
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(ethylene glycol)-b-poly(vinyl acetate) block copolymer particles with various morphologies via RAFT/MADIX aqueous emulsion PISA†
Paul
Galanopoulo
,
Pierre-Yves
Dugas
 ,
Muriel
Lansalot
* and
Franck
D'Agosto
*
,
Muriel
Lansalot
* and
Franck
D'Agosto
*
Univ Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5265, Chemistry, Catalysis, Polymers and Processes (C2P2), 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr; muriel.lansalot@univ-lyon1.fr
First published on 22nd May 2020
Abstract
The polymerization-induced self-assembly (PISA) of amphiphilic diblock copolymers of poly(ethylene glycol)-b-poly(vinyl acetate) (PEG-b-PVAc) in water was achieved through macromolecular design via interchange of xanthate (MADIX) polymerization in emulsion. Poly(ethylene glycol) with a dithiocarbonate (xanthate) end-group (PEG-X) was synthesized by post-modification of commercial poly(ethylene glycol). The chain extension of PEG-X was then conducted in water using vinyl acetate (VAc) to produce PEG-b-PVAc amphiphilic diblock copolymers. Their in situ self-assembly led to stable latex particles. The molar mass of the PVAc block was varied to trigger the formation of various morphologies including spheres, vesicles and large compound vesicles.
Introduction
Polymerization-induced self-assembly (PISA)1,2 is a concept that takes advantage of a living polymerization technique to extend preformed solvophilic polymer chains with either solvophobic (emulsion) or solvophilic (dispersion) monomer units. In both cases, the second block formed is however solvophobic, and simultaneously to its growth the resulting block copolymers can self-assemble into nanoparticles. PISA has been largely employed in recent years to produce block copolymer nanoparticles in water, under either dispersion or emulsion conditions, using mainly reversible addition–fragmentation chain transfer (RAFT).2 In addition, by varying the nature and molar mass of the two blocks and/or the solid content, different morphologies such as spheres, worms or vesicles, already depicted when self-assembling preformed block copolymers in a selective solvent of one of the two blocks,3 can also be produced by PISA in water up to a high solid content.4While these morphologies seem to be easily obtained in aqueous dispersion polymerization by simply varying the molar mass of the hydrophobic block or the targeted solid content,5 emulsion polymerization is found to be more challenging to reach higher-order morphologies different from spheres.6–19 As is now generally accepted, these morphologies result from the fusion of the original block copolymer spherical micelles and further chain rearrangements.20 In dispersion polymerization, the core monomer is soluble in water and makes its polymer swell,21 which favors fusion and rearrangement events. In emulsion polymerization,22 although the core monomer is also able to make its polymer swell, its hydrophobic character induces the presence of monomer droplets. A small fraction is also dissolved in water. Therefore, in emulsion PISA, once the first block copolymer micelles are formed the monomer has to diffuse from these droplets through the water phase to feed the particles with the monomer. This diffusion is thus allowed but also limited by the solubility of the monomer in water, which directly impacts the plasticization of the particles. This simple observation naturally led researchers to consider that hydrophobic monomers exhibiting high water solubility should favor the fusion and rearrangements of the chains mentioned above, and thus the formation of various morphologies by PISA in aqueous emulsions. This was effectively shown when using methyl methacrylate (MMA) instead of styrene (respective water solubilities at 50 °C: 15 g L−1 (ref. 23) vs. 0.45 g L−1 (ref. 24)) in a RAFT-mediated PISA system with poly(methacrylic acid-co-poly(ethylene glycol) methyl ether methacrylate) as a hydrophilic macromolecular chain transfer agent (macro-CTA). Under exactly the same conditions (pH 3.5), emulsion polymerization of styrene systematically led to spheres whatever the targeted degree of polymerization (DP, from 150 to 570) of the PS block,25 while the use of MMA allowed the formation of spheres, fibres and vesicles for DPs of 200, 300 and 500, respectively.9 This strategy was recently confirmed by Armes and coworkers who also obtained non-spherical morphologies by polymerizing in emulsion 4-hydroxybutyl methacrylate (HBMA, 20 g L−1 at 70 °C),13 glycidyl methacrylate (GlyMA, 24–25 g L−1 at 80 °C),14 and 2-methoxyethyl methacrylate (MOEMA, 19.6 g L−1 at 70 °C).16
