
Hydrogel scaffolds for tissue engineering: the importance of polymer choice
Christopher D.
Spicer
 ab
ab
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: chris.spicer@york.ac.uk
bYork Biomedical Research Institute, University of York, Heslington, York, YO10 5DD, UK
First published on 25th September 2019
Abstract
Hydrogel scaffolds that can repair or regrow damaged biological tissue have great potential for the treatment of injury and disease. These biomaterials are widely used in the tissue engineering field due to their ability to support cell proliferation, migration and differentiation, to permit oxygen and nutrient transport, and to mimic native soft tissue. Careful design of the underlying polymer scaffold is therefore vital, dictating both the physical and biological properties of a hydrogel. In this review, we will provide a critical overview of hydrogel design from the perspective of the polymer chemistry, highlighting both the advantages and limitations of particular polymer structures, properties, and architectures. In doing so, we will help equip researchers with the tools needed to design new polymer systems and hydrogel scaffolds that address current limitations in the field and hinder clinical translation.
Chris joined the University of York as a Lecturer in Chemistry in 2018. His group is interested in designing and synthesising novel biomaterial scaffolds for tissue repair, with a particular focus on materials that can recreate the spatial and temporal complexity of natural tissues. Previously, he studied Natural Sciences at the University of Cambridge, before moving to the University of Oxford to undertake a PhD with Prof. Ben Davis. Chris went on to complete postdoctoral research with Prof. Molly Stevens, first at Imperial College London and then the Karolinska Institute in Stockholm, before taking up his current position. |
1. Introduction
Millions of deaths occur worldwide each year as a consequence of injuries and diseases that cause tissue damage. The impact of tissue damage on quality of life and the associated healthcare burden are even more significant.1 The field of tissue engineering has the potential to revolutionise how we treat pathologies such as heart disease, osteoarthritis, chronic wounds, and organ failure, by repairing, regenerating, or improving the function of the damaged tissue.2 A key concept in tissue engineering is the use of biomaterials to support the growth of new cells and promote repair. Rather than being passive spectators, these materials should provide both physical scaffolding to cells and cues that direct their behaviour.3Of the many classes of material that have been used in tissue engineering, hydrogels have emerged as one of the most prominent and versatile.4,5 Hydrogels can be designed to support cell proliferation, migration, and differentiation, permit oxygen and nutrient transport, and provide cells with a 3D, highly hydrated environment that mimics native soft tissues. Critically, the properties of a hydrogel are dictated by the chemistry of the underlying polymer from which it is synthesised. In this review, we therefore aim to provide a comprehensive overview of the hydrogels currently used by the tissue engineering community from the perspective of a polymer chemist – which core polymers are most commonly used to produce hydrogel scaffolds? What are the beneficial properties of these polymers that allow them to provide gels with unique properties? What is the impact of polymerisation technique and polymer architecture on the end construct? And how can polymer architecture dictate bidirectional interactions with cells and the local environment? In doing so, we will equip researchers with the tools required to match scaffold design to a given target application. Moreover, by engaging both polymer chemists and biomaterial scientists, we hope to inspire new cross-disciplinary interactions that lead to the design of novel polymers and hydrogel scaffolds that address current limitations in the field.
1.1 Scope of the review
In this review, we will specifically focus on how polymer chemistry ultimately influences hydrogel properties and applications. After briefly discussing the gel design criteria that must be considered, we will introduce the main classes of polymer used in the tissue engineering field, providing a critical discussion of both strengths and limitations. We will then go on to discuss how dynamic and responsive polymers can be exploited to generate hydrogels with interesting properties for tissue engineering, and how polymer architecture and synthesis can affect both hydrogelation and downstream applications.While we will briefly discuss the mechanisms through which hydrogels can interact with biological systems when relevant to the discussion, for a detailed overview of this topic and how hydrogels can be used for the treatment of disease, the reader is directed to the large number of excellent reviews that have been written in the past decade.4,6–10 We wish to draw particular attention to reviews by the Su,5 Burdick,11 and Anseth12 groups. Similarly, hydrogel processing and manufacture are outside the scope of this review and the reader is instead directed to the following reference.13 Finally, we have deliberately chosen to principally focus on the contributions and properties of individual polymers in this review, to emphasise the impact of a particular structure on hydrogel properties. In practice, hydrogel blends have been produced from every conceivable mixture of multiple polymers and many of the most widely implemented hydrogels in tissue engineering are in fact composite structures. This powerful approach allows the favourable characteristics of multiple polymers to be combined, allowing the disadvantages of single component gels to be overcome. For example, by taking a polymer that is mechanically strong but inert and combining it with another which can provide sites for cell adhesion but is too weak to form stable gels on its own, a hydrogel with suitable properties for tissue engineering may be produced. However, it is important to note that the properties of these polymer blends are typically additive rather than transformative. In general, the favourable properties of two complementary polymers can be combined, however, the converse is also true, and weaknesses may also be carried through in to the final blend.
2. Polymer hydrogels for tissue engineering
2.1 Hydrogel structure and categorisation
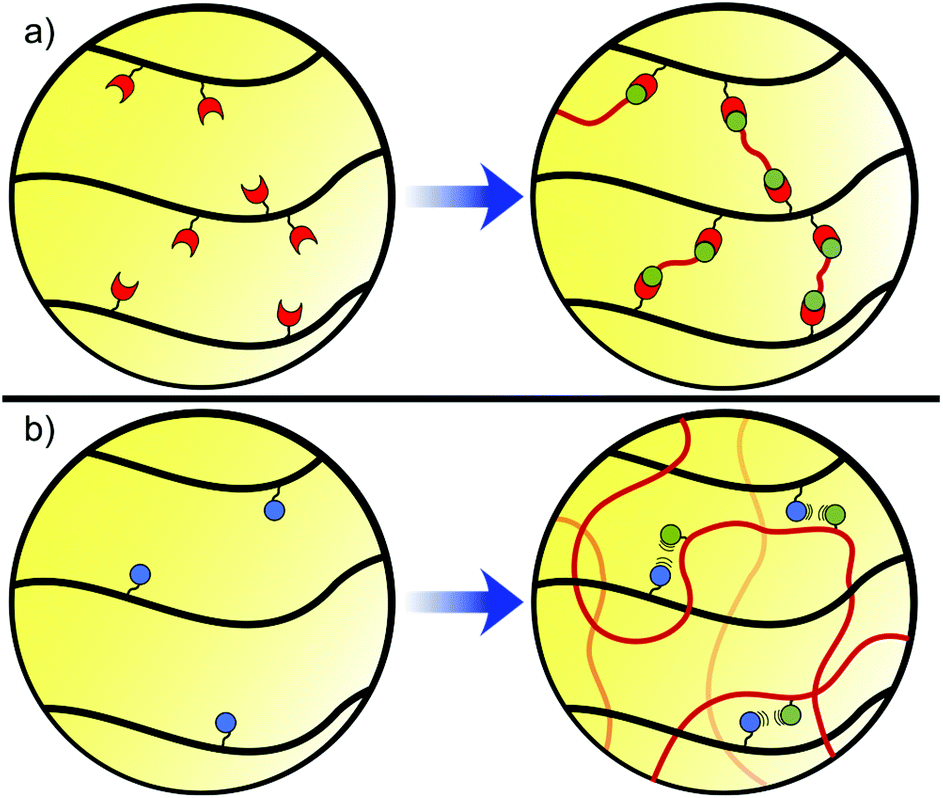
Hydrogels are 3D networks of cross-linked, hydrophilic polymers, which are able to hold large amounts of water in a swollen scaffold.14 These ‘soft’ materials display elastic behaviour that is governed by the polymer structure and architecture. While the polymer content can vary greatly, a mechanically robust gel will typically contain 0.1–10% polymer by weight, with extremes at each end of the spectrum. This leads to highly porous networks, allowing the diffusion of nutrients, oxygen, and biomolecules, while also enabling the exchange of metabolites and toxins away from cells. Porosity is often high enough to enable cell infiltration and interconnectivity, providing an excellent growth medium for tissue.13 However, densely cross-linked gels with pores <10 μm in diameter may limit cell movement, and careful design of the hydrogel network is therefore critical. Cross-linking also dictates the ability of the gel to resist expansion and maintain structure, thus contributing to the amount of water a gel can uptake (swelling ratio) and its mechanical properties.6The cross-linking within a gel can be classified into 2 categories, chemical or physical. Chemical cross-links are formed through covalent bonding between separate polymer chains, resulting in linkages that are more resistant to mechanical forces and providing gels with elastic behaviour (Fig. 1a).8 By contrast, physical cross-links rely on molecular entanglement and non-covalent interactions such as hydrogen bonding, ionic interactions, and van der Waals attractions to provide cohesion (Fig. 1b). These cross-links often allow the release of stress, providing gels with viscoelastic behaviour.8 The two classes of hydrogels result in very different properties at both the nano- and macro-scale. Material properties can be further varied through the incorporation of transiently stable covalent cross-links, and by exploiting the environmental sensitivity of non-covalent forces to create dynamic (section 5, Fig. 2a) and responsive (section 6, Fig. 2b) materials.
 | ||
| Fig. 1 (a) Chemical gels are cross-linked by covalent bonds, leading to hydrogels that can resist mechanical strain; (b) physical gels are held together by molecular entanglement and non-covalent forces, resulting in gels with viscoelastic behaviour. | ||
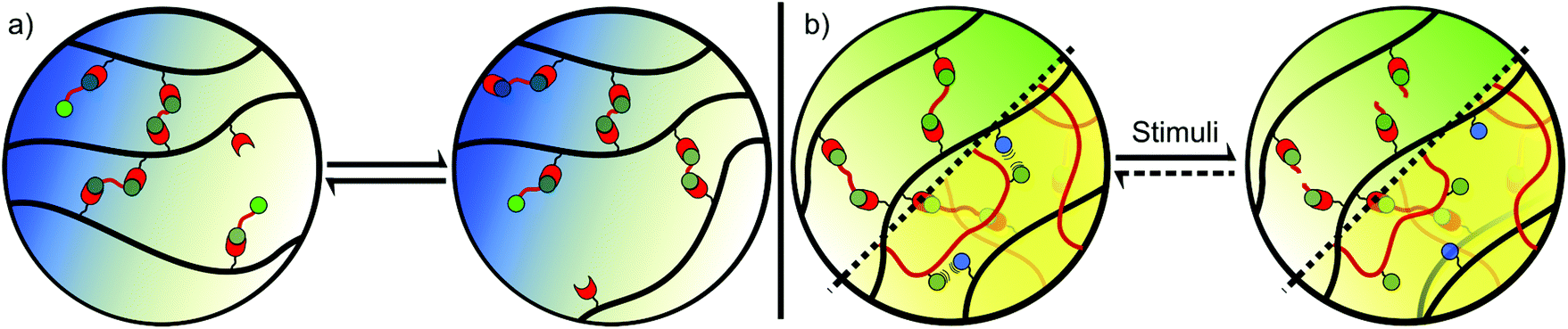 | ||
| Fig. 2 (a) Dynamic hydrogels are held together by transiently stable linkages, providing gels with a range of possible properties including self-healing and shear-thinning behaviour; (b) responsive gels undergo a change in properties upon the application of a stimulus. This change may or may not be reversible, and can result from the disruption or formation of both chemical and physical cross-links. | ||
Hydrogels can alternatively be classified into gels formed from synthetic (section 3) or natural (section 4) polymers. Each class of material has advantages and disadvantages, which will be discussed in detail later. However, more generally natural polymers possess enhanced biocompatibility and bio-instructive capabilities, with some displaying strain-stiffening behaviour that more closely resembles the native extracellular matrix (ECM).5 Synthetic polymers, on the other hand, provide predictable, precise, and tunable chemistry and versatile hydrogel properties. The choice of polymer is therefore dictated by the needs of the final scaffold.
2.2 Key polymer design considerations
There is no single ideal hydrogel that can be applied in all tissue engineering technologies. Instead, the properties must be matched to the cell type, pathology, tissue, and desired outcome in mind. For example, a hydrogel that will be used as a permanent scaffold for the growth of replacement bone will need to be very different to a gel that supplies a temporary reservoir of cells for nerve regeneration. Similarly, the bioinductive effects of a hydrogel for cartilage repair, a tissue that does not contain any blood vessels, will differ from one that will be applied in muscle, where a dense blood supply is needed for growth. Careful design of the core polymer is therefore essential to deliver the required properties. Amongst the key design considerations that must be taken into account are:(i) Will the gel be applied in vivo or used to grow tissue in vitro?
Polymer scaffolds that are well tolerated in the lab by isolated cells under controlled conditions may still have an adverse impact when implanted in the body, where they are exposed to a multitude of different cell types, and an immune system that is primed to actively attack foreign bodies.15 For example, though poly(ethylene glycol) (PEG) is widely used as a bioinert ‘stealth’ polymer in the tissue engineering community, recent studies have identified the presence of anti-PEG antibodies being generated against these materials in vivo.16 Naturally derived materials are not exempt from these effects, with the immunogenicity of protein epitopes or xeno-contamination of concern in vivo.
The stresses and strain experienced by a hydrogel implanted into a patient are also likely to be different to those experienced by a material in vitro, necessitating differing mechanical properties. Furthermore, implanted hydrogels may undergo favourable exchange with their surroundings, with metabolites being washed away by the vasculature and endogenous proteins and biomolecules being recruited from the surrounding tissue.17,18 Prior functionalisation of the scaffold with bioactive motifs may therefore prove unnecessary.
(ii) Does the gel need to be space-filling or of a defined 3D architecture?
Injectable hydrogels are favourable for in vivo applications, as they allow the gel to fill the ill-defined shape of the tissue defect, and negate the need for invasive surgery.19 However, injectability presents challenges for polymer design. One possibility is the use of a preformed hydrogel that undergoes shear-thinning or stimuli responsive behaviour, allowing injection followed by gelation in situ.20–22 Alternatively, gel precursors that undergo either spontaneous or triggered gelation upon injection can be prepared.23–25 Such systems require very rapid gelation, dictated by the polymer structure and cross-linking chemistry, to minimise the leaching of soluble components into surrounding tissues.
In a similar direction, there is increasing demand for hydrogels that possess a precisely defined 3D architecture, whether for cellular alignment, to provide pathways for vascularisation, or for the patterning of gradients that mimic native tissue. The use of pendant or backbone polymer groups that are amenable to photo-patterning is one such accessible technology.26–28 Alternatively, new technologies for 3D printing and stereolithography can be exploited, using either acellular or cell-containing ‘bio-inks’.29–31 These systems require extremely rapid gelation upon extrusion or printing to ensure spatial resolution, most commonly using the mixing of solutions of alginate and calcium salts.
(iii) What is the long-term fate of the gel?
While some hydrogels are applied as permanent implants, aimed at providing long-term scaffolding and structure to a tissue, more commonly the gel should be gradually removed from the site of action once it has served its purpose. Scaffolds composed of non-biodegradable polymers, such as those formed from vinyl polymerisation, may be poorly suited in such scenarios. However, the use of degradable cross-links may open up the possibility of bio-resorbable polymers, in which the bulk hydrogel is broken down into smaller polymer blocks which, though unable to undergo further degradation, are small enough to be excreted from the body.32,33 Recent reports have highlighted the benefits of degradable hydrogels in allowing cells to remodel their environment, providing a malleable scaffold that enables the deposition of cell-derived matrix and the formation of more mature tissue (Fig. 3).34–36
 | ||
| Fig. 3 Fluorescence microscopy images of neural progenitor cells cultured in elastin-like polypeptide-based hydrogels of varying susceptibility to degradation. An increase in hydrogel degradability results in increased proliferation (EdU) and the maintenance of stemness (nestin). Reprinted by permission from Springer Nature, Nature Materials, ‘Maintenance of neural progenitor cell stemness in 3D hydrogels requires matrix remodelling’, C. M. Madl et al., 2017.34 | ||
Even polymers able to undergo degradation may prove unsuitable if the resultant breakdown products result in a detrimental effect. For example, the build-up of glycolic and lactic acid monomers following the degradation of poly(ester)-based scaffolds has been shown to lead to a local increase in pH and resultant tissue damage.37,38 Similar effects are likely to result from the breakdown of degradable PLLA and PLGA hydrogels as discussed in section 3.3. Furthermore, degradation rates must be matched to the desired application.39 Natural polysaccharides such as hyaluronic acid are rapidly degraded by endogenous enzymes, leading to low tissue retention times.40,41 On the other hand, the polyester poly(caprolactone) (PCL) has been shown to undergo hydrolysis at a slow rate, resulting in tissue persistence on a scale of years.42 Polymer choice, the formation of composite materials, and altered polymer cross-linking density can all be used to partially tune degradation to a relevant time frame.
(iv) Should cells and proteins adhere to the polymer or should it be inert?
In most tissue engineering technologies, the hydrogel should provide an adhesive scaffold to which cells can bind. This has important implications for polymer design. The native ECM has many recognition motifs that mediate cell binding to the proteins and carbohydrates that make up the matrix. An extra layer of complexity is added by the ability of these native polymers to bind additional layers of soluble proteins, which may further mediate cell adhesion or signalling.43 In contrast, many synthetic polymers are poorly cell adhesive. For example, PEG, poly(2-hydroxyethyl methacrylate) (PHEMA), and poly(vinyl alcohol) (PVA), three of the most widely used synthetic polymers in tissue engineering, exhibit negligible cell or protein binding.6 They therefore require derivatization to mediate adhesion, either through the formation of composite scaffolds or through functionalisation with bioactive motifs.44,45 This is most commonly achieved through the attachment of synthetic peptide sequences known to mediate cell binding, such as RGDS and IKVAV. The physical properties of a polymer are also able to mediate adhesion with cells adhering to positively charged surfaces, as a result of electrostatic interactions with the negative charges present on cell membranes.46
The bioinert nature of synthetic polymers can sometimes be favourable, with the selective attachment of bioactive groups providing greater control over cell behaviour than the use of heterogeneous natural polymers which can induce promiscuous signalling. Furthermore, the bulk scaffold may be able to resist detrimental protein fouling and minimise non-specific protein adsorption. For example, it has been shown that RGD-functionalised PEG hydrogels are able to promote cell adhesion while at the same time minimising the adsorption of serum proteins. The induction of a detrimental foreign body response is therefore minimised upon implantation in vivo.47 Although less common, there are certain scenarios in which the inability of cells to bind to a polymer may also be beneficial. For example, hydrogels that are designed to deliver a bolus of cells to an area of damage, or to provide a degradable space filling material for cells to subsequently remodel, can benefit from low cell adhesion.
3. Synthetic polymers
In this section we will discuss the most widely used synthetic polymers used in the tissue engineering field. A summary of the key advantages and disadvantages of each class is provided in Table 1.| Polymer | Class | Advantages | Disadvantages |
|---|---|---|---|
| PHEMA | Polyvinyl | • High mechanical strength | • Non-degradable |
| • Generally biocompatible | • Non-adhesive | ||
| • Easily derivatized | • Potential calcification in vivo | ||
| • High monomer toxicity | |||
| PVA | Polyvinyl | • High elasticity | • Non-degradable |
| • Variable deacetylation ratios | • Non-adhesive | ||
| • High biocompatibility and hydrophilicity | |||
| PNIPAM | Polyvinyl | • Temperature responsive (LCST) | • Non-degradable |
| • Biocompatible | • Non-adhesive | ||
| • Low immunogenicity | • Monomer cyto- and neuro-toxic | ||
| • Gels have weak mechanical strength | |||
| PEG | — | • Versatile architecture and functionality | • Non-degradable |
| • ‘Blank slate’ scaffold | • Non-adhesive | ||
| • Modular gel properties | • Evidence of immunogenicity in some patients | ||
| PLA | Polyester | • Degradable by hydrolysis | • Hydrolysis products may cause inflammation |
| • Properties dependent on monomer feedstock | • Physically cross-linked gels are weak | ||
| PGA | Polyester | • Degradable by hydrolysis | • Hydrolysis products may cause inflammation |
| • Co-polymers with PLA give tunable properties | • Rapid breakdown in vivo | ||
| • Physically cross-linked gels are weak | |||
| PCL | Polyester | • Degradable by hydrolysis | • Crystallinity may slow hydrolysis beyond relevant timeframe |
| • Sensitive to degradation by lipase | |||
| • Stable hydrogels over wide concentration range | |||
| • Crystallinity provides mechanical strength |
3.1 Vinyl polymers
A vast array of functionalised vinyl monomers are either commercially available or synthetically accessible, rendering vinyl polymer-based hydrogels versatile and structurally diverse scaffolds (Fig. 4). Vinyl polymers are most commonly synthesised via free-radical polymerisation, though both anionic and cationic polymerisations can be applied to specific monomer classes.48,49 For example poly(2-isopropenyl-2-oxazoline) hydrogels, recently developed by the Jerca and Hoogenboom groups, are synthesised via living anionic polymerisation to ensure high molecular weights and low dispersity.50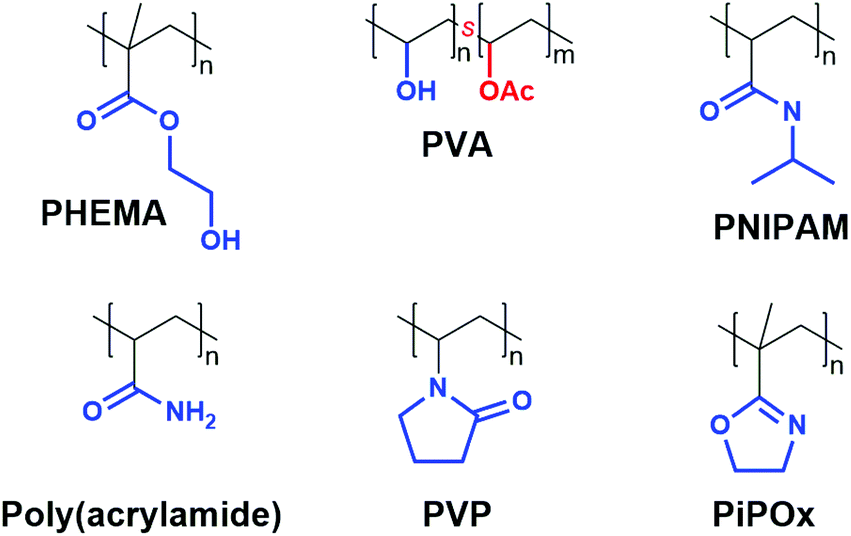 | ||
| Fig. 4 Structures of the most commonly used vinyl polymers for hydrogelation (s: statistical polymer). | ||
Through careful choice of polymerisation conditions, initiation method, and monomer feedstock, vinyl polymers can be produced with either strict or low control over molecular weight (Mw), with a high density of cross-linking or solely as linear chains, as block or random co-polymers, with versatile functional or bioactive pendant groups, and with spatial precision to produce patterned or gradient materials. In most cases, vinyl polymer backbones are non-biodegradable with subsequent limitations for in vivo applications. However, with careful monomer design degradable backbone linkages or cross-links can be incorporated into the bulk hydrogel, allowing for breakdown into resorbable macromers.32,51,52 For example, early reports by Bryant et al. demonstrated the introduction of polyester crosslinks that enable polyvinyl hydrogel breakdown via hydrolysis.51
The structure dependence of vinyl polymerisation rates have been well documented. In general, acrylates polymerise at a faster rate than analogous methacrylates, which in turn polymerise at a faster rate than the corresponding acrylamide.33 This increased reaction rate comes at a cost of increased monomer toxicity, though residual methacrylates and acrylamides are also known to cause damage in vivo.53,54 It is therefore important to reach high conversions during polymerisation, which may be challenging during chain-growth polymerisation, necessitating the use of long reaction times which in turn may be limiting for in situ or cellularised gelation (see section 8).55
Free-radical polymerisations are tolerant of functionalised vinyl monomers, providing facile access to derivatised polymers and hydrogels.44,52 Peptides, glycans, and even proteins can be modified with acrylate or methacrylate groups, and incorporated as a co-monomer during polymerisation.56–58 Similarly, monomers bearing chemically interesting pendant groups, such as the zwitterionic, phospholipid mimicking monomer 2-methacryloyloxyethyl phosphorylcholine (MPC), able to minimise stem cell activation, can be incorporated to provide vinyl polymers with added functionality.59 Jansen et al. have demonstrated that MPC can minimise serum protein adsorption in vivo, and is therefore a useful means to limit the foreign body response after implantation.60 The density of presentation can be easily tuned through alteration of the monomer feedstock, however it is important to match polymerisation rates in order to provide homogenous distributions of functional motifs. If polymerisation of the bulk polymer occurs at a significantly different rate to the co-monomer, the formation of block – rather than random – co-polymers may be produced. Vinyl groups are also commonly installed on pre-formed macromers or polymers of alternative natural or synthetic polymers, subsequently acting as covalent cross-linkers to provide hydrogels with mechanical strength or injectable solutions with ‘curability’ through the application of a radical-inducing stimuli.61–63
The high hydrophilicity of PHEMA renders it bioinert, resisting cell and protein adhesion. PHEMA hydrogels have therefore commonly been used as structural scaffolds, directing cellular growth or encapsulating cells for delivery. These properties were recently exploited by Cai et al. to photo-pattern PHEMA gels containing phenylazide co-monomers, leading to the creation of 3D cylindrical channels of adhesive collagen proteins.67 The resultant construct was able to serve as an effective nerve conduit in vivo, promoting the infiltration of neurons and the restoration of motor function within a transected spinal cord. An alternative approach to 3D patterning can be seen through the use of PHEMA-based bioinks, as developed by the Ruzzo group. They have shown that high Mw PHEMA dopants are necessary to provide sufficient viscosity to monomer inks to enable direct ink writing.68,69
To generate cell-adhesive PHEMA gels, composite structures bearing cell-adhesive co-polymers, ECM protein coatings, or adhesive peptides are often exploited.51,69–71 However, in an important recent development Hu et al. have demonstrated that high-aspect-ratio, micro-meter scale, topographical patterning of pure PHEMA gels is able to promote the adherence of human mesenchymal stem cells (hMSCs).72 In contrast to the rounded morphology adopted on flat surfaces, indicative of poor interactions with the underlying material, cells were able to spread and elongate in response to the topographical cues provided. The mechanical integrity of the PHEMA hydrogels was essential to enable patterning to be retained. This report is likely to be of great significance in the coming years, providing a powerful means to pattern and direct cell growth.
