric state of the antenna since the removal of detergent was performed on fully separated LHCII complexes, locked in the gel matrix.19,48–50 The starting assumption of our experimental design was that violaxanthin is incorporated correctly and solely in the L1 and L2 binding sites. We showed previously that the neoxanthin pool is not significantly affected in the mutant LHCII, which binds 1 neoxanthin molecule per monomer as in the WT.38 On a similar note, the V1 site binds only loosely the xanthophyll cycle carotenoids and this is lost during preparative steps in both LHCII samples.28,38 Therefore, differences visible in carotenoid absorption are likely to reflect only the changes at the lutein binding sites (L1 and L2, Fig. 2). Importantly, we found earlier that violaxanthin replaces lutein in npq1lut2 LHCII with the same stoichiometry of 2
C backbone (N = 10, lutein; N = 9, violaxanthin). This in turn results in a visible blue shift of the ground state absorption profile (85 cm−1, Fig. 1B). Lutein is structurally related to zeaxanthin, a component of the xanthophyll cycle that promotes NPQ.2 Both carotenoids display similar optical and chemical properties and were both shown to be crucial for the correct formation of NPQ in vivo.35,36 Violaxanthin, on the contrary, has been demonstrated to act antagonistically during NPQ formation.42,43 The npq1lut2 mutant, impaired in both lutein and zeaxanthin accumulation, lost completely its capacity to form the fast qE-type quenching,36 similar only to mutants lacking the protein PsbS.44 A closer inspection to the quenching inductions of the double mutant revealed that the cause of the abolished qE phenotype, might reside on its shifted ΔpH dependency when the xanthophyll composition is changed.45 Backing up this suggestion, later work showed that the chemically-induced exacerbation of the thylakoid ΔpH can restore WT amplitudes and kinetics of qE, the main component of NPQ, in npq1lut2.46 Thus, neither lutein nor zeaxanthin are strictly essential in qE development. On the contrary, it is suggested that a correct xanthophyll asset guarantees the stability of pigment-protein complexes in the thylakoids and in turn allows a fine tuning of the pK of qE, in response to physiological needs.41,45 Complexes isolated from different xanthophyll biosynthesis mutants, retain their ability to form low-fluorescent aggregates and their sensitivity to factors such as detergent and pH that mimic the changes in the membrane occurring upon light exposure.37,47 In the analysis described hereafter, we induced chemical monomerisation of WT LHCII, first isolated as trimers, to consistently study the quenched state of single lutein or violaxanthin-enriched protein complexes. The lut2 mutation, indeed, is known to induce monomerisation of the LHCII antenna, due to the lack of lutein in the L2 position, essential for trimer stability.39 The incorporation in polyacrylamide gels provided a platform to study functional changes in LHCII without affecting the monomeric state of the antenna since the removal of detergent was performed on fully separated LHCII complexes, locked in the gel matrix.19,48–50 The starting assumption of our experimental design was that violaxanthin is incorporated correctly and solely in the L1 and L2 binding sites. We showed previously that the neoxanthin pool is not significantly affected in the mutant LHCII, which binds 1 neoxanthin molecule per monomer as in the WT.38 On a similar note, the V1 site binds only loosely the xanthophyll cycle carotenoids and this is lost during preparative steps in both LHCII samples.28,38 Therefore, differences visible in carotenoid absorption are likely to reflect only the changes at the lutein binding sites (L1 and L2, Fig. 2). Importantly, we found earlier that violaxanthin replaces lutein in npq1lut2 LHCII with the same stoichiometry of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relative to neoxanthin, confirming that the L1 and L2 sites are able to accommodate both carotenoids without disrupting the LHCII stability.38 Works on recombinant LHCII showed indeed the importance of xanthophylls occupying the inner binding sites (L1 and L2) for the folding of the complexes, which doesn't occur in the absence of xanthophylls.7,51 Additionally, these and more recent studies showed that different xanthophylls can be incorporated correctly in LHCII and yield stable complexes, among these violaxanthin in lut2 and npq1lut2 mutants.31,39
:1 relative to neoxanthin, confirming that the L1 and L2 sites are able to accommodate both carotenoids without disrupting the LHCII stability.38 Works on recombinant LHCII showed indeed the importance of xanthophylls occupying the inner binding sites (L1 and L2) for the folding of the complexes, which doesn't occur in the absence of xanthophylls.7,51 Additionally, these and more recent studies showed that different xanthophylls can be incorporated correctly in LHCII and yield stable complexes, among these violaxanthin in lut2 and npq1lut2 mutants.31,39
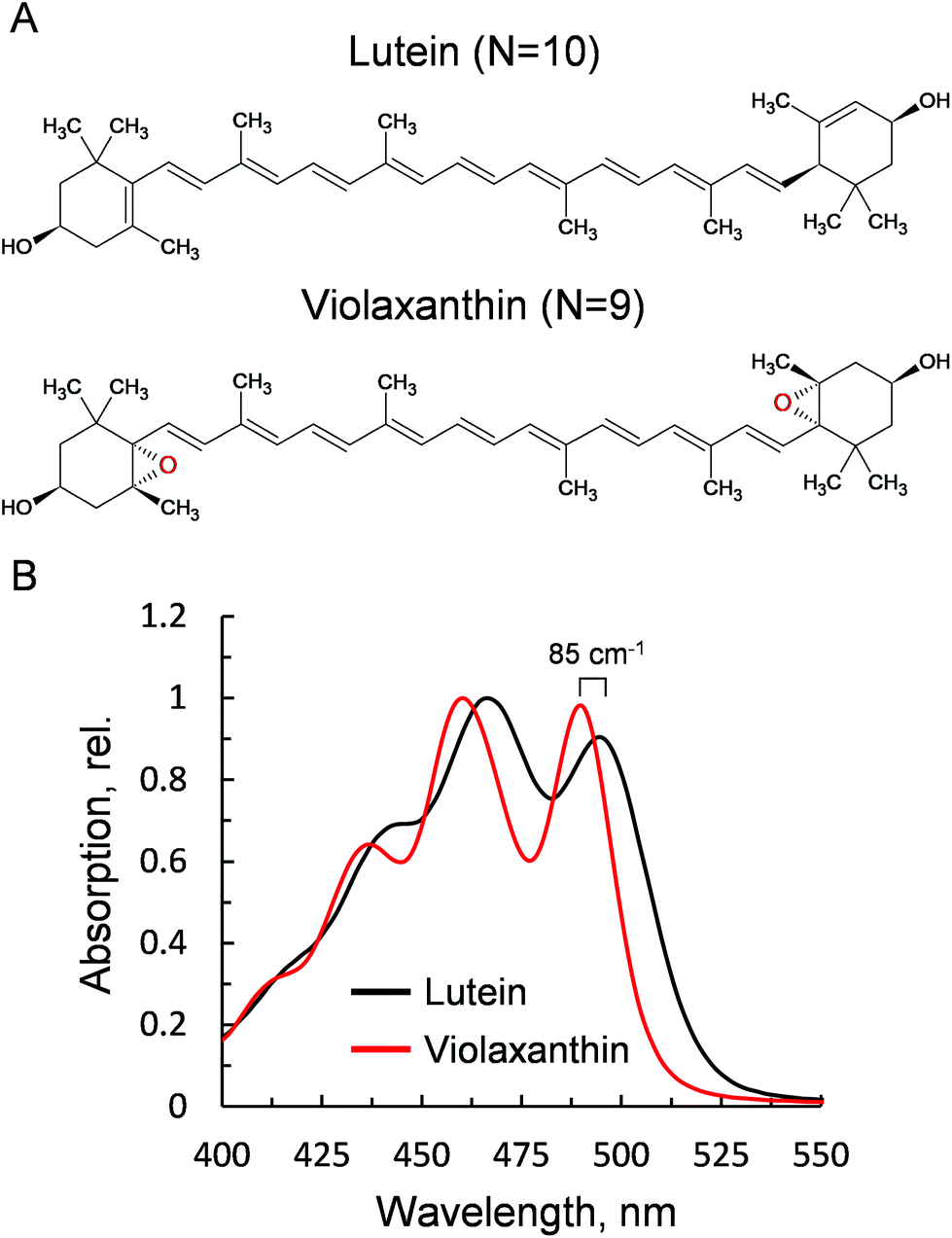 | ||
| Fig. 1 Properties of the xanthophylls lutein and violaxanthin. (A) Chemical structures. (B) Absorption spectra after HPLC purification, obtained via LHCII solubilisation in 80% acetone. | ||
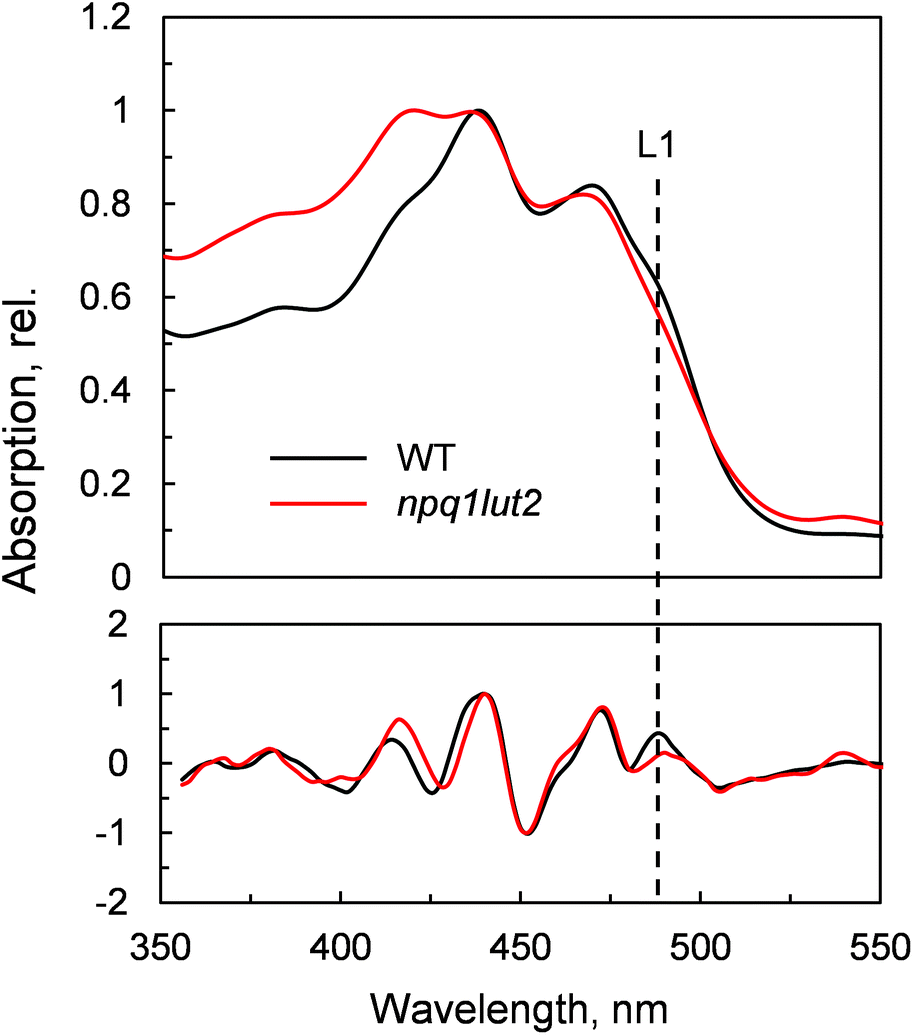 | ||
| Fig. 2 Absorption spectra of LHCII monomers in gel isolated from WT and npq1lut2 plants. Bottom panel shows the inverted second derivatives of the spectra, normalised to their maximum. The dashed line highlights the maximum of the 0–0 transition of the xanthophyll bound to the L1 site in WT LHCII. | ||
