
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intrinsic flame retardant phosphonate-based vitrimers as a recyclable alternative for commodity polymers in composite materials†
Jens C.
Markwart
 a,
Alexander
Battig
b,
Tobias
Urbaniak
c,
Katharina
Haag
c,
Katharina
Koschek
c,
Bernhard
Schartel
c and
Frederik R.
Wurm
*a
a,
Alexander
Battig
b,
Tobias
Urbaniak
c,
Katharina
Haag
c,
Katharina
Koschek
c,
Bernhard
Schartel
c and
Frederik R.
Wurm
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bBundesanstalt für Materialforschung und -prüfung (BAM), Unter den Eichen 87, 12205 Berlin, Germany
cFraunhofer Institute for Manufacturing Technology and Advanced Materials IFAM, Wiener Strasse 12, 28359 Bremen, Germany
First published on 8th June 2020
Abstract
Recycling of crosslinked fiber-reinforced polymers is difficult. Moreover, as they are often based on flammable resins, additional additives are needed. So-called “vitrimers” open the possibility of recycling and reprocessing and repairing with dynamically crosslinked chemistries. To date, vitrimer-based composites still need flame retardant additives, such as organophosphates. An additive-free vitrimer composite has not been reported. Herein, we synthesized an intrinsic flame-retardant vitrimer, relying on vinylogous polyurethanes containing covalently installed phosphonates as flame-retardant units and prepared glass-fiber-reinforced composites. We studied recycling and flame retardant properties and compared the data to phosphorus-free vitrimers and conventional epoxy resins (with and without additive flame retardant). Our phosphonate-based vitrimer proved in first tests, a flame retardant effect comparable to commercial flame retardant resins. The bending strength and bending modulus for the phosphorus-vitrimer glass fiber composites were comparable to glass fiber composites with permanently cross-linked epoxies. In summary, we were able to prove that the covalent installation of phosphonates into vitrimers allows the preparation of recyclable and intrinsic flame retardant composites that do not need flame retardant additives. We believe this concept can be expanded to other polymer networks and additives to generate recyclable and sustainable high-performance materials.
Introduction
Cross-linked-polymer composites based on epoxy resins that are currently on the market cannot be chemically recycled and often show high flammability without flame retardant additives. The development of dynamic covalent polymer networks gave access to the first composite materials with the potential for a circular economy.1 To date, however, additives such as flame retardants are still required for these materials if they are to follow safety regulations. Herein, we present a phosphonate-based and intrinsic flame-retardant dynamic polymer network based on vinylogous polyurethanes, eliminating the need for additional flame retardant additives. We show their recyclability, use in glass-fiber composites, and study their flame retardancy. We compared the materials to widely used – but not recyclable – epoxy resins, exemplarily.Due to the high flammability of epoxy resins, flame retardant additives are required to fulfill current regulatory safety requirements.2 However, the extensively used halogenated, mostly brominated, flame retardants are being phased out due to their potential negative environmental impacts and toxicity.3–6 Therefore, halogenated flame retardants are often substituted by phosphorus-based flame retardants. Due to the chemical versatility of phosphorus chemistry and high effectiveness at already low loadings, phosphorus-containing flame retardants gained recently a lot of attention in academia and industry.6
Most flame retardants are used as additives, i.e. not covalently connected to the polymer matrix. The physical blending, however, may result in leaching from the polymer matrix or alter its mechanical properties, which holds especially true for low molecular weight additives, making intrinsic flame-retardant polymeric materials more attractive.
Dynamic covalent polymer networks, which are often described as a new class of organic materials, were first reported by Leibler et al. in 2011, based on an epoxy resin.7,8 The resin was cross-linked with di- and tri-carboxylic acids. The addition of mild Lewis acid catalyst (like zinc acetate) combined the classical mechanical properties of an epoxy resin with the ability to reprocess and reshape after full curing.9 These so-called “vitrimers” have the unique characteristic that the cross-linking points exchange under thermal stimulation following an associative exchange mechanism, thus keeping the cross-linking density constant. The combination of mechanical properties and the insolubility of thermosets with a gradual change in viscosity like inorganic glasses enables processing similar to thermoplastic materials, e.g. extrusion.10–12
The first vitrimers were based on catalyzed transesterification. Since then, several different chemistries, such as olefin metathesis,13,14 aromatic disulfide metathesis,15 amine exchange of vinylogous urethanes,11,16 transalkylation of triazonium salts,17 transesterification of boronic esters,18 and diboroxolan metathesis have been reported.19
Particularly for the preparation of recyclable or reshapable composites, vitrimers could find a promising application. Composites, containing glass or carbon fibers, are gaining increased attention as lightweight and endurable alternatives to metals. These unique properties are especially interesting in structural applications like civil infrastructures or vehicles, where they are already used in Formula One or luxury cars, but it is expected that they find increased use in the automotive sector, e.g. for electric vehicles.20
However, for fiber composites, each component requires its own mold, which needs to be individually fabricated. This expensive process makes composites unsuitable for low production volumes. If it were possible to create a basic shape prepared from vitrimers, which then could be reshaped and formed to the final dimensions, a reduction in costs could be achieved.