With a solubility in water of 26 g L−1 at 50 °C,26 vinyl acetate (VAc) is one of the most hydrophilic monomers among the monomers classically employed in aqueous emulsion polymerization, and poly(vinyl acetate) (PVAc) latexes find industrial applications in various domains such as adhesives, paints, and coatings.27 The particles obtained in this process are systematically spherical. Taking advantage of the best attributes of PISA for VAc emulsion polymerization is thus very appealing in order to generate new types of non-spherical PVAc particles. Vinyl acetate is a less activated monomer and its RAFT polymerization requires the use of specific macro-CTAs carrying dithiocarbamate or dithiocarbonate (xanthate, X) chain ends.28 In this last case, the polymerization process is termed macromolecular design via interchange of xanthate (MADIX). To the best of our knowledge, only five papers depict the use of MADIX-mediated PISA for less activated monomers.17,29–32 Two of them focus on VAc emulsion polymerization, conducted with dithiocarbonate end functionalized macro-CTAs. In the first case, a dithiocarbonate end functionalized dextran (Dext-X) was used.29 The functionalization rate was rather low (ca. 30%), and when employed to mediate emulsion polymerization of VAc, spherical particles were obtained. Their formation and stabilization were possible by a fraction of in situ formed dextran-b-PVAc block copolymers. In the second study, performed in our group, emulsion polymerization of VAc was carried out in the presence of a PEG quantitatively functionalized with a dithiocarbonate end group (PEG-X).30 When using a redox initiation at 25 °C, kinetics was slow, but the control of the polymerization was shown by the increase of the molar mass upon VAc conversion and low dispersities that remained around 2, as expected. Here again, however, spherical PVAc particles composed of PEG-b-PVAc were systematically observed. The choice of the low temperature initiating conditions was conditioned by the comparative use in the same paper of poly(N-acryloylmorpholine) and poly(N-vinylpyrrolidone) macro-CTAs, the thermal instability of the latter having been demonstrated earlier.33
Building on these results, we anticipated that conducting PEG-X mediated VAc polymerization at higher temperature may not only lead to faster kinetics but also favor the softening of the particles considering both the glass transition temperature (Tg) of PVAc (ca. 30 °C)34 and the improved plasticization with VAc. In fact, this could favor fusion and rearrangements of the polymer chains to achieve the yet unattained higher-order morphologies.
Emulsion polymerizations of VAc were thus conducted in the presence of PEG-X at 70 °C using 2,2′-azobis(2-amidinopropane) dihydrochloride (AIBA) as an initiator at a solid content of 10 wt%, targeting different DPs for the PVAc block (DPtarget from 50 to 200). A systematic characterization of the macromolecular and colloidal features of the resulting latexes was performed. Particular attention was paid to the development of robust systems leading to higher-order morphologies.
Materials and methods
Materials
Vinyl acetate (VAc, 99%, Aldrich), 2,2′-azobis(2-amidinopropane) dihydrochloride (AIBA, 97%, Aldrich), poly(ethylene glycol) methyl ether (PEG-OH, Mn = 2000 g mol−1, Aldrich), O-ethyl xanthic acid potassium salt (98%, Alfa Aesar), triethylamine (99%, Aldrich), 2-bromopropionyl bromide (97%, Aldrich), dichloromethane (ACS reagent, Aldrich), ammonium chloride (99.5%, ACS reagent, Aldrich), hydrogen carbonate (ACS reagent, Aldrich), sodium chloride (99%, ACS reagent, Aldrich), and anhydrous magnesium sulfate (99.5%, Alfa Aesar) were used as received. Water was deionized before use (Purelab Classic UV, Elga LabWater).Experimental procedures
Preparation of PEG-X
General procedure for the emulsion polymerization of VAc in the presence of PEG-X
All emulsion polymerizations were performed using the following protocol. VAc, PEG-X, AIBA and deionized water were introduced into a 50 mL one-neck round bottom flask, which was immersed in an ice bath. After degassing of the medium for 15 min under argon, the flask was placed in an oil bath previously heated at 70 °C. After 90 min under stirring at about 400 rpm, the reaction was quenched by immersion of the flask in an ice bath and exposure to air. VAc consumption was followed by gravimetric analysis of samples withdrawn from the medium at different times.Analytical techniques
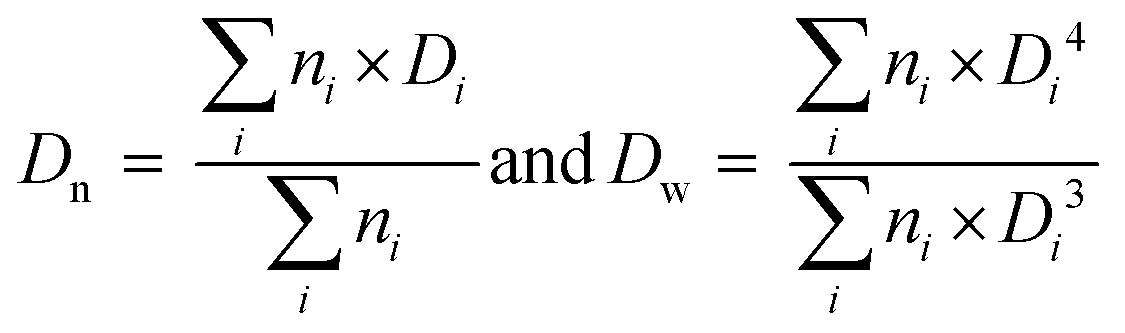
Results and discussion
PEG-X mediated emulsion polymerization of VAc for a DPtarget of 51
As mentioned in the introduction, PEG-b-PVAc spherical particles were already successfully obtained through PEG-X mediated VAc emulsion polymerization in water at 25 °C.30 Building on these preliminary results, a new set of experiments was implemented using an azo initiator (AIBA), setting the temperature at 70 °C, and targeting different DPs for the PVAc segment. The solid content was set to 10 wt%.A first experiment was conducted targeting a DP of 51 (entry A1, Table 1). As expected, and in strong contrast to our previous works for which 80% conversion was reached in 50 h, the polymerization achieved conversions higher than 90% in less than 60 min (Fig. 1a). SEC traces of samples withdrawn during the polymerization show the presence of more than one population (Fig. 1b). Accordingly, the dispersities are somewhat high and increase with the conversion (Fig. 1c).