To understand how the energy/structure of xanthophylls modulates the ability of LHCII to switch to the quenched conformation, we began our analysis investigating the xanthophyll occupancy in LHCII by means of absorption spectroscopy (Fig. 2). At a first inspection, the spectra of samples from WT and npq1lut2 plants show a similar position of the absorption bands of chlorophyll and carotenoid molecules. A peak at 418 nm has been assigned in our previous work to the presence of pheophytin as a result of the protein purification procedure.38 This may reflect the partial instability of LHCII in the absence of luteins.52 The higher absorption tail at wavelengths <400 nm, instead, arises from differences in the scattering of the gels from the two samples analysed. A second derivative analysis reveals clearly the pigment binding pattern and confirms the first observations. Unexpectedly, the LHCII from npq1lut2 mutants, do not exhibit a clear fingerprint for lutein substitution with violaxanthin (blue shift). Instead, it shows a conserved position of the absorption of the carotenoid in the inner L1 site. At most, the derivative shows a slight red shift of the absorption position in the sample with violaxanthin, visible as a decrease in the absorption at m491 nm and a shoulder appearing right-shifted 3–4 nm. The derivative analysis also shows that no absorption is present after 500 nm, except for a faint band at 540 nm in npq1lut2 belonging to pheophytin.38 This observation is in line with the evidence that the absorption of lutein in the L2 site at 510 nm originates from a distorted conformation of the xanthophyll sandwiched between protein units of the trimer and this characteristic is lost upon monomerisation.6
Having investigated the binding properties of the pigments in LHCII isolated from npq1lut2 mutants, we probed the capacity of these complexes to switch to a dissipative conformation. Contrarily to previous studies,53 the novel approach involving immobilisation in gels enabled us to probe the conformational change of solely monomeric LHCII complexes, bypassing the complexity and potential experimental drawbacks of quenched LHCII aggregates.54 The gel matrix has been exploited as a controlled and easily reproducible platform to investigate dynamic changes of LHCII.19,48,50 Most importantly, the quenching induction in gels retains closely the spectroscopic properties of NPQ in vivo.19,48 In the thylakoids, regulation of light harvesting relies on the interplay between the protein PsbS, the xanthophyll cycle, lipids and involves ΔpH formation and membrane reorganisation events.22,55–57 Therefore, all in vitro setups come at a cost regarding the dynamical aspect of NPQ, as the above-mentioned factors are needed for the quick activation and de-activation of the quenching mechanism. However, despite being a very simplified model, the pigment architecture of LHCII and the signature of quencher formation are preserved in gel, making it a useful strategy to study the changes in energy equilibration pathways while avoiding the complexity of the thylakoid membrane. Incubation of LHCII in gel with buffer devoid of detergent, resulted in a dramatic drop of the chlorophyll fluorescence yield (Fig. 3A). The quenching induction showed that in 4 hours under constant mixing the fluorescence drop was already at an extent close to the maximum experimentally obtainable. However, the complete stabilisation of the quenched conformation was achieved after 17 hours of incubation. Interestingly, the violaxanthin-enriched complexes reacted to the treatment in a very similar way to the WT. At the end of the incubation, both samples stabilised at a residual fluorescence yield corresponding roughly to one fifth of the initial value (18.4 ± 1.6%, WT; 21.6 ± 5.9, npq1lut2) Although not significant, the induction traces show differences in their trends, suggesting that vio-laxanthin in npq1lut2 exerts some resistance to the conformational switch of LHCII, as previously found for LHCII aggregates from lut2 mutant plants.53 To verify this, transient absorption was applied to gain insights into the chlorophyll excited-state lifetimes. The results shown in Fig. 3B are in agreement with the previous fluorescence lifetime data measured for isolated and aggregated LHCII complexes of npq1lut2.41,53 The mutant shows a visibly longer chlorophyll S1 lifetime compared to WT. In line with this, a correlation has been shown between fluorescence lifetime and the xanthophyll composition, with the presence of more polar carotenoids (e.g. violaxanthin) causing long-lasting chlorophyll excitation.41Table 1 shows that in the unquenched LHCII sample, the amplitude of the longest component τ3 is slightly increased compared to WT, concomitantly to a decrease of the amplitude of the fast component τ1. Confirming what preliminary suggested from the analysis of the induction traces (Fig. 3A), chlorophyll S1 lifetimes are significantly different in the quenched state, with the decay of WT sample being visibly faster than the mutant (Fig. 3B). All lifetime components in the multi-exponential fitting are found consistently longer in npq1lut2, although their amplitudes is overall similar in both samples. The calculated average lifetime value in the maximum quenching state is thus ∼200 ps for npq1lut2 against ∼79 ps for WT.