While generally considered to be biocompatible and bioinert, PHEMA has some drawbacks for in vivo applications. The hydroxyl group of HEMA has been shown to further increase monomer toxicity when compared to the corresponding alkyl methacrylate. The complete removal of residual monomer is therefore necessary to minimise cell and tissue damage.53 Furthermore, PHEMA gels implanted in vivo have been shown to potentially undergo calcification. While the mechanism and extent of this process remain unclear, further investigation is necessary due to the potential for soft tissue calcification to cause severe pathologies.73,74 Such studies are vital to enable the full potential of PHEMA as a biomedical material to be realised.
PVA exhibits high elasticity, generating hydrogels with low friction. PVA hydrogels are therefore attractive substrates for cartilage tissue engineering, acting as lubricating surfaces able to withstand the high mechanical forces imposed on joints.76,77 However, pure PVA gels are not strong enough to recreate the mechanical properties of native cartilage. To overcome this limitation Shi et al. demonstrated that the use of vinylpyrrolidone as a co-monomer can greatly improve hydrogel strength at levels as low as 1%.78
The majority of linkages within the PVA backbone generate 1,3-diols, though a small proportion of 1,2-diols are often present.79 In a similar manner to PHEMA, the polyhydroxylated nature of PVA scaffold leads to both high biocompatibility, and poor cell and protein adhesion, as a result of polymer hydrophilicity.80 PVA hydrogels therefore typically require derivatization to generate an active gel for cell encapsulation.77 This can be conveniently achieved through the chemical modification of the pendant hydroxyl groups, enabling functionalisation of the polymer chain. Alternatively, PVA can be exploited to provide inert scaffolds for cell encapsulation and storage. The Ishihara group in particular has reported a number of exciting technologies exploiting the polyhydroxyl backbone to form complexes with boronic acid-containing co-polymers, thus generating dynamic, responsive cross-links, as will be discussed later.59,81,82 These gels are able to restrict stem cell differentiation, providing increased control over cell fate through the addition of soluble factors.59 The gels can also undergo changes in mechanical properties, through the addition of competing sugars able to disrupt sugar-boronic acid binding. These hydrogels are therefore promising scaffolds for the packaging and storage of cells that can subsequently be released when required by a user-applied stimuli.
PNIPAM displays good biocompatibility and low immunogenicity in vivo.90,91 However, as for many vinyl monomers, N-isopropylacrylamide is cyto- and neuro-toxic, necessitating the complete removal of residual monomer post-polymerisation.92 Pure PNIPAM also leads to hydrogels with weak mechanical strength, held together by non-covalent interactions. While the formation of composite gels can increase mechanical strength, they may also affect the delicate balance of associative and dissociative forces that provide PNIPAM with its thermoresponsive behaviour. The LCST may be drastically altered or lost entirely as a result. For example, PNIPAM co-polymers containing 4-(hydroxybutyl)methacrylate and 6-(hydroxyhexyl)methacrylate fail to show thermoresponsive properties, whereas HEMA containing polymers do.93
In general, co-polymers with discrete phases are more likely to retain thermoresponsive behaviour, with individual polymer blocks still able to undergo a change in phase.94 Zhang et al. recently demonstrated that a PNIPAM-PHEMA co-polymer with such a discrete architecture could be used to create microfibrous hydrogels that undergo reversible changes in stiffness, up to 0.5 GPa, upon thermal cycling.95 Interestingly, hMSCs cultured under conditions of cyclic mechanical change were shown to undergo enhanced spreading and adhesion, resulting in increased osteogenic differentiation. This report therefore highlights the potential utility of dynamic gel mechanical properties as an important means to control cell fate.
Though functionalised co-monomers have been incorporated into polymers to supplement hydrogel properties, other core vinyl backbones have been less commonly exploited. For example, poly(acrylic acid) (PAA) and poly(methacrylic acid) (PMAA) have been used only rarely within hydrogels for tissue engineering, typically as part of composite materials, due to their high density of negative charge at physiological pH.101 Similarly, poly(vinylpyrrolidone) (PVP) has rarely been used to form hydrogels despite possessing low immunogenicity and being biocompatible. This is in part due to the difficulty of producing cross-linked PVP networks, due to the slow kinetics of vinylpyrrolidone polymerisation relative to the reaction of vinyl-cross-linkers doped into the system.102 Instead, vinylpyrrolidone has been more commonly applied as a co-monomer to increase the mechanical strength of polymers such as PVA, as described above.78
Poly(2-isopropenyl-2-oxazoline) (PiPOx) has recently emerged as a promising vinyl polymer for biomedical applications. The pendant groups of PiPOx possess reactive oxazoline groups, which are able to undergo efficient ring opening with carboxylic acids to generate a poly(acrylamide) backbone bearing functional esters.48,103 Functionalization with thiols and phenol groups has also been reported. Modification of the oxazoline sidechain has proved insensitive to water or oxygen, and proceeds cleanly without the generation of side-products or the need for catalysts. Though not yet reported, PiPOx therefore offers intriguing possibilities for the creation of hydrogels bearing bioactive motifs. It is likely that these exciting materials will become of increasing interest to the biomedical community in the future, though the need to undertake anionic polymerisation to achieve well defined polymer precursors may limit widespread translation to non-specialist polymer labs.
3.2 Poly(ethylene glycol), PEG
The terms poly(ethylene glycol) (PEG) and poly(ethylene oxide) (PEO) are often used interchangeably in the biomaterials community to describe the same material, regardless of traditional distinctions based on Mw or end-group functionality.104 PEG is the most widely used synthetic polymer for hydrogelation in the tissue engineering field, certainly in the academic community, if not commercially or in the clinic. This is largely due to the chemical and biological inertness of PEG, along with the ease of derivatization, the large number of polymer architectures and lengths that are accessible commercially or synthetically, and the high hydrophilicity of the polymer backbone. In effect, PEG hydrogels act as a ‘blank slate’, providing an inert structural component that can then be decorated with active functionalities at will.105,106 This allows the production of controlled and homogenous scaffolds in which individual facets of hydrogel design and their influence on cell fate can be delineated from each other, and PEG hydrogels have therefore been at the forefront of efforts to understand cell growth and behaviour within tissue engineering scaffolds. In some prominent recent examples, PEG-based hydrogels have played a critical role in enabling demonstrations that local heterogeneities in cell growth and gel degradation help retain scaffold integrity during tissue growth,107 dynamic cross-links must be balanced with bulk stability to maximise ECM deposition during cartilage growth,108 and that nascent protein deposition and remodelling plays an important role in dictating cell fate from an early stage.109PEG is commonly produced through the anionic polymerisation of ethylene oxide. Branched- or star-polymers can be readily synthesised and are often used to produce cross-linked polymer hydrogels. End-capping groups can also be easily introduced, either through the use of functional initiators or post-polymerisation functionalisation. As a result, covalent cross-linking can be achieved via a diverse range of coupling chemistries, each with distinct advantages or applications, such as nucleophilic and radical thiol–ene reactions,110,111 copper-catalysed or strain-promoted azide–alkyne cycloadditions,27,112 inverse-electron demand Diels–Alder reactions between tetrazines and strained alkenes,113 oxime and hydrazone formation,114 and many other diverse coupling chemistries44 (Fig. 5). Indeed, the versatility of PEG-end functionalisation enables the exploitation of multiple cross-linking reactions within the same hydrogel scaffold, providing hydrogels with unparalleled tunability that can be exploited to maximise cell growth. For example, the Maynard group have demonstrated that varying the ratio of oxime and hydrazone cross-linkages within a PEG hydrogel can control scaffold degradation over a period of 1–7 days.115 Richardson et al. have gone on to show that changing the ratio of benzyl- to alkyl-hydrazones within a gel, by controlling PEG end-capping functionality, enables the formation of gels with stress relaxation times ranging from seconds to months, with important implications for cell fate.108
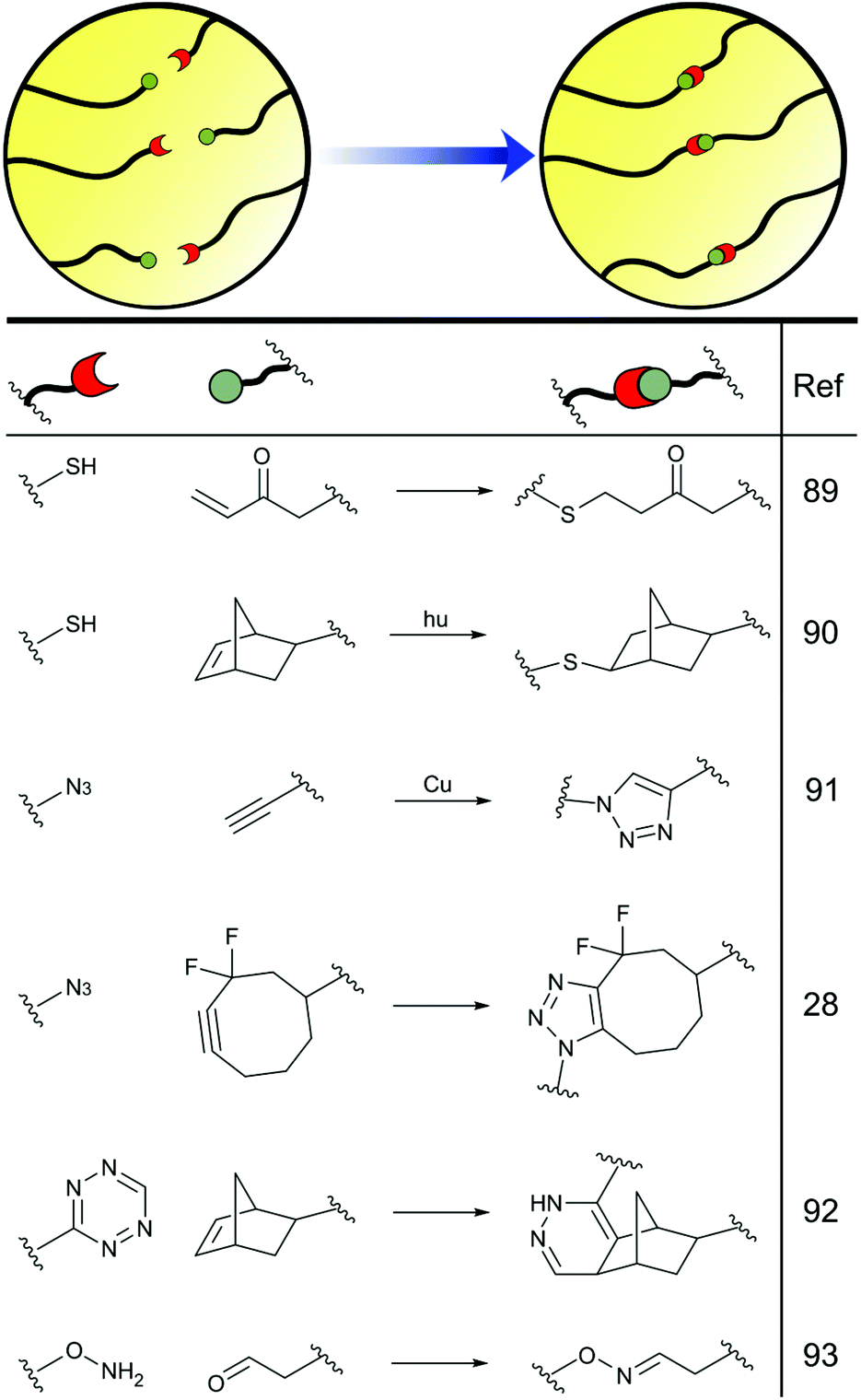 | ||
| Fig. 5 End-functionalisation of PEG allows the incorporation of diverse reactive handles that can subsequently be used for chemical cross-linking. | ||
Hydrogelation in these cases typically proceeds via the ‘step-growth’ polymerisation of functional monomers, with important implications for hydrogel properties as will be discussed in section 8. These chemistries can also be exploited for the incorporation of adhesive peptides and the formation of composites with natural polymers, via the modification of end-capping groups.27,34,111,116–119 Importantly, each ‘modification’ uses a functional handle that would otherwise have been used for cross-linking. Kim et al. have shown that this can have an important influence on hydrogel properties, with increasing densities of RGD peptide presentation within a PEG hydrogel resulting in weakened storage moduli, slower gelation times, and increased swelling ratios.120 Interestingly, these changes were significantly reduced when 8-arm PEG gel precursors were utilised rather than 4-arm, even at the same peptide and cross-linking densities, highlighting the importance of careful polymer design as will be discussed further in section 7.
As an alternative to the step-growth cross-linking chemistries discussed above, chain-growth mechanisms can also be used to generate PEG hydrogels. The use of PEG-di(meth)acrylates is particularly widespread for free-radical hydrogelation and the formation of covalently-linked composites with alternative polymers.116,117,121,122 This is particularly advantageous for in situ hydrogelation, though the limitations of such techniques will be discussed in section 8.123 It has been well documented for a number of years that the use of chain-growth vs. step-growth cross-linking has important implications for the bulk mechanical properties of PEG hydrogels and the presentation of mechanical cues to encapsulated cells, due to an increase in gel imperfections.124,125 However, in a recent study Vats et al. have also highlighted differences in hydrogel properties at the nanoscale via atomic force microscopy.126 Chain-growth polymerisation of PEG-dimethacrylate was found to generate hydrophobic pockets of poly(methacrylate) with increased stiffness, leading to high heterogeneity that influenced protein and cell adhesion. Indeed, the authors went on to show that the clustering of attached adhesive peptides during polymerisation led to a decrease in cell density when compared to homogeneously distributed peptide.
Though PEG is non-degradable, below a Mw ∼10 kDa polymers can be cleared from the body by renal excretion.127 The use of degradable linkers to generate soluble PEG macromers upon hydrogel breakdown, either via hydrolysis or enzymatic cleavage, is therefore common.34,128 However, recent reports on the generation of anti-PEG antibodies suggest that PEG-based hydrogels may not be as bioinert as previously thought. Interestingly, Chang et al. have demonstrated that immunogenicity is patient specific, with genetic markers for PEG-sensitivity being identified.129 Further research into the causes and downstream implications of PEG-immunogenicity is therefore essential.16
3.3 Polyesters
Polymer backbones containing ester groups are able to undergo biodegradation. Polyesters are therefore popular materials for the production of biomedical hydrogels. In general, polyesters are reasonably hydrophobic and are therefore poor substrates for the formation of hydrogels in isolation. Instead, they are commonly combined with a hydrophilic polymer, such as PEG, to provide amphiphilic block co-polymers able to undergo self-assembly and physical gelation. Gelation is preceded by the initial formation of intermediate micelles. As polymer concentration is increased, these micelles aggregate leading to a sol-to-gel transition dictated by the critical gel concentration (CGC), as described by Jeong et al.130 This concentration is strongly influenced by the polymer properties, including the monomer composition, amphiphilic ratio, Mw, and architecture. The importance of these parameters will be discussed in detail in section 7, however for our purposes here, in general increasing the polyester content leads to a decrease in CGC as a result of increased hydrophobic interactions which are able to drive the assembly process.131,132 Assembly is typically temperature sensitive, leading to thermoresponsive polymers with gelation temperatures that must be tuned to a physiologically relevant range.133The rate of polyester degradation strongly influences hydrogel mechanics and the ability of cells to remodel their environment. The degradability must therefore be carefully matched to the target application. For example, Kang et al. have demonstrated that polyester structure can be used to tune in vivo persistence times from days to years depending on monomer composition.42 Furthermore, the potential downstream effects of the breakdown products must also be considered. Polyesters typically degrade into monomers bearing carboxylic acids, acting to raise local pH and potentially causing tissue damage.133,134 The accumulation of acidic monomers at the site of implantation may also act to induce local calcification.
The stereochemistry of a PLA polymer has a significant effect on its properties, and thus those of an amphiphilic PEG block co-polymer and the hydrogels that it is able to form. While enantiopure PLLA leads to semi-crystalline hydrophobic aggregates upon gelation, racemic PDLLA typically adopts an amorphous structure. This in turn leads to an increased susceptibility to hydrolysis and a decrease in mechanical strength (Fig. 6).127,137 Alternatively, gelation can be greatly accelerated by combining mixtures of PLLA- and PDLA-based amphiphiles, as a result of sterocomplexation effects, as demonstrated by Hiemstra and co-workers.138,139
 | ||
| Fig. 6 Polyester structure dictates the properties of the resulting hydrogel. With increasing hydrophobicity of the carbon backbone, the critical gel concentration decreases, leading to gels that are stable over a wider concentration range. Similarly, the rate of hydrolysis also decreases, due to increased crystallinity. The increased crystallinity of regio-regular PLLA/PDLA vs. PDLLA has a similar effect. | ||
The stability of PLA-based gels can be enhanced through chemical cross-linking, most commonly achieved via the photo-polymerisation of vinyl-end groups.140,141 These modifications can also be used to cross-link composite scaffolds, incorporating natural polymers with bioactive properties.29,142 Additional cross-linking may be particularly beneficial due to the known propensity of PLA to undergo autocatalytic breakdown. The acidity of the α-hydroxy acid PLA degradation product has been shown to accelerate the interior breakdown of polyester hydrogels, leading to the generation of capsular gels with hollow interiors.143 Importantly, even after cross-linking, hydrogel properties are still highly dependent on monomer feedstock chirality. The Tuan group have demonstrated that covalently cross-linking PEG-PLLA/PDLLA results in hydrogels of differing stiffnesses, due to the increased crystallinity of PLLA.137 While these gels undergo slower degradation, they also result in decreased ECM deposition by encapsulated hMSCs.
As an alternative to the use of pure PGA, PLGA co-polymers can be synthesised, either as random or block structures, and grafted to PEG to create polyester amphiphiles able to form stable gels at physiological temperature. The increased hydrophilicity of PGA/PLGA also leads to lower crystallinity of the hydrophobic aggregate, therefore leading to an increased rate of hydrolysis when compared to pure PLA amphiphiles.131,141 As for PLA, the generation of α-hydroxy acid monomers upon hydrolysis of PLGA, in the form of glycolic acid and lactic acid, may again act to auto-accelerate polymer degradation.147
The hydrophobicity of PCL makes it an attractive component of hydrogels that must be able to withstand high mechanical stress, for example for the engineering of cartilage tissue. In such scenarios, the hydrophobic clustering of PCL domains constrains the swelling of the polymer hydrophilic regions. However, the slow degradation of PCL can then prove limiting. To elegantly address this challenge, Yin and co-workers grafted short pendant PCL chains to a hydrophilic poly(L-glutamic acid) polymer backbone.152 Though the PCL chains were able to associate and provide the hydrogels with improved mechanical properties suitable for meniscal tissue engineering, the short length prevented large scale crystallinity. The gels were therefore still able to undergo degradation over a suitable time period, resulting in the partial regeneration of meniscus tissue in vivo.
 | ||
| Fig. 7 Other polymers that are either commonly used or are emerging as promising materials for biomedical hydrogelation. | ||
3.4 Other synthetic polymers
PiPOx polymers, introduced in section 3.1.4, bear reactive oxazolines that can be used for post-polymerisation functionalisation. 2-Oxazolines can also act as monomers in their own right, undergoing cationic ring-opening polymerisation under strictly anhydrous conditions to generate a pseudo-polypeptide backbone (Fig. 7).160 The amide ‘R’ group dictates the properties of a particular construct. Poly(2-alkyl/aryl-2-oxazoline)s (PAOx) bearing multiple functional groups can be synthesised via mixed monomer feedstocks.161 For example, Farrugia et al. demonstrated that oxazoline monomers bearing reactive alkene handles for post-polymerisation modification enable the introduction of adhesive peptides that can subsequently mediate cell attachment.162 PAOxs are particularly attractive as alternative ‘stealth polymers’ to PEG, providing bioinert, stable, and biocompatible scaffolds. Indeed, even under harsh oxidative conditions able to induce cleavage events in PEG-based materials, the backbone of PAOx was found to be stable as side-chain degradation was favoured.160 Structurally related polypeptoids, bearing N-linked R-groups can also be synthesised through the ring-opening polymerisation of N-carboxyanhydrides.163 These polymers possess a similar tunability of structure and gelation properties, and the possibility to incorporate functional motifs.Polyisocyanopeptides, developed by the groups of Rowan and Kower, have recently emerged as interesting materials for tissue engineering. These polymers are able to form helical structures that undergo hierarchical assembly into fibres, and then fibrous bundles. In this way, they are able to mimic the structure and behaviour of native ECM proteins.164,165 Hydrogelation has been reported to occur at extremely low polymer concentrations, with the presence of pendant chiral peptides driving helix formation and subsequent assembly into fibres. These materials, unlike most synthetic polymer gels, display stress-stiffening behaviour that is more akin to the behaviour displayed by natural protein-based gels, allowing them to mediate stem-cell differentiation (Fig. 8).166,167
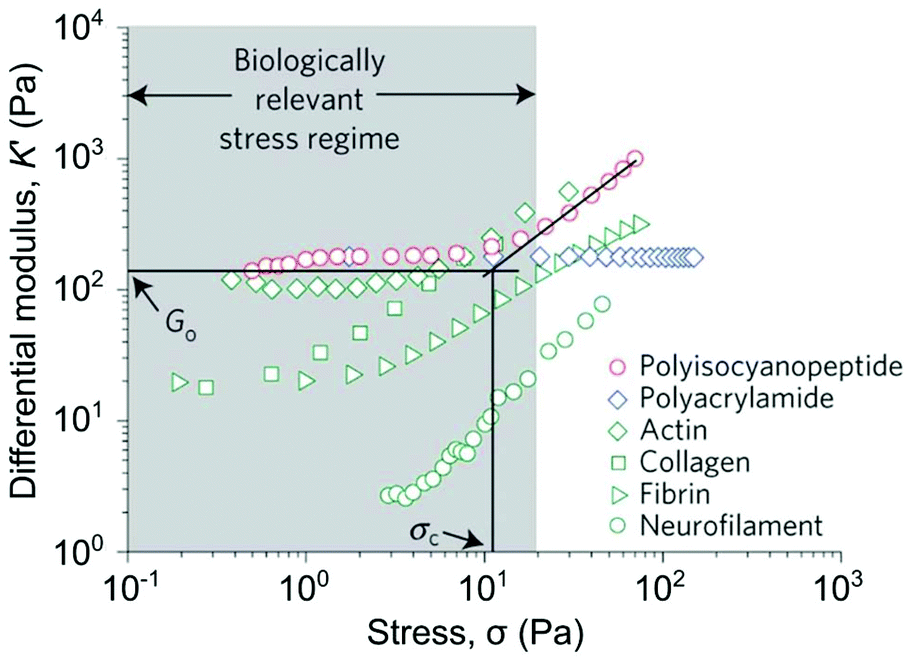 | ||
| Fig. 8 Strain-stiffening behaviour of natural and synthetic polymer gels. Of particular note, is the characteristic strain-stiffening behaviour of collagen and fibrin, which has been shown to be beneficial to stem cell survival. Polyisocyanopeptides, introduced in section 3.4, possess biomimetic stiffening behaviour, whereas a chemically cross-linked poly(acrylamide) hydrogel does not. Go indicates the equilibrium bulk stiffness and σC denotes the critical stress for the onset of stress stiffening for the polyisocyanopeptide gel. Reprinted by permission from Springer Nature, Nature Materials, ‘Stress-stiffening-mediated stem-cell commitment switch in soft responsive hydrogels’, R. K. Das et al., 2016.167 | ||
4. Naturally-derived polymers
The use of protein/peptide and polysaccharide natural polymers as substrates for hydrogelation is widespread in tissue engineering. In this section, we will discuss the key polymer classes, with a summary provided in Table 2.| Polymer | Class | Advantages | Disadvantages |
|---|---|---|---|
| Collagen | Proteinaceous | • Adhesive and bioactive | • Assembly sensitive to modification |
| • Abundant and biodegradable | • Contamination can lead to immunogenicity | ||
| • Mimics native ECM | • Mechanical stability lost during processing | ||
| Gelatin | Proteinaceous | • Adhesive and bioactive | • Mechanically weak |
| • Tolerant of functionalisation | • Contamination can lead to immunogenicity | ||
| • Abundant and biodegradable | • Requires cross-linking | ||
| Silk | Proteinaceous | • High mechanical strength and elasticity | • Slow gelation |
| • Adhesive | |||
| • Low immunogenicity | |||
| ELPs | Proteinaceous | • Thermoresponsive (LCST) | • Low stability without cross-linking |
| • Tunable structure and sequence | |||
| • Recombinant expression | |||
| Alginate | Polysaccharide | • Rapid gelation with divalent cations | • Cation leaching leads to dissolution |
| • Abundant | • Non-biodegradable | ||
| • Ease of use for 3D printing | • Poorly adhesive | ||
| • Reactive handles for functionalisation | |||
| Chitosan | Polysaccharide | • Adhesive and antimicrobial | • Poor solubility at neutral pH |
| • Abundant | |||
| • Low immunogenicity | |||
| HA | Polysaccharide | • Bioactive and biocompatible | • Low stability without cross-linking |
| • Binds growth factors and cytokines | • Rapidly degraded in vivo | ||
| • Reactive handles for functionalisation | |||
| Chondroitin sulfate | Polysaccharide | • Bioactive and biocompatible | • Low stability without cross-linking |
| • Binds growth factors and cytokines | • Rapidly degraded in vivo | ||
| • Reactive handles for functionalisation |
4.1 Peptide and protein based materials
Proteins play a vital structural and signalling role in native ECM. For example, in cartilage, 50–80% of the dry mass of the tissue is made up of triple-helical collagen fibres.6 It is therefore not surprising that peptide- and protein-based polymers have been widely used to form hydrogels for tissue engineering. These materials have an inherent, sequence-dependent ability to mediate cell signalling and adhesion, and to control tissue development. These interactions are bidirectional, with cells also able to remodel and manipulate proteinaceous hydrogels.168 Natural polymers often assemble into fibrous hydrogels, held together via non-covalent interactions. As a result, they display viscoelastic and strain-stiffening behaviour similar to that of native ECM (Fig. 8). These properties have been shown to be highly beneficial in dictating stem cell fate and survival, as recently demonstrated by the Chaudhuri and Mooney groups.35,169In principle, proteins/peptides possess low immunogenicity, as the epitopes they display are typically found in native tissues anyway. However, as protein substrates are commonly extracted from natural sources, achieving high purity is often challenging, with contamination and heterogeneous substrates limiting clinical implementation. The use of recombinant proteins partially addresses this challenge, in addition to delivering sequence flexibility and ease of largescale production. However contamination with bacterial contaminants remains a significant concern.