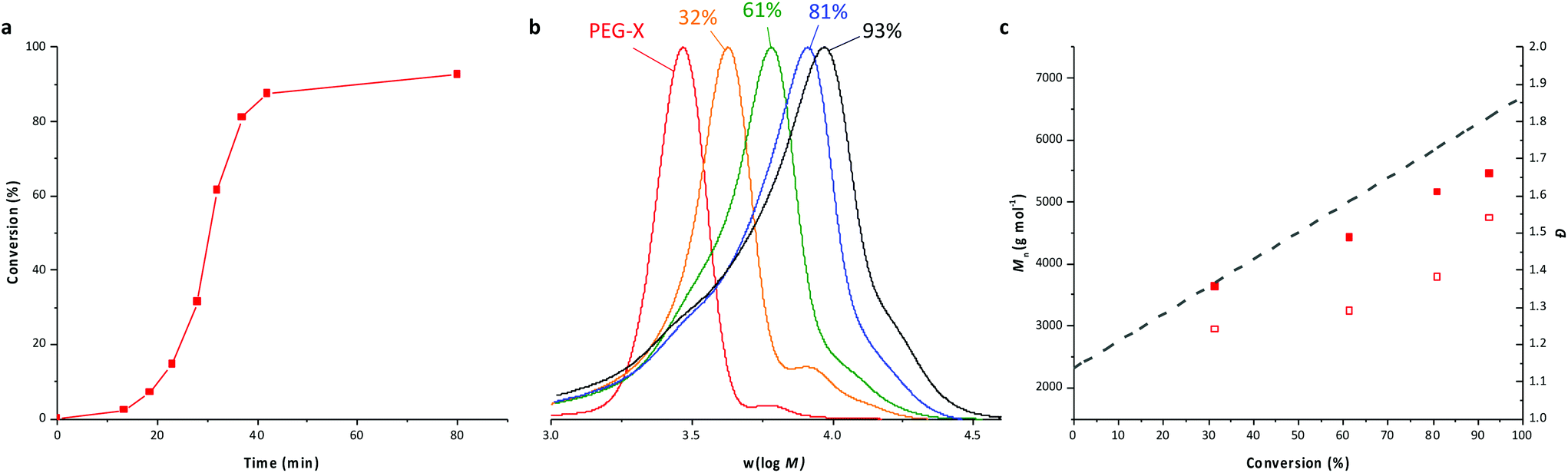 | ||
| Fig. 1 PEG-X mediated VAc emulsion polymerization for DPtarget = 51 (entry A1, Table 1). (a) Evolution of conversion versus time, (b) SEC traces (RI detection) for samples withdrawn during the polymerization. (c) Corresponding evolution of the number-average molar masses Mn (plain squares) and dispersity (Đ = Mw/Mn, hollow squares) with VAc conversion (the dashed line corresponds to the theoretical Mn values). | ||
| Exp | DPtargetb | t (min) |
τ
th![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (wt%) c (wt%) |
τ (wt%) | Conv.d (%) | DPexpe |
M
n
thf (g mol−1) |
M
n
SECg (g mol−1) |
Đ |
M
n
NMRi (g mol−1) |
Z ave (nm) PdIj | Morphologyk (Dn (nm)/Dw/Dn) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a All experiments were performed at 70 °C with [PEG-X]/[AIBA] = 3. The final pH was around 3 for each latex (for an initial pH ranging between 4.5 and 5. The slight decrease of the pH value can likely be attributed to the formation of some acetic acid resulting from the hydrolysis of VAc, which is however very limited at pH ca. 4.534). b DPtarget is defined by the experimental ratio: [PEG-X]/[VAc]. c Theoretical solid content (τth) and experimental final solid content measured by gravimetric analysis (τ). d Conversion calculated from the solid content. e Experimental DP calculated from the conversion. f Theoretical number-average molar mass of the block copolymer calculated using the conversion and the targeted DP. g Experimental number-average molar mass of the block copolymer determined by SEC-THF using a calibration based on polystyrene standards. h Dispersity of the block copolymers, Mw/Mn, determined by SEC-THF. i Experimental number-average molar mass of the block copolymers determined by 1H NMR. j Intensity-average diameter and polydispersity of the final latex from DLS. k Main morphologies based on cryo-TEM images of the final latex: S = spheres, W = worms, V = vesicles, and LCV = Large compound vesicles. For the spheres, the number-average diameter and dispersity index determined by statistical analysis of the TEM images are also indicated (Fig. S17†). l For this experiment, Zave was not relevant as the DLS showed the presence of a very low amount of larger objects that were however not spotted during TEM observations (Fig. S18†). m For this experiment, multilamellar vesicles were also observed (see A5 cryo-TEM image, Fig. 6). | ||||||||||||
| A1 | 51 | 80 | 10.4 | 9.8 | 93 | 47 | 6400 | 5460 | 1.5 | 6300 | 22 | S (15.9/1.04) |
| 0.05 | ||||||||||||
| A2 | 62 | 120 | 10.4 | 9.8 | 95 | 59 | 7400 | 8300 | 1.7 | 7230 | 65 | S + V |
| 0.50 | ||||||||||||
| A3 | 75 | 125 | 10.2 | 9.6 | 94 | 71 | 8400 | 7930 | 1.8 | 8130 | 113 | S + V |