 | ||
| Fig. 3 Quenching induction in isolated LHCII in gel. (A) Chlorophyll a fluorescence quenching monitored at 1, 2, 3, 4 and 20 hours from the incubation of LHCII gels in buffer without detergent. (B) Transient absorption kinetics monitored at 684 nm after the excitation of Chl a at 674 nm. The x-axis is displayed in logarithmic scale. In both graphs, empty/filled symbols mark no/maximum quenching, respectively, and color gradients indicate intermediate quenching states. | ||
| Sample | A1 | τ 1(ps) | A 2 | τ 2(ps) | A 3 | τ 3(ps) |
|---|---|---|---|---|---|---|
| Unquenched WT | 0.26 ± 0.01 | 6.3 ± 0.69 | 0.24 ± 0.02 | 89.29 ± 15.94 | 0.43 ± 0.02 | 1428.57 ± 204.08 |
| Unquenched npq1lut2 | 0.15 ± 0.01 | 3.48 ± 0.35 | 0.21 ± 0.01 | 66.23 ± 7.02 | 0.47 ± 0.01 | 1000 ± 79.64 |
| Quenched WT | 0.13 ± 0.03 | 1.75 ± 0.81 | 0.42 ± 0.02 | 17.76 ± 2.59 | 0.38 ± 0.02 | 188.68 ± 21.36 |
| Quenched npq1lut2 | 0.08 ± 0.01 | 3.54 ± 2.21 | 0.42 ± 0.03 | 45.66 ± 7.30 | 0.38 ± 0.03 | 476.19 ± 68.03 |
The next step we undertook was to analyse the changes occurring in the pigment binding sites upon quenching induction, probing the fingerprints of the quenched conformation (Fig. 4). In WT, a pronounced decrease in absorptivity in the carotenoid absorption region is visible by the simple inspection of the normalised spectra (Fig. 4A). Difference spectra in panel C, highlight the exact position of these changes. The results are in good agreement with what was previously observed in LHCII trimers,19 revealing that the quencher is born within single LHCII monomers, in the absence of protein aggregation. The difference spectra show that upon quenching induction, the LHCII is perturbed such that all pigments are to some degree affected. However, the most prominent changes occur in the carotenoid occupying the L1 site, as evidenced by the marked absorption loss at 490 nm. npq1lut2 shows overall a very similar picture with respect to the WT (Fig. 4B and D). However, the maximum of absorption loss presents a non-negligible 4 nm red shift in the violaxanthin-enriched samples. This is opposite to a blue-shift expected by the lower energy 0–0 transition of violaxanthin (Fig. 1B). This red shift is also slightly evidenced by the second derivative analysis of the absorption spectra in Fig. 2. This observation suggests that the protein scaffold is strongly interfering with the properties of the carotenoid in the L1 site. Carotenoid transition dipole moments are very sensitive to the polarisability of the environment and display marked variations depending on the nature of their solvent.58 In addition, they are sensitive to distortion of structural deformations of the CC backbone.59,60 A shift to lower energies transitions has been reported various times in pigment-protein complexes.6,61,62 Notable is the example of lutein 2 in trimeric LHCII, which appears to be bent at the interface between monomers, giving rise to a shift ∼630 cm−1 energy shift of its absorption band.6,63 Ultimately, the results shown in Fig. 4D reveal how the protein, acting as a “programmed solvent”,64 modulates the electronic properties of the violaxanthin in L1.
 | ||
| Fig. 4 Absorption changes in the Soret region induced upon quenching. (A) and (C), WT LHCII in gel; (B) and (D), npq1lut2 LHCII in gel. (A) and (B) show the absorption spectra normalised at the maximum chlorophyll a absorption at ∼438 nm, while (C) and (D) show the offset difference spectra (quenched-minus-unquenched) at different stages of the quenching induction. | ||
The outcome of the absorption analysis in Fig. 4 highlights the resemblance of the changes occurring to the pigment arrangement in LHCII in violaxanthin- and lutein-enriched complexes. Despite the different pigment energetics, indeed, quenching still occurs in both complexes (Fig. 3) and involves similarly in both samples the participation of the L1 xanthophyll site (Fig. 4). The L1 site is enclosed by the chlorophylls of the terminal emitter site, the main proposed energy sink in LHCII responsible for inter-complexes energy transfer.27,65 To unravel the involvement of this site in the energy quenching of both complexes studied, we performed a detailed analysis of the QY absorption band of chlorophyll a in LHCII (Fig. 5 and Fig. S1†). The position of the QY transition can identify chlorophyll a sub-populations and detect changes in pigment site energies. The absorption spectra in Fig. S1† evidence a decrease of the amplitude of the QY band during the induction (0.039 ± 0.001, unquenched; 0.034 ± 0.001, quenched; Fig. S1A†). This decrease is accompanied by a shift in the maximum absorption position, which, upon quenching, peaks 2 nm blue-shifted (674 nm, unquenched; 672 nm, quenched; Fig. S1B†). In room temperature LHCII spectra, QY is a complex convolution of the signal coming from all 8 chlorophylls a bound. A crude distinction of the different chlorophyll a sub-populations can come from a second derivative analysis of these spectra, which picks up subtle flex points in the curves (Fig. 5). From the analysis of the WT two chlorophyll populations emerge clearly, one with maximum at 672 nm and another peaking at 680 nm (Fig. 5A). The normalisation at 672 nm highlights a consistent trend of the red-most band intensity to decrease upon quenching induction. In the most quenched samples, it barely remains a shoulder of the main 672 nm band. A very similar picture occurs in npq1lut2 samples(Fig. 5B). It is interesting to note that the position of the second derivative peaks is shifted towards the blue by 2–3 nm compared to the WT sample. The pronounced loss of absorption in the red wing of the QY band suggests that the lower energy chlorophylls are the most affected during the quencher switch. This is similar to what was previously reported in isolated trimers and intact membranes.48,66 Beside confirming the involvement of terminal emitting chlorophylls in quenching, our findings show that this is independent of xanthophyll composition. This observation suggests that the conformational switch of LHCII is a remarkably robust trait that works beyond the nature of the carotenoids bound.