Another challenge, which could be overcome by vitrimers, is that with the increasing size of the fabricated composite pieces, the risks of defects and challenges in manufacturing also increase. Thus, composites cannot always be prepared from a single part but engineers are forced to split larger parts into several smaller ones, which require later assembly and fixation strategies. For thermoplastic composites, several welding techniques are known.21,22 In contrast, for thermoset composites, which mostly exhibit more desirable mechanical properties, welding and reprocessing is not an option, because of the cross-linked structure after curing. Therefore, thermoset based composites require mechanical jointing23,24 of the different parts or the use of an additional adhesive.25–29 Due to the possibility for reshaping with temperature, vitrimers represent a potential solution to this issue, as highlighted in previous work.30 Furthermore, vitrimers open the possibility of repairing, recycling, including reprocessing, and separation of the fibers from the matrix, which is almost impossible for conventional fiber-reinforced polymer composites.31
Vitrimers based on vinylogous urethanes were reported in a series of elegant papers by the Du Prez lab. They are an attractive pathway to catalyst-free vitrimers with similar mechanical properties to different commodity polymers.11,16,32 However, most of these vitrimers are highly flammable, making a phosphorus-containing vitrimer an attractive approach to intrinsic flame retardant vitrimers for composite applications. Recently, Wang et al. prepared phosphate-based vitrimers by a Schiff-base chemistry.33 Herein, we prepare a phosphonate-based flame-retardant vitrimer by the versatile vinylogous polyurethane chemistry and compare it to a phosphorus-free analog. Besides, the polymerization was conducted in the presence of glass fibres to present the first vitrimer-embedded and flame retardant composite material. We believe the additional installation of functions into recyclable vitrimers will broaden their scope for a future circular economy.
Experimental
Materials
All chemicals were purchased from commercial suppliers (Sigma Aldrich and Acros Organics) as reagent grade and used without further purification.Methods
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) to give a clear oil (yield: 91.6 g, 58%).
:7) to give a clear oil (yield: 91.6 g, 58%).
1H NMR (300 MHz, Chloroform-d, δ): 4.04 (d, 2H, c-cis), 3.94 (d, 2H, c-trans), 3.49 (d, 2H, h-cis), 3.43 (s, 2H, b), 3.42 (d, 2H, h-trans), 2.24 (s, 3H, a), 1.86–1.77 (m, 4H, d & g), 1.67–0.86 (m, 8H, e & f).
ASAP-MS: 229.2 [M + H]+, 457.5 [2M + H]+ (calculated M+: 228.14).
The crude product was purified by flash chromatography (ethyl acetate/petroleum ether 7:3) to give a clear, slight yellow oil (yield: 82.9 g, 69%).
1H NMR (300 MHz, chloroform-d, δ): 7.79–7.46 (m, 5H, i–k), 4.08–3.76 (m, 8H, c & h), 3.43 (s, 4H, b), 2.22 (s, 6H, a), 1.80 (m, 4H, d & g), 1.46–0.95 (m, 16H, e & f); 13C NMR (75 MHz, chloroform-d, δ): 31P {H} NMR (121 MHz, dichloromethane-d2, δ): 18.95 (d, 1P, 1).
ASAP-MS: 578.7 [M + H]+ (calculated M+: 578.26).