| 0.21 | ||||||||||||
| A4 | 100 | 130 | 10.0 | 9.4 | 92 | 92 | 10200 |
9450 | 2.1 | 11800 |
203 | V |
| 0.16 | ||||||||||||
| A5 | 152 | 80 | 10.2 | 9.7 | 95 | 144 | 14700 |
14360 |
2.1 | 14450 |
197 | Vm |
| 0.07 | ||||||||||||
| A6 | 199 | 90 | 10.1 | 9.1 | 94 | 187 | 18400 |
17920 |
2.3 | 18270 |
186 | LCV |
| 0.15 | ||||||||||||
| B1 | 51 | 80 | 15.4 | 14.8 | 95 | 48 | 6400 | 6930 | 1.5 | 6300 | 57 | S (17.3/1.04) |
| 0.19l | ||||||||||||
| B2 | 76 | 90 | 15.1 | 14.9 | 97 | 73 | 8600 | 8800 | 1.8 | 7780 | 241 | S + V (+W) |
| 0.56 | ||||||||||||
| B3 | 100 | 80 | 15.0 | 14.6 | 97 | 97 | 10600 |
10730 |
1.9 | 10900 |
437 | V |
| 0.40 | ||||||||||||
Two phenomena could explain these results. (i) A shoulder on the high molar mass side of the molar mass distribution (MMD) is visible. It is shifted, together with the whole distribution, towards high molar masses upon VAc conversion. It is worth mentioning here that PEG-X MMD features such a shoulder too, but of very small intensity (Fig. S4†). Indeed, this shoulder probably corresponds to contamination by dihydroxy telechelic PEG formed as a side product during anionic polymerization of ethylene oxide (traces of water initiation). The presence of these species probably led to the formation of a small amount of dixanthate telechelic PEG after the functionalization of PEG-OH (X-PEG-X), visible on SEC analyses using both RI and UV detection at 270 nm (Fig. S4†). During the VAc emulsion polymerization, these chains could thus be extended into PVAc-b-PEG-b-PVAc triblock copolymers and would correspond to the shoulder observed on the high molar mass of the distribution (Fig. S5†). (ii) The MMD also features a tailing on the low molar mass side. VAc's inclination to produce head-to-head (HH) units during the polymerization is documented in the literature and has been particularly discussed in the case of RAFT/MADIX.35–38 The corresponding chains terminated by –CH(OAc)-CH2-X are much less reactivatable than the expected –CH2-CH(OAc)-X. Indeed, the tailing observed on the low molar mass side is thus not ascribed to unreacted PEG-X (see below), although it is worth mentioning that the degradation of PEG-X during the reaction or slow reinitiation could contribute to this tailing. Rather, –CH(OAc)-CH2-X terminated block copolymers probably accumulate. This is indeed consistent with the SEC traces recorded using UV at 270 nm (Fig. S5†) that show that the chains causing this tailing also carry a xanthate moiety. All in all, either (i) or (ii) assumption gives rise to side populations that are nevertheless amphiphilic block copolymers containing PEG and PVAc segments that will be involved in the self-assembly process.
