DOI:
10.1039/D0PY00137F
(Paper)
Polym. Chem., 2020,
11, 4920-4927
Poly(alanine-nylon-alanine) as a bioplastic: chemoenzymatic synthesis, thermal properties and biological degradation effects†
Received
27th January 2020
, Accepted 21st March 2020
First published on 20th April 2020
Abstract
Poly(amino acids) such as polypeptides and proteins are attractive biomass-based polymers that potentially contribute to circular economy for plastic. In the current study, we synthesized polypeptides containing periodic nylon units to incorporate thermal plasticity into polypeptides. We successfully synthesized polypeptides containing nylon units by chemoenzymatic polymerization. In addition to nylon 4, which is of biomass origin, nylon 3, nylon 5, and nylon 6 were chosen as the second monomer units with alanine, a natural amino acid. The synthesized polypeptides showed partial melting behavior prior to their thermal degradation. It was found that the nylon unit length significantly changed the thermal properties of the resulting polypeptides, as evidenced by WAXS and DSC analysis. Furthermore, the biodegradability and environmental toxicity of the synthesized polypeptides were evaluated using biological systems. The copolymer containing nylon 4 is considered as a highly potential biopolymer in terms of the polymerization results and environmental toxicity. These results open a new door for utilizing nylon-containing peptides as biomaterials, especially as thermally processable and eco-friendly structural materials.
Introduction
For the circular economy, it is time to consider the recycling, reduction and reuse of fossil fuel-based polymer materials, including bulk plastics. To solve the one-way economy and consumption of fossil oil, many biomass-based polymers have been studied as alternatives to the present fossil plastics. Poly(amino acid)s including polypeptides and proteins are attractive biomass-based polymers that potentially contribute to the circular economy of plastics. Peptides and polypeptides emerge as important functional materials owing to their potential in biomedical applications and nanobiotechnology.1–4 However, as structural bulk material, polypeptides and proteins are potential candidates to mimic structural proteins such as spider and silkworm silks.5 Structural proteins, including silk, elastin, and collagen, are mainly composed of periodically repetitive sequences. These repetitive sequences assemble into higher-order structures with excellent mechanical and physical properties.6 To improve the physical properties of peptides, diversifying the peptide backbone by the introduction of unnatural amino acids enables novel functionalities.7 This amendment can improve the applicability of these peptides, enhancing their widespread applications, including exploiting them as structural materials.
To design and develop polypeptides with unnatural functions, we need to choose appropriate synthesis methods and strategies. Conventional solid-phase peptide synthesis and ring-opening polymerization of amino acid N-carboxyanhydrides (NCAs) are a popular technique for peptide synthesis; however, tedious multistep reactions with several protection and deprotection steps restrict the large-scale production of these materials.8 On the other hand, chemoenzymatic polymerizations have emerged as a superior method over other solution-based syntheses because of their eco-friendly nature.9–11 These facile approaches use mild aqueous media, are atom economical reactions, and are scalable to industrial size.12 We have developed various polypeptides, which include not only homopolymers but also random copolymers containing several amino acid residues.13–15 Di- or tripeptide esters are also available for use as monomers in chemoenzymatic polymerization, resulting in periodic primary structures.16 The periodic sequence with α,α-disubstituted amino acids controlled the secondary structures of resultant polypeptides.15 The chemoenzymatic polymerization of tripeptide has emerged as an effective method for the incorporation of natural and unnatural amino acids with poor affinity for proteases. Proline, which has poor affinity for papain, was incorporated between glycine and valine units, and the resulting tripeptide could be effectively copolymerized to obtain elastin-mimicking polypeptides.17 To introduce aromatic units into the peptide backbone, the tripeptide alanine-(4-aminobenzoic acid)-alanine ethyl ester was also successfully polymerized by chemoenzymatic polymerization.18
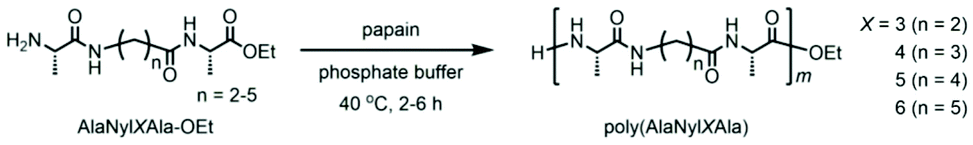
Even though they are attractive structural and functional materials, polypeptides cannot be processed thermally as typical bulk plastics, which restricts their use as structural or bulk materials such as consumable plastics. Peptides and proteins cannot be thermally processed due to strong intermolecular interactions through hydrogen bonds, resulting in thermal degradation prior to melting during the heating process. As plastics, nylons, which are similar polyamides to polypeptides, show excellent mechanical and thermal properties, high chemical resistance, and low permeability to gases. Among various nylons, nylon 4 is one of the most promising biodegradable and biobased plastics.19–21 The nylon 4 monomer was previously introduced into the polypeptide chain by chemoenzymatic polymerization of nylon and amino acid monomers.22 However, only ∼14 mol% of nylon units could be introduced due to the poor affinity of nylon monomers with enzymes, and the sequence could not be controlled.13 Here, in the current study, utilizing the same tripeptide technique, we designed and synthesized novel nylon-containing polypeptides. Herein, we aimed to increase the nylon content in the peptide backbone by adopting sequential alignment of monomers. We successfully synthesized polypeptides containing nylon units by chemoenzymatic polymerization (Scheme 1). In addition, nylon 4, nylon 3, nylon 5, and nylon 6 were chosen to be copolymerized with alanine, a natural amino acid. These polypeptides are expected to affect melting characteristics prior to their thermal degradation as well as to maintain their biodegradability and eco-friendly properties.
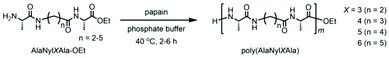 |
| | Scheme 1 Chemoenzymatic polymerization of nylon-containing tripeptides. | |
Experimental
Materials
Papain was purchased from Wako Pure Chemical Industries, Ltd (Osaka, Japan) and used as received. The activity was approximately 0.5 U g−1, where one unit is defined as the amount of enzyme needed to hydrolyze 1 mmol of N-benzoyl-DL-arginine p-nitroanilide per minute at pH 7.5 and 25 °C. N-Methyl-2-pyrrolidone (NMP) and N,N-dimethylacetamide (DMAc) were dried over 4 Å molecular sieves before use. Diethyl ether and triethylamine were dried over 4 Å molecular sieves and stored under nitrogen. All other reagents were used as received without any purification unless otherwise noted.