Polymerizations
Prepreg preparations
For poly-1: Monomer 1 (19.2 g; 33.2 mmol; 1 eq.) and the mixture of m-xylylenediamine (2.2 g; 15.9 mmol; 0.48 eq.) and tris(2-aminoethyl)amine (1.8 g; 12.6 mmol; 0.38 eq.) were weight into separate graduated cylinders and filled up to 55 mL with methanol to prepare separate stock solutions. For each prepreg, 5 mL of the two solutions were mixed quickly and 7.5 mL of this solution was then poured over twill weave glass fibres (Type 92125, Twill 2/2, 280 g m−2, P-D Interglass Technologies GmbH, Erbach, Germany) in a silicon mould. The mould with the prepreg was transferred to a preheated convection oven at 60 °C and the methanol was removed for 30 min. The prepregs were cured for 24 h at 90 °C and 1 h at 150 °C.For poly-2: 1,4-Bis(hydroxymethyl)cyclohexane bis-acetoacetate (17.3 g; 55.4 mmol; 1 eq.) and the mixture of m-xylylenediamine (3.6 g; 26.6 mmol; 0.48 eq.) and tris(2-aminoethyl)amine (3.1 g; 21.0 mmol; 0.38 eq.) were weight into separate graduated cylinders and filled up to 55 mL with methanol to prepare separate stock solutions. For each prepreg, 5 mL of the two solutions were mixed quickly and 7.5 mL of this solution were poured over twill weave glass fibres (Type 92125, Twill 2/2, 280 g m−2, P-D Interglass Technologies GmbH, Erbach, Germany) in a silicon mould. The mould with the prepreg was transferred to a preheated convection oven at 60 °C and the methanol was removed for 30 min. The prepregs were cured for 24 h at 90 °C and 1 h at 150 °C.
Plates for cone calorimetry
The vitrimer was shredded in a blender (Bestek BTBL1193). 50 g of the resulting granulate were weighed into a 12 cm × 12 cm steel form. The form with the vitrimer was heated to 150 °C in the press without pressure to prevent deformation of the Teflo sheet. Afterwards, a sheet of Teflo was placed ontop of the vitrimer and the stamp of the form was inserted. The pressure was adjusted to 20 kN at 150 °C for 30 min and to 50 kN for another 30 min. The form was allowed to cool to room temperature, while keeping the pressure at 50 kN.3-Point-bending testing
Dimensions of the composite samples of 70 × 10 × 1.5 mm3 were exactly measured using a caliper (ABS Digimatic CD-15DCX, Mitutoyo Corp., Kawasaki, JP). Experiments were performed according to DIN EN ISO 14125 in a climatised lab (23 °C/50% RH). A universal testing machine (Machine: UTS Testsysteme GmbH & Co. KG, Ulm, DE; control system: Zwick Roell AG, Ulm, DE; Software: TestXpert II, V 3.1, Zwick Roell AG, Ulm, DE) with a load cell with a maximum capacity of 1 kN was used. The distance between the support roles was 56 mm. The displacement speed during the experiments was set to 5 mm min−1.Results and discussion
A novel phosphonate-based monomer (1) for vinylogous polyurethanes was prepared by a two-step synthesis (Fig. 1a). First, 1,4-cyclohexanedimethanol was transesterified with tert-butyl acetoacetate to 1a, which was coupled with dichlorophenylphosphine oxide to compound 1. 1 was obtained as a slightly yellow wax in 40% overall yield. To prevent a Knoevenagel condensation as a side reaction, pyridine was chosen as base to neutralize the hydrochloric acid formed during synthesis, instead of the commonly used triethylamine. The aryl group was installed for two reasons: firstly, it sterically protects the phosphoester-linkages from transesterification and secondly, it can act as a char precursor to improve the flame-retardant properties. | ||
| Fig. 1 (a) Synthesis of the β-ketoester-containing phosphonate monomer 1 and the formation of the vinylogous polyurethane network by polycondensation with amines (b) and (c) (reference vitrimer poly-2). (d) 31P {H} NMR (121 MHz in CDCl3 at 298 K) of 1. (e) 31P MAS solid-state NMR of poly-1, 20 kHz MAS, at 298 K and 121.5 MHz 31P Lamor frequency. (f) Curing of poly-1 glass fiber composites at 90 °C in a convection oven. | ||
Characterization of 1 by 1H and 31P NMR proved the successful synthesis (cf. Fig. 1 and ESI†); 1 exhibited a characteristic resonance at 18.95 ppm in the 31P NMR.
Monomer 1 was copolymerized with m-xylylendiamine and tris(2-aminoethyl)amine to generate the vinylogues polyurethanes. The polycondensation of the commercially available amines with the β-ketoester was conducted at room temperature in solution or elevated temperature in bulk to lower viscosity during the reaction (Fig. 1b and c). The formulation was derived from a previous publication.32 As phosphorus-free reference vitrimer, the vinylogous polyurethane previously published by Du Prez et al. was prepared (poly-2 in Fig. 1c), of which for the first time, fire performance was investigated and compared to poly-1.32
During the spontaneous polycondensation, water is released, which needs to be removed. Therefore, the polymerized material was pressed to 1–2 mm thin plates at 90 °C by a hydraulic press to remove the generated water during the thermal curing in a convection oven. The oven was preheated to 90 °C and the temperature was increased to 150 °C after 24 h for an additional 1 h.