The 1H NMR spectrum (Fig. 2) recorded for the powder obtained after drying the latex confirms the presence of signals at about 3.15 ppm and 4.05 ppm, which are, respectively, characteristics of HH units linked to a xanthate moiety and of dead chains.36 The full assignment of this 1H NMR spectrum shows that no trace of remaining PEG-X could be detected, indicating that the majority of these chains have been chain extended. The 1H NMR assignments further allow the calculation of the final molar mass of the formed chains by the relative integration of the signal attributed to the protons f at 4.90 ppm compared to the signal attributed to the protons of the methoxy group at 3.37 ppm. It shows an almost perfect fit with the expected theoretical values (Table 1, A1). These data are additionally consistent with the molar mass data collected from SEC that show a linear increase of the number-average molar masses upon VAc conversion (Fig. 1c). A good match between the expected and experimental values is obtained although the values obtained using polystyrene calibration might be slightly flawed. Nevertheless, the consistency among the molar mass values obtained from 1H NMR, SEC and theory is a good indication that the degradation of PEG-X suspected above is low (if any), and that the majority of the chains are extended during VAc polymerization. These results confirmed that the expected amphiphilic block copolymers were formed.
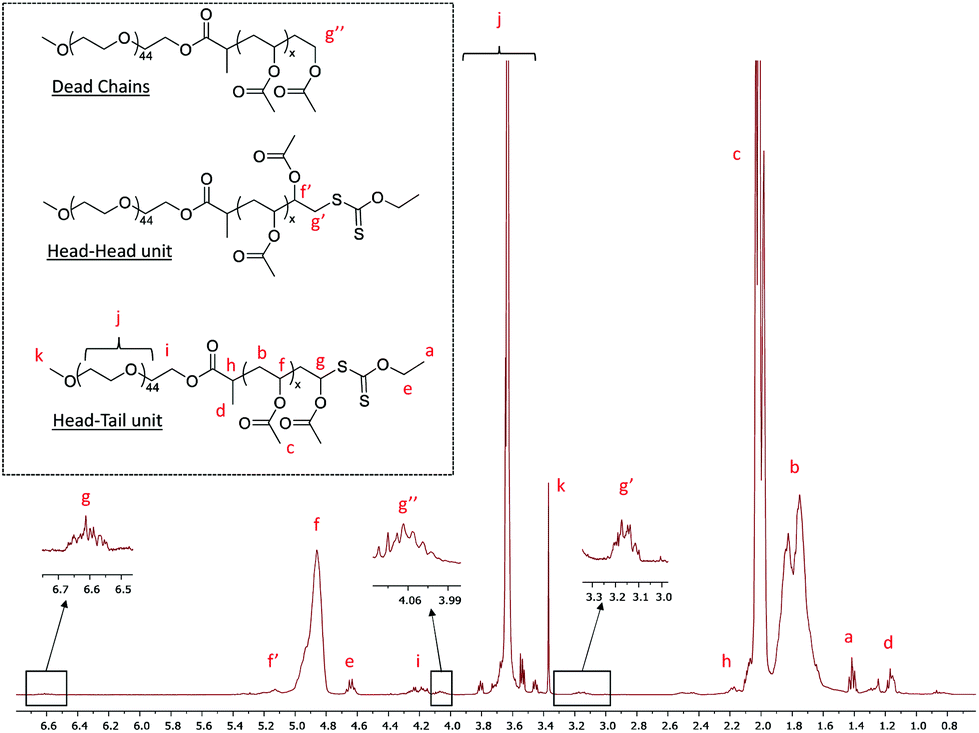 | ||
| Fig. 2 1H NMR spectrum (CDCl3, 256 scans) of the dried latex obtained from entry A1, Table 1, after 93% conversion. | ||
The polymerization A1 in Table 1 visually led to a clear solution (Fig. 3, DPexp = 47). However, as seen from the cryo-TEM image (Fig. 4) and DLS results (Table 1), the obtained medium is a latex made of isometric spherical particles with a diameter of ca. 20 nm. It can thus be acknowledged that these spherical particles are made of PEG-b-PVAc amphiphilic block copolymers resulting from their direct self-assembly, which is consistent with the expected PISA mechanism.
 | ||
| Fig. 3 Digital pictures of the crude latexes A1, A2, A3 and A6 (Table 1). | ||
 | ||
| Fig. 4 Representative cryo-TEM image of the latex obtained from entry A1 in Table 1 and the corresponding particle size distribution of the crude latex. | ||
Influence of the targeted DP of the PVAc block
The DPtarget was then increased to investigate the impact on the PEG-b-PVAc self-assembly. A set of experiments was thus performed varying the DPtarget of the PVAc block between 50 and 200 (entries A1 to A6, Table 1). The total solid content was kept at 10 wt%.For all the aforementioned syntheses, as exemplarily shown in Fig. 5a, the polymerization proceeded to almost completion (>92%) in less than 60 min. It remains however difficult to compare the kinetics of the different experiments with each other in detail since the initiator, monomer and PEG-X concentrations were modified simultaneously to maintain the solid content constant. Nevertheless, the very similar profiles obtained are consistent with a rapid compartmentalization of the reaction within particles (nucleation).
 | ||
| Fig. 5 PEG-X mediated VAc emulsion polymerizations when targeting different DPs for the PVAc block. Evolution of (a) conversion versus time and (b) number-average molar masses Mn (plain squares) and dispersity (Đ = Mw/Mn, hollow squares) with VAc conversion (the dashed lines correspond to the theoretical Mn values). See Table 1 for details. | ||
Overall, all the experiments showed a good blocking efficiency as shown by the linear increases of number-average molar masses with the conversion (Fig. 5b). Once again, the molar masses obtained using SEC do not fully match with the expected ones probably due to the use of polystyrene standards, which are not well suited for the particular nature of the PEG-b-PVAc block copolymers. However, the molar masses calculated according to the 1H NMR spectra of the final latexes are in good agreement with the theoretical ones (Table 1).