Characterization procedures
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Varian NMR System 500 (Varian Medical Systems, Palo Alto, CA) at 25 °C and at frequencies of 500 and 125 MHz, respectively. Dimethylsulfoxide-d6 (DMSO-d6) with trifluoroacetic acid-d (TFA-d) (5/1 in volume) was used as the solvent for the polypeptides with tetramethylsilane as the internal standard. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis (MALDI-TOF MS) was conducted using an ultrafleXtreme MALDI-TOF spectrophotometer (Bruker Daltonics, Billerica, MA) operating in reflection mode at an accelerating voltage of 15 kV. The sample was dissolved in water/acetonitrile (0.80 mg mL−1) containing 0.1% TFA, mixed with a solution of α-cyano-4-hydroxycinnamic acid (CHCA) in water/acetonitrile (10 mg mL−1), and deposited on an MTP 384 ground steel BC target plate. The infrared (IR) spectra of the samples were recorded by an IRPrestige-21 Fourier transform infrared spectrophotometer (Shimadzu Corporation, Kyoto, Japan) with a MIRacle A single-reflection ATR unit using a Ge prism. Thermal gravimetric analysis (TGA) was performed using a TGA/DSC2 (Mettler Toledo, Columbus, OH, USA). The polypeptide samples (5.0 mg) were weighed on an aluminum pan and heated with an empty reference cell at a heating rate of 20 °C min−1 from 30 to 500 °C under nitrogen. Differential scanning calorimetry (DSC) measurements were conducted by using a DSC 8500 (PerkinElmer, MA, USA). The polymer sample of 3–5 mg was weighed and sealed in an aluminum pan, which was heated and cooled at 20 °C min−1 from −10 °C to 150 °C under a nitrogen atmosphere. The device was calibrated with an empty cell to establish a baseline and with indium to characterize the heat flow and temperature of the system. The synchrotron wide angle X-ray scattering (WAXS) measurements were performed on the BL45XU beam line of SPring-8, Harima, Japan, using an X-ray energy of 12.4 keV (wavelength, 0.1 nm); a beam with a diameter of approximately 45 μm was employed, according to a previous study.23 Temperature-controlled measurements were also performed according to a previous study.24 The sample-to-detector distance during the WAXS measurements was approximately 180 mm. The obtained two-dimensional (2D) scattering patterns were converted into one-dimensional (1D) profiles by using Fit2D software.25
Synthetic procedures
Synthesis of Nyl3Ala-OEt HCl salt.
N-Boc-Nylon3 was synthesized according to a previously reported procedure.26 To a flask equipped with an addition funnel and a stir bar were added N-Boc-Nylon3 (25.3 g, 134 mmol), L-alanine ethyl ester hydrochloride (20.5 g, 134 mmol), 1-hydroxybenzotriazole (HOBt) monohydrate (18.0 g, 134 mmol), triethylamine (18.6 mL, 134 mmol) and chloroform (125 mL) at −10 °C under nitrogen. A solution of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (water-soluble carbodiimide, WSCI) hydrochloride (25.6 g, 134 mmol) in chloroform (125 mL) was added dropwise over 30 min, and the resulting mixture was stirred at −10 °C for 30 min and then at 25 °C for 24 h. The mixture was washed sequentially with water, 5% NaHCO3 aq., and brine. The organic layer was dried with Na2SO4 and concentrated using a rotary evaporator. The product was dried in vacuo to give Boc-Nyl3Ala-OEt with a yield of 36.2 g (94.0%). The obtained Boc-Nyl3Ala-OEt was then subjected to deprotection of the Boc group. To the solution of Boc-Nyl3Ala-OEt (34.0 g, 118 mmol) in dichloromethane (84.0 mL) was slowly added trifluoroacetic acid (45.1 mL, 590 mmol) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 10 min and then at 25 °C for 24 h. After the solvent was removed under reduced pressure, the crude product was dissolved in dioxane/HCl (4.0 M, 68.0 mL). The solution was poured into diethyl ether. The precipitate was filtered, washed with diethyl ether, and dried in vacuo to afford Nyl3Ala-OEt as a white hygroscopic solid. The yield was 21.5 g (97.0%).
Synthesis of AlaNyl3Ala HCl salt.
To a flask equipped with an addition funnel and a stir bar were added N-Boc-L-alanine (25.2 g, 133 mmol), Nyl3Ala-OEt hydrochloride (25.1 g, 133 mmol), HOBt monohydrate (18.0 g, 133 mmol), triethylamine (18.6 mL, 133 mmol) and chloroform (75 mL) at −10 °C under nitrogen. A solution of WSCI hydrochloride (25.6 g, 78.9 mmol) in chloroform (75.0 mL) was added dropwise over 30 min, and the resulting mixture was stirred at −10 °C for 30 min and then at 25 °C for 24 h. The mixture was washed sequentially with water, aq. 5% NaHCO3 and brine. The organic layer was dried with Na2SO4 and concentrated using a rotary evaporator. The product was dried in vacuo to give Boc-AlaNyl3Ala in a yield of 45.5 g (95.3%). The obtained Boc-AlaNyl3Ala was subsequently subjected to deprotection of the Boc group. To the solution of Boc-AlaNyl3Ala (10.5 g, 29.3 mmol) in dichloromethane (20.0 mL) was slowly added trifluoroacetic acid (11.2 mL, 146 mmol) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 10 min and then at 25 °C for 24 h. After the solvent was removed under reduced pressure, the crude product was dissolved in dioxane/HCl (4.0 M, 20.0 mL). The solution was poured into diethyl ether. The precipitate was filtered, washed with diethyl ether, and dried in vacuo to afford AlaNyl3Ala as a white hygroscopic solid. The yield was 7.05 g (93.0%). All other nylons containing the alanine tripeptide ethyl ester AlaNylXAla (X = 4, 5, and 6) were prepared using the same experimental procedure.
Chemoenzymatic synthesis of poly(AlaNylXAla).
A solution of AlaNyl6Ala hydrochloride (1.07 g, 3.54 mmol) in phosphate buffer (12.4 mL, 1.0 M, pH = 8.0) was placed in a glass tube equipped with a stirring bar. To this solution, papain (0.708 g) was added. The final concentrations of AlaNyl6Ala and papain were 0.25 M and 50 mg mL−1, respectively. The mixture was stirred at 40 °C at 800 rpm for 2 h using an EYELA ChemiStation PPS-5511 (Tokyo Rikakikai Co. Ltd, Tokyo, Japan). After 2 h, the reaction mixture was cooled to room temperature; the precipitate was collected by centrifuging at 9000 rpm and 20 °C for 20 min. The non-reacted monomer remained in the reaction buffer and was removed by the centrifugation step. The crude product was washed twice with deionized water, centrifuged, and lyophilized to obtain poly(AlaNyl6Ala) as a white powder. The yield was 27%. The non-reacted monomers and dimers were removed with these washing process. The lyophilized powders were used for further characterizations. All other nylons containing the alanine polypeptide poly(AlaNylXAla) were prepared using the same experimental procedure.