The 31P MAS solid-state NMR of poly-1 revealed a single resonance at 16.77 ppm, showing no sign of degradation or transesterification (Fig. 1e). IR spectroscopy of poly-1 proved the formation of the vinylogous polyurethane network with the distinct vibrations at ca. 1605 cm−1 and 1645 cm−1 for the successful reaction of β-ketoesters with amines, as reported in literature for other vinylogous polyurethanes (Fig. S17†).32 Differential scanning calorimetry (DSC) with a heating rate of 10 K min−1 revealed a glass-transition temperature (Tg) of 70 °C, which is similar to the conventional fiber reinforced polymers matrix polyether-amine cured Bisphenol A diglycidyl ether (DGEBA) (Tg of 88 °C).34 The thermal stability of the resulting network was measured by thermogravimetric analysis in a nitrogen atmosphere and a heating rate at 10 K min−1. The material was stable beyond 200 °C and showed a 5% onset degradation temperature of 285 °C. After heating to 700 °C, a char residue of approx. 22 wt% for the phosphorus-containing vitrimer was found, while the non-phosphorus-based vitrimer (poly-2) proved a lower char yield of approx. 14 wt%.
A key characteristic of vitrimers is their recyclability while sustaining material properties. Therefore, dynamic mechanical analysis was utilized to study the recycling of the phosphonate-based vitrimer (poly-1). The vinylogous polyurethane network was cut into small pieces and then loaded into a mold, where the pieces were compressed at 10 kN for 30 min at 150 °C. After cooling to room temperature while keeping the pressure constant, the samples were removed from the mold and measured by a rheometer in a plate-plate geometry. The recycled poly-1 exhibited a storage modulus (G′) of 0.36 GPa at 50 °C, while poly-2 has a storage modulus of 2.4 GPa at 50 °C32 (in comparison: DGEBA-based epoxy resins exhibited storage moduli of 4.0–8.5 GPa at −25 °C).35 Repeating the recycling a second and a third time, proved materials with similar properties as the as-prepared sample (Fig. 2b and c). Furthermore, the storage modulus (G′) dominated the loss modulus (G′′), which is typical for cross-linked networks. In addition, the max loss modulus in dynamic mechanical analysis confirmed the Tg of 70 °C obtained from DSC measurements, which could probably be altered by changing the amine crosslinker during network preparation.
 | ||
| Fig. 2 (a) Preparation of the plates (poly-1) with a size of 12 × 12 cm2, which are cut to 10 × 10 cm2, and used for cone calorimetry. (b) and (c) Dynamical mechanical behavior of poly-1, showing identical values after recycling. (d) Heat release rate (HRR) over time of vitrimers poly-1 compared to the reference vitrimer (poly-2) and neat epoxy resin and epoxy resin loaded with BDP. (e) Total heat evolved (THE) plotted against the ratio of peak heat release rate to the time of ignition (PHRR/tig) of both vitrimers compared to the epoxy resins (“Petrella plot”). (f) and (g) Char residues of poly-1 and poly-2 after cone calorimeter test. | ||
The intrinsic flame-retardant properties of poly-1 were investigated by forced flaming using the cone calorimeter, which is widely used for assessing the fire behavior of polymer materials.36 The cone calorimeter provides information on the heat release rate (HRR), total heat released (THR), time to ignition (TTI), mass loss rate, smoke emission rate, and average CO and CO2 emissions of a given sample, which represent the most important parameters to characterize fires.37 The heat release curve from cone calorimetry gives information about the thermal thickness of a material and its charring behavior.38 The THR is the integral of HRR with respect to time. The THR at the end of the test is the total heat evolved (THE), which represents the fire load of a material.38 The effective heat of combustion (EHC) is the total heat evolved divided by the total mass loss; it represents the heat available per unit of mass loss in the cone calorimeter. A reduction of EHC is indicative of the gas phase activity of an FR, i.e. flame dilution or flame inhibition.39
The specimen plates for cone calorimeter measurements were prepared by grinding the vinylogous urethane network to a powder using a kitchen blender. 50 g of the powder was transferred to a mold sized 12 × 12 cm2 and compressed at 150 °C with 20 kN for 30 min and 50 kN for another 30 min to form the final plates as shown in Fig. 2a. The mold was allowed to reach 50 °C while keeping the pressure of 50 kN before removing the sample. Of each material, three plates were prepared and cut to the final shape of 10 × 10 cm2. The plates had a thickness of approx. 3 mm. The phosphonate-based vitrimer poly-1 exhibited a clear reduction in THE by 27% (from 89 MJ m−2 to 65 MJ m−2) and PHRR by 33% (from 2346 kW m−2 to 1566 kW m−2) compared to the phosphorus-free vitrimer (poly-2). The reduction in THE and PHRR is explained by the increased residue from 10 wt% to 23 wt% in cone calorimetry, which is in agreement with TGA measurements, of the phosphonate-based vitrimer poly-1: the fixation of fuel in the char decreased the fire load, and the combination of increased residue yield and gas emission led to an intumescent behavior in fire, exhibiting an increased protective layer effect, which lowered the PHRR (Fig. 2f and g show photos of the chars obtained after cone calorimetry). The protective layer effect was further evidenced by the plateau-like shape of the HRR curve resulting from the strong intumescent behavior. The intumescent behavior was supported by the phosphorus in poly-1 since it acted as an acid source, promoting the formation of a strong and voluminous multicellular char, which acted as a thermal insulator (Fig. 2f and g).