The SEC traces obtained for all these experiments are similar to those of latex A1 (Fig. S6–S10†). However, the dispersity tends to increase with both the conversion and the targeted degree of polymerization of the PVAc block. This might be related to the above-mentioned side formation of PVAC-b-PEG-b-PVAC triblock copolymers, the impact of which on the dispersity is probably exacerbated when the DP of the PVAc segment increases.
The obtained latexes showed different aspects depending on the DPexp of the PVAc segment. As mentioned above, the synthesis with the lowest DP (A1, DPexp = 47) led to a completely transparent dispersion. However, as the DP increases, the latex becomes slightly translucent (A2, DPexp = 59) before showing the aspect of a classical milky dispersion (A3, DPexp = 71 to A6, DPexp = 187) (Fig. 3). DLS analysis results of these dispersions are quite consistent with these observations, with a particle diameter that increases with the final DP (Table 1). However, as shown by the polydispersity index, the DLS results tend to show that the objects present in the latexes are either very dispersed in size or that non spherical particles are also present.
Further analyses of the latexes by cryo-TEM however show that the in situ formed amphiphilic block copolymers self-assemble into nano-objects of various sizes and morphologies (Fig. 6). As the DP of the hydrophobic PVAc block increases, the morphology of the particles shifts from isometric spherical particles (A1, DPexp = 47) to vesicles (A4, DPexp = 92) and then to higher-order morphologies (A6, DPexp = 187). For a DPexp between 59 and 92 (A2, A3 and A4), the latexes are composed of vesicles and spherical particles, with the size and proportion of vesicles increasing with the DPexp. Further increase of the hydrophobic block molar mass gives access to higher-order morphologies such as distorted or multilamellar vesicles (A5, DPexp = 144) and eventually to “large compound vesicles” (LCVs, A6, DPexp = 187) (see additional cryo-TEM images in Fig. S11†).
 | ||
| Fig. 6 Representative cryo-TEM images of the crude latexes A1–A6 (Table 1). | ||
Morphology prediction in PISA is difficult since the mechanisms involved are complex and depend on many parameters. Nonetheless, a reproducible pattern for the morphological transitions has now been established through various studies.2,20,39–41 It is agreed that initial spherical particles merge to form worm-like particles which themselves give rise to branched “octopus-like” structures as a transition state to vesicles. The morphologies observed match with this established pattern, and morphologies of even higher order than those of vesicles are observed. However, according to this pattern, the worm/fibre morphology that is not seen here should be formed for a DP between 47 (A1, spheres) and 59 (A2, spheres + vesicles). The narrowness of this DP window is consistent with the PISA literature reports concerning the formation of worm/fibre particles.42
To the best of our knowledge, these experiments are the first depicting the obtention of non-spherical morphologies by VAc PISA in emulsion. Indeed, as mentioned above, the morphologies obtained in emulsion are most of the time restrained to kinetically trapped spheres, and the number of reports of higher-order morphologies remain limited.2 The formation of these morphologies in PISA is the result of particle collision/fusion phenomena (vide supra).20 As such, their formation is directly linked to the mobility of the polymer chains. In this specific system, several factors may be involved. In particular, the solubility of VAc in water is most certainly crucial. As mentioned in the introduction, the importance of the monomer solubility in water for the formation of non-spherical objects in PISA emulsion-based systems has recently been highlighted. Cockram et al. obtained unusual “monkey nut” particles through RAFT-mediated emulsion polymerization in water using a core forming monomer, namely HBMA, with a water solubility higher than those usually employed (20 g L−1 at 70 °C).13 The authors postulated that a higher monomer aqueous solubility would lead to a more efficient plasticization of the hydrophobic core, which would facilitate the rearrangement of the polymer chains. The same group then obtained well-defined worms by emulsion polymerization of GlyMA, which has a similar aqueous solubility (24–25 g L−1 at 80 °C),14 and both worms and vesicles with MOEMA (19.6 g L−1 at 70 °C).16 With a water solubility of 26 g L−1 (at 50 °C), VAc provides a system with comparable features. Thus, the results obtained through this work tend to confirm the determining role of this parameter to trigger the formation of particles with higher-order morphologies.