Though more rarely used, collagen II is also attractive for tissue engineering. In particular, collagen II has been shown to be more effective at inducing chondrogenesis than type I. However, collagen II also has a higher density of glycosylation sites which act to reduce fibrillation capacity, leading to hydrogels with very weak mechanical properties. Vázquez-Portalatín et al. have demonstrated that the use of collagen type I/II blends allows partial recovery of hydrogel strength.173 Alternatively, Yang et al. have employed photo cross-linking of methacrylated collagen II to generate homopolymer hydrogels for the first time, with a resultant upregulation of chondrogenesis when compared to analogous collagen I scaffolds.174
Though only a small subset of the population display an immune response to type I collagen, the common use of mammalian sources to derive protein for gelation leads to a high risk of immunogenicity and challenges associated with heterogeneity.175 More recently, recombinant human collagen has become commercially available, with a resultant improvement in biocompatibility and reproducibility. The use of bacterial collagens is also becoming increasingly attractive and offers a viable alternative to mammalian proteins. In a series of papers, the Ramshaw and Stevens groups have demonstrated that the Streptococcal collagen-like 2 (Scl2) proteins provide the advantageous triple helical structure and mechanical strength of human collagen, while also being non-immunogenic, non-toxic, and amenable to reproducible large-scale expression.176–178 Importantly, these bacterial collagens lack bioactive epitopes, providing a ‘blank-slate’ into which adhesive, signalling, and degradable motifs can be recombinantly introduced. The authors have therefore demonstrated that Scl2-derived hydrogels overcome the heterogeneity of mammalian collagens, providing precise control over chondrogenesis.178
As an alternative to full length collagen, collagen-mimetic peptides have also been reported to undergo hydrogelation. In their native form these short peptides are often unable to form stable helices. As will be discussed further in section 5, assembly can, however, be controlled through a careful balance of hydrophobic and hydrophilic content. In an early report, Yu et al. reported that lipidation of a collagen-mimetic increased amphiphilicity and drove helix formation.179 In an alternative approach, Luo and Tong demonstrated that collagen epitopes can be displayed on the surfaces of β-sheet forming peptides.180 Although this peptide does not form a triple helix, it is able to aggregate into fibrillar hydrogels which are able to partially recapitulate the properties of native collagen.
The hydrogelation of silk fibroin is driven by β-sheet assembly. While spontaneous in solution, this process can be slow and physically cross-linked silk is therefore a poor choice of material for injectable hydrogels.196 However, fibroin does display shear thinning behaviour, and so has found use as part of composite printable and injectable mixtures, and has also been widely functionalised to allow rapid chemical curing.22,197,198 Interestingly, the Mandal group demonstrated in 2018 that combining two fibroins from different species can also greatly enhance gelation rates, attributed to accelerated assembly due to differences in hydrophobicity (Fig. 9).196,199 Alternatively, Cheng et al. have reported that accelerated cooperative assembly can be triggered through the addition of small molecule peptide-gelators. The resultant hydrogels can form at fibroin concentrations as low as 0.1%, and have been shown to trigger angiogenesis in vivo.200
 | ||
| Fig. 9 Fabrication of silk hydrogels using a blend of fibroins from different species. Gelation is monitored by following absorbance at 550 nm. Using either B. mori or A. assama fibroins in isolation leads to very slow gelation, whereas mixtures of the two rapidly undergo gelation through accelerated assembly. Adapted from Biomaterials, vol. 187, M. Kumar et al., ‘Immunomodulatory injectable silk hydrogels maintaining functional islets and promoting anti-inflammatory M2 macrophage polarization’ 1–17, Copyright 2018, with permission from Elsevier.196 | ||
Fibroin hydrogel degradation is significantly slower than many other protein-based materials, as a result of the high crystallinity generated during β-sheet assembly, offering great benefits for stability and longer term structure. Recombinant silks have also emerged in recent years, providing opportunities to incorporate additional bioactive motifs, thermoresponsive behaviour, and tunable degradation rates.195,201–203
A major beneficial property of silk hydrogels are their lack of immunogenicity in vivo. Indeed, silk sutures have been used in medicine for centuries, eliciting only a very minimal foreign body response.194 Though often thought to be less immunogenic than other proteinaceous polymers, there are in fact few studies that make direct comparisons to materials such as collagen, with often conflicting results.204 The benefits of silk instead appear to derive from the purity with which gel precursors can be obtained, in contrast to the common contamination of other proteins. In recent years, it has further become apparent that although raw silks containing both fibroin-sericin are pro-inflammatory, materials composed solely of sericin are in fact also well tolerated, as reported by Jiao et al.205 This has prompted an exciting reinvestigation of sericin-based hydrogels in recent years, due to its cell adhesive and pro-angiogenic properties.206,207
ELPs exhibit thermoresponsive behaviour, with an LCST above which protein assembly can be induced, providing opportunities for responsive hydrogels as will be discussed in section 6. The temperature transition displays hysteresis upon reversal, providing an increased temperature window in which gels are stable for biological applications.209 The LCST is strongly influenced by the nature of the guest residue – hydrophobic residues promote the collapse and aggregation of the protein to form gels at lower temperatures, while hydrophilic residues stabilise the solvated and extended conformation that exists below the LCST. Assembly typically results in a protein coacervate, with subsequent cross-linking often necessary to form stable hydrogels.210 The incorporation of nucleophilic or enzymatically sensitive guest residues can therefore provide reactive handles to mediate direct or indirect chemical cross-linking.210–212 Cross-linking also helps to accelerate the assembly process, which can otherwise be slow.212 Wang et al. recently demonstrated that cross-linking soluble proteins prior to assembly can mediate the resultant hydrogel properties when the temperature is raised above the LCST, giving diverse materials from the same ELP construct.213 By varying cross-linking stoichiometry prior to assembly, gels with elastic moduli ranging from 50–890 Pa could be generated. This report therefore offers intriguing possibilities for tuning hydrogel properties, and thus cellular interactions, from a common protein precursor.
The similarity of the ELP primary sequence to naturally occurring elastin results in minimal immunogenicity and a low sensitivity to protease activity.208 Furthermore, the versatility of expression enables the incorporation of additional peptide motifs that are able to enhance the bioactivity of ELP hydrogels. For example, RGD peptides have been integrated into the primary structure of ELP constructs by the Heilshorn group, and shown to enhance hydrogel cell adhesion (Fig. 10).34,214,215 Composite structures can also be created, most commonly in the form of silk-elastin like polypeptides (SELPs) that combine the mechanical strength and cell adhesive properties of silk with the thermoresponsive properties of ELPs.216 To overcome the slow formation of silk domains into β-sheets, which can otherwise hinder the injectability of SELPS, Cipriani et al. have reported that pre-annealing below the LCST of the ELP construct can accelerate assembly prior to gelation, enabling rapid formation of stable SELP hydrogels following injection.203 These gels were able to promote chondrogensis in an ex vivo model of cartilage regeneration, and their future application in vivo is therefore intriguing.
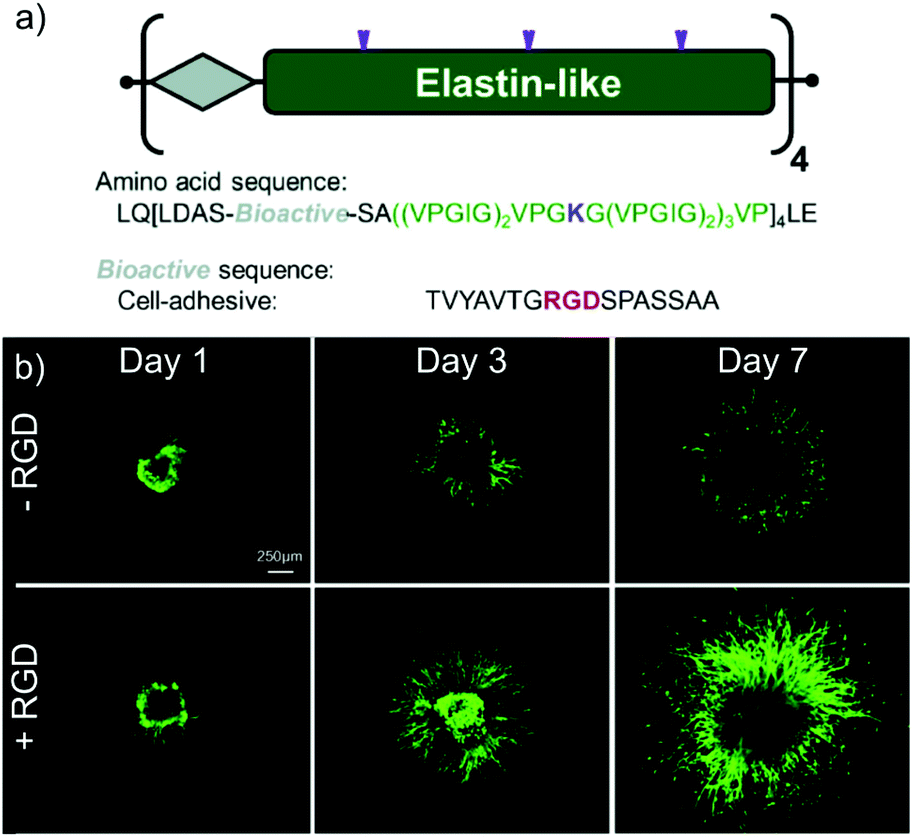 | ||
| Fig. 10 (a) Schematic of an ELP construct containing cell-adhesive RGD sequences; (b) fluorescence microscopy images of explanted chick dorsal root ganglia encapsulated within ELP hydrogels over 7 days in culture, in the presence or absence of RGD motifs. Neurite outgrowth was greatly enhanced in the presence of the adhesive peptide. Adapted from Acta Biomaterialia, vol. 9, K. J. Lampe, A. L. Antaris, and S. C. Heilshorn, ‘Design of three-dimensional engineered protein hydrogels for tailored control of neurite growth’, 5590–5599, Copyright 2013, with permission from Elsevier.170 | ||
A number of ECM derived protein gels have also found use, with perhaps the most common being Matrigel, a commercially available, soluble, and sterile protein extract from mouse Engelbreth-Holm Swarm (EHS) tumours. Matrigel is mostly composed of native ECM proteins, including collagen and laminin, but also contains significant quantities of proteoglycans, heparin sulfate, and cell-instructive growth factors. It is the heterogeneous composition of Matrigel that is both its biggest strength and limitation – the ability of Matrigel-based hydrogels to closely recapitulate native ECM allows potent modulation of cell behaviour.218 However these effects are often uncontrolled and the benefits have been shown to quickly diminish as transient signalling molecules are lost.219 Combined with its origin in cancerous mouse tissue, the heterogeneity of Matrigel also prevents in vivo application. Alternatively, decellularised ECM scaffolds have generated increasing interest due to their inherent similarity to native cellular environments. Moreover, tissue-specific hydrogels can be generated for the pathology they will be used to treat.220,221 However, progress in this field is currently limited by the need to obtain donor tissue and to match the immunological properties in order to limit the need for immunosuppression.
4.2 Polysaccharide polymers
Polysaccharide-based hydrogels are a highly attractive and widely used class of natural materials for tissue engineering. While some polysaccharides are naturally present in the ECM, playing an important role in mediating cell behaviour and protein adsorption, others are derived from highly abundant natural sources in plants, algae, and animals.7 The properties of a particular polysaccharide are dictated by the substitution patterns on individual sugar building blocks, which are then linked through O-glycosidic bonds to form either linear or branched polymers with high Mw (Fig. 11). These polymers are typically highly polydisperse, both in terms of structure and sequence, and high batch-to-batch variability can be problematic.222 However, when compared to protein-based materials, polysaccharide hydrogels exhibit greatly diminished immunogenicity, high solubility and hydrophilicity that results in high swelling gels, and abundant reactive handles for chemical modification and cross-linking.223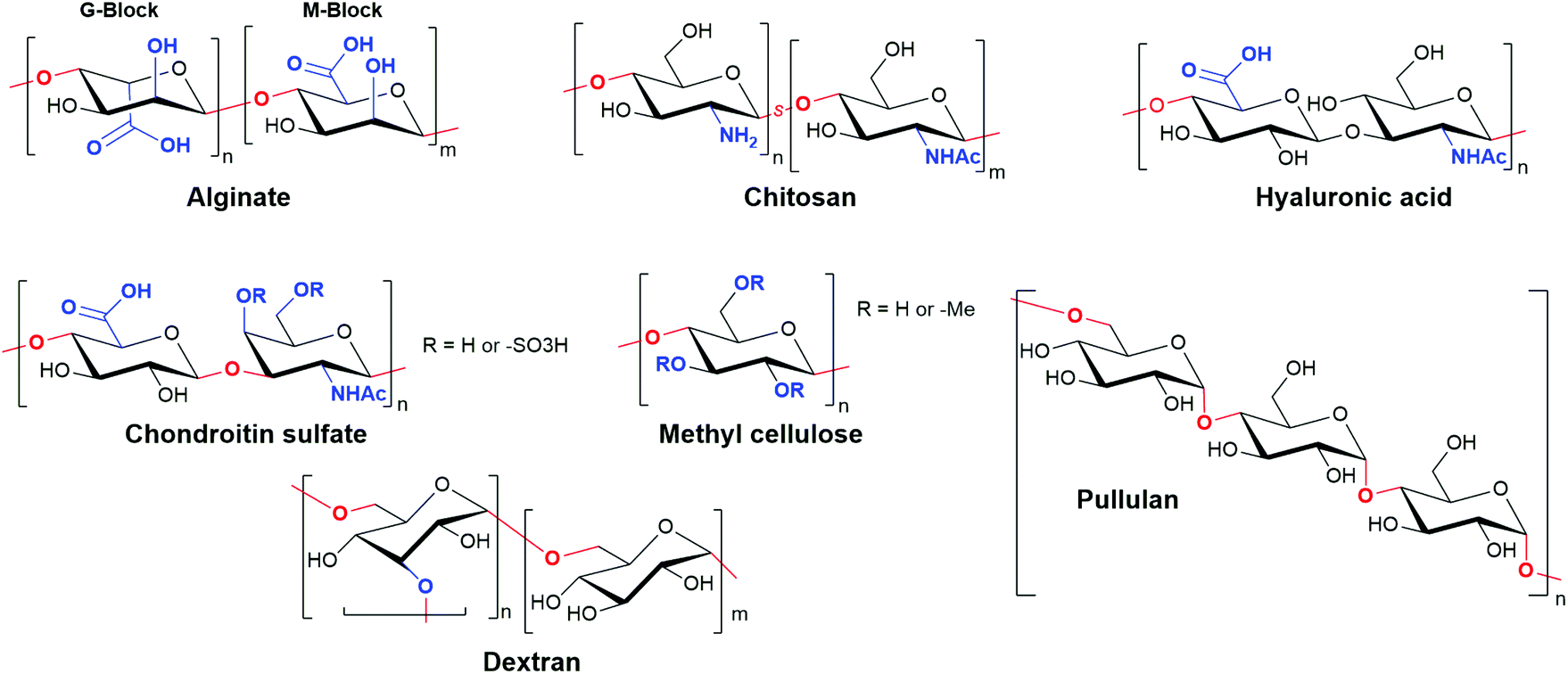 | ||
| Fig. 11 Structures of the most commonly used polysaccharides used to form hydrogels for tissue engineering applications. Deviations from the monosaccharide structure of glucose are highlighted in blue, and linkages between monosaccharides in red (s: statistical polymer). | ||
The popularity of alginate is largely a consequence of the ease with which it can be gelated through the addition of divalent cations. The ionic nature of cross-linking provides alginate hydrogels with elasticity and the ability to relax under strain, in contrast to covalently cross-linked materials.35 It is thought that only G-blocks are able to ionically bond cations, inducing gelation and providing mechanical strength. Alginates containing regions of high G content therefore lead to gels with increased stiffness.225 Gelation is currently thought to occur through an ‘egg-box’ model, first proposed by Grant et al., whereby calcium ions sit within junctions between a corrugated ‘egg-box’ of poly-G sequences (Fig. 12).226 Concentrations of Ca2+ as low as 100 μM cause very rapid gelation. Alginate has therefore been widely used as part of printable substrates through in situ exposure to calcium-containing solutions, either in isolation or to provide structure to a composite material.30,227–229 The rapid evolution of microfabrication technologies has enabled the production of alginate hydrogels with increasingly complex structures. For example, Jia et al. have recently produced superhelical, hollow hydrogel microfibers, able to recreate the complex architecture of helical blood vessels, using microfluidics.230 These hydrogels were able to be produced with impressive μm resolution and used to support the tubular growth of human endothelium.
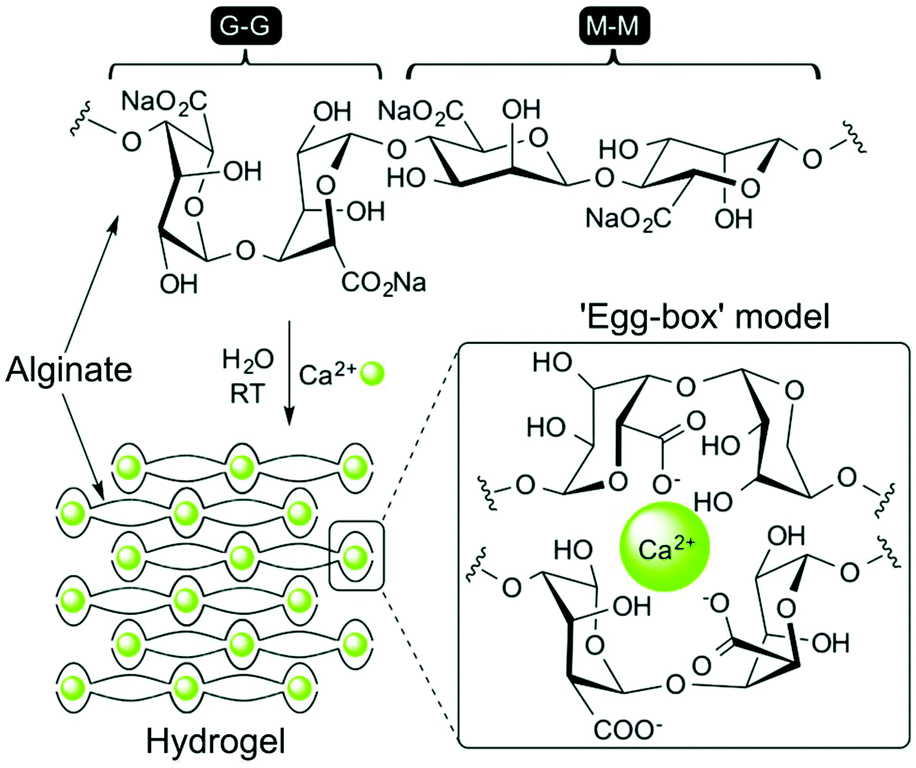 | ||
| Fig. 12 Structure of sodium alginate, and the formation of an ‘egg-box’ structure in the presence of Ca2+ ions that induces rapid hydrogelation. Reproduced with permission from New Journal of Chemistry, vol. 40, J. V. Alegre-Requena et al., ‘Regulatory parameters of self-healing alginate hydrogel networks prepared via mussel-inspired dynamic chemistry’, 8493–8501, Copyright 2016 – Published by The Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS) and the RSC.343 | ||
Gelation speed is dependent on the calcium source used, with calcium(II) chloride having been shown to greatly increase gelation rates when compared to alternatives such as calcium(II) sulfate.23 Interestingly, it has been reported that rapid gelation can be problematic in certain instances, precluding adequate mixing and leading to gels with heterogeneous structures and properties. The use of calcium(II) carbonate as an alternative calcium source is therefore intriguing, due to its pH dependent dissociation. The use of glucono-δ-lactone as a mediator that hydrolyses gradually to slowly create an acidic pH can therefore be used, as reported by the Sell group, to enable controlled gelation and the production of hydrogels with consistent and homogeneous properties.231
Though most widely applied, calcium ions are not unique in being able to induce hydrogel formation. Indeed, divalent cations of lead, copper, and cadmium have a far higher affinity for alginate, leading to stiffer gels. However, the toxicity of these ions prevents their use in biotechnology.232 The use of strontium and zinc containing hydrogels, as previously reported by Place et al., has become increasingly attractive though, due to the beneficial osteogenic properties of these ions.233 Similarly, barium-induced gelation has also been exploited in tissue engineering, as Ba2+ leads to the creation of stronger gels than Ca2+.234,235
Alginate hydrogels are non-biodegradable, however they are able to undergo dissolution following cation leaching into the surrounding media. This leaching can be problematic to long-term gel stability and the resultant soluble polymers may also remain too large for efficient tissue clearance.224 Partially oxidised alginate can be used to promote polymer clearance, by generating sporadic open chain polymers which are able to undergo hydrolysis at a rate that depends on the levels of oxidation, as first reported by Bouhadir et al.236 Furthermore, leached metals can interfere with the local tissue. For example, though calcium leaching may be favourable for inducing osteogenesis, in alternative tissues it may cause downstream problems arising from tissue calcification.237 Similarly, barium is known to block potassium channels at high concentrations, though the increased affinity of Ba2+ for alginate allows lower cation concentrations to be utilised.234 The long term fate of alginate hydrogels in vivo should therefore be carefully considered.
While alginate itself induces limited cell adhesion, the presence of carboxyl groups on each individual sugar unit provides a versatile handle for functionalisation. The derivatization of alginate both pre- and post-gelation with adhesive and bioactive motifs, the formation of composite hydrogels, and chemical cross-linking to enhance stability are all therefore common.238–240
In addition to being able to mediate cell adhesion, chitosan also possesses inherent antimicrobial activity.242 It is well tolerated in vivo with limited immunogenicity. Furthermore, chitosan readily forms complexes with negatively charged polysaccharides, such as hyaluronic acid and chondroitin sulfate, making it an attractive structural component of composite gels that exploit the bioactive properties of these native ECM polysaccharides described below.40,243,244
The low solubility of chitosan at neutral pH can be problematic for polymer processing. Solutions of chitosan are most commonly prepared at acidic pH, leading to glucosamine protonation, followed by gelation upon pH adjustment. The need to work at low pH limits homogenous cell encapsulation prior to gelation. Chitosan derivatives that possess solubility at neutral pH, either through appropriate choice of counterion,244,245 or through chemical derivitisation,246–248 are therefore attractive for the formation of cellularised hydrogels. Carboxymethyl chitosans are a particularly prominent class of modified chitosans, with control over N- or O-functionalisation dictating the solubility range of a particular construct.249 For example, Müller et al. utilised N,O-carboxymethyl chitosan to produce water soluble polymers able to complex polyphosphate, and form composite, printable hydrogels with alginate when exposed to Ca2+ ions.250 The presence of polyphosphate was seen to promote osteogenesis of implanted gels in vivo, making these scaffolds promising materials for bone tissue engineering. More recently, Zhao et al. designed O-carboxymethyl chitosan-amorphous calcium phosphate nanoparticles able to undergo gelation upon a reduction in pH from 11 to 7.5.251 The resultant gels were also found to be osteoconductive in vivo promoting the formation of uniformly mineralized and mature bone. These gels could be both printed and injected, making them interesting candidates for bone engineering in the future.