Biodegradation test
The biochemical oxygen demand (BOD) test was performed to estimate the biodegradability of the samples in sea water (Chemicals Evaluation and Research Institute, Japan) with an Oxitop IS-6 (WTW GmbH, Weilheim in Oberbayern, Germany) according to previous literature.21,27 The biodegradability was calculated from the theoretical oxygen demand (ThOD) and BOD as follows:
| Biodegradability (%) = BOD/ThOD × 100 |
The poly(AlaNylXAla) sample (approximately 10 mg) was immersed in 43.5 mL of seawater at 25 °C for 5 days. The seawater used in this study was obtained from Odaiba Seaside Park in Tokyo Bay (Tokyo, Japan), which had a pH of 8.0 and salinity of 3.6%.
Environmental toxicity test
The environmental toxicity of the samples was tested according to the OECD test guideline 202, acute immobilization test (OECD), with slight modification.28,29 Briefly, 30 or 60 Daphnia magna, which were not more than 24 h old at the beginning of the test, were used for each test sample and control. For each treatment, the animals were divided into six or 12 groups of five animals, and each group was kept in 10 mL of synthetic M4 medium containing one of the test samples (or no additives as a control) for one week at a water temperature of 20 ± 1 °C and a light–dark cycle of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 h. The final concentration of the samples was set at 100 mg L−1, which was the higher limit provided by the test guidelines. The powder samples were dispersed well by sonication just before use. Survival curves of each treatment were produced based on daily observations during the test period. The effects of the samples on their survival were evaluated by comparing the Kaplan–Meier survival curves using the log-rank test followed by Bonferroni correction.
:8 h. The final concentration of the samples was set at 100 mg L−1, which was the higher limit provided by the test guidelines. The powder samples were dispersed well by sonication just before use. Survival curves of each treatment were produced based on daily observations during the test period. The effects of the samples on their survival were evaluated by comparing the Kaplan–Meier survival curves using the log-rank test followed by Bonferroni correction.
Statistical analysis
The significance of differences in studies of biodegradation and environmental toxicity tests were determined by unpaired t-tests with a two-tailed distribution. Differences were considered statistically significant at p < 0.01 (*).
Results and discussion
Chemoenzymatic polymerization
We synthesized tripeptide ethyl esters where the ω-aminoalkanoic acid unit (nylon monomer unit) was sandwiched between two alanine residues, one of the amino acids with high affinity for papain (Scheme 1). The resultant tripeptide AlaNylXAla (X = 3, 4, 5, and 6) was polymerized by papain to obtain polypeptides containing periodic nylon units. The chemoenzymatic polymerization of the tripeptide was carried out using 1 M phosphate buffer solution (PBS) of pH 8.0 at 40 °C; the results are listed in Table 1. The reaction was carried out at different time intervals and at varied concentrations. The reaction at a 1.0 M concentration of the tripeptide ethyl esters (Runs 1, 5, 9, 13) resulted in a poor yield. Additionally, the reaction was