Influence of the solid content
To investigate further this system and identify conditions for which worms/fibres could be obtained, three additional syntheses were performed at a higher solid contents, that is 15 wt%, targeting DP value of 51, 76 and 100 for the PVAc block (entries B1, B2 and B3 in Table 1, respectively). The same good control (Fig. S12–S16†) over the chain extension of PEG-X with VAc is observed as attested by the good match between the expected and experimental molar mass values obtained upon VAc conversions, and the rather low dispersities (Table 1). In addition, the morphology evolution as the DP increases is similar to the one reported in the previous series of experiments (10 wt% solid content), with a transition from spherical particles (B1, DPexp = 48) to vesicles (B3, DPexp = 97) as shown by the cryo-TEM images (Fig. 7). However, the increase in the solid content allowed triggering the formation of short worms along with the spheres and vesicles in B2 (DPexp = 73).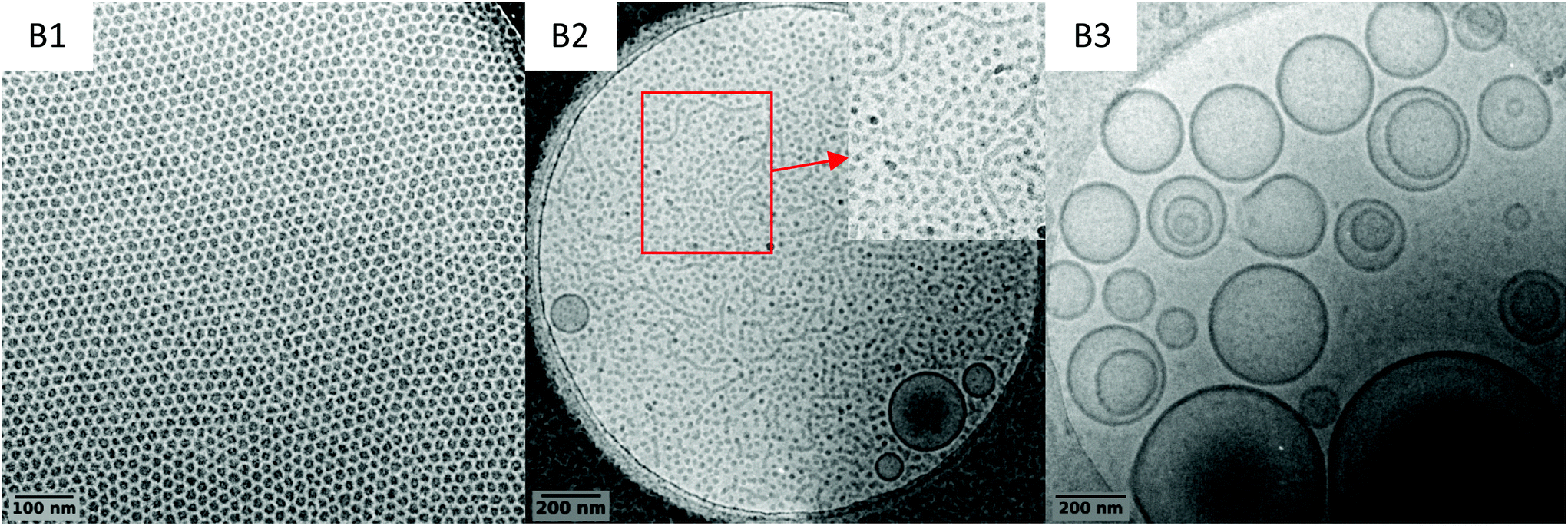 | ||
| Fig. 7 Representative cryo-TEM images of the crude latexes B1–B3 (Table 1). | ||
The coexistence of the three morphologies indicates that the experimental window (i.e. the DP) for which the worm/fibre morphology can be exclusively attained is very narrow. The difficulty in obtaining a pure worm/fibre morphology in this system probably lies on the fact that the underpinning formation mechanisms are intricate. Indeed, in addition to VAc hydrophilicity, PVAc's low Tg (ca. 30 °C) also plays a significant role. The synthesis temperature is fixed way above the Tg of the PVAc, which implies that the hydrophobic block maintains a certain mobility ensuring a labile structure for the self-assemblies. This provides more opportunities for morphological transitions that we did not achieve in our seminal works on PEG-X mediated VAc emulsion polymerization, where the polymerizations were performed at 25 °C. Eventually, the nature of the hydrophilic block plays an important role in the self-assembly and rearrangement mechanism. More specifically, the relatively low steric repulsion between the PEG corona compared to other hydrophilic macro-CTAs, including charged ones, would enhance the effective collision of the assemblies and thus promote their rearrangement.43 The fine tuning of these parameters should allow finding conditions leading to pure worm/fibre morphologies. These investigations are underway is our laboratories.