As a key component of human ECM, HA-based gels are naturally well tolerated, with high biocompatibility. However, physically cross-linked networks also display poor mechanical properties as a result of their high water content and swelling. Furthermore, HA is susceptible to rapid degradation due to the prevalence in native tissues of hyaluronidase enzymes.252 HA gels therefore typically require chemical cross-linking for tissue engineering applications, or are incorporated as bioactive components of composite scaffolds.244,256–259 The use of fast covalent cross-linking reactions, such as the inverse-electron demand Diels–Alder reaction between complementary tetrazine- and trans-cyclooctene-functionalised HA derivatives as reported by Park et al., enable the formation of injectable HA preparations that undergo rapid gelation in vivo.260 The Burdick group have also reported that supramolecular cross-links, as discussed further in section 5, can be used to produce shear-thinning hydrogels, that can be used for 3D printing of stable HA scaffolds.261 Finally, as an exciting alternative to these technologies, the Appel and Woo groups have recently disclosed a novel approach to form injectable HA scaffolds, by exploiting non-covalent interactions between a hydrophobically-modified HA and PEG-PLA nanoparticles.262 Gels of varying strength could be generated by varying the functionalisation density of the HA, without affecting its biochemical signalling capabilities. Importantly, this polymer-nanoparticle hydrogel strategy can potentially be translated to a wide range of alternative natural and synthetic polymers, and is therefore likely to be the subject of increasing interest in the future.
In a similar manner to HA-based hydrogels, physically cross-linked chondroitin sulfate gels have a very high water content as a result of their high charge density. These gels are mechanically weak as a result. Although less sensitive than HA to enzymatic degradation, chondroitin sulfate is also broken down by hyaluronidase enzymes.41 Chondroitin sulfate is therefore most commonly applied in cross-linked form or as part of composite materials with enhanced mechanical stability.263,265–267
Chondroitin sulfate-based hydrogels have been particularly widely studied for chondrogenic applications. The Bian and Bryant groups have recently demonstrated that chondroitin sulfate can direct MSCs towards a chondrogenic phenotype, while importantly inhibiting detrimental hypertrophy that can lead to mineralisation.268,269 However, Kim et al. have also shown that chondroitin sulfate-containing hydrogels can also induce an osteogenic MSC fate when integrated into bone defects, with Ca2+ ions binding to the negatively charged sulfate groups, leading to an ion-rich environment which promoted mineralisation.270
By contrast, cellulose is formed exclusively of α-1,4-linked D-glucose. Despite being the most abundant bio-polymer on the planet, cellulose is poorly suited as a polymer for hydrogelation due to its low water solubility. However, methylation to form methyl cellulose leads to the generation of a polysaccharide with reverse thermoresponsive behaviour, with an LCST for gelation that is dependent on the level of methylation.272 This has led to increasing interest in the use of methyl cellulose as an abundant material for the 3D printing of hydrogel scaffolds.273 In a notable recent report, Cochis et al. have demonstrated that methyl cellulose is well tolerated in vivo, and moreover is able to induce hMSC chondrogenesis under mechanical strain.274
Finally, pullulan, composed of α-1,6-linked maltotriose units (triglucose linked by α-1,4 glycosidic bonds), is derived from fungal metabolism of starch. It is biodegradable, non-immunogenic, and FDA approved, making it an attractive material for tissue engineering.24 Though not inherently cell adhesive, it has particularly found use in composite materials that provide the motifs necessary for cell attachment.24,275,276 Pullulan hydrogels are therefore an emerging class of substrates for tissue engineering.
5. Dynamic and supramolecular materials
Hydrogels with reversible or non-covalent cross-links, or composed of supramolecular polymers or assemblies, display unique properties for tissue engineering. The synthesis, properties, and applications of these gels have been extensively reviewed, and the reader is directed to the following references for excellent recent overviews of the area by the Gelain, Bowman, and Smith groups.277–280 Here, we will briefly summarise some of the key material classes and the advantages offered by these technologies.Self-assembled hydrogels are typically formed from low molecular weight gelators (LMWGs). These substrates undergo spontaneous and reversible formation of organised structures, while retaining the chemical versatility and ease of synthesis of small molecules. The formation of hydrogels from these LMWGs is on a knife-edge of stability – small perturbations in the delicate balance of attractive and dissociative forces can lead to the formation of precipitates or dissolution of the gelator.280 Assembly is often triggered by an applied stimuli. It is important to consider the effects of this stimuli, as common triggers such as changes in pH or temperature may result in pre- or post-gelation conditions that are damaging to cells. Alternative triggers, such as the use of protein–protein interactions as reported by Wong Po Foo et al., are therefore attractive.20
Hydrogels formed from LMWGs benefit from the capacity to self-heal, display shear-thinning behaviour, and undergo localised reassembly to adapt to stress and dissipate forces exerted by cells.281,282 However, they also possess weak mechanical properties as a result of the non-covalent forces that lead to assembly and hold together the scaffold. Recent reports on multi-component gels are therefore attractive.280 For example, Vieira et al. recently demonstrated that a LMWG, DBS-CONHNH2, could be used to generate hydrogels for cell growth. However, the addition of a polymeric component, in the form of agarose, enabled an order of magnitude improvement in gel mechanical properties to be achieved.283 In doing so, the authors combined the dynamic properties of a LMWG with the mechanical strength of a traditional polymer gelator.
Self-assembling peptides have been particularly widely used to produce biomedical hydrogels. Short peptides can routinely be synthesised on solid phase with high modularity, and are degraded into amino acids making them highly bio-compatible materials.284 Assembling peptides are typically amphiphilic, with either hydrophobic amino acids or non-natural alkyl or aromatic motifs being used to drive assembly into fibrillar architectures. Importantly, since pioneering work by the Stupp group in 2004,285 it has been widely demonstrated that self-assembling peptides can be engineered to enable the display of bioactive sequences at high density, making them attractive scaffolds for tissue engineering.286–288 Assembly into β-sheets is common, leading to higher mechanical strength than amorphous aggregates. For example, Tang et al. have recently reported a series of related pentapeptides, based around the parent sequence KYFIL, able to rapidly form stable hydrogels via the assembly of β-sheet nanofibers.282 These gels display sheer thinning and self-healing behaviour, with single amino acid substitutions enabling the Young's modulus of the materials to be tuned over two orders of magnitude. Fibre morphology and pH responsive properties were also shown to be highly sequence dependent. This work therefore highlights one of the key benefits of self-assembling peptides, with the diversity and versatility of molecular changes having a profound impact on hydrogel properties.
Dynamic or supramolecular cross-linking can also be exploited to create hydrogels that display self-healing and shear-thinning behaviour, from both synthetic and natural polymer precursors. For example, the complexation of boronic acids with diols to form boronic esters results in dynamic covalent bonds that can undergo exchange.289,290 Complexation is generally favoured at pH > pKa of the boronic acid. The use of benzoxaborole or phenylboronic acids bearing electron withdrawing groups is therefore required to bring the pKa into a physiologically relevant range (Fig. 13).291–293 However, Yesilyurt et al. have demonstrated that if the pKa is lowered too far, bonding becomes too stable and the dynamic properties of the gel are lost.291 The authors showed that exploiting the complexation of a boronic acid with a pKa ∼6.5 and glucose led to strong gels that were rigid across a biologically relevant pH range. By contrast, fluorophenyl boronic acids, with a pKa ∼7.2, were seen to produce shear-thinning, injectable hydrogels.
 | ||
| Fig. 13 (a) Schematic of a self-healing hydrogel, able to undergo dynamic exchange of chemical cross-links; (b) fluorination of phenyl boronic acid is necessary to lower the pKa of the boronic acid into a physiologically relevant range, allowing it to form dynamic bonds with D-gluconic acid linked polymers; (c) the use of this chemistry can be used to form self-healing PEG hydrogels. Separate gels can be placed together and fused to form a single, stretchable gel, through which the encapsulated methylene blue dye can diffuse. Part C reproduced from Advanced Materials, vol. 28, V. Yesilyurt et al., ‘Injectable Self-Healing Glucose-Responsive Hydrogels with pH-Regulated Mechanical Properties’, 86–91, Copyright 2016, with permission from John Wiley and Sons.291 | ||
Host–guest complexation has also been exploited to form self-healing gels, particularly through the use of cucurbit[n]uril and β-cyclodextrin hosts, as first reported by the Scherman and Stupp groups respectively.294–297 The reversible nature of host–guest interactions enables the modulation of hydrogel stiffness, as recently demonstrated by Shih and Lin. PEG hydrogels functionalised with β-cylodextrin were shown to form stabilising crosslinks with adamantane-derivatised multi-arm PEGs. The addition of competitive, soluble β-cyclodextrin led to reversal of this stiffening, and this process could be repeated multiple times with little change in response. As highlighted earlier, hydrogel stiffness is a critical determinant of stem cell fate, and such reversible systems are therefore likely to be of increasing interest in the near future.
Finally, reversible hydrazone bonds have been increasingly widely studied as a means to impart hydrogels with dynamic properties. In these systems, the equilibrium constant of hydrazone formation/hydrolysis is critical, with benzyl aldehydes leading to more stable linkages than their aliphatic analogues, thus dictating the mechanical properties of the gel.108 In an interesting demonstration of the importance of hydrogel dynamic properties on cell fate, the Anseth group recently showed that a balance of benzyl and alkyl aldehydes was critical to maximise ECM deposition by encapsulated chondrocytes within a PEG hydrogel.108 Unsurprisingly, weak gels quickly lost network connectivity. However, in contrast if the gel was too stable chondrogenesis was found to be stifled and matrix deposition constrained.
6. Responsive hydrogels
The static nature of many hydrogels is in stark contrast to the dynamic environment experienced by cells in their native extracellular niche. Gels that can respond to external stimuli and undergo a change in structure or properties are therefore attractive materials for tissue engineering. In this section, we will provide a brief overview of the key design features that can lead to responsive hydrogels and their applications.Thermoresponsive gels have been particularly widely used, predominantly as injectable materials that undergo gelation when applied in vivo. This enables minimally invasive delivery to be achieved through the injection of liquid precursors that gelate in situ, along with the ability to adapt to the size or shape of the cavity in which the gel is being applied.19 Two categories of thermoresponsive behaviour are theoretically suitable for such applications – the use of polymers which gel when cooled to body temperature, or alternatively polymers which possess an LCST and gelate when heated to body temperature. In practice the use of polymers with an LCST is particularly attractive, as it negates the risk of damage from the injection of heated solutions.19 Assembly above the LCST of a polymer is predominantly driven by entropic contributions. PNIPAM, introduced in section 3.1.3, is the archetype of a thermoresponsive polymer, possessing an LCST ∼32 °C. Below this temperature, the polymer chains are highly solvated. However, when the temperature is raised, aggregation of the hydrophobic iso-propyl groups takes place, leading to an increase in entropy as the solvation shell is released.83 Many other synthetic polymers also possess LCSTs within a biologically relevant range and have therefore found use as components of injectable hydrogels, including poly(oligo(ethylene glycol)methyl ether methacrylate) (POEGMA)298 and pluronics (PEG-poly(propylene oxide) co-polymers).299 POEGMA is particularly attractive as a thermoresponsive polymer as its LCST and gelation time can be tuned based on the number of ethylene oxide repeat units across a wide temperature range (20–90 °C). Importantly, the Hoare group have recently demonstrated that by mixing different POEGMA constructs, rational tuning of these properties can be achieved in an ‘off-the-shelf’ manner.298 In a series of papers they have subsequently gone on to exploit injectable POEGMA-based hydrogels to produce 3D-aligned myotubes for cardiac tissue engineering,300 adhesive hydrogels for in vivo fibroblast penetration,301 and degradable scaffolds for epithelial proliferation.302 Alternatively, naturally-derived polymers can be modified to provide LCST behaviour, such as chitosan-β-glycerophosphate blends,303 ELPs,214 methyl cellulose,273 hydroxybutyl chitosan,304 and galactose-modified xyloglucan.305,306 Of relevance to biomedical hydrogels, LCSTs are dependent on both the concentration and nature of the salt content, with the potential to alter the applicable temperature range of a particular thermoresponsive polymer as a result.307,308
Enzymatic responsiveness can also be imparted to hydrogel scaffolds. Indeed, the ability of cells to remodel their environment through the action of extracellular enzymes has been shown to be highly beneficial to tissue development, as described in section 2.2.34–36 Within the family of naturally derived polymers, sensitivity to enzymatic degradation is inherent to their biological origins. Degradation by endogenous enzymes may prove problematic, as highlighted by the instability of HA-based gels due to the activity of hyaluronidases.252 In contrast, within synthetic polymers enzymatic sensitivity must be built into the hydrogel scaffold. This is most commonly achieved via cross-linking with protease-sensitive peptides, enabling cleavage through the action of enzymes such as those of the matrix metalloprotease (MMP) family.27,34,44
The use of light has emerged as a potent means to modulate hydrogel properties with both spatial and temporal control. Light-responsive hydrogels typically exploit photo-cleavage or -cross-linking to alter the scaffold architecture or functionality (Fig. 14).28,309 The use of light to mediate changes results in a fast response time, with micron-scale patterning possible in 3D through the use of 2-photon irradiation.309 However, light penetration is quickly attenuated with sample thickness, and the use of UV irradiation in particular can be damaging to cells, as described in section 8. The recent work of Lunzer et al., exploiting a small molecule 2-photon sensitizer to greatly reduce the irradiation times and intensities needed to achieve robust 3D patterning, is therefore particularly exciting.310
 | ||
| Fig. 14 (a) Schematic of a responsive hydrogel, where chemical cross-links are broken in response to a light stimuli; (b) nitrobenzyl ethers undergo photocleavage in response to either single (λ = 365 nm) or 2-photon (λ = 740 nm) irradiation; (c) 3D patterning of channels in a PEG-based hydrogel by a combination of photodegradation and photo thiol–ene modification with RGD motifs, allowing the encapsulation and outgrowth of 3T3 fibroblasts. Inset is a top-down projection of the channels. Red – hydrogel; green – F-actin; blue – cell nuclei, scale bar 100 μm. Reprinted by permission from Springer Nature, Nature Chemistry, ‘Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions’, C. A. DeForest and K. S. Anseth, 2011.344 | ||
With photo-sensitive groups that respond to orthogonal wavelengths of light, it is possible to modulate multiple gel properties in isolation, allowing precise control over gel stiffness or bioactivity within the same scaffold, as demonstrated by Rosales et al.311,312 The authors synthesised a HA-based hydrogel that could undergo softening through the cleavage of nitrobenzyl-crosslinkers with 365 nm UV irradiation. Alternatively, the gel could be stiffened via methacrylate crosslinking following addition of a photoinitiator and irradiation at 400–500 nm. Further developments in spectrally resolved photochemistry will enable increasing complexity to be built into these systems, with interesting opportunities for creating temporally responsive scaffolds.
Other means of triggering hydrogel responsiveness are of less relevance to tissue engineering. For example, pH sensitive gels have been widely exploited in drug delivery, but changes in the acidity or basicity of a gel are also likely to lead to cellular damage. Even gels that exhibit changes in morphology over very narrow pH ranges require cells to be exposed to pH regimes that are unfavourable for tissue growth.313 Similarly, while disulfide linkages have been used to generate gels that are sensitive to reductive environments, particularly for intracellular drug delivery, extracellular reduction has been less commonly implemented.
7. The influence of polymer architecture
Careful control over polymer design can be critical to dictate hydrogel properties. Two polymers with the same monomer composition, but different architectures, can generate gels with drastically different characteristics. This is typified by the assembly and hydrogelation of amphiphilic block co-polymers. For example, ABA polymers (where A is hydrophobic and B is hydrophilic e.g. PCL-PEG) undergo gelation at lower concentrations (i.e. lower CGC) and produce stronger gels than an AB polymer of the same composition. This can be rationalised through the ability of the ABA polymer to form intra- as well as inter-polymer interactions, promoting the aggregation of micelles to form bulk hydrogels.148 Similarly, ABA type polymers undergo more effective gelation than the analogous BAB construct due to the ability of the hydrophobic blocks to form loops and bridge micelles, accelerating assembly.314 Importantly, block length must also be carefully considered alongside total content of a particular hydrophobic polymer. Zhang et al. recently demonstrated that short PCL chains, grafted to a poly(glutamic acid) backbone, were unable to form crystalline domains, even though presented at high density. The amorphous aggregates were able to provide robust mechanical strength, while enabling hydrolysis to occur on a relevant timeframe in vivo.152 As a result, the authors were able to address the disadvantages of PCL as discussed in section 3.3.3.The situation is further complicated for non-linear polymers, with an increase in arms in star-polymers leading to a drop in CGC.315 As demonstrated by Li et al., hyperbranched polymers possess a greatly reduced CGC as a result of increased interactions between polymer branches, in contrast to the singular interactions imposed by linear architectures, leading to hydrogels with increased mechanical stability.316,317 Similarly A-g-B graft co-polymers behave very differently to the analogous B-g-A construct.318 These trends in assembly are also reflected in the rate of hydrolysis in polyester-based hydrogels, with the structure of polymer precursors greatly influencing degradation rate. For example, Jeong et al. have reported that the hydrolysis of PLGA side chains in PEG-graft co-polymers results in drastically faster gel degradation in vivo than the analogous polymer bearing a PLGA backbone and PEG sidechains.318 The authors hypothesise that the generated short chain hydrolysis products are easily washed away from the site of implantation, in contrast to the long chain PLGA-g-PEG products that result from backbone cleavage.
Within chemically cross-linked gels, properties can still be dictated by the design of both polymers and macromer precursors. While many models for the hydrogelation of macromers assume ‘ideal’ functionality, whereby all end-groups form productive cross-links, experimental results deviate from this model with partially reacted cross-linkers, intramolecular loop formation, and chain entanglements acting to alter the hydrogel network structure.319,320 In theory, to maximise ‘ideality’ gelation should be undertaken at an optimal concentration at which polymer chains are starting to come into contact, as recently highlighted by Wang et al.321 Below this concentration, cross-linking is less efficient, promoting loop formation, while at higher concentrations entanglement limits diffusion. Importantly, the authors showed that the use of star polymers was more amenable to identifying this optimal concentration, due to the known propensity of star-systems to form elongated rather than coiled structures, that behave more like predictable tetrahedra and producing a more ‘crystal like’ network structure.
When star-macromers are utilised to form hydrogels, the consequences of each imperfection also become less significant with increasing number of arms, as a relatively lower proportion of cross-linking opportunities are lost.320 Kim et al. have also shown that sacrificial modification of end-groups, for example with adhesive peptides, is better tolerated.120 However, the chances of forming imperfections in the first place also increases, and macromer design can therefore strongly influence hydrogel mechanical properties, pore size, and degradation.322,323 Interestingly, the Johnson and Olsen groups have used a combination of modelling and experimental studies to further demonstrate that gels formed from macromers bearing an odd number of junction-forming arms result in a higher proportion of loops and cyclic defects than star polymers with even functionality.324
The impact of chain imperfections on hydrogel mechanical properties and homogeneity is widely acknowledged. However, in a recent report Schneider et al. have highlighted that areas of network heterogeneity may in fact promote the formation of mature tissue.107 In order for cells to deposit their own matrix, hydrogel breakdown must occur simultaneously to provide space in which to do so. Areas of heterogeneity provide localised pockets of low crosslinking density able to support this growth, whilst maintaining bulk structural integrity. Gels must therefore provide a delicate balance between supporting ECM production and preventing reverse gelation.
In recent years, dendrimeric polymers have emerged as promising building blocks for forming hydrogels for tissue engineering. In 2006, the Grinstaff group first reported that conjugation of a dendrimeric polyester to the termini of a linear PEG chain, in effect creating a dendrimer–linear–dendrimer ABA-type polymer, enabled the formation of degradable hydrogels able to support chondrocyte proliferation.325 This structure is key to the success of dendrimeric hydrogels, as the linear component provides sufficient pore size to enable cell encapsulation and proliferation. On the other hand, the dendrimers provide opportunities for dense pockets of cross-linking, that deliver mechanical strength at low polymer concentrations and enhanced gelation rates.326–328 Importantly, since dendrimers can be precisely synthesised they offer opportunities for increased reproducibility during hydrogelation. Hodgson et al. recently demonstrated that dendrimer-capped PEG macromers enabled hydrogels with near identical Young's moduli to be produced over multiple batches, in contrast to gels formed from linear PEG macromers which varied by almost 200%.329 Finally, the Bitton and Matson groups have recently demonstrated that dendrimers of ELP can also be produced and used to form hydrogels.330,331 Di- or tri-ELP repeats can be incorporated into these dendrimers and the resultant constructs have been shown to retain LCST behaviour. However, the transition temperature is increased when compared to the analogous linear construct, which the authors hypothesise is a result of changes in solvent accessibility.
8. Implications of polymerisation and hydrogelation method
The method used to induce polymerisation or cross-linking can have major implications on the properties or application of a hydrogel scaffold. Here, we will discuss some of the most important considerations that must be taken into account during hydrogel design.The use of free-radical polymerisation or cross-linking is particularly widespread, and often used to encapsulate cells in situ. At the earliest stage, the method of initiation has important downstream implications. While in traditional polymer synthesis the thermal activation of initiators such as AIBN is common, the need to heat the activator may be limiting in the presence of cells or tissue.32,332,333 Similarly, ionising irradiation and redox-initiators are useful means to generate acellular hydrogels or polymer precursors, but are damaging to cells.334 The use of photo-activation has therefore found increasing popularity as a means of initiation. Light provides fast activation that can be temporally controlled and spatially defined, making it particularly attractive for the patterning of 3D gels.309 However, the choice of initiator and activation wavelength is important. Many early reports exploited the UV-activation of initiators such as Igracure 651, which are poorly cytocompatible.335 There has been a subsequent shift towards initiators with reduced cytotoxicity, and that can be activated with less damaging visible light. These techniques have been increasingly commonly used to initiate hydrogelation in vivo, though the penetration of light through thick samples and tissue limits the size of gels that can be produced.25
Once generated, highly reactive free radicals continue to be damaging to cells and tissues. These detrimental processes in fact act to inhibit polymerisation by consuming radicals, as demonstrated by Chu et al.336 When this is coupled with the high sensitivity of vinyl polymerisations to oxygen, large amounts of radicals are required to achieve high conversion, with a corresponding increase in irradiation time necessary to reach gelation.125 Furthermore, vinyl polymerisations are exothermic, leading to heat generation that may be damaging to tissue.337 This leads to a conflict between the need for rapid polymerisations at the site of application, to limit the leaching of soluble components into surrounding tissue, and the need to minimise damaging radical concentrations and local heating. In contrast, radical-mediated thiol–ene reactions display greatly reduced oxygen sensitivity and occur at a faster rate, reducing the concentration of radicals necessary to achieve gelation and also negating the need for potentially toxic initiatiors.338 Indeed, in an important recent paper Ruskowitz and DeForest demonstrated that low doses of 365 nm UV light, able to induce thiol–ene gelation, led to negligible damage to cells at the proteomic level (Fig. 15).339
 | ||
| Fig. 15 Quantification of cell apoptotic activity following irradiation at different wavelengths and intensities (10 min for λ = 365 nm, 0.5 min for λ = 254 nm), by monitoring Caspase 3/7 activation and normalising to cell number, in (a) NIH3T3 mouse fibroblasts; or (b) human mesenchymal stem cells (hMSCs); (c) representative fluorescence microscopy images, staining for nuclei (blue) and caspase activity (green). Adapted with permission from ACS Biomaterials Science and Engineering, vol. 5, 2111–2116, E. R. Ruskowitz and C. A. DeForest, ‘Proteome-side analysis of cellular response to ultraviolet light for biomaterials synthesis and modification’. Copyright 2019 American Chemical Society.339 | ||
Chemically cross-linked hydrogels can be formed via either chain-growth or step-growth mechanisms, with significant implications for downstream applications. Chain-growth polymerisations are able to generate high Mw polymers early on. However, as solutions become increasingly viscous and eventually undergo gelation, monomer diffusion is slowed and achieving complete consumption becomes challenging.55 As described above, these residual monomers are often toxic, presenting challenges for hydrogel biocompatibility. Though the use of functionalised macromers for chain-growth cross-linking can address this limitation, they provide their own challenges due to the possible for hydrogel heterogeneity. Vats et al. have recently demonstrated that the hydrogelation of PEG-dimethacrylate results in the formation of stiff, hydrophobic pockets at the points of crosslinking, detectable at the nanoscale by atomic force microscopy, as discussed in section 3.2.126 This heterogeneity also impacted on cell adhesion, with the clustering of conjugated adhesive peptides leading to lower cell densities than analogous gels with a homogeneous distribution.
In contrast, step-growth reactions, such as thiol–ene polymerisations, can be used to generate polymers with complete monomer conversion. However, they require high conversions and careful control over stoichiometry to achieve the high Mw polymers necessary for gelation.340 Furthermore, the number of ‘imperfections’ generated in a hydrogel network have been shown to be higher in gels formed via step-growth rather than chain-growth mechanisms.319,320 To address the limitations of each polymerisation mode for hydrogelation, two approaches have been described – the first is to exploit mixed-mode polymerisations, using both chain- and step-growth polymerisations within a single system, as first reported by Salinas and Anseth.340 Alternatively, the step-growth cross-linking of pre-formed high Mw macromer precursors has found increasing popularity. These macromers can be tuned in size to enable excretion, and cross-linked via a variety of different means to generate responsive, degradable, or stable hydrogels.55
Finally, the influence of polymer dispersity (Đ) should be considered. While Đ has less impact on hydrogel properties than it does on those of discrete polymer populations, Đ can still be influential. For example, polymers with a narrow distribution exhibit sharper phase transitions from sol-to-gel, and also lead to more precisely defined pore sizes within chemically cross-linked gels.341 Furthermore, since renal excretion is Mw dependent, controlled polymerisations are able to produce homogeneous degradation products that are able to be efficiently cleared from the site of action.32 Finally, as described above, variability in polymer precursors resulting from high Đ can lead to poor reproducibility and batch-to-batch variation in hydrogel properties, as highlighted by Hodgson et al.329 The use of highly controlled polymerisation techniques is therefore of increasing importance to the tissue engineering community.