carried out at lower concentrations (0.1, 0.25, and 0.5 M). In the case of the reactions with 0.1 M of the tripeptide ethyl esters (Runs 4, 8, 12, 16), the product yield was highest. However, the reaction often yielded a high percentage of undesired product (contaminated with alanine due to transamidation15), as shown in Fig. 1. In the reactions with 0.5 M tripeptide ethyl esters, the reaction yields were relatively higher than those of the reactions with 1.0 M tripeptide ethyl esters (Runs 2, 6, 10, 14). The reaction at 0.25 M of the tripeptide ethyl esters (Runs 3, 7, 11, and 15) showed relatively better yields and reduced the formation of undesired products. Hence, this optimized condition at 0.25 M AlaNylXAla was employed in synthesizing polypeptides containing nylon units.
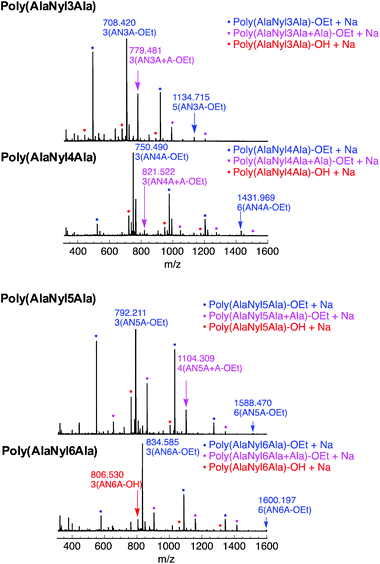 |
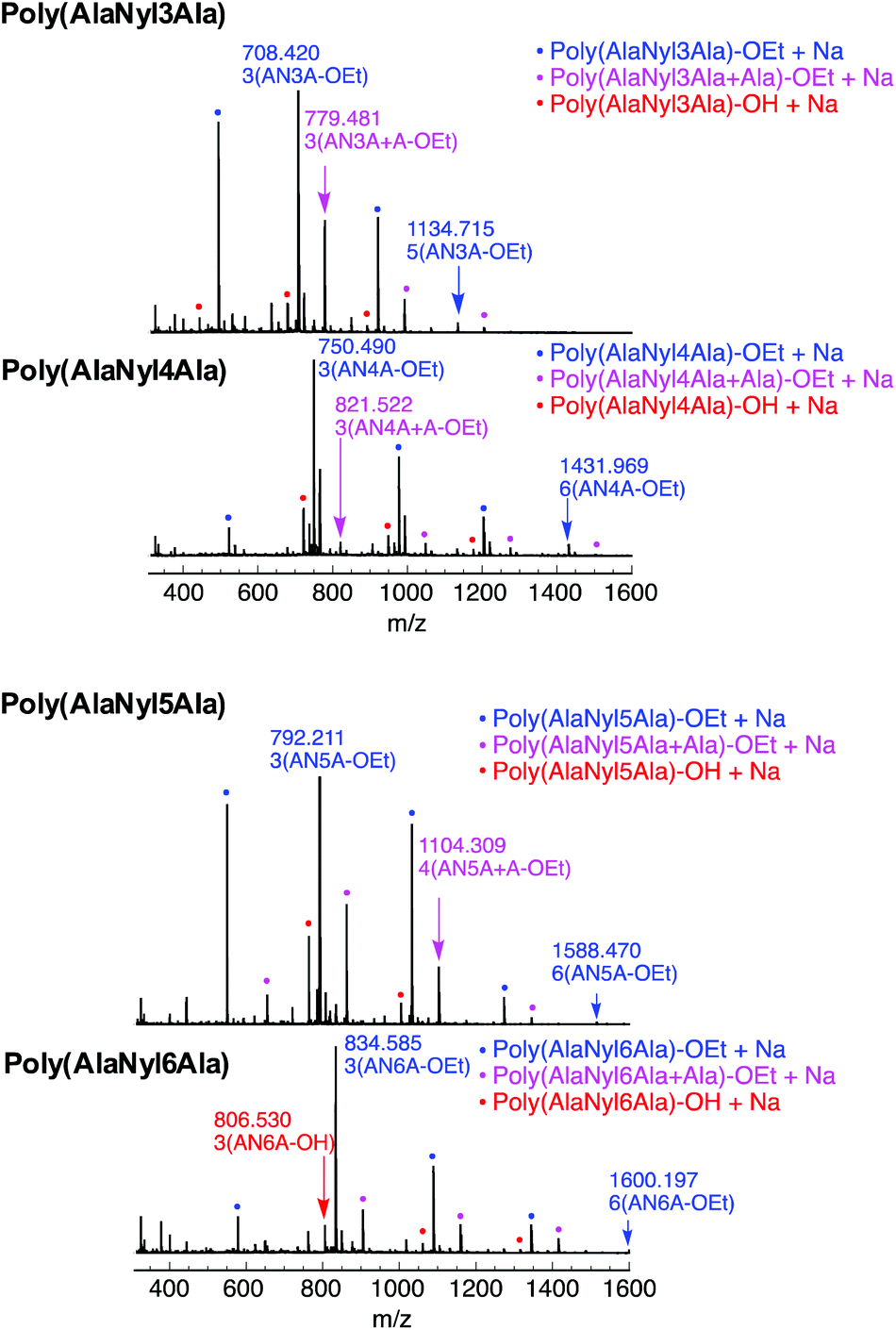
| | Fig. 1 MALDI-TOF mass spectra of poly(AlaNylXAla) obtained by papain-catalyzed polymerization. | |
Table 1 Chemoenzymatic polymerizations with AlaNylXAla tripeptides
| Runa |
Monomer |
Conc. (M) |
Yieldb (%) |
Time (h) |
DPmaxc |
|
Polymerization was carried out using monomer (HCl salt) and papain (50 mg mL−1) in phosphate buffer (1 M, pH 8.0) at 40 °C for 2–6 h.
Precipitation was collected by centrifugation, washed with water, and lyophilized.
Maximum degree of polymerization of AlaNylXAla determined by MALDI-TOF mass.
|
| 1 |
AlaNyl3Ala |
1.0 |
4.4 |
2 |
3 |
| 2 |
AlaNyl3Ala |
0.5 |
12 |
2 |
4 |
| 3 |
AlaNyl3Ala |
0.25 |
35 |
2 |
5 |
| 4 |
AlaNyl3Ala |
0.1 |
34 |
2 |
5 |
| 5 |
AlaNyl4Ala |
1.0 |
10 |
6 |
5 |
| 6 |
AlaNyl4Ala |
0.5 |
20 |
2 |
6 |
| 7 |
AlaNyl4Ala |
0.25 |
39 |
2 |
6 |
| 8 |
AlaNyl4Ala |
0.1 |
35 |
2 |
6 |
| 9 |
AlaNyl5Ala |
1.0 |
4.3 |
2 |
3 |
| 10 |
AlaNyl5Ala |
0.5 |
13 |
2 |
5 |
| 11 |
AlaNyl5Ala |
0.25 |
43 |
3 |
5 |
| 12 |
AlaNyl5Ala |
0.1 |
40 |
2 |
6 |
| 13 |
AlaNyl6Ala |
1.0 |
16 |
2 |
3 |
| 14 |
AlaNyl6Ala |
0.5 |
17 |
2 |
4 |
| 15 |
AlaNyl6Ala |
0.25 |
30 |
2 |
6 |
| 16 |
AlaNyl6Ala |
0.1 |
42 |
2 |
6 |
Chemical structure analyses by MALDI-TOF MS and NMR spectroscopy
The chemical structures of all these products were confirmed by MALDI-TOF MS and 1H NMR. MALDI-TOF MS spectra of poly(AlaNylXAla) are shown in Fig. 1. Poly(AlaNyl3Ala) exhibited three series of products, one series with a terminal ethyl ester and another with carboxylic acid at the C-terminal end. Interestingly, a new series of peaks were observed that were attributed to alanine units along with tripeptide monomer units. The formation of this product was found to increase at lower concentrations. The series of peaks with an interval of 213 m/z, which corresponds to the AlaNyl3Ala tripeptide unit, indicates the successful polymerization of AlaNyl3Ala. Similar sets of peaks were detected for AlaNyl4Ala, AlaNyl5Ala, and AlaNyl6Ala with up to 6-mer chain lengths. The molar mass of poly(AlaNylXAla) was in the range from 400 to 1600 Da. The maximum degree of polymerization (DPmax) was determined with MALDI-TOF mass as listed in Table 1. The reaction conditions for higher yields gave relatively higher DPmax. The negative control reactions without enzyme were also performed (Fig. S1†), resulting that no polymerization of tripeptide was detected. Overall, unexpected impurities were not detected as major components.