Conclusions
The aqueous MADIX-mediated emulsion polymerization of vinyl acetate was conducted using a poly(ethylene glycol) bearing a xanthate chain-end as a hydrophilic macro-CTA. The study focused mainly on synthesis performed at 70 °C with a targeted solid content of 10 wt%, which led to relatively well controlled polymerizations, with a good blocking efficiency, and the formation of stable latexes. Increasing the DP of the PVAc block allowed triggering of morphological transitions of the self-assembled PEG-b-PVAc nano-objects, from spherical particles for a DP of 47 to a mixture of spheres and vesicles for the DP comprised between 62 and 100, and eventually to higher-order morphologies, notably large compound vesicles for a DP of 187. On increasing the solid content to 15 wt%, the worm morphology was obtained for a DPexp of 73, however coexisting with spheres and vesicles.The access to non-spherical morphologies depicted here for the first time for VAc emulsion polymerization was interpreted as the result of the high water solubility of VAc compared to other hydrophobic monomers classically employed in emulsion polymerization. Although the overall picture of the phenomena impacting the formation of a specific morphology is yet to be provided, adjusting the polymerization temperature and solid content may allow the isolation of pure PVAc worm/fibre particles and eventually establish a phase diagram for this system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Agence Nationale de la Recherche (grant number ANR-18-CE06-0014 CKAPART).Notes and references
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950 RSC.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- I. Chaduc, E. Reynaud, L. Dumas, L. Albertin, F. D'Agosto and M. Lansalot, Polymer, 2016, 106, 218–228 CrossRef CAS.
- J. Lesage de la Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS PubMed.
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2017, 6, 1013–1019 CrossRef CAS.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- F. L. Hatton, A. M. Park, Y. Zhang, G. D. Fuchs, C. K. Ober and S. P. Armes, Polym. Chem., 2019, 10, 194–200 RSC.
- J. Tan, X. Dai, Y. Zhang, L. Yu, H. Sun and L. Zhang, ACS Macro Lett., 2019, 8, 205–212 CrossRef CAS.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS PubMed.
- J. Siirilä, S. Häkkinen and H. Tenhu, Polym. Chem., 2019, 10, 766–775 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 Search PubMed.
- J. Zhou, H. Yao and J. Ma, Polym. Chem., 2018, 9, 2532–2561 Search PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. M. Van Herk and M. Monteiro, in Handbook of Radical Polymerization, ed. K. Matyjaszewski and T. P. Davis, 2002, pp. 301–331, DOI:10.1002/0471220450.ch6.
- Chemistry and Technology of Emulsion Polymerisation, ed. A. M. Van Herk, John Wiley & Sons, Ltd, 2nd edn, 2013 Search PubMed.
- M. J. Ballard, D. H. Napper and R. G. Gilbert, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 3225–3253 CrossRef CAS.
- W. H. Lane, Ind. Eng. Chem., Anal. Ed., 1946, 18, 295–296 CrossRef CAS.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- H. D. Bruyn, R. G. Gilbert and B. S. Hawkett, Polymer, 2000, 41, 8633–8639 CrossRef CAS.
- H. Y. Erbil, Vinyl Acetate Emulsion Polymerization and Copolymerization with Acrylic Monomers, CRC Press, Boca Raton, FA, 2000 Search PubMed.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2845–2857 CrossRef CAS.
- S. Binauld, L. Delafresnaye, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2014, 47, 3461–3472 CrossRef CAS.
- L. Etchenausia, E. Deniau, A. Brûlet, J. Forcada and M. Save, Macromolecules, 2018, 51, 2551–2563 CrossRef CAS.
- L. Etchenausia, A. Khoukh, E. Deniau Lejeune and M. Save, Polym. Chem., 2017, 8, 2244–2256 RSC.
- A. Guinaudeau, S. Mazieres, D. J. Wilson and M. Destarac, Polym. Chem., 2012, 3, 81–84 RSC.
- C. F. Cordeiro and F. P. Petrocelli, in Kirk-Othmer Encyclopedia of Chemical Technology, 2005, DOI:10.1002/0471238961.2209142503151804.a01.pub2.
- S. Harrisson, X. Liu, J.-N. Ollagnier, O. Coutelier, J.-D. Marty and M. Destarac, Polymers, 2014, 6, 1437 CrossRef.
- M. Guerre, M. Semsarilar, F. Godiard, B. Ameduri and V. Ladmiral, Polym. Chem., 2017, 8, 1477–1487 RSC.
- A. Castanon Rodriguez, PhD, University of Nottingham/Université Claude Bernard Lyon 1, 2018.
- N. De Rybel, P. H. M. Van Steenberge, M.-F. Reyniers, D. R. D'Hooge and G. B. Marin, Macromolecules, 2019, 52, 4555–4569 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1800438 Search PubMed.
- G. Mellot, J.-M. Guigner, L. Bouteiller, F. Stoffelbach and J. Rieger, Angew. Chem., 2019, 131, 3205–3209 CrossRef.
- F. Brunel, J. Lesage de la Haye, M. Lansalot and F. D'Agosto, J. Phys. Chem. B, 2019, 123, 6609–6617 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00467g |
| This journal is © The Royal Society of Chemistry 2020 |