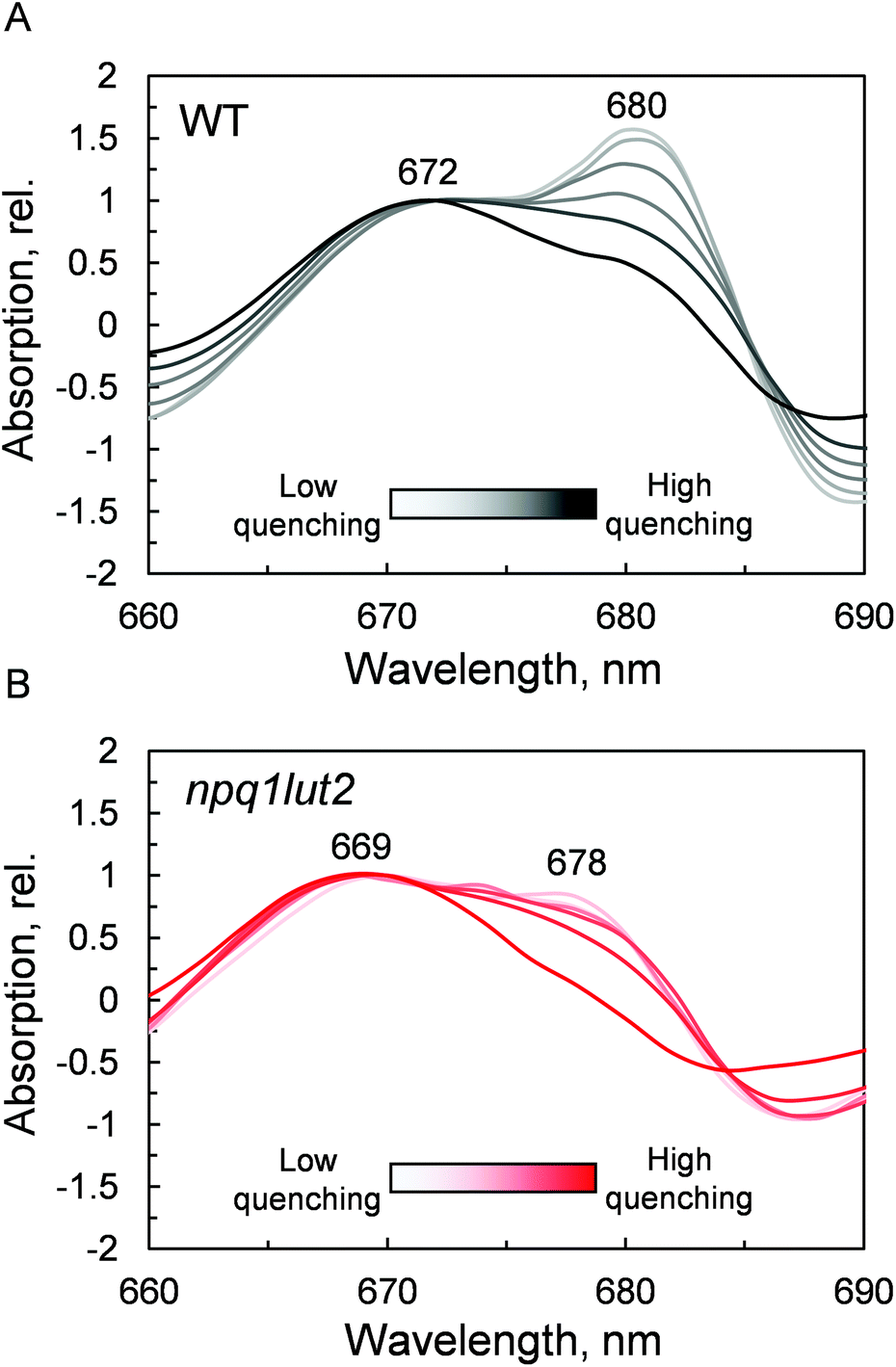 | ||
| Fig. 5 Quenching-induced changes of chlorophyll a QY absorption band. Inverted second derivatives of the absorption spectra of LHCII in gel, normalised at 672 nm for WT (A) and 669 nm for npq1lut2 (B). | ||
Ultimately, having revealed the common involvement of the terminal emitting site during quenching in WT and mutant LHCII, we investigated the changes occurring in this site upon the conformational switch. Low temperature fluorescence measurements can yield information about the lower-energy chlorophyll clusters in LHCII and their relative coupling.67 From the emission spectra, it emerges that npq1lut2 complexes have reduced emission in the red region of the main fluorescent peak compared to WT, with maximum signal loss located at ∼686 nm (Fig. S2†). Concomitantly, they exhibit a slight increase of the vibrionic far-red band and a blue-shift of the whole spectrum. The featureless emission below 670 nm partly originates from the free pheophytin present in mutant LHCII (cf.Fig. 2). These data, together with the blue shift of the QY absorption band (Fig. 5B), suggest a loosening of strong excitonic coupling in terminal emitting chlorophylls. It is worth noting that LHCII emission spectra can easily deviate from a narrow and symmetric spectral distribution by modifying the relative orientation, and thus coupling, of chlorophylls a 611 and 612 (using notation of ref. 23).67,68
Spectra were then recorded at different phases of the quenching induction (Fig. 6). Intriguingly, the changes observed are minimal, despite the 5-fold difference in fluorescence yield (Fig. 3A). The comparison with previous data highlights differences between the signature of quenched LHCII immobilised in gels and the aggregated complexes in solution.69 While the latter exhibits a marked emission peak centred roughly at 700 nm, the former displays a smaller and featureless increase in the intensity of far-red emission.19 Instead, the most evident change occurring is a gradual red-shift upon quenching. Spectral broadening is likely to arise from the heterogeneous contribution of several conformational states. Due to its average nature, bulk spectroscopic measurements can't discriminate precisely between these different conformations, which is instead possible in single-molecule studies.68 The quenching induction could thus increase the number of states accessible, leading to a broader fluorescence peak. Generally, small spectral shifts can identify variations of the chlorophyll site energies or their excitonic coupling, since the magnitude of both effects is similar.70 Concomitantly to the red-shift and broadening, a blue-shifted emission at around 670 nm is slightly enhanced in the quenched LHCII from mutants, while no change is detected in the WT (Fig. 6B). The position of this emission matches closely the blue-most emission contribution to the steady-state unquenched spectrum in npq1lut2 that is missing in WT (Fig. S2†). This again may result from an effect of the lutein-violaxanthin substitution, structurally affecting the mutual orientation of chlorophylls and therefore disrupting, to some degree, specific excitonic clusters. This could lead to the observed heterogeneity in the emission spectra. Upon quenching induction, however, the “bluer” chlorophylls are only marginally affected, compared to the main red-most emitting chlorophyll cluster. Spectral fluctuations are indicative of a high degree of disorder in the protein environment. LHCII is characterised by conformational plasticity and a high degree of static disorder characterises the terminal emitter site.71 A combination of pigment site energies variations and changes in excitonic coupling at the level of the terminal emitter are likely to generate the dissipative interactions in