In pyrolysis combustion flow calorimetry (PCFC) measurements, similar heat release capacities (HRC) for both materials were determined (287 ± 2 and 284 ± 6 J g−1 K−1 for the reference poly-2 and the phosphonate-based vitrimer poly-1, respectively). As PCFC cannot detect flame poisoning, the nearly identical HRC values indicated a gas phase mechanism via flame-poisoning for the phosphonate-based vitrimer poly-1 as a major mode of action during the fire. This assumption was further supported by a reduction of EHC by 14% as seen in the cone calorimetry data (from 28 MJ kg−1 to 24 MJ kg−1). In addition, the THE-values measured by PCFC of 24.65 kJ g−1 for the reference poly-2 and 24.6 kJ g−1 for the phosphonate-based vitrimer poly-1, were very similar, which suggests no additional contribution to THE from the phosphorus species. Both UL-94 and LOI measurments proved the flame-retardant effect of the phosphonate groups in poly-1 compared to the phosphorus-free analogue of poly-2 (Table S4†). In UL-94 vertical tests, poly-2 burned intensely after ignition and burned to the clamp, leading to a N.R. rating. The material further exhibited strong burning dripping. Contrarily, poly-1 attained a V-2 rating: the material became viscous almost immediately after ignition, causing some initial burning dripping. However, the dripping also caused the specimen to extinguish. The material could not be further ignited, as its low viscosity could not hold a flame. Therefore, poly-1 exhibited a much better fire behavior than the phosphorus-free poly-2, as its strong melt-dripping characteristic protected the bulk of the material from further combustion. Dripping or melt-flow in fires is common for thermoplastics, and although it may lead to higher burning rates or pool fires in some cases, it also removes fuel and heat from the pyrolysis front or fire source by dripping away, as is the case for poly-1.40 Poly-2 exhibited an oxygen index (OI) of 26.1% due to its tendency to char, while poly-1 displayed a lower OI at 21.3% owing to its tendency to drip along the edges of the sample. Reaction-to-small-fire tests highlighted the ability of poly-1 to act via melt-dripping with small flames rather than through the formation of residue like poly-2. This dripping behavior led to a V-2 classification for poly-1, but its lower temperature to become liquid also led to a lower OI compared to poly-1. Further flame retardant studies on a systematic library of different phosphorus-based vitrimers is currently under way.
Epoxy resins are often used in fiber-reinforced composites. Therefore, a comparison of the flammability of epoxy resins with both vitrimers is interesting. According to our recent publication,41 a DGEBA/2,2′-dimethyl-4,4′-methylene-bis-(cyclohexylamine)-based epoxy resin exhibited a THE of 108 MJ m−2 and a PHRR/tig of 36 kW m−2 s−1 (Fig. 2e). The fire-load, which is indicated by the THE, was significantly reduced for both vitrimers compared to the epoxy resin. Adding 10 wt% BDP to the epoxy resin lowered the THE and PHRR significantly, but the THE with 87 MJ m−2 was on a similar level to the reference vitrimer poly-2 but still considerably higher than the phosphonate-based vitrimer poly-1. However, the fire growth index (PHRR/tig) was higher than 90 kW m−2 s−1 for both vitrimers as seen in Fig. 2e and thus higher as the fire growth index of the neat epoxy (36 kW m−2 s−1) and the epoxy loaded with 10 wt% BDP (29 kW m−2 s−1). The above-mentioned data underlines the promising application of intrinsic flame-retardant vitrimers in composite materials. In conventional composites epoxides are often used due to their good mechanical properties. Therefore, we also assessed the mechanical properties in composites of our material.