9. Conclusions
In this review, we have summarised the use of hydrogels for tissue engineering from the perspective of polymer chemistry. The properties of a gel, its mechanical strength, ability to influence cell growth, and potential to cause immunogenicity and cytotoxicity, are all dictated by the structure of the underlying polymer. By considering each class of polymer, we have been able to highlight the major advantages and disadvantages of each individual material, and it is clear that there is no one ‘magic bullet’ polymer that is ideal in every scenario. Indeed, even for a particular clinical application there is rarely one polymer that can provide all of the desirable properties. Creating gels that combine tissue-dependent mechanical strength, cell adhesion and signalling, and biodegradability on a relevant timescale is highly challenging. The design of hybrid, composite gels that combine the beneficial properties of one polymer with those of a complementary partner is therefore common, such as supplementing the bioactivity of hyaluronic acid with the mechanical strength of covalently cross-linked PDLLA-PEG.29 However, though blending two polymers can deliver great benefits, this approach rarely leads to new properties that were not present in the original components. Indeed, these properties often ‘meet in the middle’, and combining a mechanically strong polymer with one that is weak will commonly result in a hydrogel of intermediate strength. There is therefore a delicate balance to be reached when designing such composite materials, and multi-factor ‘design-of-experiments’ studies, such as that recently disclosed by Kaiser et al. for cardiac tissue engineering, will prove key in the future for the identification of optimal compositions for tissue development.342The incredible diversity of polymer structure and function that can be delivered by polymer chemists has had a vast impact on the materials sciences. However, it is noticeable that the majority of hydrogels exploited by the tissue engineering community continue to make use of the same limited subset of materials. Though one could hypothesise that we already have access to polymers which provide optimal characteristics, it is far more likely that there is a reluctance to embrace new materials which break the mould. That new polymers are developed and proposed to be suitable materials for tissue engineering, with only a minimum demonstration that they are non-toxic to cells required, only serves to exacerbate this problem. There is therefore a pressing need for enhanced collaborations between polymer chemists and biomedical engineers, to provide a platform for testing emerging polymers under more rigorous conditions. Perhaps more importantly, there is also a need for more cross-disciplinary dialogue, so that polymer chemists can create the next generation of polymer hydrogels with features that fit the needs of the biomedical community. This trend can already be seen in recently emerging materials, such as the strain-stiffening polyisocyanopeptides and tunable PiPOx and polypeptoid structures introduced in section 3.4. These materials deliver real advantages to the tissue engineering community and are likely to be of increasing interest in coming years. With continued innovation, polymer chemistry will become increasingly influential, driving the design and clinical translation of new hydrogels, and leading to tangible patient benefit in the coming years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CDS is grateful to the Royal Society of Chemistry (RF19-8128) and the Royal Society (RGS\R1\191210) for their generous support. V. Bloom and Dr M. Thomas are thanked for critical evaluation of the manuscript.References
- WHO Methods and Data Sources for Global Burden of Disease Estimates 2000–2015, 2017.
- R. Langer and J. P. Vacanti, Tissue Engineering, Science, 1993, 260, 920–926 CrossRef CAS.
- F. J. O'Brien, Biomaterials & Scaffolds for Tissue Engineering, Mater. Today, 2011, 14(3), 88–95 CrossRef.
- J. A. Hunt, R. Chen, T. van Veen and N. Bryan, Hydrogels for Tissue Engineering and Regenerative Medicine, J. Mater. Chem. B, 2014, 2, 5319–5338 RSC.
- L. Wang, M. Neumann, T. Fu, W. Li, X. Cheng and B. L. Su, Porous and Responsive Hydrogels for Cell Therapy, Curr. Opin. Colloid Interface Sci., 2018, 38, 135–157 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Hydrogels in Regenerative Medicine, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS.
- W. Zhao, X. Jin, Y. Cong and J. Fu, Degradable Natural Polymer Hydrogels for Articular Cartilage Tissue Engineering, J. Chem. Technol. Biotechnol., 2013, 88, 327–339 CrossRef CAS.
- Q. Huang, Y. Zou, M. C. Arno, S. Chen, T. Wang, J. Gao, A. P. Dove and J. Du, Hydrogel Scaffolds for Differentiation of Adipose-Derived Stem Cells, Chem. Soc. Rev., 2017, 46(20), 6255–6275 RSC.
- X. Li, Q. Sun, Q. Li, N. Kawazoe and G. Chen, Functional Hydrogels With Tunable Structures and Properties for Tissue Engineering Applications, Front. Chem., 2018, 6, 499 CrossRef CAS.
- A. S. Mao and D. J. Mooney, Regenerative Medicine: Current Therapies and Future Directions, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(47), 14452–14459 CrossRef CAS.
- S. R. Caliari and J. A. Burdick, A Practical Guide to Hydrogels for Cell Culture, Nat. Methods, 2016, 13(5), 405–414 CrossRef CAS.
- C. M. Magin, D. L. Alge and K. S. Anseth, Bio-Inspired 3D Microenvironments: A New Dimension in Tissue Engineering, Biomed. Mater., 2016, 11(2), 022001 CrossRef.
- F. Khan, M. Tanaka and S. R. Ahmad, Fabrication of Polymeric Biomaterials: A Strategy for Tissue Engineering and Medical Devices, J. Mater. Chem. B, 2015, 3(42), 8224–8249 RSC.
- I. M. El-Sherbiny and M. H. Yacoub, Hydrogel Scaffolds for Tissue Engineering: Progress and Challenges, Glob. Cardiol. Sci. Pract., 2013, 38, 317–342 Search PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Foreign Body Reaction to Biomaterials, Semin. Immunol., 2008, 20(2), 86–100 CrossRef CAS PubMed.
- Q. Yang and S. K. Lai, Anti-PEG Immunity: Emergence, Characteristics, and Unaddressed Questions, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7(5), 655–677 CAS.
- Z. Wang, Z. Wang, W. W. Lu, W. Zhen, D. Yang and S. Peng, Novel Biomaterial Strategies for Controlled Growth Factor Delivery for Biomedical Applications, NPG Asia Mater., 2017, 9, e435 CrossRef CAS.
- S. I. S. Hendrikse, S. Spaans, E. W. Meijer and P. Y. W. Dankers, Supramolecular Platform Stabilizing Growth Factors, Biomacromolecules, 2018, 19, 2610–2617 CrossRef CAS.
- L. Yu and J. Ding, Injectable Hydrogels as Unique Biomedical Materials, Chem. Soc. Rev., 2008, 37(8), 1473–1481 RSC.
- W. P. Foo, C. T. S. Lee, J. S. Mulyasasmita, W. Parisi-Amon, A. Heilshorn and S. C, Two-Component Protein-Engineered Physical Hydrogels for Cell Encapsulation, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(52), 22067–22072 CrossRef.
- Y. Wang, L. Li, Y. Kotsuchibashi, S. Vshyvenko, Y. Liu, D. Hall, H. Zeng and R. Narain, Self-Healing and Injectable Shear Thinning Hydrogels Based on Dynamic Oxaborole-Diol Covalent Cross-Linking, ACS Biomater. Sci. Eng., 2016, 2(12), 2315–2323 CrossRef CAS.
- M. J. Rodriguez, J. Brown, J. Giordano, S. J. Lin, F. G. Omenetto and D. L. Kaplan, Silk Based Bioinks for Soft Tissue Reconstruction Using 3-Dimensional (3D) Printing with in Vitro and in Vivo Assessments, Biomaterials, 2017, 117, 105–115 CrossRef CAS.
- M. M. Stevens, H. F. Qanadilo, R. Langer and V. P. Shastri, A Rapid-Curing Alginate Gel System: Utility in Periosteum-Derived Cartilage Tissue Engineering, Biomaterials, 2004, 25(5), 887–894 CrossRef CAS.
- F. Chen, S. Yu, B. Liu, Y. Ni, C. Yu, Y. Su, X. Zhu, X. Yu, Y. Zhou and D. Yan, An Injectable Enzymatically Crosslinked Carboxymethylated Pullulan/Chondroitin Sulfate Hydrogel for Cartilage Tissue Engineering, Sci. Rep., 2016, 6, 20014 CrossRef CAS.
- R.-Z. Lin, Y.-C. Chen, R. Moreno-Luna, A. Khademhosseini and J. M. Melero-Martin, Transdermal Regulation of Vascular Network Bioengineering Using a Photopolymerizable Methacrylated Gelatin Hydrogel, Biomaterials, 2013, 34(28), 6785–6796 CrossRef CAS.
- Y. Luo and M. S. Shoichet, A Photolabile Hydrogel for Guided Three-Dimensional Cell Growth and Migration, Nat. Mater., 2004, 3(4), 249–253 CrossRef CAS.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Sequential Click Reactions for Synthesizing and Patterning Three-Dimensional Cell Microenvironments, Nat. Mater., 2009, 8(8), 659–664 CrossRef CAS.
- C. A. DeForest and D. A. Tirrell, A Photoreversible Protein-Patterning Approach for Guiding Stem Cell Fate in Three-Dimensional Gels, Nat. Mater., 2015, 14, 523–531 CrossRef CAS.
- A. X. Sun, H. Lin, A. M. Beck, E. J. Kilroy and R. S. Tuan, Projection Stereolithographic Fabrication of Human Adipose Stem Cell-Incorporated Biodegradable Scaffolds for Cartilage Tissue Engineering, Front. Bioeng. Biotechnol., 2015, 3, 115 Search PubMed.
- R. Xiong, Z. Zhang, W. Chai, Y. Huang and D. B. Chrisey, Freeform Drop-on-Demand Laser Printing of 3D Alginate and Cellular Constructs, Biofabrication, 2015, 7(4), 045011 CrossRef.
- Z. Liu, M. Tang, J. Zhao, R. Chai and J. Kang, Looking into the Future: Toward Advanced 3D Biomaterials for Stem-Cell-Based Regenerative Medicine, Adv. Mater., 2018, 30(17), 1705388 CrossRef.
- H. Macková, Z. Plichta, H. Hlídková, O. Sedláček, R. Konefal, Z. Sadakbayeva, M. Dušková-Smrčková, D. Horák and Š. Kubinová, Reductively Degradable Poly(2-Hydroxyethyl Methacrylate) Hydrogels with Oriented Porosity for Tissue Engineering Applications, ACS Appl. Mater. Interfaces, 2017, 9(12), 10544–10553 CrossRef.
- C. R. Nuttelman, M. A. Rice, A. E. Rydholm, C. N. Salinas, D. N. Shah and K. S. Anseth, Macromolecular Monomers for the Synthesis of Hydrogel Niches and Their Application in Cell Encapsulation and Tissue Engineering, Prog. Polym. Sci., 2008, 33(2), 167–179 CrossRef CAS.
- C. M. Madl, B. L. Lesavage, R. E. Dewi, C. B. Dinh, R. S. Stowers, M. Khariton, K. J. Lampe, D. Nguyen, O. Chaudhuri and A. Enejder, et al., Maintenance of Neural Progenitor Cell Stemness in 3D Hydrogels Requires Matrix Remodelling, Nat. Mater., 2017, 16(12), 1233–1242 CrossRef CAS PubMed.
- H. Lee, L. Gu, D. J. Mooney, M. E. Levenston and O. Chaudhuri, Mechanical Confinement Regulates Cartilage Matrix Formation by Chondrocytes, Nat. Mater., 2017, 16(12), 1243–1251 CrossRef CAS PubMed.
- E. A. Aisenbrey and S. J. Bryant, A MMP7-Sensitive Photoclickable Biomimetic Hydrogel for MSC Encapsulation towards Engineering Human Cartilage, J. Biomed. Mater. Res., Part A, 2018, 2344–2355 CrossRef CAS.
- K. Ceonzo, A. Gaynor, L. Shaffer, K. Kojima, C. A. Vacanti and G. L. Stahl, Polyglycolic Acid-Induced Inflammation : Role of Hydrolysis and Resulting Complement Activation, Tissue Eng., 2006, 12(2), 301–308 CrossRef CAS.
- D. Chen, Z. Su, L. Weng, L. Cao, C. Chen, S. Zeng, S. Zhang, T. Wu, Q. Hu and J. Xiao, Effect of Inflammation on Endothelial Cells Induced by Poly-L-Lactic Acid Degradation in Vitro and in Vivo, J. Biomater. Sci., Polym. Ed., 2018, 29(15), 1909–1919 CrossRef CAS.
- F. Brandl, F. Sommer and A. Goepferich, Rational Design of Hydrogels for Tissue Engineering: Impact of Physical Factors on Cell Behavior, Biomaterials, 2007, 28(2), 134–146 CrossRef CAS.
- L. Li, N. Wang, X. Jin, R. Deng, S. Nie, L. Sun, Q. Wu, Y. Wei and C. Gong, Biodegradable and Injectable in Situ Cross-Linking Chitosan-Hyaluronic Acid Based Hydrogels for Postoperative Adhesion Prevention, Biomaterials, 2014, 35(12), 3903–3917 CrossRef CAS.
- L. Karumbaiah, S. F. Enam, A. C. Brown, T. Saxena, M. I. Betancur, T. H. Barker and R. V. Bellamkonda, Chondroitin Sulfate Glycosaminoglycan Hydrogels Create Endogenous Niches for Neural Stem Cells, Bioconjugate Chem., 2015, 26(12), 2336–2349 CrossRef CAS.
- Y. M. Kang, S. H. Lee, J. Y. Lee, J. S. Son, B. S. Kim, B. Lee, H. J. Chun, B. H. Min, J. H. Kim and M. S. Kim, A Biodegradable, Injectable, Gel System Based on MPEG-b-(PCL-Ran-PLLA) Diblock Copolymers with an Adjustable Therapeutic Window, Biomaterials, 2010, 31(9), 2453–2460 CrossRef CAS.
- U. Blache and M. Ehrbar, Inspired by Nature: Hydrogels as Versatile Tools for Vascular Engineering, Adv. Wound Care, 2017, 7, 232–246 CrossRef.
- C. D. Spicer, E. T. Pashuck and M. M. Stevens, Achieving Controlled Biomolecule-Biomaterial Conjugation, Chem. Rev., 2018, 118(16), 7702–7743 CrossRef CAS PubMed.
- S. A. Fisher, A. E. G. Baker and M. S. Shoichet, Designing Peptide and Protein Modified Hydrogels: Selecting the Optimal Conjugation Strategy, J. Am. Chem. Soc., 2017, 139(22), 7416–7427 CrossRef CAS.
- G. B. Schneider, A. English, M. Abraham, R. Zaharias, C. Stanford and J. Keller, The Effect of Hydrogel Charge Density on Cell Attachment, Biomaterials, 2004, 25(15), 3023–3028 CrossRef CAS.
- M. D. Swartzlander, C. A. Barnes, A. K. Blakney, J. L. Kaar, T. R. Kyriakides and S. J. Bryant, Linking the Foreign Body Response and Protein Adsorption to PEG-Based Hydrogels Using Proteomics, Biomaterials, 2015, 41, 26–36 CrossRef CAS.
- F. A. Jerca, A. M. Anghelache, E. Ghibu, S. Cecoltan, I. C. Stancu, R. Trusca, E. Vasile, M. Teodorescu, D. M. Vuluga and R. Hoogenboom, et al., Poly(2-Isopropenyl-2-Oxazoline) Hydrogels for Biomedical Applications, Chem. Mater., 2018, 30(21), 7938–7949 CrossRef CAS.
- S. Aoshima and S. Kanaoka, A Renaissance in Living Cationic Polymerization, Chem. Rev., 2009, 109(11), 5245–5287 CrossRef CAS.
- F. A. Jerca, V. V. Jerca, A. M. Anghelache, D. M. Vuluga and R. Hoogenboom, Poly(2-Isopropenyl-2-Oxazoline) as a Versatile Platform towards Thermoresponsive Copolymers, Polym. Chem., 2018, 9(25), 3473–3478 RSC.
- S. J. Bryant, J. L. Cuy, K. D. Hauch and B. D. Ratner, Photo-Patterning of Porous Hydrogels for Tissue Engineering, Biomaterials, 2007, 28, 2978–2986 CrossRef CAS.
- V. Delplace and J. Nicolas, Degradable Vinyl Polymers for Biomedical Applications, Nat. Chem., 2015, 7, 771–784 CrossRef CAS.
- E. Yoshii, Cytotoxic Effects of Acrylates and Methacrylates: Relationships of Monomer Structures and Cytotoxicity, J. Biomed. Mater. Res., 1997, 37(4), 517–524 CrossRef CAS.
- J. Winter, S. Shiga and A. Islur, The Complications of Polyacrylamide Hydrogel Augmentation Mammoplasty: A Case Report and Review of the Literature, Plast. Surg. Case Stud., 2016, 2(3), 47–50 CrossRef.
- S. Q. Liu, R. Tay, M. Khan, P. L. Rachel Ee, J. L. Hedrick and Y. Y. Yang, Synthetic Hydrogels for Controlled Stem Cell Differentiation, Soft Matter, 2009, 6(1), 67–81 RSC.
- J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou and S. Daunert, Genetically Engineered Protein in Hydrogels Tailors Stimuli-Responsive Characteristics, Nat. Mater., 2005, 4(4), 298–302 CrossRef CAS PubMed.
- X. He, J. Ma and E. Jabbari, Effect of Grafting RGD and BMP-2 Protein-Derived Peptides to a Hydrogel Substrate on Osteogenic Differentiation of Marrow Stromal Cells, Langmuir, 2008, 24(16), 12508–12516 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties, Science, 2009, 324, 59–63 CrossRef CAS.
- H. Oda, T. Konno and K. Ishihara, Efficient Differentiation of Stem Cells Encapsulated in a Cytocompatible Phospholipid Polymer Hydrogel with Tunable Physical Properties, Biomaterials, 2015, 56, 86–91 CrossRef CAS.
- L. E. Jansen, L. D. Amer, E. Y. T. Chen, T. V. Nguyen, L. S. Saleh, T. Emrick, W. F. Liu, S. J. Bryant and S. R. Peyton, Zwitterionic PEG-PC Hydrogels Modulate the Foreign Body Response in a Modulus-Dependent Manner, Biomacromolecules, 2018, 19(7), 2880–2888 CrossRef CAS.
- L. Bian, M. Guvendiren, R. L. Mauck and J. A. Burdick, Hydrogels That Mimic Developmentally Relevant Matrix and N-Cadherin Interactions Enhance MSC Chondrogenesis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(25), 10117–10122 CrossRef CAS.
- D. Hanjaya-Putra, K. T. Wong, K. Hirotsu, S. Khetan, J. A. Burdick and S. Gerecht, Spatial Control of Cell-Mediated Degradation to Regulate Vasculogenesis and Angiogenesis in Hyaluronan Hydrogels, Biomaterials, 2012, 33(26), 6123–6131 CrossRef CAS.
- R. Ravichandran, M. M. Islam, E. I. Alarcon, A. Samanta, S. Wang, P. Lundström, J. Hilborn, M. Griffith and J. Phopase, Functionalised Type-I Collagen as a Hydrogel Building Block for Bio-Orthogonal Tissue Engineering Applications, J. Mater. Chem. B, 2015, 4(2), 318–326 RSC.
- O. Wichterle and D. Lim, Hydrogels for Biological Use, Nature, 1960, 185(4706), 117–118 CrossRef.
- S. Atzet, S. Curtin, P. Trinh, S. Bryant and B. Ratner, Degradable Poly(2-Hydroxyethyl Methacrylate)-Co-Polycaprolactone Hydrogels for Tissue Engineering Scaffolds, Biomacromolecules, 2008, 9(12), 3370–3377 CrossRef CAS.
- Y. Wang, Q. Chen, M. Chen, Y. Guan and Y. Zhang, PHEMA Hydrogel Films Crosslinked with Dynamic Disulfide Bonds: Synthesis, Swelling-Induced Mechanical Instability and Self-Healing, Polym. Chem., 2019, 10, 4844–4851 RSC.
- Z. Cai, Y. Gan, C. Bao, W. Wu, X. Wang, Z. Zhang, Q. Zhou, Q. Lin, Y. Yang and L. Zhu, Photosensitive Hydrogel Creates Favorable Biologic Niches to Promote Spinal Cord Injury Repair, Adv. Healthcare Mater., 2019, 8, 1900013 CrossRef.
- J. M. McCracken, A. Badea, M. E. Kandel, A. S. Gladman, D. J. Wetzel, G. Popescu, J. A. Lewis and R. G. Nuzzo, Programming Mechanical and Physicochemical Properties of 3D Hydrogel Cellular Microcultures via Direct Ink Writing, Adv. Healthcare Mater., 2016, 5(9), 1025–1039 CrossRef CAS PubMed.
- A. Badea, J. M. McCracken, E. G. Tillmaand, M. E. Kandel, A. W. Oraham, M. B. Mevis, S. S. Rubakhin, G. Popescu, J. V. Sweedler and R. G. Nuzzo, 3D-Printed PHEMA Materials for Topographical and Biochemical Modulation of Dorsal Root Ganglion Cell Response, ACS Appl. Mater. Interfaces, 2017, 9(36), 30318–30328 CrossRef CAS.
- Š. Kubinová, D. Horák and E. Syková, Cholesterol-Modified Superporous Poly(2-Hydroxyethyl Methacrylate) Scaffolds for Tissue Engineering, Biomaterials, 2009, 30(27), 4601–4609 CrossRef.
- J. S. Belkas, C. A. Munro, M. S. Shoichet, M. Johnston and R. Midha, Long-Term in Vivo Biomechanical Properties and Biocompatibility of Poly(2-Hydroxyethyl Methacrylate-Co-Methyl Methacrylate) Nerve Conduits, Biomaterials, 2005, 26, 1741–1749 CrossRef CAS.
- Y. Hu, J. O. You and J. Aizenberg, Micropatterned Hydrogel Surface with High-Aspect-Ratio Features for Cell Guidance and Tissue Growth, ACS Appl. Mater. Interfaces, 2016, 8(34), 21939–21945 CrossRef CAS.
- S. Vijayasekaran, T. V. Chirila, T. A. Robertson, X. Lou, H. Fitton, C. R. Hicks and I. J. Constable, Calcification of Poly (2-Hydroxyethyl Methacrylate) Hydrogel Sponges Implanted in the Rabbit Cornea : A 3-Month Study, J. Biomater. Sci., Polym. Ed., 2000, 11(6), 599–615 CrossRef CAS.
- D. J. T. Zainuddin Hill, T. V. Chirila, A. K. Whittaker and A. Kemp, Experimental Calcification on HEMA-Based Hydrogels in the Presence of Albumin and a Comparison to the in Vivo Calcification, Biomacromolecules, 2006, 7(6), 1758–1765 CrossRef.
- A. Kumar and S. S. Han, PVA-Based Hydrogels for Tissue Engineering: A Review, Int. J. Polym. Mater. Polym. Biomater., 2017, 66(4), 159–182 CrossRef CAS.
- Y. Pan, D.-S. Xiong and R.-Y. Ma, A Study on the Friction Properties of Poly (Vinyl Alcohol) Hydrogel as Articular Cartilage against Titanium Alloy, Wear, 2007, 262, 1021–1025 CrossRef CAS.
- T. Miao, E. J. Miller, C. McKenzie and R. A. Oldinski, Physically Crosslinked Polyvinyl Alcohol and Gelatin Interpenetrating Polymer Network Theta-Gels for Cartilage Regeneration, J. Mater. Chem. B, 2015, 3(48), 9242–9249 RSC.
- Y. Shi, D. Xiong, Y. Liu, N. Wang and X. Zhao, Swelling, Mechanical and Friction Properties of PVA/PVP Hydrogels after Swelling in Osmotic Pressure Solution, Mater. Sci. Eng., C, 2016, 65, 172–180 CrossRef CAS.
- N. Ben Halima, Poly,(Vinyl Alcohol): Review of Its Promising Applications and Insights into Biodegradation, RSC Adv., 2016, 6(46), 39823–39832 RSC.
- R. N. Charles, J. M. Derek, M. H. Scott and S. A. Kristi, Attachment of Fibronectin to Poly(Vinyl Alcohol) Hydrogels Promotes NIH3T3 Cell Adhesion, Proliferation, and Migration, J. Biomed. Mater. Res., 2001, 57(2), 217–223 CrossRef.
- T. Konno and K. Ishihara, Temporal and Spatially Controllable Cell Encapsulation Using a Water-Soluble Phospholipid Polymer with Phenylboronic Acid Moiety, Biomaterials, 2007, 28(10), 1770–1777 CrossRef CAS.
- Y. Xu, K. Mawatari, T. Konno, T. Kitamori and K. Ishihara, Spontaneous Packaging and Hypothermic Storage of Mammalian Cells with a Cell-Membrane-Mimetic Polymer Hydrogel in a Microchip, ACS Appl. Mater. Interfaces, 2015, 7(41), 23089–23097 CrossRef CAS.
- M. A. Haq, Y. Su and D. Wang, Mechanical Properties of PNIPAM Based Hydrogels: A Review, Mater. Sci. Eng., C, 2017, 70, 842–855 CrossRef.