quenched LHCII in gel. Energy transfer models can explain red-shifts lower than 695 nm considering variations in pigment site energies and without invoking the mixing of excitonic and charge transfer states.18,27 On the contrary, considerable emission >695 nm in LHCII aggregates and thylakoid membranes likely arises from the formation of charge transfer states.72–74 While these have been found to correlate with NPQ and have been proposed to play a direct role in energy dissipation,75 our data suggest that these states do not occur significantly in LHCII monomers, as similarly found in isolated CP29 complexes,15 and are therefore not involved as energy quenchers. Carotenoids, instead, are good candidates for the quencher in virtue of their complex excited states photodynamics, which result in a ladder of intermediate short-lived states.76–78 The perturbation of the local interaction with chlorophylls open up an energy transfer channel that ends with a fast internal conversion to the ground state.15,16
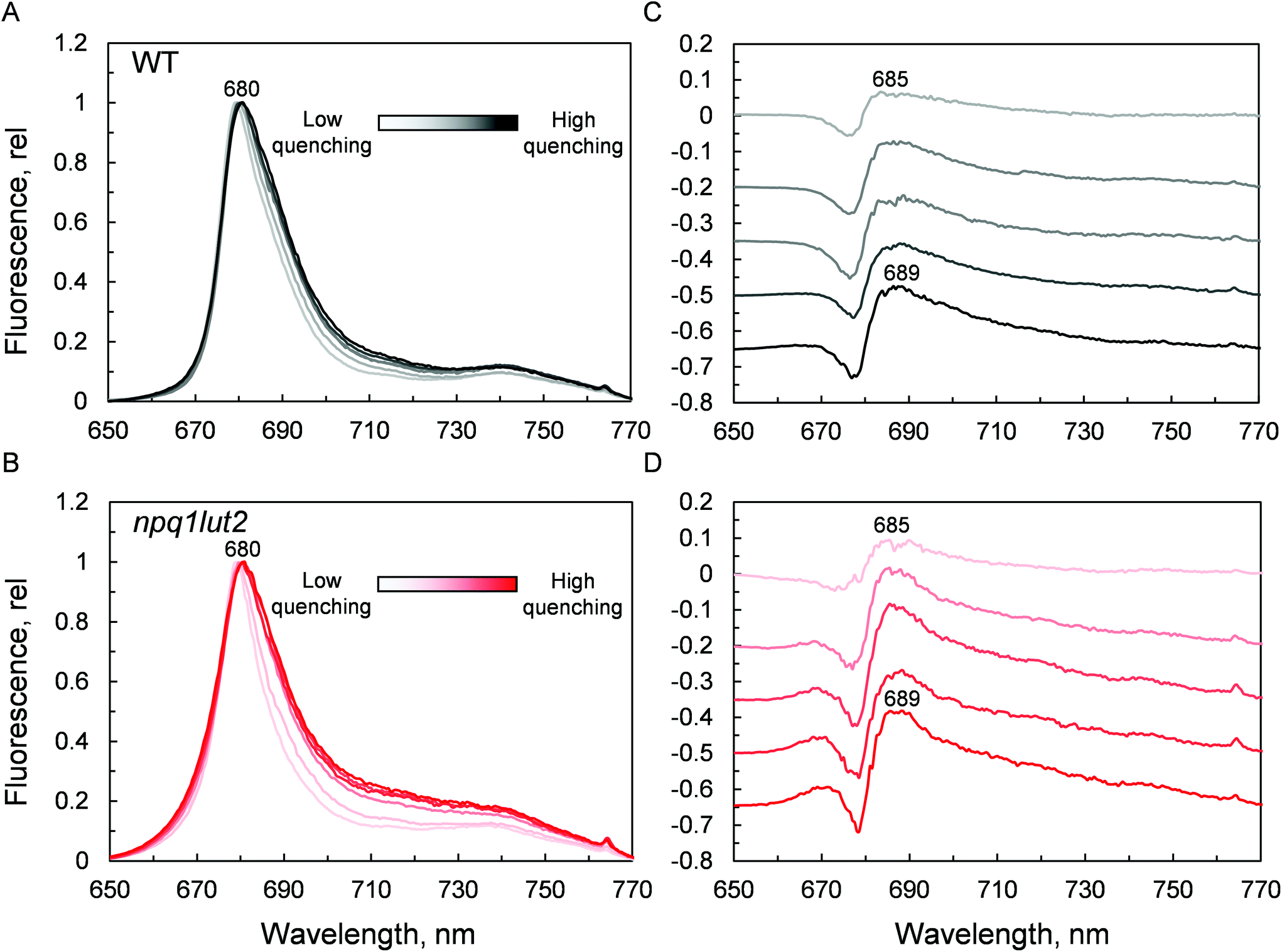 | ||
| Fig. 6 Low temperature fluorescence analysis during quenching induction. (A) and (C), WT LHCII in gel; (B) and (D), npq1lut2 LHCII in gel. (A) and (B) show the 77 K normalised fluorescence spectra, while (C) and (D) show the offset difference spectra (quenched-minus-unquenched) at different stages of the quenching induction. | ||
To summarise, our data on major LHCII in monomeric form obtained from different xanthophyll mutants showed that the carotenoid chemical structure and properties do not hinder the capacity of the complexes to undergo dissipative conformational changes. Both lutein and violaxanthin, that affect NPQ in vivo in seemingly opposite ways,36 not only can bind to the inner L1 and L2 sites,39 but also display redundancy in their possible function as energy quenchers. But how, then, is it possible that pigments with so different energies and polarity retain the same function when bound to specific LHCII pockets? Several works have revealed the strong homogenising effect on the spectroscopic properties of carotenoids in light harvesting complexes.30,31,38 On this aspect, it is interesting to note that early-diverging phototrophic organisms, such as higher plants and diatoms, retain certain conserved features in their respective light-harvesting complexes. These complexes exhibit a remarkable conservation of the α-helices in the proximity of the L1 and L2 carotenoid binding sites.79,80 This results in the high degree of superimposition of carotenoids and chlorophylls occupying them, that retain similar orientation and distance in different organisms.80 Therefore, despite being so diverse in thylakoid chromophore composition and organisation, key structural details such as the conservation of the L1 and L2 sites are maintained, underlying their pivotal role in light harvesting and photoprotection. Our data show in particular an extreme plasticity of the carotenoid and chlorophylls bound to in the terminal emitting site (L1). The presence of two hydroxyl groups in the end rings of lutein is exploited by the protein to anchor the pigment via hydrogen bonds with specific amino acids (R162, L162, Q197, P205, Fig. 7). Both lutein and violaxanthin possess these functional groups in their end rings. Since also the length of the carotenoids in their planar configuration is similar (Fig. 1), the occupancy of violaxanthin in the L1 site is not unexpected. However, this work highlights the high degree of modulation of the spectroscopic properties of the xanthophyll bound to L1 by the surrounding protein environment. The colour of the carotenoid is tuned by the residues forming the binding pocket of the chromophore (Fig. 2 and 7). The robustness of the light harvesting architecture is an essential trait for the subsistence