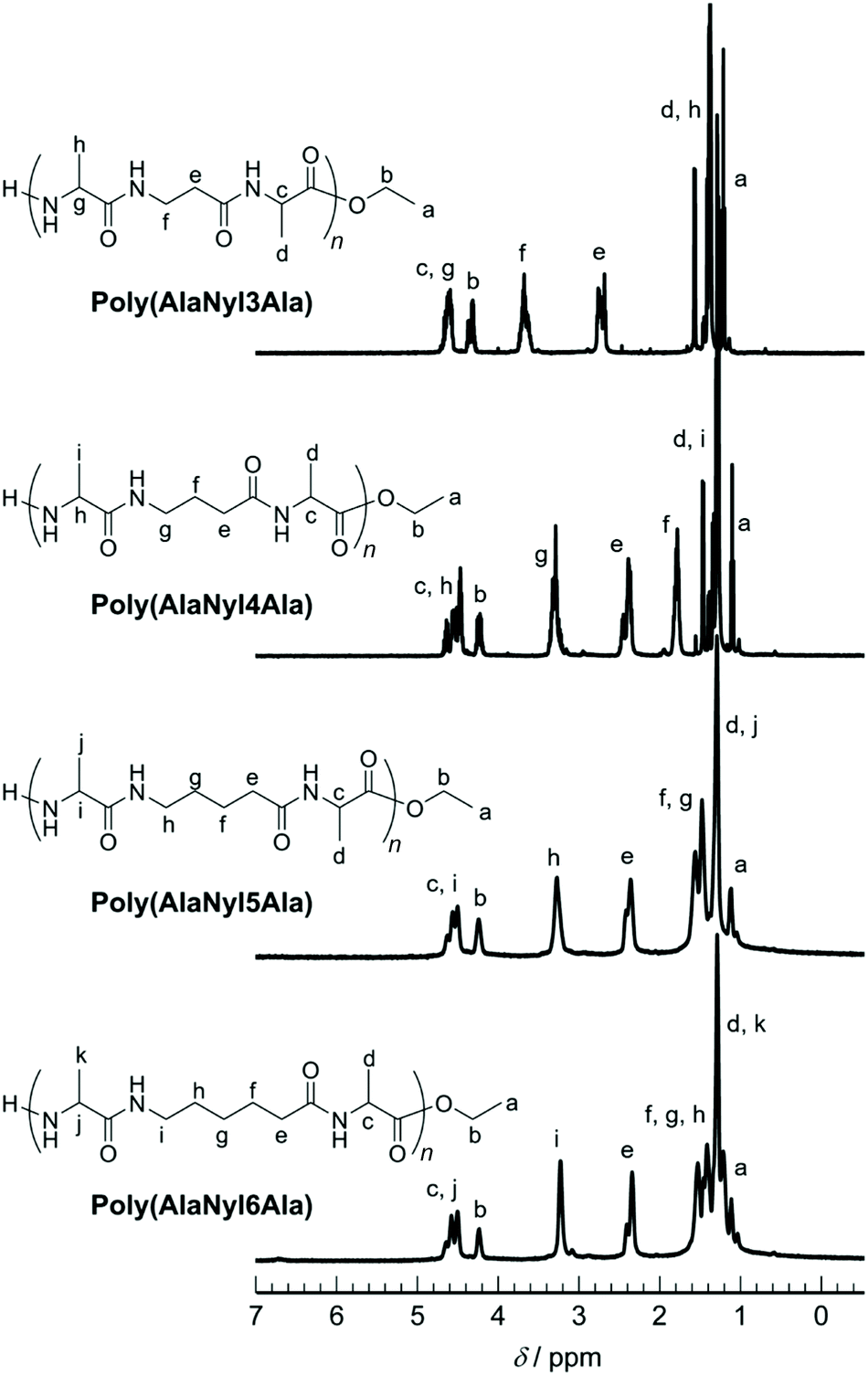
The poly(AlaNylXAla) was characterized by both 1H and 13C NMR using TFA-d as a solvent (Fig. S2†). The methyl peaks originating from alanine appeared at 1.2–1.4 ppm. The nylon peaks appeared at 1.5–4.0 ppm. The quartet peak corresponding to the methine proton for poly(AlaNylXAla) appeared at 3.5–5.0 ppm. The quartet corresponding to the methylene proton of the terminal ester group appeared at 4.2–5.0 ppm for poly(AlaNylXAla) (Fig. 2).
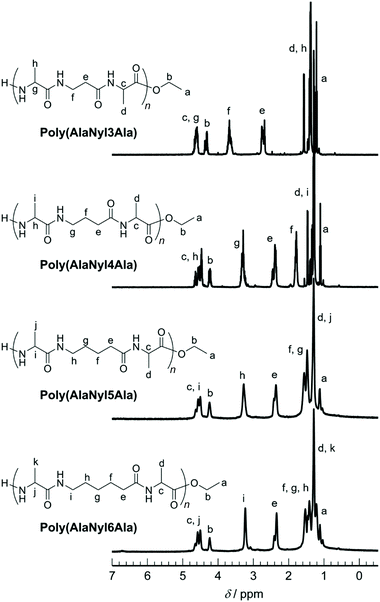 |
| | Fig. 2
1H NMR spectra of poly(AlaNylXAla) obtained by papain-catalyzed polymerization. | |
Thermal analysis
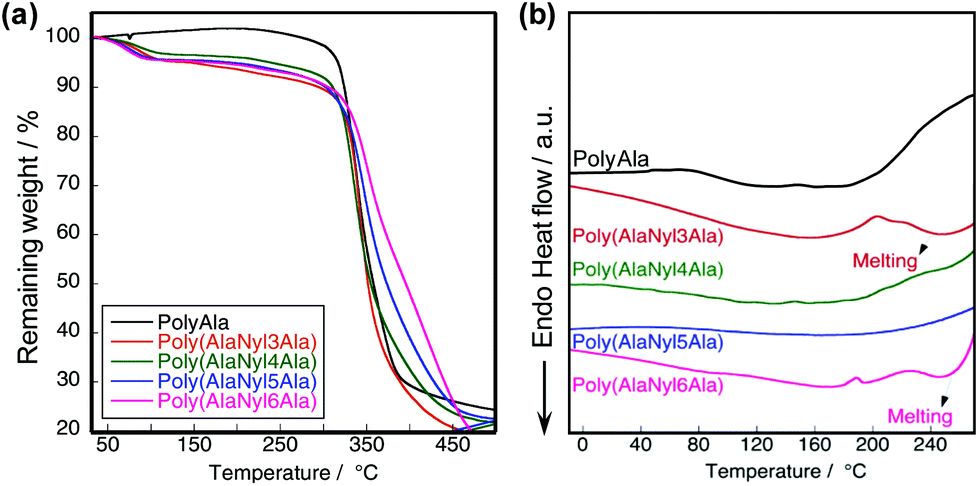
To characterize the difference in the thermal behavior of poly(AlaNylXAla), we performed TGA and DSC measurements. The lyophilized powder samples after removal of the non-reacted monomers and dimers were used for the measurements. All the samples were supposed to be crystallized via alanine motifs by the lyophilization.30 The TGA profiles of poly(AlaNylXAla) showed a 5% degradation temperature (Td5) of approximately 190 °C regardless of the nylon chain length (Fig. 3a). PolyAla was used as a negative control, which did not contain any nylon units. The thermal stability of all poly(AlaNylXAla) was found to be relatively the same, meaning that the carbon chain length of the nylon unit does not contribute to the thermal properties. DSC profiles of the first heating scans are shown in Fig. 3b. The first heating scan indicated an endothermic peak ranging from 50 °C to 120 °C, which corresponded to water removal from the peptide based on the TGA results and previous literature.24 The endothermic peak observed from 240 °C to 260 °C could be due to the melting behavior. This result was probably due to the inhibition of the intermolecular hydrogen bonds in poly(AlaNylXAla) with approximately 30 mol% nylon units, and thus, the thermal stability of poly(AlaNylXAla) decreased. Poly(AlaNyl3Ala) and poly(AlaNyl6Ala) demonstrated crystallization around 180–220 °C, and hence melting behaviors were more clear than other samples. Among 4 types of poly(AlaNylXAla) samples, poly(AlaNyl5Ala) did not show obvious melting behavior, similar to polyAla. The chain-end structures, such as hydroxyl group or ethyl ester, are known to affect the thermal properties, especially, in the case of lower molecular weight polymers.31 In the current study, we detected two types of the C-terminal structures, namely, carboxylic acid and ethyl ester ends. However, the most of the C-terminal end was ethyl ester, and hence the effect of the chain end structure was not significant. The thermal degradation temperatures and the melting region were too close to be characterized in the second heating profile.