Poly-1 and poly-2 were used for fiber reinforced polymer preparation with glass fabrics: the pre-impregnated fibers (“prepregs”) were prepared by polycondensation of the β-ketoester monomers of the reference or the phosphonate-based vitrimer (1) and the amines directly on the glass fiber textiles. The monomers were dissolved separately in methanol and for each prepreg 5 mL of the stock solutions were mixed quickly and approx. 7.5 mL were poured over single layers of twill weave glass fibers in a silicon mold. The mold with the prepreg was transferred to a preheated convection oven at 60 °C and the methanol was evaporated. The prepregs were cured for 24 h at 90 °C and 1 h at 150 °C.
Optimum heat press parameters for the manufacturing the glass fiber reinforced polymers (GFRP) were determined in preliminary experiments performing peel tests. With decreasing thickness of spacers (from 0.6 to 0.0 mm), the quality of the composite increases as indicated by higher peel forces. The composites were manufactured by stacking six prepreg layers and consolidating them using a preheated hot press at 150 °C and 10 kN for 1 h. No polymer loss from the prepregs was observed during hot pressing and the glass fabric layers were well impregnated (see Fig. 3c and e). The fiber mass content of the composite was 62.2% for the herein presented vitrimer poly-1 and 58.6% for the reference system poly-2 as determined by TGA of the composites and the unreinforced polymers (Tables S1–S3†). From the resulting composites, 5–6 specimens with the dimensions 10 × 70 mm2 were cut using guillotine shears (Fig. 3e). The samples were conditioned in a desiccator with a dry atmosphere to exclude effects of humidity uptake until testing.
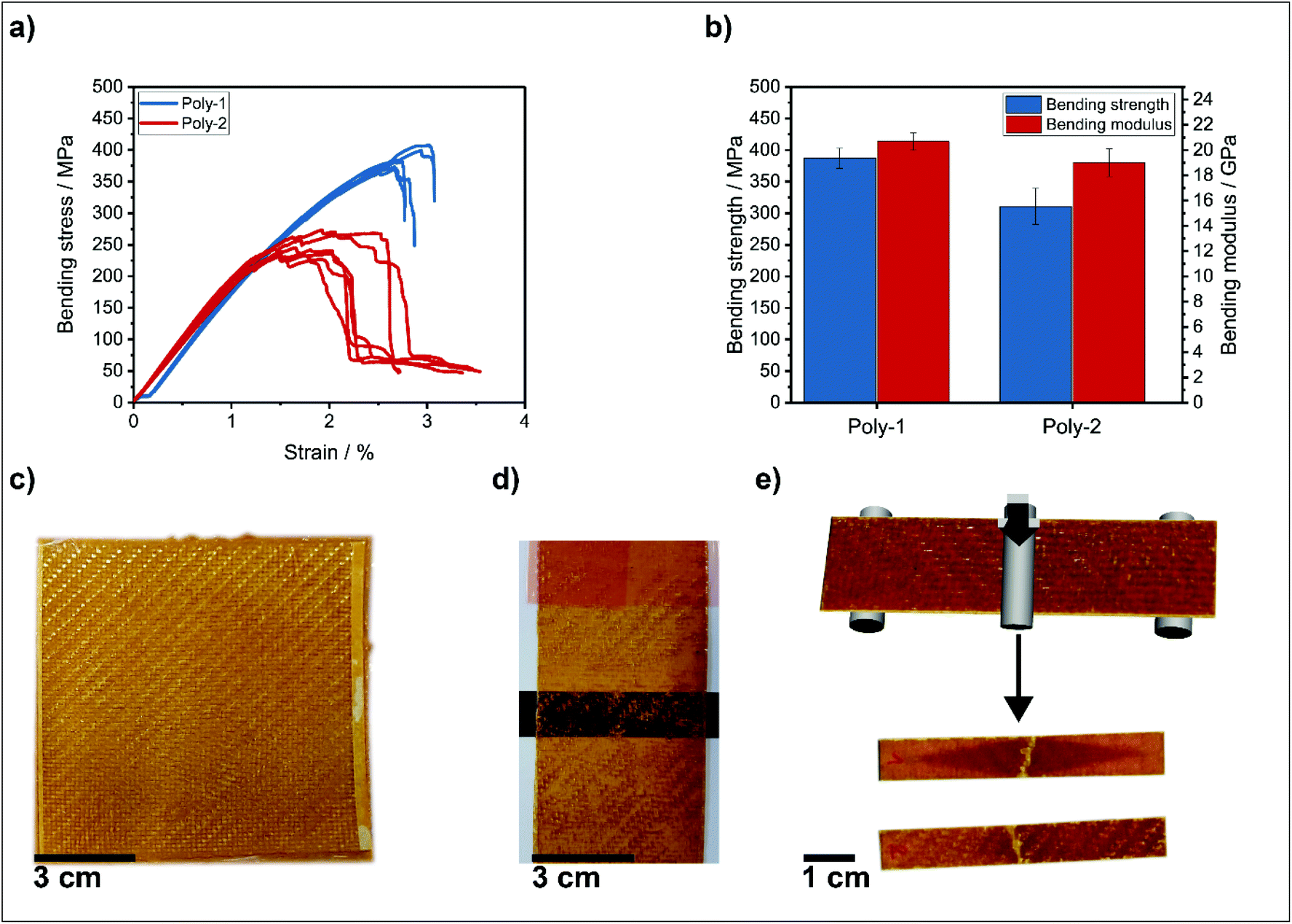 | ||
| Fig. 3 (a) Stress–strain curves from the bending experiments. Blue curves correspond to the composites with poly-1, red curves to the reference poly-2. (b) Bending strength and bending modulus of the composites with poly-1 and poly-2. (c) Prepreg of poly-1 after curing. (d) 2 prepregs were stacked and compression-molded with a separation layer on top for delamination experiments. (e) From the composite, test specimens were cut and mechanical properties were determined in 3-point bending experiments. | ||
The mechanical properties of the composites determined by 3-point-bending tests were performed according to DIN EN ISO 14125 in an air-conditioned test lab (23 °C, 50% RH) and are shown in Fig. 3a and b. The good mechanical properties proved the high potential of the vitrimers for composite applications: a bending strength of 387 ± 16 MPa and a bending modulus of 20.7 ± 0.7 GPa for poly-1 composites were determined. These values for the phosphorous-containing vitrimer were proven within the range of permanently cross-linked epoxy resins reinforced with the same glass fibers and processed via vacuum infusion.42 The specimens showed fiber failure under tension at the bottom sides and without larger scale delamination of the layers (Fig. 3e). This failure behavior indicates adequate load transfer from the polymer to the fibers and explains the high mechanical values.