of photosynthetic organisms. One of the most fundamental characteristics underlying sunlight harvesting, is the programmed pigment architecture in antenna complexes.81 The diversity of pigment types and the controlled interactions between them and with the protein ensure efficient light harvesting in a noisy environment.82,83 Thus, the protein scaffold seems not only to fix tightly the relative orientations between chromophores in the L1 site,80 but also to modulate their site energies, to maximise both light-harvesting and photoprotective roles. In this context, carotenoid composition becomes dispensable for the quenching function of LHCII.30,31,37,39,47 By contrast, it emerges that structural cues predominantly dictate the functional aspects of antenna components.16,84 By examining the intermediate stages of quenching formation, we also disclosed that quenching is not an on/off switch, but rather relies in shifting the equilibrium of LHCII towards dissipative conformations. Controlled protein disorder, modulated by factors that are related to NPQ in vivo such as pH and zeaxanthin, underlies this equilibrium shift.20 It is likely that a gradual conformational change varies the orientation and coupling of chlorophylls and the L1 xanthophyll, enabling the formation of dissipative interactions.57,63,85,86 The allosteric modulation by zeaxanthin and PsbS and the cooperative nature of the quenching switch grant the controlled modulation of the light harvesting function of LHCII in vivo.22
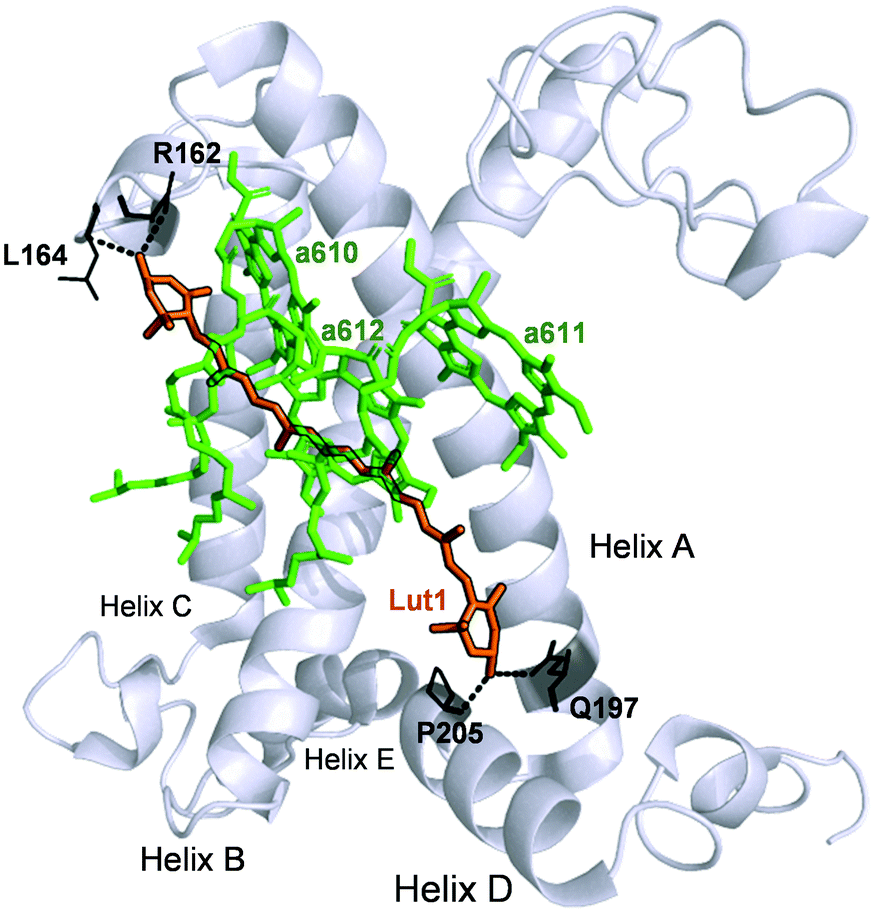 | ||
| Fig. 7 Structure of the WT LHCII monomer. Lutein in position L1 (Lut1) is shown in orange. The amino acids involved in its binding are shown in black and the dashed lines mark hydrogen bonds. Chlorophylls a of the terminal emitter site (610, 611 and 612 in the notation of Liu et al.23) are displayed in green. | ||
4. Conclusions
In this work, we investigated the role of the LHCII protein structure in the context of the mechanism of excited-state energy quenching (NPQ). Comparing isolated monomers of major LHCII complexes of WT or npq1lut2 plants we were able to identify the terminal emitting chlorophylls and the xanthophyll in the L1 pocket as the primary site for energy quenching. The qualitatively and quantitatively similar spectroscopic changes, regardless of the differential binding of either lutein or violaxanthin, suggest that the LHCII protein scaffold evolved to be both flexible, allowing functional conformational switches, and robust, allowing redundancy in the nature of the xanthophylls bound.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 675006. TP and MD thanks the Czech Science Foundation grant 19-28323X for financial support.References
- D. Siefermann-Harms, Physiol. Plant., 1987, 69, 561–568 CrossRef CAS.
- B. Demmig-Adams and W. W. Adams, Trends Plant Sci., 1996, 1, 21–26 CrossRef.
- M. Guerin, M. E. Huntley and M. Olaizola, Trends Biotechnol., 2003, 21, 210–216 CrossRef CAS.
- N. I. Krinsky, J. T. Landrum and R. A. Bone, Annu. Rev. Nutr., 2003, 23, 171–201 CrossRef CAS PubMed.
- A. V. Ruban, A. J. Young, A. A. Pascal and P. Horton, Plant Physiol., 1994, 104, 227–234 CrossRef CAS PubMed.
- A. V. Ruban, A. A. Pascal and B. Robert, FEBS Lett., 2000, 477, 181–185 CrossRef CAS PubMed.
- R. Croce, S. Weiss and R. Bassi, J. Biol. Chem., 1999, 274, 29613–29623 CrossRef CAS PubMed.
- H. A. Frank and R. J. Cogdell, Photochem. Photobiol., 1996, 63, 257–264 CrossRef CAS PubMed.
- R. Croce, M. G. Müller, R. Bassi and A. R. Holzwarth, Biophys. J., 2001, 80, 901–915 CrossRef CAS PubMed.
- T. Polívka and H. A. Frank, Acc. Chem. Res., 2010, 43, 1125–1134 CrossRef PubMed.
- E. J. G. Peterman, C. C. Gradinaru, F. Calkoen, J. C. Borst, R. van Grondelle and H. van Amerongen, Biochemistry, 1997, 36, 12208–12215 CrossRef CAS PubMed.