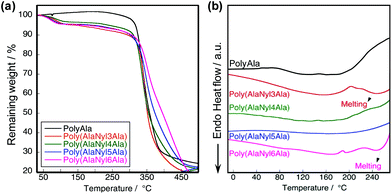 |
| | Fig. 3 Thermal properties of poly(AlaNylXAla). TGA (a) and DSC profiles (b) at 20 °C min−1. | |
Structural characterizations
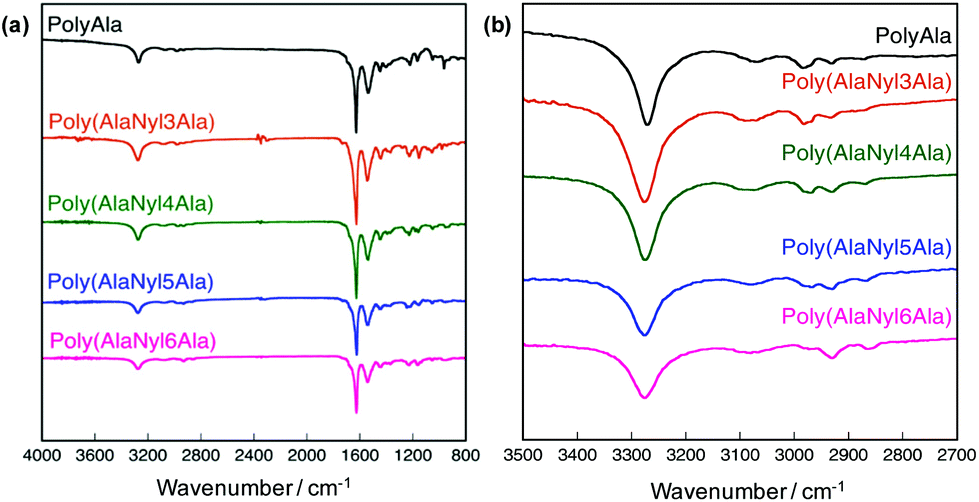
The FTIR spectra of poly(AlaNylXAla) were obtained to evaluate the effect of nylon monomers on hydrogen bond formation (Fig. 4). In the FTIR spectra of polyAla, which is a control sample without nylon units, a strong peak in the amide I region (1700–1600 cm−1) with a major contribution of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration coupled with the C–N stretching vibration was detected. In particular, the peak at 1628 cm−1 corresponding to β-sheet structures was also confirmed.32 The FTIR spectra of the nylon-containing polypeptides, poly(AlaNylXAla), showed a slight shift to lower wavenumbers in the amide I region. The amide II region (1550–1530 cm−1), resulting from C–N stretching and N–H bending modes in amide bonds, also showed a slight shift to lower wavenumbers for nylon-containing polypeptides. On the other hand, the peak at approximately 3280 cm−1, which corresponds to the N–H stretching vibration that reflects the hydrogen bonding between amide groups in the peptide backbone,13 was clearly detected. As the chain length of the nylon increased, the intensity of this peak decreased, indicating that the presence of longer nylon units successfully reduced more inter- and intra-molecular hydrogen bonding. A new peak was recognized at approximately 2860 cm−1 assignable to the C–H stretching vibration, which became significant with increasing nylon chain length.
O stretching vibration coupled with the C–N stretching vibration was detected. In particular, the peak at 1628 cm−1 corresponding to β-sheet structures was also confirmed.32 The FTIR spectra of the nylon-containing polypeptides, poly(AlaNylXAla), showed a slight shift to lower wavenumbers in the amide I region. The amide II region (1550–1530 cm−1), resulting from C–N stretching and N–H bending modes in amide bonds, also showed a slight shift to lower wavenumbers for nylon-containing polypeptides. On the other hand, the peak at approximately 3280 cm−1, which corresponds to the N–H stretching vibration that reflects the hydrogen bonding between amide groups in the peptide backbone,13 was clearly detected. As the chain length of the nylon increased, the intensity of this peak decreased, indicating that the presence of longer nylon units successfully reduced more inter- and intra-molecular hydrogen bonding. A new peak was recognized at approximately 2860 cm−1 assignable to the C–H stretching vibration, which became significant with increasing nylon chain length.
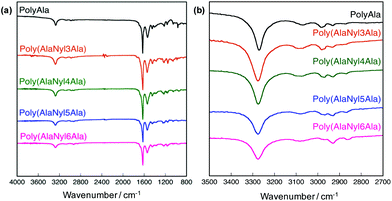 |
| | Fig. 4 FTIR spectra of poly(AlaNylXAla) and polyAla. (a) Overall spectra from 800 to 4000 cm−1. (b) Enlarged spectra from 2700 to 3500 cm−1. | |
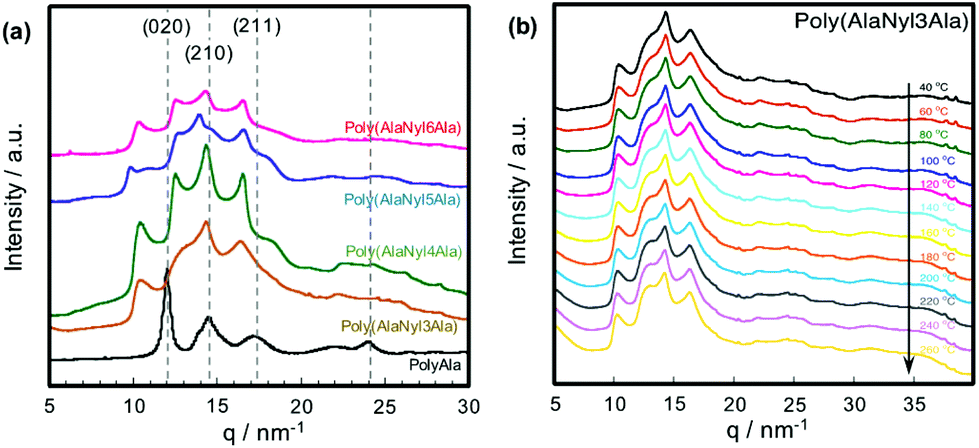
To investigate the effect of the nylon units on the secondary and crystal structures of poly(AlaNylXAla), we performed WAXS measurements using polyAla as a control sample (Fig. 5a). WAXS 1D profiles of polyAla show peaks originating from the polyAla antiparallel β-sheet structure (020), (210) and (211),5,23,33 which did not closely match the other profiles of poly(AlaNylXAla). Furthermore, the reported nylon structures were not identical to the current d-spacing. This result indicates that the crystal structures of poly(AlaNylXAla) were more affected by nylon units than by alanine residues. The different d-spacing values for poly(AlaNylXAla) also suggested that the crystal structures of poly(AlaNylXAla) were dependent on the type of nylon units. However, we could not assign the peaks, and hence did not evaluate more details of the crystalline state such as crystallinity and orientation. To characterize the effect of the nylon units on the thermal properties of poly(AlaNylXAla), we performed temperature-dependent WAXS with a heating rate of 20 °C min−1 (Fig. 5b and Fig. S3†). The WAXS results of poly(AlaNyl3Ala) and poly(AlaNyl5Ala) showed a decrease in peak intensity, especially with a d-spacing value of 0.20 nm, as the temperature increased. A similar tendency was observed in the case of poly(AlaNyl4Ala) and poly(AlaNyl6Ala). However, the crystal structures were still maintained even at 260 °C, demonstrating that the melting behaviors detected by DSC were caused by semicrystalline regions of the polypeptides. Furthermore, the predominant structure of poly(AlaNylXAla) was beta-sheet-like crystalline state. In other words, the tightly packed crystalline region was not melted in the DSC experiments.