While a more or less ductile stepwise failure was observed for the reference poly-2 (higher strain at fracture compared to strain at maximum strength), the phosphorus-containing vitrimer poly-1 had a more brittle failure when reaching the maximum bending strength (see Fig. 3a).
Summary
To avoid flame retardant additives in recyclable composite materials, we prepared a phosphonate-based and intrinsic flame retardant vitrimer and investigated its applicability in glass fiber reinforced composites. Vinylogous polyurethane chemistry was used to prepare the phosphonate-based vitrimer and an analog phosphorus-free vitrimer for comparison. Poly-1 was successfully recycled, which was proven by dynamic mechanical analysis after compressing the blended flakes. In cone calorimeter tests, poly-1 exhibited a clear reduction of the THE by 27% and PHRR by 33% compared to the reference poly-2. The reduction in THE and PHRR can be explained by the increased residue from 10 wt% to 23 wt% (obtained by cone calorimetry) of the phosphonate-based vitrimer poly-1 and the, therefore, fixation of fuel in the char, which decreased the fire load. The combination of increased residue yield and gas emission led to an intumescent behavior in fire.Successful preparation of glass-fiber reinforced composites of poly-1 proved values for the bending strength and bending modulus for poly-1 similar to permanently cross-linked epoxy resins reinforced with the same glass fibers.42 The specimens exhibited fiber failure under tension at the bottom sides and without larger scale delamination of the layers. This failure behavior indicates adequate load transfer from the polymer to the fibers and explains the high mechanical values. Such materials could be sustainable and recyclable alternatives to currently used fiber-reinforced polymers and broaden the scope of available vitrimers to polyphosphonates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG WU 750/8-1; SCHA 730/15-1) for funding. F. R. Wurm and Jens C. Markwart thank Prof. Dr Katharina Landfester (MPI-P, Germany) for support. We thank Dr Kaloian Koynov and Andreas Hanewald for their support with dynamic mechanical analysis. The authors thank Katharina Maisenbacher (MPIP) for assistance with graphical design. The authors K. Haag, T. Urbaniak and K. Koschek gratefully acknowledge the financial support from the Bundesministerium für Bildung und Forschung (BMBF) through the NanoMatFutur award (DuroCycleFVK 03XP0001). We further thank Dr Hannes Schäfer for technical support. Open Access funding provided by the Max Planck Society.References
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- B. Perret, B. Schartel, K. Stöβ, M. Ciesielski, J. Diederichs, M. Döring, J. Krämer and V. Altstädt, Eur. Polym. J., 2011, 47, 1081–1089 CrossRef CAS.