- M. Havaux and K. K. Niyogi, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8762–8767 CrossRef CAS PubMed.
- L. Dall'Osto, S. Cazzaniga, M. Havaux and R. Bassi, Mol. Plant, 2010, 3, 576–593 CrossRef PubMed.
- A. V. Ruban, R. Berera, C. Ilioaia, I. H. van Stokkum, J. T. M. Kennis, A. A. Pascal, H. van Amerongen, B. Robert, P. Horton and R. van Grondelle, Nature, 2007, 450, 575–578 CrossRef CAS PubMed.
- V. Mascoli, N. Liguori, P. Xu, L. M. Roy, I. H. van Stokkum and R. Croce, Chem, 2019, 5, 2900–2912 CAS.
- M. Son, A. Pinnola, S. C. Gordon, R. Bassi and G. S. Schlau-Cohen, Nat. Commun., 2020, 11, 1295 CrossRef CAS PubMed.
- A. V. Ruban, Plant Physiol., 2016, 170, 1903–1916 CrossRef CAS PubMed.
- T. P. J. Krüger, V. I. Novoderezhkin, C. Ilioaia and R. Van Grondelle, Biophys. J., 2010, 98, 3093–3101 CrossRef PubMed.
- C. Ilioaia, M. P. Johnson, P. Horton and A. V. Ruban, J. Biol. Chem., 2008, 283, 29505–29512 CrossRef CAS PubMed.
- T. P. J. Krüger, C. Ilioaia, M. P. Johnson, E. Belgio, P. Horton, A. V. Ruban and R. Van Grondelle, Biophys. J., 2013, 105, 1018–1026 CrossRef PubMed.
- G. S. Schlau-Cohen, H. Y. Yang, T. P. Krüger, P. Xu, M. Gwizdala, R. Van Grondelle, R. Croce and W. E. Moerner, J. Phys. Chem. Lett., 2015, 6, 860–867 CrossRef CAS PubMed.
- F. Saccon, V. Giovagnetti, M. K. Shukla and A. V. Ruban, J. Exp. Bot., 2020, 1–12 Search PubMed.
- Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- R. Croce, R. Remelli, C. Varotto, J. Breton and R. Bassi, FEBS Lett., 1999, 456, 1–6 CrossRef CAS PubMed.
- M. Mozzo, L. Dall'Osto, R. Hienerwadel, R. Bassi and R. Croce, J. Biol. Chem., 2008, 283, 6184–6192 CrossRef CAS PubMed.
- M. Son, A. Pinnola, R. Bassi and G. S. Schlau-Cohen, Chem, 2019, 5, 575–584 CAS.
- V. I. Novoderezhkin, M. A. Palacios, H. Van Amerongen and R. Van Grondelle, J. Phys. Chem. B, 2005, 109, 10493–10504 CrossRef CAS PubMed.
- A. V. Ruban, P. J. Lee, M. Wentworth, A. J. Young and P. Horton, J. Biol. Chem., 1999, 274, 10458–10465 CrossRef CAS PubMed.
- P. Jahns, D. Latowski and K. Strzalka, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 3–14 CrossRef CAS PubMed.
- T. Polívka, D. Zigmantas, V. Sundström, E. Formaggio, G. Cinque and R. Bassi, Biochemistry, 2002, 41, 439–450 CrossRef PubMed.
- M. Fuciman, M. M. Enriquez, T. Polívka, L. Dallosto, R. Bassi and H. A. Frank, J. Phys. Chem. B, 2012, 116, 3834–3849 CrossRef CAS PubMed.
- N. Liguori, P. Xu, I. H. van Stokkum, B. van Oort, Y. Lu, D. Karcher, R. Bock and R. Croce, Nat. Commun., 2017, 8, 1994 CrossRef PubMed.
- N. E. Holt, D. Zigmantas, L. Valkunas, X. P. Li, K. K. Niyogi and G. R. Fleming, Science, 2005, 307, 433–436 CrossRef CAS PubMed.
- S. Park, A. L. Fischer, C. J. Steen, M. Iwai, J. M. Morris, P. J. Walla, K. K. Niyogi and G. R. Fleming, J. Am. Chem. Soc., 2018, 140, 11965–11973 CrossRef CAS PubMed.
- B. J. Pogson, K. K. Niyogi, O. Bjorkman and D. DellaPenna, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13324–13329 CrossRef CAS PubMed.
- K. Niyogi, C. Shih, C. Soon, B. Pogson, D. Dellapenna and O. Björkman, Photosynth. Res., 2001, 67, 139–145 CrossRef CAS PubMed.
- M. P. Johnson, A. Zia, P. Horton and A. V. Ruban, Chem. Phys., 2010, 373, 23–32 CrossRef CAS.
- F. Saccon, M. Durchan, R. Kana, O. Prášil, A. V. Ruban and T. Polívka, J. Phys. Chem. B, 2019, 123, 9312–9320 CrossRef CAS PubMed.
- L. Dall'Osto, C. Lico, J. Alric, G. Giuliano, M. Havaux and R. Bassi, BMC Plant Biol., 2006, 6, 1–20 CrossRef PubMed.
- M. Wentworth, A. V. Ruban and P. Horton, Biochemistry, 2004, 43, 501–509 CrossRef CAS PubMed.
- A. V. Ruban and M. P. Johnson, Arch. Biochem. Biophys., 2010, 504, 78–85 CrossRef CAS PubMed.
- H. Ocampo-Alvarez, E. García-Mendoza and Govindjee, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 427–437 CrossRef CAS PubMed.
- R. Kana, E. Kotabová, J. Kopečná, E. Trsková, E. Belgio, R. Sobotka and A. V. Ruban, FEBS Lett., 2016, 590, 1076–1085 CrossRef CAS PubMed.
- X. P. Li, O. Björkman, C. Shih, A. R. Grossman, M. Rosenquist, S. Jansson and K. K. Niyogi, Nature, 2000, 403, 391–395 CrossRef CAS PubMed.
- M. L. Pérez-Bueno, M. P. Johnson, A. Zia, A. V. Ruban and P. Horton, FEBS Lett., 2008, 582, 1477–1482 CrossRef PubMed.
- M. P. Johnson, A. Zia and A. V. Ruban, Planta, 2012, 235, 193–204 CrossRef CAS PubMed.
- M. Tutkus, J. Chmeliov, D. Rutkauskas, A. V. Ruban and L. Valkunas, J. Phys. Chem. Lett., 2017, 8, 5898–5906 CrossRef CAS PubMed.
- C. Ilioaia, M. P. Johnson, P.-N. Liao, A. A. Pascal, R. van Grondelle, P. J. Walla, A. V. Ruban and B. Robert, J. Biol. Chem., 2011, 286, 27247–27254 CrossRef CAS PubMed.
- D. Rutkauskas, J. Chmeliov, M. Johnson, A. Ruban and L. Valkunas, Chem. Phys., 2012, 404, 123–128 CrossRef CAS.
- P. Akhtar, M. Dorogi, K. Pawlak, L. Kovács, A. Bóta, T. Kiss, G. Garab and P. H. Lambrev, J. Biol. Chem., 2015, 290, 4877–4886 CrossRef CAS PubMed.
- F. G. Plumley and G. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 146–150 CrossRef CAS PubMed.
- H. Lokstein, L. Tian, J. E. Polle and D. DellaPenna, Biochim. Biophys. Acta, Bioenerg., 2002, 1553, 309–319 CrossRef CAS.
- M. P. Johnson, M. L. Pérez-Bueno, A. Zia, P. Horton and A. V. Ruban, Plant Physiol., 2009, 149, 1061–1075 CrossRef CAS PubMed.
- B. van Oort, L. M. Roy, P. Xu, Y. Lu, D. Karcher, R. Bock and R. Croce, J. Phys. Chem. Lett., 2018, 9, 346–352 CrossRef CAS PubMed.
- M. P. Johnson, J. Exp. Bot., 2020, 71, 3380–3382 CrossRef PubMed.
- S. Tietz, M. Leuenberger, R. Höhner, A. H. Olson, G. R. Fleming and H. Kirchhoff, J. Biol. Chem., 2020, 295, 1857–1866 CrossRef CAS PubMed.
- H. Li, Y. Wang, M. Ye, S. Li, D. Li, H. Ren, M. Wang, L. Du, H. Li, G. Veglia, J. Gao and Y. Weng, Sci. China: Chem., 2020, 63, 1121–1133 CrossRef CAS.
- P. O. Andersson, T. Gillbro, L. Ferguson and R. J. Cogdell, Photochem. Photobiol., 1991, 54, 353–360 CrossRef CAS.
- M. Macernis, J. Sulskus, C. D. P. Duffy, A. V. Ruban and L. Valkunas, J. Phys. Chem. A, 2012, 116, 9843–9853 CrossRef CAS PubMed.
- M. J. Llansola-Portoles, R. Sobotka, E. Kish, M. K. Shukla, A. A. Pascal, T. Polívka and B. Robert, J. Biol. Chem., 2017, 292, 1396–1403 CrossRef CAS PubMed.
- R. J. van Dorssen, J. Breton, J. J. Plijter, K. Satoh, H. J. van Gorkom and J. Amesz, Biochim. Biophys. Acta, Bioenerg., 1987, 893, 267–274 CrossRef CAS.
- M. M. Mendes-Pinto, D. Galzerano, A. Telfer, A. A. Pascal, B. Robert and C. Ilioaia, J. Biol. Chem., 2013, 288, 18758–18765 CrossRef CAS PubMed.
- H. Yan, P. Zhang, C. Wang, Z. Liu and W. Chang, Biochem. Biophys. Res. Commun., 2007, 355, 457–463 CrossRef CAS PubMed.
- G. D. Scholes, C. Curutchet, B. Mennucci, R. Cammi and J. Tomasi, J. Phys. Chem. B, 2007, 111, 6978–6982 CrossRef CAS PubMed.
- F. Müh, M. E. A. Madjet and T. Renger, J. Phys. Chem. B, 2010, 114, 13517–13535 CrossRef PubMed.
- M. P. Johnson and A. V. Ruban, J. Biol. Chem., 2009, 284, 23592–23601 CrossRef CAS PubMed.
- C. Ramanan, J. M. Gruber, P. Malý, M. Negretti, V. Novoderezhkin, T. P. Krüger, T. Mančal, R. Croce and R. Van Grondelle, Biophys. J., 2015, 108, 1047–1056 CrossRef CAS PubMed.
- T. P. J. Krüger, P. Malý, M. T. A. Alexandre, T. Mančal, C. Büchel and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11063–E11071 CrossRef PubMed.
- A. Natali, J. M. Gruber, L. Dietzel, M. C. Stuart, R. Van Grondelle and R. Croce, J. Biol. Chem., 2016, 291, 16730–16739 CrossRef CAS PubMed.
- J. Adolphs and T. Renger, Biophys. J., 2006, 91, 2778–2797 CrossRef CAS PubMed.
- N. Liguori, X. Periole, S. J. Marrink and R. Croce, Sci. Rep., 2015, 5, 15661 CrossRef CAS PubMed.
- J. Chmeliov, A. Gelzinis, E. Songaila, R. Augulis, C. D. Duffy, A. V. Ruban and L. Valkunas, Nat. Plants, 2016, 2, 16045 CrossRef CAS PubMed.
- J. Chmeliov, A. Gelzinis, M. Franckevičius, M. Tutkus, F. Saccon, A. V. Ruban and L. Valkunas, J. Phys. Chem. Lett., 2019, 10, 7340–7346 CrossRef CAS PubMed.
- V. Mascoli, A. Gelzinis, J. Chmeliov, L. Valkunas and R. Croce, Chem. Sci., 2020, 11, 5697–5709 RSC.
- K. Pawlak, S. Paul, C. Liu, M. Reus, C. Yang and A. R. Holzwarth, Photosynth. Res., 2020, 144, 195–208 CrossRef CAS PubMed.
- E. J. Taffet, B. G. Lee, Z. S. Toa, N. Pace, G. Rumbles, J. Southall, R. J. Cogdell and G. D. Scholes, J. Phys. Chem. B, 2019, 123, 8628–8643 CrossRef CAS PubMed.
- C. C. Gradinaru, J. T. Kennis, E. Papagiannakis, I. H. Van Stokkum, R. J. Cogdell, G. R. Fleming, R. A. Niederman and R. Van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2364–2369 CrossRef CAS PubMed.
- T. Polívka and V. Sundström, Chem. Phys. Lett., 2009, 477, 1–11 CrossRef.
- D. Carbonera, A. Agostini, M. Di Valentin, C. Gerotto, S. Basso, G. M. Giacometti and T. Morosinotto, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1235–1246 CrossRef CAS PubMed.
- W. Wang, L. J. Yu, C. Xu, T. Tomizaki, S. Zhao, Y. Umena, X. Chen, X. Qin, Y. Xin, M. Suga, G. Han, T. Kuang and J. R. Shen, Science, 2019, 363, eaav0365 CrossRef CAS PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. Van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- P. Malý, A. T. Gardiner, R. J. Cogdell, R. van Grondelle and T. Mančal, Phys. Chem. Chem. Phys., 2018, 20, 4360–4372 RSC.
- T. B. Arp, J. Kistner-Morris, V. Aji, R. J. Cogdell, R. van Grondelle and N. M. Gabor, Science, 2020, 368, 1490–1495 CrossRef CAS PubMed.
- M. J. Llansola-Portoles, F. Li, P. Xu, S. Streckaite, C. Ilioaia, C. Yang, A. Gall, A. A. Pascal, R. Croce and B. Robert, Biochim. Biophys. Acta, Bioenerg., 2020, 1861, 148078 CrossRef CAS PubMed.
- C. D. P. Duffy, J. Chmeliov, M. Macernis, J. Sulskus, L. Valkunas and A. V. Ruban, J. Phys. Chem. B, 2013, 117, 10974–10986 CrossRef CAS PubMed.
- K. F. Fox, V. Balevičius, J. Chmeliov, L. Valkunas, A. V. Ruban and C. D. P. Duffy, Phys. Chem. Chem. Phys., 2017, 19, 22957–22968 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Chlorophyll QY absorption analysis; low-temperature fluorescence spectra of unquenched LHCII. See DOI: 10.1039/D0PP00174K |
| This journal is © The Royal Society of Chemistry and Owner Societies 2020 |