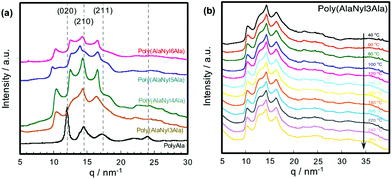 |
| | Fig. 5 WAXS 1D profiles of poly(AlaNylXAla) and polyAla. (a) WAXS 1D profiles at 25 °C. The peaks originated from the beta-sheet structure of polyAla are assigned. (b) WAXS 1D profiles of poly(AlaNyl3Ala) at elevated temperatures from 40 to 260 °C. | |
Biodegradability and degradation product assay
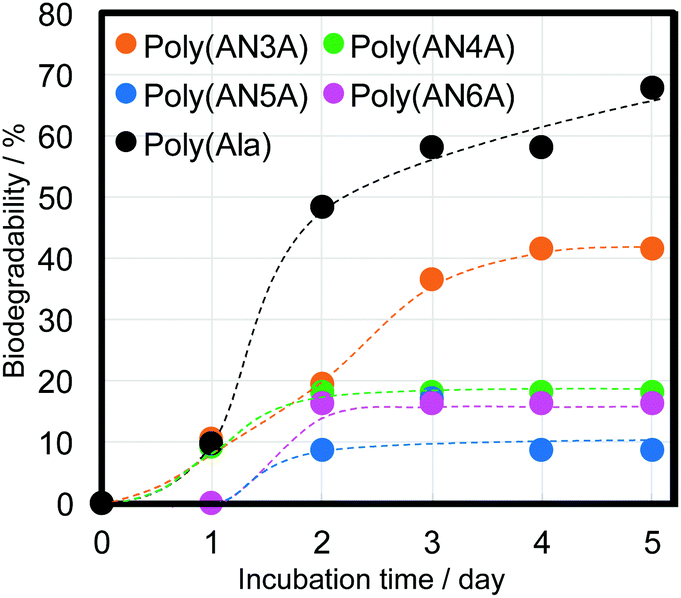
Polypeptides composed of natural amino acids are considered to show biodegradability in natural environments; however, polypeptides containing nylon such as poly(AlaNylXAla) need to be studied for biodegradability. To confirm the biodegradation of poly(AlaNylXAla), we performed a BOD test using poly(AlaNylXAla) powder samples under seawater conditions at 25 °C (Fig. 6). The biodegradability of the sample was calculated from the ThOD and experimental BOD values as described in the Experimental section. As a result, the significant biodegradability of poly(AlaNylXAla) was confirmed, even though polyAla showed higher biodegradability. The biodegradability based on the BOD tests did not indicate significant differences among the poly(AlaNylXAla) samples.
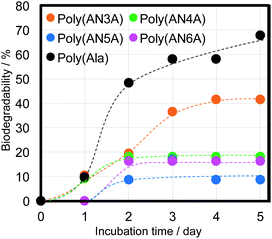 |
| | Fig. 6 BOD tests of poly(AlaNylXAla) and polyAla in seawater at 25 °C. | |
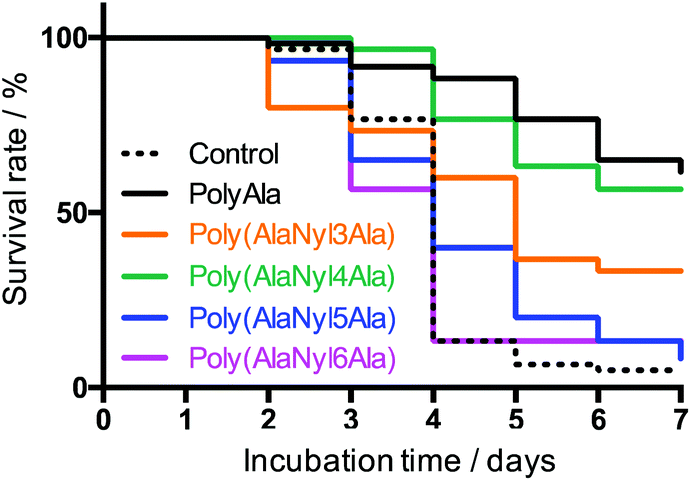
We performed environmental toxicity tests of poly(AlaNylXAla) and polyAla using D. magna, and no reduction in survival was observed in any of the conditions compared to the control (Fig. 7). In contrast, polyAla, poly(AlaNyl3Ala) and poly(AlaNyl4Ala) significantly improved their survival compared to the control, in which most animals experienced death from starvation during the test period (p < 0.05), suggesting that they may act as a nutrient rather than a toxicant for daphnids. These results with D. magna demonstrate that poly(AlaNylXAla) and its degradation products did not provide toxic effects on the natural model animal.