- S. V. Levchik and E. D. Weil, J. Fire Sci., 2006, 24, 345–364 CrossRef CAS.
- S.-Y. Lu and I. Hamerton, Prog. Polym. Sci., 2002, 27, 1661–1712 CrossRef CAS.
- S. D. Shaw, A. Blum, R. Weber, K. Kannan, D. Rich, D. Lucas, C. P. Koshland, D. Dobraca, S. Hanson and L. S. Birnbaum, Rev. Environ. Health, 2010, 25, 261–305 CAS.
- M. M. Velencoso, A. Battig, J. C. Markwart, B. Schartel and F. R. Wurm, Angew. Chem., Int. Ed., 2018, 57, 10450–10467 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- Y.-X. Lu, F. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- L. Nicolais, M. Meo and E. Milella, Composite Materials: A Vision for the Future, Springer, London, 2011 Search PubMed.
- C. Ageorges, L. Ye and M. Hou, Composites, Part A, 2001, 32, 839–857 CrossRef.
- D. Stavrov and H. E. N. Bersee, Composites, Part A, 2005, 36, 39–54 CrossRef.
- E. W. Godwin and F. L. Matthews, Composites, 1980, 11, 155–160 CrossRef.
- M. J. Troughton, Handbook of Plastics Joining: A Practical Guide, Elsevier Science, 2008 Search PubMed.
- N. A. de Bruyne, Aircr. Eng. Aerosp. Technol., 1944, 16, 115–118 CrossRef.
- F. L. Matthews, P. F. Kilty and E. W. Godwin, Composites, 1982, 13, 29–37 CrossRef CAS.
- J. R. J. Wingfield, Int. J. Adhes. Adhes., 1993, 13, 151–156 CrossRef CAS.
- H. S. da Costa Mattos, E. M. Sampaio and A. H. Monteiro, Int. J. Adhes. Adhes., 2011, 31, 446–454 CrossRef CAS.
- H. S. da Costa Mattos, A. H. Monteiro and R. Palazzetti, Mater. Des., 2012, 33, 242–247 CrossRef CAS.
- E. Chabert, J. Vial, J.-P. Cauchois, M. Mihaluta and F. Tournilhac, Soft Matter, 2016, 12, 4838–4845 RSC.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Macromolecules, 2018, 51, 8001–8012 CrossRef CAS.
- T. H. Hsieh, A. J. Kinloch, K. Masania, A. C. Taylor and S. Sprenger, Polymer, 2010, 51, 6284–6294 CrossRef CAS.
- C. R. Amaral, R. J. S. Rodriguez, F. G. Garcia, L. P. B. Junior and E. A. Carvalho, Polym. Eng. Sci., 2014, 54, 2132–2138 CrossRef CAS.
- V. Babrauskas, Fire Mater., 1984, 8, 81–95 CrossRef CAS.
- SO 5660-1:2015, Reaction to fire tests: heat release, smoke production and mass loss rate—part 1: heat release rate (cone calorimeter method) and smoke production rate (dynamic measurement). International Organization for Standardization, Geneva, 2015.
- B. Schartel and T. R. Hull, Fire Mater., 2007, 31, 327–354 CrossRef CAS.
- B. Schartel, B. Perret, B. Dittrich, M. Ciesielski, J. Krämer, P. Müller, V. Altstädt, L. Zang and M. Döring, Macromol. Mater. Eng., 2015, 301, 9–35 CrossRef.
- F. Kempel, B. Schartel, J. M. Marti, K. M. Butler, R. Rossi, S. R. Idelsohn, E. Oñate and A. Hofmann, Fire Mater., 2015, 39, 570–584 CrossRef CAS.
- J. C. Markwart, A. Battig, L. Zimmermann, M. Wagner, J. Fischer, B. Schartel and F. R. Wurm, ACS Appl. Polym. Mater., 2019, 1, 1118–1128 CrossRef CAS.
- K. Haag, J. Deitschun, D. Godlinski, V. Zoellmer and K. Koschek, Presented in part at the ECCM18 – 18th European Conference on Composite Materials, Athens, Greece, 24–28th June 2018, 2018.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00275e |
| This journal is © The Royal Society of Chemistry 2020 |