- M. A. Cooperstein and H. E. Canavan, Biological Cell Detachment from Poly(N-Isopropyl Acrylamide) and Its Applications, Langmuir, 2010, 26(11), 7695–7707 CrossRef CAS.
- R. Ravichandran, C. Astrand, H. K. Patra, A. P. F. Turner, V. Chotteau and J. Phopase, Intelligent ECM Mimetic Injectable Scaffolds Based on Functional Collagen Building Blocks for Tissue Engineering and Biomedical Applications, RSC Adv., 2017, 7(34), 21068–21078 RSC.
- Y. Chen, Y. Gao, L. P. Da Silva, R. P. Pirraco, M. Ma, L. Yang, R. L. Reis and J. Chen, A Thermo-/PH-Responsive Hydrogel (PNIPAM-PDMA-PAA) with Diverse Nanostructures and Gel Behaviors as a General Drug Carrier for Drug Release, Polym. Chem., 2018, 9(29), 4063–4072 RSC.
- Y. Lei and D. V. Schaffer, A Fully Defined and Scalable 3D Culture System for Human Pluripotent Stem Cell Expansion and Differentiation, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(52), E5039–E5048 CrossRef CAS.
- M. M. Adil, G. M. C. Rodrigues, R. U. Kulkarni, A. T. Rao, N. E. Chernavsky, E. W. Miller and D. V. Schaffer, Efficient Generation of HPSC-Derived Midbrain Dopaminergic Neurons in a Fully Defined, Scalable, 3D Biomaterial Platform, Sci. Rep., 2017, 7, 40573 CrossRef CAS.
- B. Guo, G. Pan, Q. Guo, C. Zhu, W. Cui, B. Li and H. Yang, Saccharides and Temperature Dual-Responsive Hydrogel Layers for Harvesting Cell Sheets, Chem. Commun., 2015, 51(4), 644–647 RSC.
- R. P. Dumitriu, A. Oprea, C. N. Cheaburu, M. Nistor, O. Novac, C. M. Ghiciuc, L. Profire and C. Vasile, Biocompatible and Biodegradable Alginate/Poly(N -Isopropylacrylamide) Hydrogels for Sustained Theophylline Release, J. Appl. Polym. Sci., 2014, 131(17), 40733 CrossRef.
- A. A. Thorpe, C. Freeman, P. Farthing, J. Callaghan, V. Paul, I. M. Brook, C. Sammon, C. Lyn and L. Maitre, In Vivo Safety and Efficacy Testing of a Thermally Triggered Injectable Hydrogel Scaffold for Bone Regeneration and Augmentation in a Rat Model, Oncotarget, 2018, 9(26), 18277–18295 CrossRef.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Cytotoxicity of Thermosensitive Polymers Poly(N-Isopropylacrylamide), Poly(N-Vinylcaprolactam) and Amphiphilically Modified Poly(N-Vinylcaprolactam), Biomaterials, 2005, 26(16), 3055–3064 CrossRef CAS.
- D. S. Jones, C. P. Lorimer, C. P. McCoy and S. P. Gorman, Characterization of the Physicochemical, Antimicrobial, and Drug Release Properties of Thermoresponsive Hydrogel Copolymers Designed for Medical Device Applications, J. Biomed. Mater. Res., Part B, 2008, 85(2), 417–426 CrossRef.
- D. S. Jones, G. P. Andrews, D. L. Caldwell, C. Lorimer, S. P. Gorman and C. P. McCoy, Novel Semi-Interpenetrating Hydrogel Networks with Enhanced Mechanical Properties and Thermoresponsive Engineered Drug Delivery, Designed as Bioactive Endotracheal Tube Biomaterials, Eur. J. Pharm. Biopharm., 2012, 82(3), 563–571 CrossRef CAS.
- J. Zhang, C. Cheng, J. L. Cuellar-Camacho, M. Li, Y. Xia, W. Li and R. Haag, Thermally Responsive Microfibers Mediated Stem Cell Fate via Reversibly Dynamic Mechanical Stimulation, Adv. Funct. Mater., 2018, 28, 1804773 CrossRef.
- M. Tonbul, M. Adas, T. Bekmezci and A. D. Kara, Intra-Articular Polyacrylamide Hydrogel Injections Are Not Innocent, Case Rep. Orthop., 2014, 2014, 150709 Search PubMed.
- A. R. Cameron, J. E. Frith and J. J. Cooper-White, The Influence of Substrate Creep on Mesenchymal Stem Cell Behaviour and Phenotype, Biomaterials, 2011, 32(26), 5979–5993 CrossRef CAS.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Highly Stretchable and Tough Hydrogels, Nature, 2012, 489(7414), 133–136 CrossRef CAS.
- G. E. Giammanco, B. Carrion, R. M. Coleman and A. D. Ostrowski, Photoresponsive Polysaccharide-Based Hydrogels with Tunable Mechanical Properties for Cartilage Tissue Engineering, ACS Appl. Mater. Interfaces, 2016, 8(23), 14423–14429 CrossRef CAS PubMed.
- J. Deng, C. Cheng, Y. Teng, C. Nie and C. Zhao, Mussel-Inspired Post-Heparinization of a Stretchable Hollow Hydrogel Tube and Its Potential Application as an Artificial Blood Vessel, Polym. Chem., 2017, 8(14), 2266–2275 RSC.
- N. Rahimi, D. G. Molin, T. J. Cleij, M. A. Van Zandvoort and M. J. Post, Electrosensitive Polyacrylic Acid/Fibrin Hydrogel Facilitates Cell Seeding and Alignment, Biomacromolecules, 2012, 13(5), 1448–1457 CrossRef CAS.
- L. E. Smith, S. Rimmer and S. MacNeil, Examination of the Effects of Poly(N-Vinylpyrrolidinone) Hydrogels in Direct and Indirect Contact with Cells, Biomaterials, 2006, 27, 2806–2812 CrossRef CAS.
- Z. Kroneková, M. Mikulec, N. Petrenčíková, E. Paulovičová, L. Paulovičová, V. Jančinová, R. Nosál, P. S. Reddy, G. D. Shimoga and D. Chorvát Jr., et al., Ex Vivo and In Vitro Studies on the Cytotoxicity and Immunomodulative Properties of Poly (2-Isopropenyl-2-Oxazoline) as a New Type of Biomedical Polymer, Macromol. Biosci., 2016, 16, 1200–1211 CrossRef.
- N. Badi, Non-Linear PEG-Based Thermoresponsive Polymer Systems, Prog. Polym. Sci., 2017, 66, 54–79 CrossRef CAS.
- K. S. Masters and K. S. Anseth, Cell-Material Interactions, in Advances in Chemical Engineering: Molecular and Cellular Foundations of Biomaterials, ed. N. A. Peppas and M. V. Sefton, Elsevier Inc., San Diego, 2004 Search PubMed.
- J. Zhu, Bioactive Modification of Poly(Ethylene Glycol) Hydrogels for Tissue Engineering, Biomaterials, 2010, 31(17), 4639–4656 CrossRef CAS.
- M. C. Schneider, S. Chu, S. L. Sridhar, G. De Roucy, F. J. Vernerey and S. J. Bryant, Local Heterogeneities Improve Matrix Connectivity in Degradable and Photoclickable Poly(Ethylene Glycol) Hydrogels for Applications in Tissue Engineering, ACS Biomater. Sci. Eng., 2017, 3(10), 2480–2492 CrossRef CAS PubMed.
- B. M. Richardson, D. G. Wilcox, M. A. Randolph and K. S. Anseth, Hydrazone Covalent Adaptable Networks Modulate Extracellular Matrix Deposition for Cartilage Tissue Engineering, Acta Biomater., 2018, 83, 71–82 CrossRef.
- C. Loebel, R. L. Mauck and J. A. Burdick, Local Nascent Protein Deposition and Remodelling Guide Mesenchymal Stromal Cell Mechanosensing and Fate in Three-Dimensional Hydrogels, Nat. Mater., 2019, 18, 883–891 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, a. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Synthetic Matrix Metalloproteinase-Sensitive Hydrogels for the Conduction of Tissue Regeneration: Engineering Cell-Invasion Characteristics, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(9), 5413–5418 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization, Adv. Mater., 2009, 21(48), 5005–5010 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank and K. Kingsbury, et al., Synthesis of Well-Defined Hydrogel Networks Using Click Chemistry, Chem. Commun., 2006, 26(26), 2774–2776 RSC.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Synthetically Tractable Click Hydrogels for Three-Dimensional Cell Culture Formed Using Tetrazine-Norbornene Chemistry, Biomacromolecules, 2013, 14(4), 949–953 CrossRef CAS.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biocompatible Hydrogels by Oxime Click Chemistry, Biomacromolecules, 2012, 13(10), 3013–3017 CrossRef CAS PubMed.
- N. Boehnke, C. Cam, E. Bat, T. Segura and H. D. Maynard, Imine Hydrogels with Tunable Degradability for Tissue Engineering, Biomacromolecules, 2015, 16(7), 2101–2108 CrossRef CAS PubMed.
- J. A. Burdick and K. S. Anseth, Photoencapsulation of Osteoblasts in Injectable RGD-Modified PEG Hydrogels for Bone Tissue Engineering, 2002, vol. 23, pp. 4315–4323 Search PubMed.
- J. C. Hoffmann and J. L. West, Three-Dimensional Photolithographic Patterning of Multiple Bioactive Ligands in Poly(Ethylene Glycol) Hydrogels, Soft Matter, 2010, 6(20), 5056–5063 RSC.
- G. Sun, X. Zhang, Y.-I. Shen, R. Sebastian, L. E. Dickinson, K. Fox-Talbot, M. Reinblatt, C. Steenbergen, J. W. Harmon and S. Gerecht, Dextran Hydrogel Scaffolds Enhance Angiogenic Responses and Promote Complete Skin Regeneration during Burn Wound Healing, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(52), 20976–20981 CrossRef CAS PubMed.
- Y. Fu, K. Xu, X. Zheng, A. J. Giacomin, A. W. Mix and W. J. Kao, 3D Cell Entrapment in Crosslinked Thiolated Gelatin-Poly(Ethylene Glycol) Diacrylate Hydrogels, Biomaterials, 2012, 33(1), 48–58 CrossRef CAS.
- J. Kim, Y. P. Kong, S. M. Niedzielski, R. K. Singh, A. J. Putnam and A. Shikanov, Characterization of the Crosslinking Kinetics of Multi-Arm Poly(Ethylene Glycol) Hydrogels Formed via Michael-Type Addition, Soft Matter, 2016, 12(7), 2076–2085 RSC.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Photoinitiated Polymerization of PEG-Diacrylate with Lithium Phenyl-2,4,6-Trimethylbenzoylphosphinate: Polymerization Rate and Cytocompatibility, Biomaterials, 2009, 30(35), 6702–6707 CrossRef CAS.
- S. Khetan and J. A. Burdick, Patterning Network Structure to Spatially Control Cellular Remodeling and Stem Cell Fate within 3-Dimensional Hydrogels, Biomaterials, 2010, 31(32), 8228–8234 CrossRef CAS.
- J. Elisseeff, K. Anseth, D. Sims, W. McIntosh, M. Randolph and R. Langer, Transdermal Photopolymerization for Minimally Invasive Implantation, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(March), 3104–3107 CrossRef CAS.
- A. Metters and J. Hubbell, Network Formation and Degradation Behavior of Hydrogels Formed by Michael-Type Addition Reactions, Biomacromolecules, 2005, 6(1), 290–301 CrossRef CAS.
- J. J. Roberts and S. J. Bryant, Comparison of Photopolymerizable Thiol-Ene PEG and Acrylate-Based PEG Hydrogels for Cartilage Development, Biomaterials, 2013, 34(38), 9969–9979 CrossRef CAS.
- K. Vats, G. Marsh, K. Harding, I. Zampetakis, R. E. Waugh and D. S. W. Benoit, Nanoscale Physicochemical Properties of Chain- and Step-Growth Polymerized PEG Hydrogels Affect Cell-Material Interactions, J. Biomed. Mater. Res., Part A, 2017, 105(4), 1112–1122 CrossRef CAS.
- B. Jeong, D. S. Lee, J. I. Shon, Y. H. Bae and S. W. Kim, Thermoreversible Gelation of Poly(Ethylene Oxide) Biodegradable Polyester Block Copolymers, J. Polym. Sci., Part A: Polym. Chem., 1999, 37(6), 751–760 CrossRef CAS.
- S. P. Zustiak and J. B. Leach, Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties, Biomacromolecules, 2010, 11, 1348–1357 CrossRef CAS.
- C. J. Chang, C. H. Chen, B. M. Chen, Y. C. Su, Y. T. Chen, M. S. Hershfield, M. T. M. Lee, T. L. Cheng, Y. T. Chen and S. R. Roffler, et al., A Genome-Wide Association Study Identifies a Novel Susceptibility Locus for the Immunogenicity of Polyethylene Glycol, Nat. Commun., 2017, 8(1), 522 CrossRef.
- B. Jeong, Y. H. Bae and S. W. Kim, Thermoreversible Gelation of PEG-PLGA-PEG Triblock Copolymer Aqueous Solutions, Macromolecules, 1999, 32(21), 7064–7069 CrossRef CAS.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Chang, Novel Thermoreversible Gelation of Biodegradable PLGA-Block-PEO-Block-PLGA Triblock Copolymers in Aqueous Solution, Macromol. Rapid Commun., 2001, 22(8), 587–592 CrossRef CAS.
- K. A. Aamer, H. Sardinha, S. R. Bhatia and G. N. Tew, Rheological Studies of PLLA-PEO-PLLA Triblock Copolymer Hydrogels, Biomaterials, 2004, 25(6), 1087–1093 CrossRef CAS.
- J. I. Kim, D. Y. Kim, D. Y. Kwon, H. J. Kang, J. H. Kim, B. H. Min and M. S. Kim, An Injectable Biodegradable Temperature-Responsive Gel with an Adjustable Persistence Window, Biomaterials, 2012, 33(10), 2823–2834 CrossRef CAS.
- M. S. Kim, H. H. Ahn, Y. N. Shin, M. H. Cho, G. Khang and H. B. Lee, An in Vivo Study of the Host Tissue Response to Subcutaneous Implantation of PLGA- and/or Porcine Small Intestinal Submucosa-Based Scaffolds, Biomaterials, 2007, 28(34), 5137–5143 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Stereocontrolled Ring-Opening Polymerisation of Lactide, Chem. Soc. Rev., 2010, 39(2), 486–494 RSC.
- V. E. G. Diederich, T. Villiger, G. Storti and M. Lattuada, Modeling of the Degradation of Poly(Ethylene Glycol)-Co-(Lactic Acid)-Dimethacrylate Hydrogels, Macromolecules, 2017, 50(14), 5527–5538 CrossRef CAS.
- A. X. Sun, H. Lin, M. R. Fritch, H. Shen, P. G. Alexander, M. DeHart and R. S. Tuan, Chondrogenesis of Human Bone Marrow Mesenchymal Stem Cells in 3-Dimensional, Photocrosslinked Hydrogel Constructs: Effect of Cell Seeding Density and Material Stiffness, Acta Biomater., 2017, 58, 302–311 CrossRef CAS.
- C. Hiemstra, Z. Zhong, P. J. Dijkstra and J. Feijen, Stereocomplex Mediated Gelation of PEG-(PLA)2 and PEG-(PLA)8 Block Copolymers, Macromol. Symp., 2005, 224, 119–131 CrossRef CAS.
- C. Hiemstra, W. Zhou, Z. Zhong, M. Wouters and J. Feijen, Rapidly in Situ Forming Biodegradable Robust Hydrogels by Combining Stereocomplexation and Photopolymerization, J. Am. Chem. Soc., 2007, 129(32), 9918–9926 CrossRef CAS.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Bioerodible Hydrogels Based on Photopolymerized Poly(Ethylene Glycol)-Co-Poly (α-Hydroxy Acid) Diacrylate Macromers, Macromolecules, 1993, 26(4), 581–587 CrossRef CAS.
- M. J. Mahoney and K. S. Anseth, Three-Dimensional Growth and Function of Neural Tissue in Degradable Polyethylene Glycol Hydrogels, Biomaterials, 2006, 27(10), 2265–2274 CrossRef CAS.
- Y. Deng, A. X. Sun, K. J. Overholt, G. Z. Yu, M. R. Fritch, P. G. Alexander, H. Shen, R. S. Tuan and H. Lin, Enhancing Chondrogenesis and Mechanical Strength Retention in Physiologically Relevant Hydrogels with Incorporation of Hyaluronic Acid and Direct Loading of TGF-β, Acta Biomater., 2018, 83, 167–176 CrossRef.
- G. L. Siparsky, Degradation Kinetics of Poly(Hydroxy) Acids: PLA and PCL, in Polymers from Renewable Resources, 2001, pp. 230–251 Search PubMed.
- S. A. Bencherif, A. Srinivasan, J. A. Sheehan, L. M. Walker, C. Gayathri, R. Gil, J. O. Hollinger, K. Matyjaszewski and N. R. Washburn, End-Group Effects on the Properties of PEG-Co-PGA Hydrogels, Acta Biomater., 2009, 5(6), 1872–1883 CrossRef CAS.
- J. L. Guo, Y. S. Kim, V. Y. Xie, B. T. Smith, E. Watson, J. Lam, H. A. Pearce, P. S. Engel and A. G. Mikos, Modular, Tissue-Specific, and Biodegradable Hydrogel Cross-Linkers for Tissue Engineering, Sci. Adv., 2019, 5(6), eaaw7396 CrossRef CAS.
- P. Destito, J. R. Couceiro, H. Faustino, F. López and J. L. Mascareñas, Ruthenium-Catalyzed Azide–Thioalkyne Cycloadditions in Aqueous Media: A Mild, Orthogonal, and Biocompatible Chemical Ligation, Angew. Chem., Int. Ed., 2017, 56(36), 10766–10770 CrossRef CAS.
- S. M. Li, H. Garreau and M. Vert, Structure-Property Relationships in the Case of the Degradation of Massive Poly-(α-Hydroxy Acids) in Aqueous Media, J. Mater. Sci. Mater. Med., 1990, 1, 123–130 CrossRef CAS.
- S. J. Bae, M. K. Joo, Y. Jeong, S. W. Kim, W. K. Lee, Y. S. Sohn and B. Jeong, Gelation Behavior of Poly(Ethylene Glycol) and Polycaprolactone Triblock and Multiblock Copolymer Aqueous Solutions, Macromolecules, 2006, 39(14), 4873–4879 CrossRef CAS.
- H. Hyun, Y. H. Kim, I. B. Song, J. W. Lee, M. S. Kim, G. Khang, K. Park and H. B. Lee, In Vitro and in Vivo Release of Albumin Using a Biodegradable MPEG-PCL Diblock Copolymer as an in Situ Gel-Forming Carrier, Biomacromolecules, 2007, 8, 1093–1100 CrossRef CAS.
- K. A. Davis, J. A. Burdick and K. S. Anseth, Photoinitiated Crosslinked Degradable Copolymer Networks for Tissue Engineering Applications, Biomaterials, 2003, 24(14), 2485–2495 CrossRef CAS.
- M. A. Rice, J. Sanchez-Adams and K. S. Anseth, Exogenously Triggered, Enzymatic Degradation of Photopolymerized Hydrogels with Polycaprolactone Subunits: Experimental Observation and Modeling of Mass Loss Behavior, Biomacromolecules, 2006, 7(6), 1968–1975 CrossRef CAS.
- K. Zhang, J. Wu, W. Zhang, S. Yan, J. Ding, X. Chen, L. Cui and J. Yin, In Situ Formation of Hydrophobic Clusters to Enhance Mechanical Performance of Biodegradable Poly(l-Glutamic Acid)/Poly(ε-Caprolactone) Hydrogel towards Meniscus Tissue Engineering, J. Mater. Chem. B, 2018, 6(47), 7822–7833 RSC.
- H. Shin, P. Quinten Ruhé, A. G. Mikos and J. A. Jansen, In Vivo Bone and Soft Tissue Response to Injectable, Biodegradable Oligo(Poly(Ethylene Glycol) Fumarate) Hydrogels, Biomaterials, 2003, 24(19), 3201–3211 CrossRef CAS.
- E. Behravesh and A. G. Mikos, Three-Dimensional Culture of Differentiating Marrow Stromal Osteoblasts in Biomimetic Poly(Propylene Fumarate-Co-Ethylene Glycol)-Based Macroporous Hydrogels, J. Biomed. Mater. Res., Part A, 2003, 66(3), 698–706 CrossRef PubMed.
- J. S. Temenoff, H. Park, E. Jabbari, T. L. Sheffield, R. G. LeBaron, C. G. Ambrose and A. G. Mikos, In Vitro Osteogenic Differentiation of Marrow Stromal Cells Encapsulated in Biodegradable Hydrogels, J. Biomed. Mater. Res., Part A, 2004, 70(2), 235–244 CrossRef.
- S. He, J. Yaszemski, M. Yasko, A. W. Engel, P. S. Mikos and A. G, Injectable Biodegradable Polymer Composites Based on Poly(Propylene Fumarate) Crosslinked with Poly(Ethylene Glycol)-Dimethacrylate, Biomaterials, 2000, 21(23), 2389–2394 CrossRef CAS.
- M. Dadsetan, J. P. Szatkowski, M. J. Yaszemski and L. Lu, Characterization of Photo-Cross-Linked Oli[Poly(Ethylene Glycol) Fumarate] Hydrogels for Cartilage Tissue Engineering, Biomacromolecules, 2007, 8(5), 1702–1709 CrossRef CAS.
- D.-A. Wang, C. G. Williams, F. Yang, N. Cher, H. Lee and J. H. Elisseeff, Bioresponsive Phosphoester Hydrogels for Bone Tissue Engineering, Tissue Eng., 2005, 11(1/2), 201–213 CrossRef CAS.
- Q. Li, J. Wang, S. Shahani, D. D. N. Sun, B. Sharma, J. H. Elisseeff and K. W. Leong, Biodegradable and Photocrosslinkable Polyphosphoester Hydrogel, Biomaterials, 2006, 27, 1027–1034 CrossRef CAS.
- T. R. Dargaville, J. R. Park and R. Hoogenboom, Poly(2-Oxazoline) Hydrogels: State-of-the-Art and Emerging Applications, Macromol. Biosci., 2018, 18(6), 1800070 CrossRef.
- M. Hartlieb, K. Kempe and U. S. Schubert, Covalently Cross-Linked Poly(2-Oxazoline) Materials for Biomedical Applications-from Hydrogels to Self-Assembled and Templated Structures, J. Mater. Chem. B, 2015, 3(4), 526–538 RSC.
- B. L. Farrugia, K. Kempe, U. S. Schubert, R. Hoogenboom and T. R. Dargaville, Poly(2-Oxazoline) Hydrogels for Controlled Fibroblast Attachment, Biomacromolecules, 2013, 14, 2724–2732 CrossRef CAS PubMed.
- S. Xuan, C. U. Lee, C. Chen, A. B. Doyle, Y. Zhang, L. Guo, V. T. John, D. Hayes and D. Zhang, Thermoreversible and Injectable ABC Polypeptoid Hydrogels: Controlling the Hydrogel Properties through Molecular Design, Chem. Mater., 2016, 28(3), 727–737 CrossRef CAS.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. Van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto and R. Hoogenboom, et al., Responsive Biomimetic Networks from Polyisocyanopeptide Hydrogels, Nature, 2013, 493(7434), 651–655 CrossRef CAS.
- M. Jaspers, A. C. H. Pape, I. K. Voets, A. E. Rowan, G. Portale and P. H. J. Kouwer, Bundle Formation in Biomimetic Hydrogels, Biomacromolecules, 2016, 17(8), 2642–2649 CrossRef CAS.
- M. Jaspers, M. Dennison, M. F. J. Mabesoone, F. C. MacKintosh, A. E. Rowan and P. H. J. Kouwer, Ultra-Responsive Soft Matter from Strain-Stiffening Hydrogels, Nat. Commun., 2014, 5, 5808 CrossRef.
- R. K. Das, V. Gocheva, R. Hammink, O. F. Zouani and A. E. Rowan, Stress-Stiffening-Mediated Stem-Cell Commitment Switch in Soft Responsive Hydrogels, Nat. Mater., 2016, 15(3), 318–325 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Synthetic Biomaterials as Instructive Extracellular Microenvironments for Morphogenesis in Tissue Engineering, Nat. Biotechnol., 2005, 23(1), 47–55 CrossRef CAS.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. Lee, E. Lippens and G. N. Duda, et al., Hydrogels with Tunable Stress Relaxation Regulate Stem Cell Fate and Activity, Nat. Mater., 2016, 15(3), 326–334 CrossRef CAS.
- J. Zhu and R. E. Marchant, Design Properties of Hydrogel Tissue-Engineering Scaffolds, Expert Rev. Med. Devices, 2011, 8(5), 607–626 CrossRef CAS.
- G. Tronci, S. J. Russell and D. J. Wood, Photo-Active Collagen Systems with Controlled Triple Helix Architecture, J. Mater. Chem. B, 2013, 1(30), 3705–3715 RSC.
- L. Chen, K. Yang, H. Zhao, A. Liu, W. Tu, C. Wu, S. Chen, Z. Guo, H. Luo and J. Sun, et al., Biomineralized Hydrogel with Enhanced Toughness by Chemical Bonding of Alkaline Phosphatase and Vinylphosphonic Acid in Collagen Framework, ACS Biomater. Sci. Eng., 2019, 5(3), 1405–1415 CrossRef CAS.