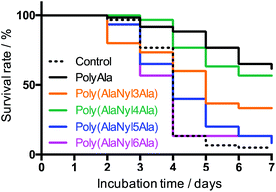 |
| | Fig. 7 Kaplan–Meier survival curves to evaluate the effects of poly(AlaNylXAla) and polyAla on the aquatic model animal Daphnia magna at 20 °C. Control denotes the background values without any test samples. Number of sample daphnids (n) = 30 (for poly(AlaNyl3Ala), poly(AlaNyl4Ala), poly(AlaNyl6Ala)) or 60 (for control, polyAla, poly(AlaNyl5Ala)). | |
Considering that the types of nylon units in poly(AlaNylXAla) showed different thermal and structural properties, as shown in Fig. 3 and 5, the biodegradability and environmental toxicity also show different behaviors among the different poly(AlaNylXAla) samples. In particular, similar to poly(Ala), poly(AlaNyl4Ala) showed higher survival rate in average.
Conclusions
In conclusion, we successfully synthesized polypeptides containing periodic nylon units by sandwiching a nylon unit between two alanine residues. We successfully established optimized chemoenzymatic polymerization conditions for the synthesis of poly(AlaNylXAla), even though the molecular weight was not so sufficient for bulk applications. The nylon content (approximately 30 mol%) within the polypeptide chain could be enhanced by utilizing the tripeptide strategy. The current nylon content was significantly enhanced in comparison to the previous study on the random copolymerization of nylon and amino acid monomers.13 The resulting nylon-containing polypeptides showed different melting behaviors prior to degradation depending on the length of the nylon units, which opens a new door for utilizing polypeptides as thermally processable biomaterials, which can be processed with 3D printers. Lower molecular weight products are supposed to be degraded more easily, and thus the resultant polypeptides can be potential candidate for fast-degradable materials. Utilizing enzymes for the reaction not only helps in obtaining stereospecific and sequential amino acid sequences but also provides an eco-friendly and sustainable synthetic approach. Future aspects include variation in amino acid sequences and employing longer-chain nylon units, which could provide more interesting biological polymers.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was financially supported by the Impulsing Paradigm Change through Disruptive Technologies Program (ImPACT) of the Japan Science and Technology Agency (JST), RIKEN Engineering Network, and JST ERATO (Grant Number JPMJER1602).
Notes and references
- S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
- K. Numata and D. L. Kaplan, Adv. Drug Delivery Rev., 2010, 62, 1497–1508 CrossRef CAS PubMed.
- K. Tsuchiya and K. Numata, ACS Macro Lett., 2017, 6, 103–106 CrossRef CAS.
- C. Holland, K. Numata, J. Rnjak-Kovacina and F. P. Seib, Adv. Healthcare Mater., 2019, 8, e1800465 CrossRef PubMed.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
-
R. B. Merrifield, Advances in Enzymology and Related Areas of Molecular Biology, John Wiley & Sons, New Jersey, 1969, pp. 221–296 Search PubMed.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2017, 17, 1700177 CrossRef PubMed.
- K. Yazawa and K. Numata, Molecules, 2014, 19, 13755–13774 CrossRef PubMed.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- J. Gimenez-Dejoz, K. Tsuchiya and K. Numata, ACS Chem. Biol., 2019, 14, 1280–1292 CrossRef CAS PubMed.
- K. Yazawa, J. Gimenez-Dejoz, H. Masunaga, T. Hikima and K. Numata, Polym. Chem., 2017, 8, 4172–4176 RSC.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2016, 16, 1001–1008 CrossRef CAS PubMed.
- K. Tsuchiya and K. Numata, Chem. Commun., 2017, 53, 7318–7321 RSC.
- X. Qin, A. C. Khuong, Z. Yu, W. Z. Du, J. Decatur and R. A. Gross, Chem. Commun., 2013, 49, 385–387 RSC.
- P. G. Gudeangadi, K. Tsuchiya, T. Sakai and K. Numata, Polym. Chem., 2018, 9, 2336–2344 RSC.
- K. Tsuchiya, N. Kurokawa, J. Gimenez-Dejoz, P. G. Gudeangadi, H. Masunaga and K. Numata, Polym. J., 2019, 51, 1287–1298 CrossRef CAS.
- K. Hashimoto, T. Hamano and M. Okada, J. Appl. Polym. Sci., 1994, 54, 1579–1583 CrossRef CAS.
- N. Yamano, A. Nakayama, N. Kawasaki, N. Yamamoto and S. Aiba, J. Polym. Environ., 2008, 16, 141–146 CrossRef CAS.
- K. Tachibana, Y. Urano and K. Numata, Polym. Degrad. Stab., 2013, 98, 1847–1851 CrossRef CAS.
- K. Yazawa and K. Numata, Polymer, 2016, 8, 194 Search PubMed.
- K. Numata, R. Sato, K. Yazawa, T. Hikima and H. Masunaga, Polymer, 2015, 77, 87–94 CrossRef CAS.
- K. Yazawa, K. Ishida, H. Masunaga, T. Hikima and K. Numata, Biomacromolecules, 2016, 17, 1057–1066 CrossRef CAS PubMed.
-
A. P. Hammersley, European Synchrotron Radiation Facility Internal Report, 1997, ESRF97HA02T Search PubMed.
- S. K. Maji, R. Banerjee, D. Velmurugan, A. Razak, H. K. Fun and A. Banerjee, J. Org. Chem., 2002, 67, 633–639 CrossRef CAS PubMed.
- K. Numata, N. Ifuku, H. Masunaga, T. Hikima and T. Sakai, Biomacromolecules, 2017, 18, 1937–1946 CrossRef CAS PubMed.
- R. Abe, K. Toyota, H. Miyakawa, H. Watanabe, T. Oka, S. Miyagawa, H. Nishide, I. Uchiyama, K. E. Tollefsen, T. Iguchi and N. Tatarazako, Aquat. Toxicol., 2015, 159, 44–51 CrossRef CAS PubMed.
- B. P. Elendt and W. R. Bias, Water Res., 1990, 24, 1157–1167 CrossRef CAS.
- P. J. Baker and K. Numata, Biomacromolecules, 2012, 13, 947–951 CrossRef CAS PubMed.
- L. H. Zhang, J. A. Marsiglio, T. Lan and J. M. Torkelson, Macromolecules, 2016, 49, 2387–2398 CrossRef CAS.
- O. S. Rabotyagova, P. Cebe and D. L. Kaplan, Biomacromolecules, 2009, 10, 229–236 CrossRef CAS PubMed.
- C. Riekel, M. Muller and F. Vollrath, Macromolecules, 1999, 32, 4464–4466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: DETAILS. See DOI: 10.1039/d0py00137f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kei
Uchida
b,
Ayaka
Tateishi
a,
Kayo
Terada
a,
Hiroyasu
Masunaga
c,
Kousuke
Tsuchiya
a,
Kei
Uchida
b,
Ayaka
Tateishi
a,
Kayo
Terada
a,
Hiroyasu
Masunaga
c,
Kousuke
Tsuchiya