- N. Vázquez-Portalatín, C. E. Kilmer, A. Panitch and J. C. Liu, Characterization of Collagen Type I and II Blended Hydrogels for Articular Cartilage Tissue Engineering, Biomacromolecules, 2016, 17(10), 3145–3152 CrossRef.
- K. Yang, J. Sun, D. Wei, L. Yuan, J. Yang, L. Guo, H. Fan and X. Zhang, Photo-Crosslinked Mono-Component Type II Collagen Hydrogel as a Matrix to Induce Chondrogenic Differentiation of Bone Marrow Mesenchymal Stem Cells, J. Mater. Chem. B, 2017, 5(44), 8707–8718 RSC.
- R. Parenteau-Bareil, R. Gauvin and F. Berthod, Collagen-Based Biomaterials for Tissue Engineering Applications, Materials, 2010, 3, 1863–1887 CrossRef CAS.
- P. A. Parmar, L. W. Chow, J.-P. St-Pierre, C.-M. Horejs, Y. Y. Peng, J. A. Werkmeister, J. A. M. Ramshaw and M. M. Stevens, Collagen-Mimetic Peptide-Modifiable Hydrogels for Articular Cartilage Regeneration, Biomaterials, 2015, 54, 213–225 CrossRef CAS.
- P. A. Parmar, S. C. Skaalure, L. W. Chow, J.-P. St-Pierre, V. Stoichevska, Y. Y. Peng, J. A. Werkmeister, J. A. M. Ramshaw and M. M. Stevens, Temporally Degradable Collagen–Mimetic Hydrogels Tuned to Chondrogenesis of Human Mesenchymal Stem Cells, Biomaterials, 2016, 99, 56–71 CrossRef CAS PubMed.
- P. A. Parmar, J. St-Pierre, L. W. Chow, C. D. Spicer, V. Stoichevska, Y. Y. Peng, J. A. Werkmeister, J. A. M. Ramshaw and M. M. Stevens, Enhanced Articular Cartilage by Human Mesenchymal Stem Cells in Enzymatically Mediated Transiently RGDS–Functionalized Collagen–Mimetic Hydrogels, Acta Biomater., 2017, 51, 75–88 CrossRef CAS PubMed.
- Y. C. Yu, M. Tirrell and G. B. Fields, Minimal Lipidation Stabilizes Protein-like Molecular Architecture, J. Am. Chem. Soc., 1998, 120(39), 9979–9987 CrossRef CAS.
- J. Luo and Y. W. Tong, Self-Assembly of Collagen-Mimetic Peptide Amphiphiles into Biofunctional Nanofiber, ACS Nano, 2011, 5(10), 7739–7747 CrossRef CAS.
- P. Jaipan, A. Nguyen and R. J. Narayan, Gelatin-Based Hydrogels for Biomedical Applications, MRS Commun., 2017, 7(3), 416–426 CrossRef CAS.
- C. Tondera, S. Hauser, A. Krüger-Genge, F. Jung, A. T. Neffe, A. Lendlein, R. Klopfleisch, J. Steinbach, C. Neuber and J. Pietzsch, Gelatin-Based Hydrogel Degradation and Tissue Interaction in Vivo: Insights from Multimodal Preclinical Imaging in Immunocompetent Nude Mice, Theranostics, 2016, 6(12), 2114–2128 CrossRef CAS.
- J. B. Rose, S. Pacelli, A. J. El Haj, H. S. Dua, A. Hopkinson, L. J. White and F. R. A. J. Rose, Gelatin-Based Materials in Ocular Tissue Engineering, Materials, 2014, 7(4), 3106–3135 CrossRef CAS.
- W. Shi, M. Sun, X. Hu, B. Ren, J. Cheng, C. Li, X. Duan, X. Fu, J. Zhang and H. Chen, et al., Structurally and Functionally Optimized Silk-Fibroin–Gelatin Scaffold Using 3D Printing to Repair Cartilage Injury In Vitro and In Vivo, Adv. Mater., 2017, 29, 1701089 CrossRef.
- P. Angele, R. Müller, D. Schumann, C. Englert, J. Zellner, B. Johnstone, J. Yoo, J. Hammer, J. Fierlbeck and M. K. Angele, et al., Characterization of Esterified Hyaluronan-Gelatin Polymer Composites Suitable for Chondrogenic Differentiation of Mesenchymal Stem Cells, J. Biomed. Mater. Res., Part A, 2009, 91(2), 416–427 CrossRef.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Cell-Laden Microengineered Gelatin Methacrylate Hydrogels, Biomaterials, 2010, 31(21), 5536–5544 CrossRef CAS PubMed.
- N. Celikkin, S. Mastrogiacomo, J. Jaroszewicz, X. F. Walboomers and W. Swieszkowski, Gelatin Methacrylate Scaffold for Bone Tissue Engineering: The Influence of Polymer Concentration, J. Biomed. Mater. Res., Part A, 2018, 106(1), 201–209 CrossRef CAS PubMed.
- Y. Sun, R. Deng, X. Ren, K. Zhang and J. Li, 2D Gelatin Methacrylate Hydrogels with Tunable Stiffness for Investigating Cell Behaviors, ACS Appl. Bio Mater., 2019, 2(1), 570–576 CrossRef CAS.
- S. Xiao, T. Zhao, J. Wang, C. Wang, J. Du, L. Ying, J. Lin, C. Zhang, W. Hu and L. Wang, et al., Gelatin Methacrylate (GelMA) -Based Hydrogels for Cell Transplantation : An Effective Strategy for Tissue Engineering, Stem Cell Rev. Rep., 2019, 15(5), 664–679 CrossRef.
- K. Yue, G. Trujillo-de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Synthesis, Properties, and Biomedical Applications of Gelatin Methacryloyl (GelMA) Hydrogels, Biomaterials, 2015, 73, 254–271 CrossRef CAS.
- F. A. Pennacchio, C. Casale, F. Urciuolo, G. Imparato, R. Vecchione and P. A. Netti, Controlling the Orientation of a Cell-Synthesized Extracellular Matrix by Using Engineered Gelatin-Based Building Blocks, Biomater. Sci., 2018, 6(8), 2084–2091 RSC.
- N. Monteiro, W. He, C. M. Franca, A. Athirasala and L. E. Bertassoni, Engineering Microvascular Networks in LED Light-Cured Cell-Laden Hydrogels, ACS Biomater. Sci. Eng., 2018, 4(7), 2563–2570 CrossRef CAS.
- M. Zhu, Y. Wang, G. Ferracci, J. Zheng, N. J. Cho and B. H. Lee, Gelatin Methacryloyl and Its Hydrogels with an Exceptional Degree of Controllability and Batch-to-Batch Consistency, Sci. Rep., 2019, 9, 6863 CrossRef.
- S. Kapoor and S. C. Kundu, Silk Protein-Based Hydrogels: Promising Advanced Materials for Biomedical Applications, Acta Biomater., 2016, 31, 17–32 CrossRef CAS.
- K. Schacht and T. Scheibel, Controlled Hydrogel Formation of a Recombinant Spider Silk Protein, Biomacromolecules, 2011, 12(7), 2488–2495 CrossRef CAS.
- M. Kumar, P. Gupta, S. Bhattacharjee, S. K. Nandi and B. B. Mandal, Immunomodulatory Injectable Silk Hydrogels Maintaining Functional Islets and Promoting Anti-Inflammatory M2 Macrophage Polarization, Biomaterials, 2018, 187, 1–17 CrossRef CAS PubMed.
- X. Wang, B. Partlow, J. Liu, Z. Zheng, B. Su, Y. Wang and D. L. Kaplan, Injectable Silk-Polyethylene Glycol Hydrogels, Acta Biomater., 2015, 12(1), 51–61 CrossRef CAS.
- S. H. Kim, Y. K. Yeon, J. M. Lee, J. R. Chao, Y. J. Lee, Y. B. Seo, M. T. Sultan, O. J. Lee, J. S. Lee and S. I. Yoon, et al., Precisely Printable and Biocompatible Silk Fibroin Bioink for Digital Light Processing 3D Printing, Nat. Commun., 2018, 9, 1620 CrossRef PubMed.
- D. Chouhan, T. u. Lohe, P. K. Samudrala and B. B. Mandal, In Situ Forming Injectable Silk Fibroin Hydrogel Promotes Skin Regeneration in Full Thickness Burn Wounds, Adv. Healthcare Mater., 2018, 7(24), 1801092 CrossRef PubMed.
- B. Cheng, Y. Yan, J. Qi, L. Deng, Z. W. Shao, K. Q. Zhang, B. Li, Z. Sun and X. Li, Cooperative Assembly of a Peptide Gelator and Silk Fibroin Afford an Injectable Hydrogel for Tissue Engineering, ACS Appl. Mater. Interfaces, 2018, 10(15), 12474–12484 CrossRef CAS.
- R. Price, A. Poursaid, J. Cappello and H. Ghandehari, Effect of Shear on Physicochemical Properties of Matrix Metalloproteinase Responsive Silk-Elastinlike Hydrogels, J. Controlled Release, 2014, 195, 92–98 CrossRef CAS.
- K. J. Isaacson, M. M. Jensen, A. H. Watanabe, B. E. Green, M. A. Correa, J. Cappello and H. Ghandehari, Self-Assembly of Thermoresponsive Recombinant Silk-Elastinlike Nanogels, Macromol. Biosci., 2018, 18, 1700192 CrossRef.
- F. Cipriani, M. Krüger, I. G. De Torre, L. Q. Sierra, M. A. Rodrigo, L. Kock and J. C. Rodriguez-Cabello, Cartilage Regeneration in Preannealed Silk Elastin-Like Co-Recombinamers Injectable Hydrogel Embedded with Mature Chondrocytes in an Ex Vivo Culture Platform, Biomacromolecules, 2018, 19(11), 4333–4347 CrossRef CAS.
- A. E. Thurber, F. G. Omenetto and D. L. Kaplan, In Vivo Bioresponses to Silk Proteins, Biomaterials, 2015, 71, 145–157 CrossRef CAS PubMed.
- Z. Jiao, Y. Song, Y. Jin, C. Zhang, D. Peng, Z. Chen, P. Chang, S. C. Kundu, G. Wang and Z. Wang, et al., In Vivo Characterizations of the Immune Properties of Sericin: An Ancient Material with Emerging Value in Biomedical Applications, Macromol. Biosci., 2017, 17, 1700229 CrossRef.
- Z. Wang, Y. Zhang, J. Zhang, L. Huang, J. Liu, Y. Li, G. Zhang, S. C. Kundu and L. Wang, Exploring Natural Silk Protein Sericin for Regenerative Medicine: An Injectable, Photoluminescent, Cell-Adhesive 3D Hydrogel, Sci. Rep., 2014, 4, 1–11 Search PubMed.
- Y. Song, C. Zhang, J. Zhang, N. Sun, K. Huang, H. Li, Z. Wang, K. Huang and L. Wang, An Injectable Silk Sericin Hydrogel Promotes Cardiac Functional Recovery after Ischemic Myocardial Infarction, Acta Biomater., 2016, 41, 210–223 CrossRef CAS PubMed.
- D. L. Nettles, A. Chilkoti and L. A. Setton, Applications of Elastin-like Polypeptides in Tissue Engineering, Adv. Drug Delivery Rev., 2010, 62(15), 1479–1485 CrossRef CAS.
- S. Roberts, M. Dzuricky and A. Chilkoti, Elastin-like Polypeptides as Models of Intrinsically Disordered Proteins, FEBS Lett., 2015, 589(19), 2477–2486 CrossRef CAS.
- Y. N. Zhang, R. K. Avery, Q. Vallmajo-Martin, A. Assmann, A. Vegh, A. Memic, B. D. Olsen, N. Annabi and A. Khademhosseini, A Highly Elastic and Rapidly Crosslinkable Elastin-Like Polypeptide-Based Hydrogel for Biomedical Applications, Adv. Funct. Mater., 2015, 25(30), 4814–4826 CrossRef CAS.
- K. Nagapudi, W. T. Brinkman, J. E. Leisen, L. Huang, R. A. McMillan, R. P. Apkarian, V. P. Conticello and E. L. Chaikof, Photomediated Solid-State Cross-Linking of an Elastin-Mimetic Recombinant Protein Polymer, Macromolecules, 2002, 35(5), 1730–1737 CrossRef CAS.
- M. K. McHale, L. A. Setton and A. Chilkoti, Synthesis and in Vitro Evaluation of Enzymatically Cross-Linked Elastin-like Polypeptide Gels for Cartilaginous Tissue Repair, Tissue Eng., 2005, 11(11/12), 1768–1779 CrossRef CAS.
- H. Wang, A. Paul, D. Nguyen, A. Enejder and S. C. Heilshorn, Tunable Control of Hydrogel Microstructure by Kinetic Competition between Self-Assembly and Crosslinking of Elastin-like Proteins, ACS Appl. Mater. Interfaces, 2018, 10, 21808–21815 CrossRef CAS.
- K. S. Straley and S. C. Heilshorn, Independent Tuning of Multiple Biomaterial Properties Using Protein Engineering, Soft Matter, 2009, 5(1), 114–124 RSC.
- K. J. Lampe, A. L. Antaris and S. C. Heilshorn, Design of Three-Dimensional Engineered Protein Hydrogels for Tailored Control of Neurite Growth, Acta Biomater., 2013, 9(3), 5590–5599 CrossRef CAS.
- X. X. Xia, Q. Xu, X. Hu, G. Qin and D. L. Kaplan, Tunable Self-Assembly of Genetically Engineered Silk-Elastin-like Protein Polymers, Biomacromolecules, 2011, 12(11), 3844–3850 CrossRef CAS.
- M. Keating, M. Lim, Q. Hu and E. Botvinick, Selective Stiffening of Fibrin Hydrogels with Micron Resolution via Photocrosslinking, Acta Biomater., 2019, 87, 88–96 CrossRef CAS.
- B.-J. Kang, H.-H. Ryu, S.-S. Park, Y. Kim, H.-M. Woo, W. H. Kim and O.-K. Kweon, Effect of Matrigel on the Osteogenic Potential of Canine Adipose Tissue-Derived Mesenchymal Stem Cells, J. Vet. Med. Sci., 2012, 74(7), 827–836 CrossRef CAS.
- S. Koutsopoulos and S. Zhang, Long-Term Three-Dimensional Neural Tissue Cultures in Functionalized Self-Assembling Peptide Hydrogels, Matrigel and Collagen I, Acta Biomater., 2013, 9(2), 5162–5169 CrossRef CAS.
- M. J. Hernandez, R. Gaetani, V. M. Pieters, N. W. Ng, A. E. Chang, T. R. Martin, E. van Ingen, E. A. Mol, M. Dzieciatkowska and K. C. Hansen, et al., Decellularized Extracellular Matrix Hydrogels as a Delivery Platform for MicroRNA and Extracellular Vesicle Therapeutics, Adv. Ther., 2018, 1(3), 1800032 CrossRef.
- Y. S. Kim, M. Majid, A. J. Melchiorri and A. G. Mikos, Applications of Decellularized Extracellular Matrix in Bone and Cartilage Tissue Engineering, Bioeng. Transl. Med., 2019, 4(1), 83–95 CrossRef PubMed.
- M. J. Majcher and T. Hoare, Hydrogel Synthesis and Design, in Functional Biopolymers, ed. M. A. Jafar Mazumder, H. Sheardown and A. Al-Ahmed, Springer International Publishing, Cham, 2018, pp. 1–41 Search PubMed.
- J. Radhakrishnan, A. Subramanian, U. M. Krishnan and S. Sethuraman, Injectable and 3D Bioprinted Polysaccharide Hydrogels: From Cartilage to Osteochondral Tissue Engineering, Biomacromolecules, 2017, 18(1), 1–26 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Alginate: Properties and Biomedical Applications, Prog. Polym. Sci., 2012, 37(1), 106–126 CrossRef CAS.
- R. Russo, M. Malinconico and G. Santagata, Effect of Cross-Linking with Calcium Ions on the Physical Properties of Alginate Films, Biomacromolecules, 2007, 8(10), 3193–3197 CrossRef CAS.
- G. T. Grant, E. R. Morris, D. A. Rees, P. J. C. Smith and D. Thom, Biological Interactions between Polysaccharides and Divalent Cations: The Egg-Box Model, FEBS Lett., 1973, 32(1), 195–198 CrossRef CAS.
- T. Boland, X. Tao, B. J. Damon, B. Manley, P. Kesari, S. Jalota and S. Bhaduri, Drop-on-Demand Printing of Cells and Materials for Designer Tissue Constructs, Mater. Sci. Eng., C, 2007, 27(3), 372–376 CrossRef CAS.
- J. Leppiniemi, P. Lahtinen, A. Paajanen, R. Mahlberg, S. Metsä-Kortelainen, T. Pinomaa, H. Pajari, I. Vikholm-Lundin, P. Pursula and V. P. Hytönen, 3D-Printable Bioactivated Nanocellulose-Alginate Hydrogels, ACS Appl. Mater. Interfaces, 2017, 9(26), 21959–21970 CrossRef CAS.
- F. E. Freeman and D. J. Kelly, Tuning Alginate Bioink Stiffness and Composition for Controlled Growth Factor Delivery and to Spatially Direct MSC Fate within Bioprinted Tissues, Sci. Rep., 2017, 7, 17042 CrossRef.
- L. Jia, F. Han, H. Yang, G. Turnbull, J. Wang, J. Clarke, W. Shu, M. Guo and B. Li, Microfluidic Fabrication of Biomimetic Helical Hydrogel Microfibers for Blood-Vessel-on-a-Chip Applications, Adv. Healthcare Mater., 2019, 8, 1900435 CrossRef.
- G. Kalaf, E. A. Flores, R. Bledsoe, J. G. Sell and S. A, Characterization of Slow-Gelling Alginate Hydrogels for Intervertebral Disc Tissue-Engineering Applications, Mater. Sci. Eng., C, 2016, 63, 198–210 CrossRef.
- O. Smidsrød and G. Skjåk-Bræk, Alginate as Immobilization Matrix for Cells, Trends Biotechnol., 1990, 8, 71–78 CrossRef.
- E. S. Place, L. Rojo, E. Gentleman, J. P. Sardinha and M. M. Stevens, Strontium- and Zinc-Alginate Hydrogels for Bone Tissue Engineering, Tissue Eng., Part A, 2011, 17(21–22), 2713–2722 CrossRef CAS PubMed.
- Ý. A Mørch, I. Donati, B. L. Strand and G. Skjåk-Bræk, Effect of Ca2+, Ba2+, and Sr2+ on Alginate Microbeads, Biomacromolecules, 2006, 7(5), 1471–1480 CrossRef.
- P. Gaetani, M. L. Torre, M. Klinger, M. Faustini, F. Crovato, M. Bucco, M. Marazzi, T. Chlapanidas, D. Levi and F. Tancioni, et al., Adipose-Derived Stem Cell Therapy for Intervertebral Disc Regeneration: An In Vitro Reconstructed Tissue in Alginate Capsules, Tissue Eng., Part A, 2008, 14(8), 1415–1423 CrossRef CAS.
- K. H. Bouhadir, K. Y. Lee, E. Alsberg, K. L. Damm, K. W. Anderson and D. J. Mooney, Degradation of Partially Oxidized Alginate and Its Potential Application for Tissue Engineering, Biotechnol. Prog., 2001, 17(5), 945–950 CrossRef CAS.
- C. S. D. Lee, H. R. Moyer, I. R. A. Gittens, J. K. Williams, A. L. Boskey, B. D. Boyan and Z. Schwartz, Regulating in Vivo Calcification of Alginate Microbeads, Biomaterials, 2010, 31(18), 4926–4934 CrossRef CAS PubMed.
- S.-W. Kang, B.-H. Cha, H. Park, K.-S. Park, K. Y. Lee and S.-H. Lee, The Effect of Conjugating RGD into 3D Alginate Hydrogels on Adipogenic Differentiation of Human Adipose-Derived Stromal Cells, Macromol. Biosci., 2011, 11(5), 673–679 CrossRef CAS.
- C. Lee, J. Shin, J. S. Lee, E. Byun, J. H. Ryu, S. H. Um, D.-I. Kim, H. Lee and S.-W. Cho, Bioinspired, Calcium-Free Alginate Hydrogels with Tunable Physical and Mechanical Properties and Improved Biocompatibility, Biomacromolecules, 2013, 14(6), 2004–2013 CrossRef CAS PubMed.
- N. C. Hunt, D. Hallam, A. Karimi, C. B. Mellough, J. Chen, D. H. W. Steel and M. Lako, 3D Culture of Human Pluripotent Stem Cells in RGD-Alginate Hydrogel Improves Retinal Tissue Development, Acta Biomater., 2017, 49, 329–343 CrossRef CAS.
- T. Freier, H. S. Koh, K. Kazazian and M. S. Shoichet, Controlling Cell Adhesion and Degradation of Chitosan Films by N-Acetylation, Biomaterials, 2005, 26(29), 5872–5878 CrossRef CAS PubMed.
- E. I. Rabea, M. E.-T. Badawy, C. V. Stevens, G. Smagghe and W. Steurbaut, Chitosan as Antimicrobial Agent: Applications and Mode of Action, Biomacromolecules, 2003, 4(6), 1457–1465 CrossRef CAS.
- H. Tan, C. R. Chu, K. A. Payne and K. G. Marra, Injectable in Situ Forming Biodegradable Chitosan-Hyaluronic Acid Based Hydrogels for Cartilage Tissue Engineering, Biomaterials, 2009, 30(13), 2499–2506 CrossRef CAS PubMed.
- Y. Deng, J. Ren, G. Chen, G. Li, X. Wu, G. Wang, G. Gu and J. Li, Injectable in Situ Cross-Linking Chitosan-Hyaluronic Acid Based Hydrogels for Abdominal Tissue Regeneration, Sci. Rep., 2017, 7, 2699 CrossRef PubMed.
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Novel Injectable Neutral Solutions of Chitosan Form Biodegradable Gels in Situ, Biomaterials, 2000, 21(21), 2155–2161 CrossRef CAS PubMed.
- K. Kim, J. H. Ryu, D. Y. Lee and H. Lee, Bio-Inspired Catechol Conjugation Converts Water-Insoluble Chitosan into a Highly Water-Soluble, Adhesive Chitosan Derivative for Hydrogels and LbL Assembly, Biomater. Sci., 2013, 1(7), 783–790 RSC.
- R. Jin, C. Lin and A. Cao, Enzyme-Mediated Fast Injectable Hydrogels Based on Chitosan-Glycolic Acid/Tyrosine: Preparation, Characterization, and Chondrocyte Culture, Polym. Chem., 2014, 5(2), 391–398 RSC.
- B. d. Silva, S. Krolicka, M. van den Broek, L. A. M. Frissen, A. E. Boeriu and C. G, Water-Soluble Chitosan Derivatives and PH-Responsive Hydrogels by Selective C-6 Oxidation Mediated by TEMPO-Laccase Redox System, Carbohydr. Polym., 2018, 186, 299–309 CrossRef PubMed.
- B. Fonseca-Santos and M. Chorilli, An Overview of Carboxymethyl Derivatives of Chitosan: Their Use as Biomaterials and Drug Delivery Systems, Mater. Sci. Eng., C, 2017, 77, 1349–1362 CrossRef CAS PubMed.
- W. E. G. Müller, E. Tolba, H. C. Schröder, M. Neufurth, S. Wang, T. Link, B. Al-Nawas and X. Wang, A New Printable and Durable N,O-Carboxymethyl Chitosan-Ca2+-Polyphosphate Complex with Morphogenetic Activity, J. Mater. Chem. B, 2015, 3(8), 1722–1730 RSC.
- C. Zhao, N. T. Qazvini, M. Sadati, Z. Zeng, S. Huang, A. L. De La Lastra, L. Zhang, Y. Feng, W. Liu and B. Huang, et al., A PH-Triggered, Self-Assembled, and Bioprintable Hybrid Hydrogel Scaffold for Mesenchymal Stem Cell Based Bone Tissue Engineering, ACS Appl. Mater. Interfaces, 2019, 11(9), 8749–8762 CrossRef CAS PubMed.
- X. Xu, A. K. Jha, D. A. Harrington, M. C. Farach-Carson and X. Jia, Hyaluronic Acid-Based Hydrogels: From a Natural Polysaccharide to Complex Networks, Soft Matter, 2012, 8(12), 3280–3294 RSC.
- Y. Luo, K. R. Kirker and G. D. Prestwich, Cross-Linked Hyaluronic Acid Hydrogel Films: New Biomaterials for Drug Delivery, J. Controlled Release, 2000, 69(1), 169–184 CrossRef CAS PubMed.
- C. B. Highley, G. D. Prestwich and J. A. Burdick, Recent Advances in Hyaluronic Acid Hydrogels for Biomedical Applications, Curr. Opin. Biotechnol., 2016, 40, 35–40 CrossRef CAS PubMed.
- J. Lam, N. F. Truong and T. Segura, Design of Cell-Matrix Interactions in Hyaluronic Acid Hydrogel Scaffolds, Acta Biomater., 2014, 10(4), 1571–1580 CrossRef CAS PubMed.
- J. A. Burdick, C. Chung, X. Jia, M. A. Randolph and R. Langer, Controlled Degradation and Mechanical Behavior of Photopolymerized Hyaluronic Acid Networks, Biomacromolecules, 2005, 6(1), 386–391 CrossRef CAS PubMed.
- J. Patterson, R. Siew, S. W. Herring, A. S. P. Lin, R. Guldberg and P. S. Stayton, Hyaluronic Acid Hydrogels with Controlled Degradation Properties for Oriented Bone Regeneration, Biomaterials, 2010, 31(26), 6772–6781 CrossRef CAS PubMed.
- S. S. Han, H. Y. Yoon, J. Y. Yhee, M. O. Cho, H. E. Shim, J. E. Jeong, D. E. Lee, K. Kim, H. Guim and J. H. Lee, et al., In Situ Cross-Linkable Hyaluronic Acid Hydrogels Using Copper Free Click Chemistry for Cartilage Tissue Engineering, Polym. Chem., 2018, 9(1), 20–27 RSC.
- Z. Guo, S. Mi and W. Sun, The Multifaceted Nature of Catechol Chemistry: Bioinspired PH-Initiated Hyaluronic Acid Hydrogels with Tunable Cohesive and Adhesive Properties, J. Mater. Chem. B, 2018, 6(39), 6234–6244 RSC.
- S. H. Park, J. Y. Seo, J. Y. Park, Y. B. Ji, K. Kim, H. S. Choi, S. Choi, J. H. Kim, B. H. Min and M. S. Kim, An Injectable, Click-Crosslinked, Cytomodulin-Modified Hyaluronic Acid Hydrogel for Cartilage Tissue Engineering, NPG Asia Mater., 2019, 11, 30 CrossRef.
- L. Ouyang, C. B. Highley, C. B. Rodell, W. Sun and J. A. Burdick, 3D Printing of Shear-Thinning Hyaluronic Acid Hydrogels with Secondary Cross-Linking, ACS Biomater. Sci. Eng., 2016, 2(10), 1743–1751 CrossRef CAS.
- A. N. Steele, L. M. Stapleton, J. M. Farry, H. J. Lucian, M. J. Paulsen, A. Eskandari, C. E. Hironaka, A. D. Thakore, H. Wang and A. C. Yu, et al., A Biocompatible Therapeutic Catheter-Deliverable Hydrogel for In Situ Tissue Engineering, Adv. Healthcare Mater., 2019, 8, 1801147 CrossRef PubMed.
- Y. Alinejad, A. Adoungotchodo, E. Hui, F. Zehtabi and S. Lerouge, An Injectable Chitosan/Chondroitin Sulfate Hydrogel with Tunable Mechanical Properties for Cell Therapy/Tissue Engineering, Int. J. Biol. Macromol., 2018, 113, 132–141 CrossRef CAS PubMed.
- T. Miller, M. C. Goude, T. C. Mcdevitt and J. S. Temenoff, Molecular Engineering of Glycosaminoglycan Chemistry for Biomolecule Delivery, Acta Biomater., 2014, 10(4), 1705–1719 CrossRef CAS PubMed.
- S. Jo, S. Kim and I. Noh, Synthesis of in Situ Chondroitin Sulfate Hydrogel through Phosphine-Mediated Michael Type Addition Reaction, Macromol. Res., 2012, 20(9), 968–976 CrossRef CAS.
- F. Ma, X. Pang and B. Tang, Alginate/Chondroitin Sulfate Based Hybrid Hydrogel with Different Molecular Weight and Its Capacity to Regulate Chondrocytes Activity, Carbohydr. Polym., 2019, 206, 229–237 CrossRef CAS PubMed.
- K. J. Ornell, D. Lozada, N. V. Phan and J. M. Coburn, Controlling Methacryloyl Substitution of Chondroitin Sulfate: Injectable Hydrogels with Tunable Long-Term Drug Release Profiles, J. Mater. Chem. B, 2019, 7(13), 2151–2161 RSC.
- M. Zhu, Q. Feng, Y. Sun, G. Li and L. Bian, Effect of Cartilaginous Matrix Components on the Chondrogenesis and Hypertrophy of Mesenchymal Stem Cells in Hyaluronic Acid Hydrogels, J. Biomed. Mater. Res., Part B, 2017, 105(8), 2292–2300 CrossRef CAS PubMed.
- E. A. Aisenbrey and S. J. Bryant, The Role of Chondroitin Sulfate in Regulating Hypertrophy during MSC Chondrogenesis in a Cartilage Mimetic Hydrogel under Dynamic Loading, Biomaterials, 2019, 190–191, 51–62 CrossRef CAS PubMed.
- H. D. Kim, E. A. Lee, Y. H. An, S. L. Kim, S. S. Lee, S. J. Yu, H. L. Jang, K. T. Nam, S. G. Im and N. S. Hwang, Chondroitin Sulfate-Based Biomineralizing Surface Hydrogels for Bone Tissue Engineering, ACS Appl. Mater. Interfaces, 2017, 9(26), 21639–21650 CrossRef CAS PubMed.
- G. Sun, Y. I. Shen, C. C. Ho, S. Kusuma and S. Gerecht, Functional Groups Affect Physical and Biological Properties of Dextran-Based Hydrogels, J. Biomed. Mater. Res., Part A, 2010, 93(3), 1080–1090 Search PubMed.
- N. Contessi, L. Altomare, A. Filipponi and S. Farè, Thermo-Responsive Properties of Methylcellulose Hydrogels for Cell Sheet Engineering, Mater. Lett., 2017, 207, 157–160 CrossRef CAS.
- T. Ahlfeld, T. Köhler, C. Czichy, A. Lode and M. Gelinsky, A Methylcellulose Hydrogel as Support for 3D Plotting of Complex Shaped Calcium Phosphate Scaffolds, Gels, 2018, 4(3), 68 CrossRef CAS PubMed.
- A. Cochis, S. Grad, M. J. Stoddart, S. Farè, L. Altomare, B. Azzimonti, M. Alini and L. Rimondini, Bioreactor Mechanically Guided 3D Mesenchymal Stem Cell Chondrogenesis Using a Biocompatible Novel Thermo-Reversible Methylcellulose-Based Hydrogel, Sci. Rep., 2017, 7, 45018 CrossRef CAS PubMed.
- V. W. Wong, K. C. Rustad, M. G. Galvez, E. Neofytou, J. P. Glotzbach, M. Januszyk, M. R. Major, M. Sorkin, M. T. Longaker and J. Rajadas, et al., Engineered Pullulan–Collagen Composite Dermal Hydrogels Improve Early Cutaneous Wound Healing, Tissue Eng., Part A, 2011, 17, 631–644 CrossRef CAS PubMed.
- S. Iswariya, A. V. Bhanukeerthi, P. Velswamy, T. S. Uma and P. T. Perumal, Design and Development of a Piscine Collagen Blended Pullulan Hydrogel for Skin Tissue Engineering, RSC Adv., 2016, 6(63), 57863–57871 RSC.
- R. Pugliese and F. Gelain, Peptidic Biomaterials: From Self-Assembling to Regenerative Medicine, Trends Biotechnol., 2017, 35(2), 145–158 CrossRef CAS PubMed.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Enabling Applications of Covalent Adaptable Networks, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- P. R. A. Chivers and D. K. Smith, Shaping and Structuring Supramolecular Gels, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- D. J. Cornwell and D. K. Smith, Expanding the Scope of Gels - Combining Polymers with Low-Molecular-Weight Gelators to Yield Modified Self-Assembling Smart Materials with High-Tech Applications, Mater. Horiz., 2015, 2(3), 279–293 RSC.
- H. Yokoi, T. Kinoshita and S. Zhang, Dynamic Reassembly of Peptide RADA16 Nanofiber Scaffold, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(24), 8414–8419 CrossRef CAS PubMed.
- J. D. Tang, C. Mura and K. J. Lampe, Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering, J. Am. Chem. Soc., 2019, 141(12), 4886–4899 CrossRef CAS PubMed.
- V. M. P. Vieira, A. C. Lima, M. de Jong and D. K. Smith, Commercially Relevant Orthogonal Multi-Component Supramolecular Hydrogels for Programmed Cell Growth, Chem. – Eur. J., 2018, 24(56), 15112–15118 CrossRef CAS PubMed.
- R. G. Ellis-Behnke, Y.-X. Liang, S.-W. You, D. K. C. Tay, S. Zhang, K.-F. So and G. E. Schneider, Nano Neuro Knitting: Peptide Nanofiber Scaffold for Brain Repair and Axon Regeneration with Functional Return of Vision, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(13), 5054–5059 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler, S. I. Stupp, G. A. Silva, C. Czeisler and K. L. Niece, et al., Selective Differentiation of Neural Progenitor Cells by High – Epitope Density Nanofibers, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- H. Hosseinkhani, M. Hosseinkhani, A. Khademhosseini, H. Kobayashi and Y. Tabata, Enhanced Angiogenesis through Controlled Release of Basic Fibroblast Growth Factor from Peptide Amphiphile for Tissue Regeneration, Biomaterials, 2006, 27(34), 5836–5844 CrossRef CAS PubMed.
- E. T. Pashuck, Designing Self-Assembling Biomaterials with Controlled Mechanical and Biological Performance, in Self-Assembling Biomaterials - Molecular Design, Characterization and Application in Biology and Medicine, ed. H. S. Azevedo and R. M. P. da Silva, Woodhead Publishing, 2018, pp. 7–26 Search PubMed.
- R. Pugliese and F. Gelain, Peptidic Biomaterials: From Self-Assembling to Regenerative Medicine, Trends Biotechnol., 2017, 35(2), 145–158 CrossRef CAS PubMed.
- W. L. A. Brooks and B. S. Sumerlin, Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine, Chem. Rev., 2016, 116(3), 1375–1397 CrossRef CAS PubMed.
- J. P. M. António, R. Russo, C. P. Carvalho, P. M. S. D. Cal and P. M. P. Gois, Boronic Acids as Building Blocks for the Construction of Therapeutically Useful Bioconjugates, Chem. Soc. Rev., 2019, 48, 3513–3536 RSC.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Injectable Self-Healing Glucose-Responsive Hydrogels with PH-Regulated Mechanical Properties, Adv. Mater., 2016, 28(1), 86–91 CrossRef CAS PubMed.
- Y. Chen, D. Diaz-Dussan, D. Wu, W. Wang, Y. Y. Peng, A. B. Asha, D. G. Hall, K. Ishihara and R. Narain, Bioinspired Self-Healing Hydrogel Based on Benzoxaborole-Catechol Dynamic Covalent Chemistry for 3D Cell Encapsulation, ACS Macro Lett., 2018, 7(8), 904–908 CrossRef CAS.
- Y. Chen, W. Wang, D. Wu, M. Nagao, D. G. Hall, T. Thundat and R. Narain, Injectable Self-Healing Zwitterionic Hydrogels Based on Dynamic Benzoxaborole-Sugar Interactions with Tunable Mechanical Properties, Biomacromolecules, 2018, 19(2), 596–605 CrossRef CAS PubMed.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, Supramolecular Cross-Linked Networks via Host - Guest Complexation with Cucurbit[8]Uril, J. Am. Chem. Soc., 2010, 132(16), 14251–14260 CrossRef CAS PubMed.
- M. J. Rowland, E. A. Appel, R. J. Coulston and O. A. Scherman, Dynamically Crosslinked Materials via Recognition of Amino Acids by Cucurbit[8]Uril, J. Mater. Chem. B, 2013, 1(23), 2904–2910 RSC.
- J. Boekhoven, C. M. Rubert Pérez, S. Sur, A. Worthy and S. I. Stupp, Dynamic Display of Bioactivity through Host-Guest Chemistry, Angew. Chem., Int. Ed., 2013, 52(46), 12077–12080 CrossRef CAS PubMed.
- M. J. Rowland, C. C. Parkins, J. H. McAbee, A. K. Kolb, R. Hein, X. J. Loh, C. Watts and O. A. Scherman, An Adherent Tissue-Inspired Hydrogel Delivery Vehicle Utilised in Primary Human Glioma Models, Biomaterials, 2018, 179, 199–208 CrossRef CAS PubMed.
- E. Bakaic, N. M. B. Smeets, H. Dorrington and T. Hoare, “Off-the-Shelf” Thermoresponsive Hydrogel Design: Tuning Hydrogel Properties by Mixing Precursor Polymers with Different Lower-Critical Solution Temperatures, RSC Adv., 2015, 5(42), 33364–33376 RSC.
- D. Y. Ko, M. Patel, H. J. Lee and B. Jeong, Coordinating Thermogel for Stem Cell Spheroids and Their Cyto-Effectiveness, Adv. Funct. Mater., 2018, 28(7), 1706286 CrossRef.
- K. J. De France, K. G. Yager, K. J. W. Chan, B. Corbett, E. D. Cranston and T. Hoare, Injectable Anisotropic Nanocomposite Hydrogels Direct in Situ Growth and Alignment of Myotubes, Nano Lett., 2017, 17(10), 6487–6495 CrossRef CAS PubMed.
- K. J. De France, M. Badv, J. Dorogin, E. Siebers, V. Panchal, M. Babi, J. Moran-Mirabal, M. Lawlor, E. D. Cranston and T. Hoare, Tissue Response and Biodistribution of Injectable Cellulose Nanocrystal Composite Hydrogels, ACS Biomater. Sci. Eng., 2019, 5(5), 2235–2246 CrossRef CAS.
- E. Bakaic, N. M. B. Smeets, M. Badv, M. Dodd, O. Barrigar, E. Siebers, M. Lawlor, H. Sheardown and T. Hoare, Injectable and Degradable Poly(Oligoethylene Glycol Methacrylate) Hydrogels with Tunable Charge Densities as Adhesive Peptide-Free Cell Scaffolds, ACS Biomater. Sci. Eng., 2018, 4(11), 3713–3725 CrossRef CAS.
- L. Wang and J. P. Stegemann, Thermogelling Chitosan and Collagen Composite Hydrogels Initiated with β-Glycerophosphate for Bone Tissue Engineering, Biomaterials, 2010, 31(14), 3976–3985 CrossRef CAS PubMed.
- C.-Z. Wei, C.-L. Hou, Q.-S. Gu, L.-X. Jiang, B. Zhu and A.-L. Sheng, A Thermosensitive Chitosan-Based Hydrogel Barrier for Post-Operative Adhesions’ Prevention, Biomaterials, 2009, 30(29), 5534–5540 CrossRef CAS PubMed.
- E. Zhang, J. Li, Y. Zhou, P. Che, B. Ren, Z. Qin, L. Ma, J. Cui, H. Sun and F. Yao, Biodegradable and Injectable Thermoreversible Xyloglucan Based Hydrogel for Prevention of Postoperative Adhesion, Acta Biomater., 2017, 55, 420–433 CrossRef CAS PubMed.
- E. Zhang, Q. Guo, F. Ji, X. Tian, J. Cui, Y. Song, H. Sun, J. Li and F. Yao, Thermoresponsive Polysaccharide-Based Composite Hydrogel with Antibacterial and Healing-Promoting Activities for Preventing Recurrent Adhesion after Adhesiolysis, Acta Biomater., 2018, 74, 439–453 CrossRef CAS PubMed.
- Y. K. Jhon, R. R. Bhat, C. Jeong, O. J. Rojas, I. Szleifer and J. Genzer, Salt-Induced Depression of Lower Critical Solution Temperature in a Surface-Grafted Neutral Thermoresponsive Polymer, Macromol. Rapid Commun., 2006, 27(9), 697–701 CrossRef CAS.
- S. Ahn, E. C. Monge and S. C. Song, Ion and PH Effect on the Lower Critical Solution Temperature Phase Behavior in Neutral and Acidic Poly(Organophosphazene) Counterparts, Langmuir, 2009, 25(4), 2407–2418 CrossRef CAS PubMed.
- R. Y. Tam, L. J. Smith and M. S. Shoichet, Engineering Cellular Microenvironments with Photo- and Enzymatically Responsive Hydrogels: Toward Biomimetic 3D Cell Culture Models, Acc. Chem. Res., 2017, 50(4), 703–713 CrossRef CAS PubMed.
- M. Lunzer, L. Shi, O. G. Andriotis, P. Gruber, M. Markovic, P. J. Thurner, D. Ossipov, R. Liska and A. Ovsianikov, A Modular Approach to Sensitized Two-Photon Patterning of Photodegradable Hydrogels, Angew. Chem., Int. Ed., 2018, 57(46), 15122–15127 CrossRef CAS PubMed.
- A. M. Rosales, S. L. Vega, F. W. DelRio, J. A. Burdick and K. S. Anseth, Hydrogels with Reversible Mechanics to Probe Dynamic Cell Microenvironments, Angew. Chem., Int. Ed., 2017, 56(40), 12132–12136 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, Wavelength-Controlled Photocleavage for the Orthogonal and Sequential Release of Multiple Proteins, Angew. Chem., Int. Ed., 2013, 52(51), 13803–13807 CrossRef CAS PubMed.
- H. Y. Yoshikawa, F. F. Rossetti, S. Kaufmann, T. Kaindl, J. Madsen, U. Engel, A. L. Lewis, S. P. Armes and M. Tanaka, Quantitative Evaluation of Mechanosensing of Cells on Dynamically Tunable Hydrogels, J. Am. Chem. Soc., 2011, 133, 1367–1374 CrossRef CAS PubMed.
- S. J. Bae, J. M. Suh, Y. S. Sohn, Y. H. Bae, S. W. Kim and B. Jeong, Thermogelling Poly(Caprolactone-b-Ethylene Glycol-b-Caprolactone) Aqueous Solutions, Macromolecules, 2005, 38(12), 5260–5265 CrossRef CAS.
- S. U. J. Lee, B. O. R. Han, S. Y. Park, D. K. Han and S. C. Kim, Sol-Gel Transition Behavior of Biodegradable Three-Arm and Four-Arm Star-Shaped PLGA-PEG Block Copolymer Aqueous Solution, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(2), 888–899 CrossRef CAS.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biodegradable Hyperbranched Amphiphilic Polyurethane Multiblock Copolymers Consisting of Poly(Propylene Glycol), Poly(Ethylene Glycol), and Polycaprolactone as in Situ Thermogels, Biomacromolecules, 2012, 13(12), 3977–3989 CrossRef CAS PubMed.
- Z. Li and J. Li, Control of Hyperbranched Structure of Polycaprolactone/Poly(Ethylene Glycol) Polyurethane Block Copolymers by Glycerol and Their Hydrogels for Potential Cell Delivery, J. Phys. Chem. B, 2013, 117(47), 14763–14774 CrossRef CAS PubMed.
- B. Jeong, K. M. Lee, A. Gutowska and Y. H. An, Thermogelling Biodegradable Copolymer Aqueous Solutions for Injectable Protein Delivery and Tissue Engineering, Biomacromolecules, 2002, 3(4), 865–868 CrossRef CAS PubMed.
- A. T. Metters, C. N. Bowman and K. S. Anseth, A Statistical Kinetic Model for the Bulk Degradation of PLA-b-PEG-b-PLA Hydrogel Networks, J. Phys. Chem. B, 2002, 104(30), 7043–7049 CrossRef.
- A. Metters and J. Hubbell, Network Formation and Degradation Behavior of Hydrogels Formed by Michael-Type Addition Reactions, Biomacromolecules, 2005, 290–301 CrossRef CAS PubMed.
- J. Wang, F. Zhang, W. P. Tsang, C. Wan and C. Wu, Fabrication of Injectable High Strength Hydrogel Based on 4-Arm Star PEG for Cartilage Tissue Engineering, Biomaterials, 2017, 120, 11–21 CrossRef CAS PubMed.
- K. Kawamoto, M. Zhong, R. Wang, B. D. Olsen and J. A. Johnson, Loops versus Branch Functionality in Model Click Hydrogels, Macromolecules, 2015, 48, 8980–8988 CrossRef CAS.
- H. Zhou, E. M. Schön, M. Wang, M. J. Glassman, J. Liu, M. Zhong, D. Díaz Díaz, B. D. Olsen and J. A. Johnson, Crossover Experiments Applied to Network Formation Reactions: Improved Strategies for Counting Elastically Inactive Molecular Defects in PEG Gels and Hyperbranched Polymers, J. Am. Chem. Soc., 2014, 136(26), 9464–9470 CrossRef CAS PubMed.
- R. Wang, J. A. Johnson and B. D. Olsen, Odd-Even Effect of Junction Functionality on the Topology and Elasticity of Polymer Networks, Macromolecules, 2017, 50(6), 2556–2564 CrossRef CAS.
- S. H. M. Söntjens, D. L. Nettles, M. A. Carnahan, L. A. Setton and M. W. Grinstaff, Biodendrimer-Based Hydrogel Scaffolds for Cartilage Tissue Repair, Biomacromolecules, 2006, 7(1), 310–316 CrossRef PubMed.
- Y. Wang, Q. Zhao, H. Zhang, S. Yang and X. Jia, A Novel Poly(Amido Amine)-Dendrimer-Based Hydrogel as a Mimic for the Extracellular Matrix, Adv. Mater., 2014, 26(24), 4163–4167 CrossRef CAS PubMed.
- C. Ghobril, E. K. Rodriguez, A. Nazarian and M. W. Grinstaff, Recent Advances in Dendritic Macromonomers for Hydrogel Formation and Their Medical Applications, Biomacromolecules, 2016, 17(4), 1235–1252 CrossRef CAS PubMed.
- J. Wang, H. He, R. C. Cooper and H. Yang, In Situ-Forming Polyamidoamine Dendrimer Hydrogels with Tunable Properties Prepared via Aza-Michael Addition Reaction, ACS Appl. Mater. Interfaces, 2017, 9(12), 10494–10503 CrossRef CAS PubMed.
- S. M. Hodgson, S. A. McNelles, L. Abdullahu, I. A. Marozas, K. S. Anseth and A. Adronov, Reproducible Dendronized PEG Hydrogels via SPAAC Cross-Linking, Biomacromolecules, 2017, 18(12), 4054–4059 CrossRef CAS PubMed.
- Y. Navon, M. Zhou, J. B. Matson and R. Bitton, Dendritic Elastin-like Peptides: The Effect of Branching on Thermoresponsiveness, Biomacromolecules, 2016, 17(1), 262–270 CrossRef CAS PubMed.
- Y. Shmidov, M. Zhou, G. Yosefi, R. Bitton and J. B. Matson, Hydrogels Composed of Hyaluronic Acid and Dendritic ELPs: Hierarchical Structure and Physical Properties, Soft Matter, 2019, 15(5), 917–925 RSC.
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Control of Hierarchical Polymer Mechanics with Bioinspired Metal-Coordination Dynamics, Nat. Mater., 2015, 14(12), 1210–1216 CrossRef CAS.
- Y. Jiao, Z. Liu, S. Ding, L. Li and C. Zhou, Preparation of Biodegradable Crosslinking Agents and Application in PVP Hydrogel, J. Appl. Polym. Sci., 2006, 101(3), 1515–1521 CrossRef CAS.
- E. Jabbari and S. Nozari, Swelling Behavior of Acrylic Acid Hydrogels Prepared by γ-Radiation Crosslinking of Polyacrylic Acid in Aqueous Solution, Eur. Polym. J., 2000, 36(12), 2685–2692 CrossRef CAS.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, Cytocompatibility of UV and Visible Light Photoinitiating Systems on Cultured NIH/3T3 Fibroblasts in Vitro, J. Biomater. Sci., Polym. Ed., 2000, 11(5), 439–457 CrossRef CAS.
- S. Chu, S. C. Skaalure, S. J. Bryant, S. L. Sridhar, U. Akalp and F. J. Vernerey, Understanding the Spatiotemporal Degradation Behavior of Aggrecanase-Sensitive Poly(Ethylene Glycol) Hydrogels for Use in Cartilage Tissue Engineering, Tissue Eng., Part A, 2017, 23(15–16), 795–810 CrossRef CAS PubMed.
- C. A. Rice, J. Riehl, K. Broman, J. W. Soukup and W. R. Gengier, Comparing the Degree of Exothermic Polymerization in Commonly Used Acrylic and Provisional Composite Resins for Intraoral Appliances, J. Vet. Dent., 2012, 29(2), 78–83 CrossRef CAS PubMed.
- J. D. McCall and K. S. Anseth, Thiol-Ene Photopolymerizations Provide a Facile Method to Encapsulate Proteins and Maintain Their Bioactivity, Biomacromolecules, 2012, 13(8), 2410–2417 CrossRef CAS PubMed.
- E. R. Ruskowitz and C. A. DeForest, Proteome-Side Analysis of Cellular Response to Ultraviolet Light for Biomaterials Synthesis and Modification, ACS Biomater. Sci. Eng., 2019, 5, 2111–2116 CrossRef CAS.
- C. N. Salinas and K. S. Anseth, Mixed Mode Thiol-Acrylate Photopolymerizations for the Synthesis of PEG-Peptide Hydrogels, Macromolecules, 2008, 41, 6019–6026 CrossRef CAS.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, Pluronic Block Copolymers as Novel Polymer Therapeutics for Drug and Gene Delivery, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS PubMed.
- N. J. Kaiser, R. J. Kant, A. J. Minor and K. L. K. Coulombe, Optimizing Blended Collagen-Fibrin Hydrogels for Cardiac Tissue Engineering with Human IPSC-Derived Cardiomyocytes, ACS Biomater. Sci. Eng., 2019, 5(2), 887–899 CrossRef CAS PubMed.
- J. V. Alegre-Requena, M. Häring, R. P. Herrera and D. Díaz Díaz, Regulatory parameters of self-healing alginate hydrogel networks prepared via mussel-inspired dynamic chemistry, New. J. Chem., 2016, 50, 8493–8501 RSC.
- C. A. DeForest and K. S. Anseth, Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions, Nat. Chem., 2011, 3(12), 925–931 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |