
A dual drug-based hyperbranched polymer with methotrexate and chlorambucil moieties for synergistic cancer chemotherapy†
Chengfei
Liu
a,
Huixin
Li
a,
Pengxiang
Li
a,
Caiping
Liu
b,
Yang
Bai
 *b,
Jun
Pang
a,
Jingxia
Wang
a and
Wei
Tian
*a
*b,
Jun
Pang
a,
Jingxia
Wang
a and
Wei
Tian
*a
aShaanxi Key Laboratory of Macromolecular Science and Technology, MOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an, 710072, Shaanxi, P. R. China. E-mail: happytw_3000@nwpu.edu.cn
bShaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China. E-mail: baiyang@sust.edu.cn
First published on 14th August 2020
Abstract
Drug-based hyperbranched polymers have attracted broad interest from scientists due to their attractive and distinctive characteristics in cancer chemotherapy. However, the only use of the single-drug treatment modality could lead to low or unsatisfactory anticancer activity because of the limited treatment strategy and multidrug resistance of tumour cells. In this work, a novel dual drug-based hyperbranched polymer (DHBP) simultaneously containing methotrexate (MTX) and chlorambucil (Cb) was proposed to realize synergistic cancer chemotherapy. After modification with PEG, the obtained DHBP-g-PEG self-assembled into hyperbranched polymer micelles (HBPMs) in aqueous solution with a high drug loading capacity and stable drug transport. Notably, HBPMs with excellent controlled drug release in tumour cells exhibited an obviously superior synergistic effect and possessed significant therapeutic efficiency and in vitro anticancer activity, as compared to either the individual drug or mixture drug treatment group. All these results demonstrate that the unique advantage of HBPMs may have significant potential for application in the field of synergistic cancer chemotherapy.
Introduction
Hyperbranched polymers (HBPs) as highly branched three-dimensional (3D) macromolecules have attracted growing research efforts because of their unique structures and attractive characteristics, including improved physicochemical properties, excellent multi-functionalities, special three dimensional globular structures and potential for application in many fields such as catalysts, biomaterials, medicine and chemistry.1 In particular, some distinctive advantages such as convenient synthesis through one-pot reactions, favorable biocompatibility and biodegradability, a large number of surface groups, and unusual self-assembly ability make HBPs ideal nanocarriers for drug and gene delivery in biological and biomedical systems and devices.2 However, most of these HBPs are typically inert excipients, only serving as carriers, leading to low drug loading capacity and potential premature drug leakage during preparation and transportation processes.3 Furthermore, the poor stability and batch-to-batch variability in drug loading and release kinetics may also cause unpredictable therapeutic efficacy of the treatment.4Recently, a new concept of directly using drug molecules as building blocks to conjugate hyperbranched polymer nanodrug delivery systems has been designed.5 The obtained drug-based hyperbranched polymers could effectively enhance the drug loading capacity with minimum use of inert materials and increase the stability of drug transport by suppressing premature burst release. However, the only use of one drug molecule could lead to poor inhibition against cancer cells because of the limited treatment strategies and multidrug resistance of tumour cells, which greatly reduced the therapeutic efficacy.6 Moreover, the single-drug treatment strategy often fails to maintain long-lasting and effective anticancer activity because of the rapid development of drug resistance in tumour cells.6b Alternatively, the synergistic effect of two anticancer drugs with different anticancer sites and mechanisms could significantly render a great therapeutic effect through overcoming the multidrug resistance of tumour cells and maximizing the therapeutic efficacy by combining the respective advantages of different drugs.7 Therefore, it is very necessary to design drug-based hyperbranched polymers simultaneously conjugating two anticancer drugs to suppress the disadvantages of the traditional single-drug treatment modality and enhance synergistic cancer chemotherapy.
Herein, we constructed a dual drug-based hyperbranched polymer (DHBP) on the basis of the amidation reaction between bis(amine-disulfur)-methotrexate (A2 monomer) and tri(carboxylic acid-ester)-chlorambucil (B3 monomer) (Scheme 1a). According to the literature,8 methotrexate (MTX) can interrupt DNA and RNA synthesis while chlorambucil (Cb) is one of the DNA-alkylating anticancer drugs. The above two drugs have different drug activities but complementary anticancer mechanisms. Meanwhile, two redox-responsive disulfide bonds were incorporated into the A2 monomer for the reduction response by rich GSH, and a pH/esterase-responsive ester bond was introduced into the B3 monomer for hydrolysis in the special tumour microenvironment with enzymes and low pH values. In addition, in order to improve the solubility and biocompatibility of the DHBP, the hydrophilic polyethylene glycol (PEG), approved by the FDA9 for its excellent biocompatibility, was grafted to the end of the hydrophobic DHBP. Then, the obtained DHBP-g-PEG could self-assemble into hyperbranched polymer micelles (HBPMs) in water (Scheme 1a), serving as both carries and therapeutic components. After internalization of the HBPMs into tumour cells, the active MTX and Cb ingredients were controllably released on the basis of the degradation of the disulfide bonds in A2 and disintegration of the ester bond in B3, achieving the synergistic effect in anticancer activity (Scheme 1b). In vitro studies demonstrated that HBPMs display excellent synergistic cancer chemotherapy and may be a potential emerging and promising candidate in drug self-delivery systems and cancer chemotherapy.
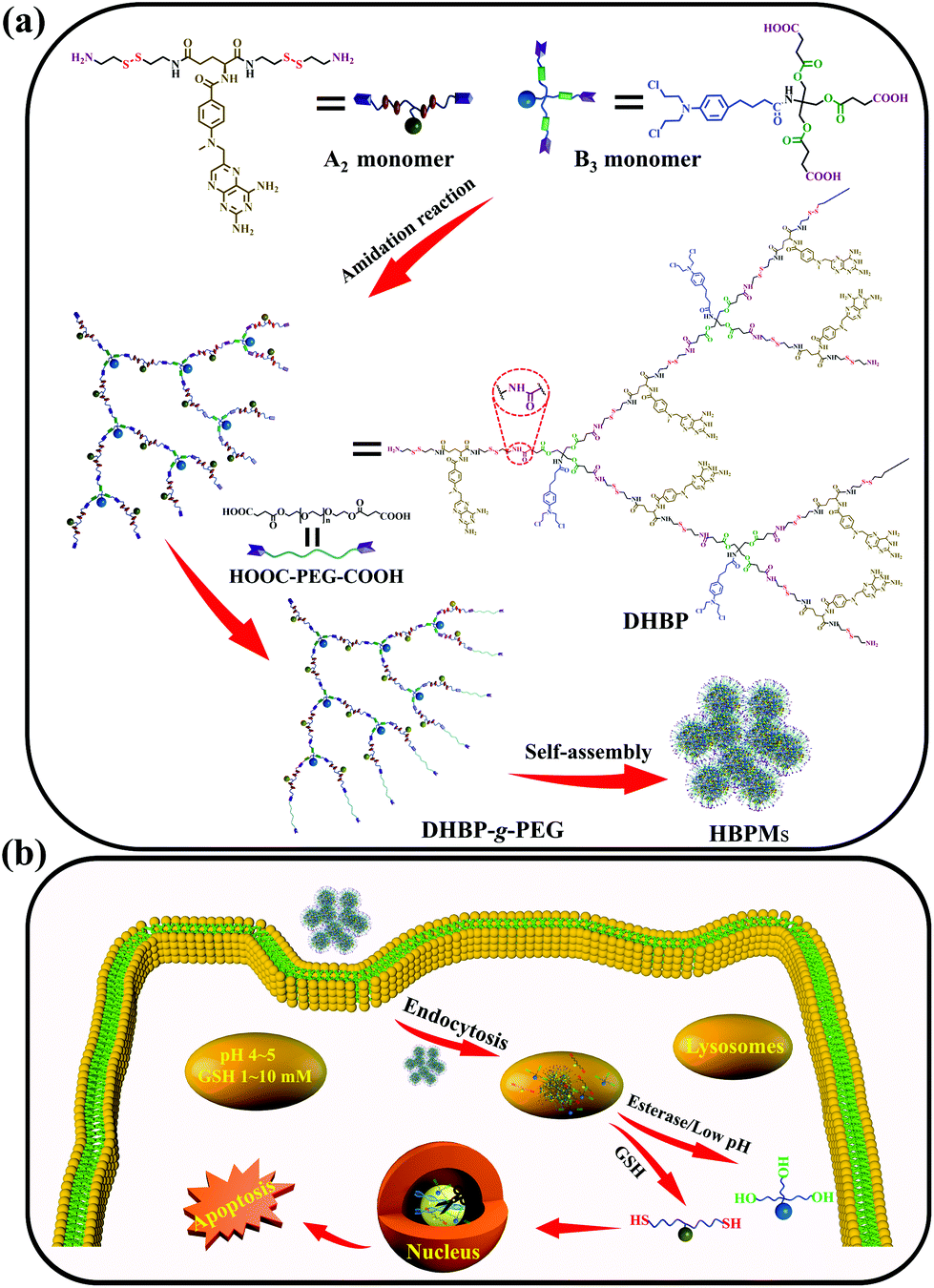 | ||
| Scheme 1 Schematic illustration of construction of the DHBP and HBPMs for cancer therapy. (a) Construction of the DHBP, DHBP-g-PEG, and HBPMs; (b) controlled drug release and synergistic cancer chemotherapy of HBPMs in the special physiological microenvironment of tumour cells. | ||
Results and discussion
Synthesis and modification of the dual drug-based hyperbranched polymer
To obtain the targeted DHBP, two main building blocks, A2 monomer and B3 monomer, were first synthesized according to the routes (Schemes S1 and S2†). The A2 monomer, consisting of one methotrexate molecule and two reduction-responsive disulfide bonds as well as two amino end groups (MTX-(SS-NH2)2), was synthesized via the deprotection of MTX-(SS-Boc)2 under trifluoroacetic acid conditions (Scheme S1†). On the other aspect, the B3 monomer, composed of one chlorambucil molecule and a pH/esterase-responsive ester bond with three carboxyl end groups, was prepared by rationally connecting succinic anhydride and Cb-(OH)3via an esterification reaction (Scheme S2†). The structure characterization results including 1H NMR, 13C NMR and ESI-TOF MS are shown in Fig. S1–S15.† The DHBP was then synthesized by an amidation reaction between A2 and B3 with a molar ratio of 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 3 days at room temperature (Schemes 1a and S3†).
:1 for 3 days at room temperature (Schemes 1a and S3†).
The chemical structure of the DHBP was first confirmed by 1H NMR and 13C NMR, which clearly displayed the characteristic peaks arising from both A2 monomer and B3 monomer (Fig. 1a and b). Compared with the 1H NMR spectrum of B3 (Fig. S13†), the peak at 12.15 ppm related to the carboxylic acid proton disappeared completely, whereas the new chemical shift at 7 (δ 2.35) belonging to the methylene (–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2COOH) of B3 appeared in the 1H NMR spectrum of the DHBP, indicating the successful amidation reaction between A2 and B3. In addition, as compared with the 13C NMR spectrum of B3 (Fig. S14†), the appearance of new characteristic peaks at 15 (δ 171.23) and 19 (δ 29.11) corresponding to the carbonyl group (–
2COOH) of B3 appeared in the 1H NMR spectrum of the DHBP, indicating the successful amidation reaction between A2 and B3. In addition, as compared with the 13C NMR spectrum of B3 (Fig. S14†), the appearance of new characteristic peaks at 15 (δ 171.23) and 19 (δ 29.11) corresponding to the carbonyl group (–![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) NH–) and the methylene (–2COOH) of B3 in the 13C NMR spectrum of the DHBP further confirmed the successful amidation reaction. SEC/MALLS results of the DHBP are shown in Table 1. The average molecular weight (Mw) of the DHBP was 133.1 kDa and the polydispersity index (PDI = Mw/Mn) was 2.90. Additionally, compared to the UV/Vis absorption of the free A2 monomer at 388 nm, a blue-shift was observed in the absorption of the DHBP in DMF at 385 nm (Fig. S16†). The maximum fluorescence emission of the DHBP red-shifts from 342 to 350 nm and 460 to 468 nm compared with A2 in DMF (Fig. S17†). All the above experimental results demonstrated that the DHBP has been synthesized successfully.
NH–) and the methylene (–2COOH) of B3 in the 13C NMR spectrum of the DHBP further confirmed the successful amidation reaction. SEC/MALLS results of the DHBP are shown in Table 1. The average molecular weight (Mw) of the DHBP was 133.1 kDa and the polydispersity index (PDI = Mw/Mn) was 2.90. Additionally, compared to the UV/Vis absorption of the free A2 monomer at 388 nm, a blue-shift was observed in the absorption of the DHBP in DMF at 385 nm (Fig. S16†). The maximum fluorescence emission of the DHBP red-shifts from 342 to 350 nm and 460 to 468 nm compared with A2 in DMF (Fig. S17†). All the above experimental results demonstrated that the DHBP has been synthesized successfully.
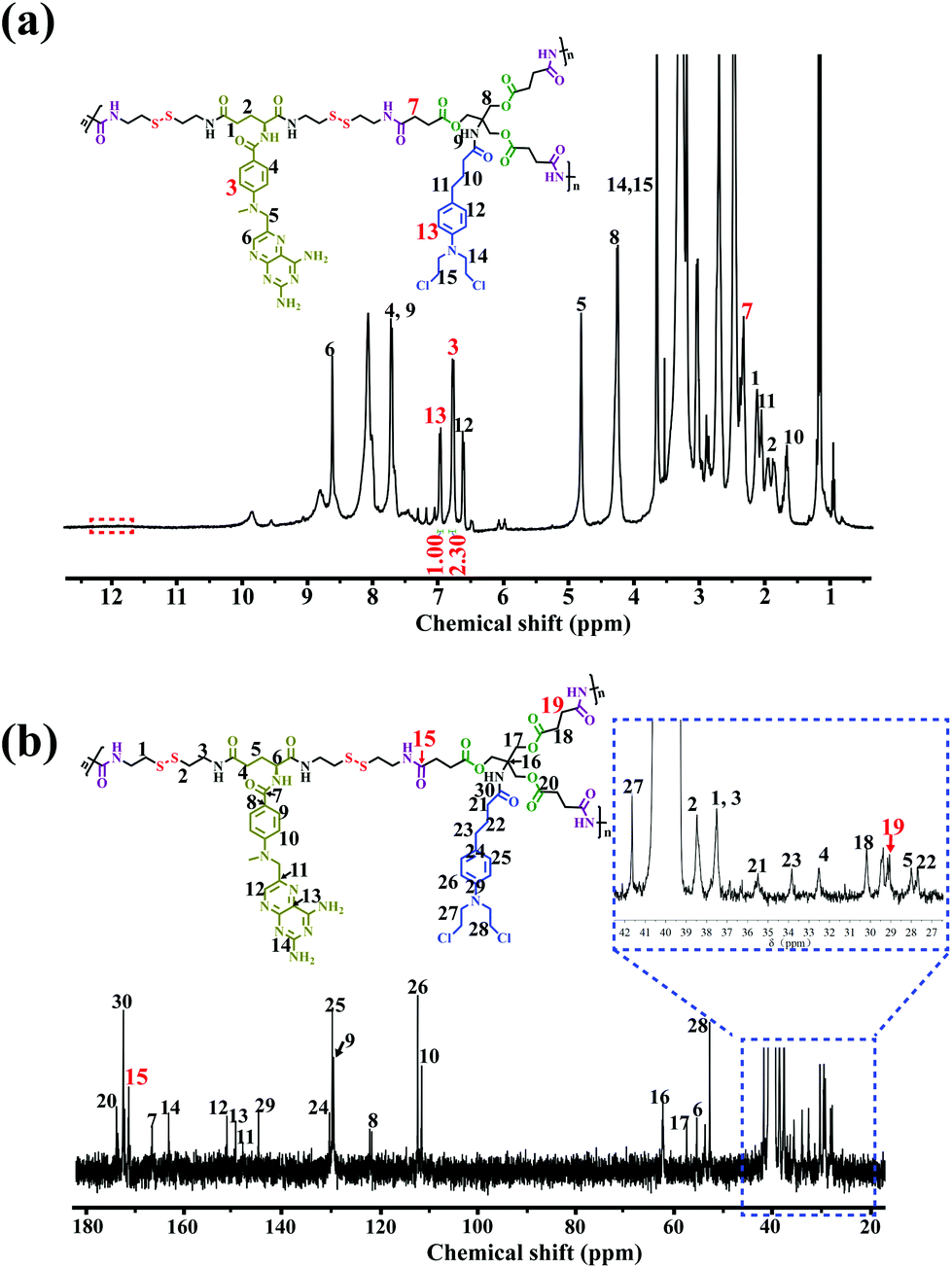 | ||
| Fig. 1 Structural characterization of DHBP. (a) 1H NMR and (b) 13C NMR spectra of the DHBP in DMSO-d6. | ||
| Sample | A2/B3 (molar ratio) | MTX/Cb (molar ratio)a |
M
nb (kDa) |
M
wb (kDa) |
PDIb | Grafting ratioc | dn/dcb |
|---|---|---|---|---|---|---|---|
| a The molar ratio of MTX to Cb in the DHBP was quantified via integrating the characteristic peaks of A2 and B3. b Molecular weight, molecular weight distribution (PDI) and refractive index increment (dn/dc) were evaluated by SEC/MALLS with DMF as the solvent. MW = weight-average molecular weight, Mn = number-average molecular weight, PDI = MW/Mn. c The grafting ratio of PEG in DHBP-g-PEG was determined by integrating the characteristic peaks of the DHBP and HOOC-PEG-COOH in the 1H NMR spectrum of DHBP-g-PEG.1i | |||||||
| DHBP | 2.5:1.0 |
2.3:1.0 |
45.8 | 133.1 | 2.90 | — | 0.129 |
| DHBP-g-PEG | — | — | 205.2 | 317.5 | 1.55 | 0.47 | 0.103 |
On the other hand, the precise molar ratio of combined drugs has made a profound impact on whether anticancer drug combinations act synergistically or antagonistically.4a Moreover, uncertain ratios between the two components may lead to low or unsatisfactory therapeutic efficacy in the cancer treatment. Thus, in order to accurately quantify the molar ratio of MTX to Cb in the DHBP, we integrated the characteristic peaks of A2 at 3 (δ 6.80) and B3 at 13 (δ 7.00) in the 1H NMR spectrum of the DHBP (Fig. 1a). According to the 1H NMR integration, their molar ratio is about 2.3:1, which was consistent with the feed ratio of A2 to B3 (2.5:1), suggesting a controllable molar ratio of the combined drugs in the DHBP.
DHBP-g-PEG was then synthesized by grafting the carboxyl-terminated poly(ethylene glycol) (HOOC-PEG-COOH) onto the surfaces of the DHBP through an amidation reaction, with a feed molar ratio of 1:5 between DHBP and HOOC-PEG-COOH in DMF (Scheme S5†). HOOC-PEG-COOH was first synthesized by reacting PEG with succinic anhydride in dichloromethane, according to a previous report (Scheme S4 and Fig. S18 and 19†).10 The obtained DHBP-g-PEG was purified by dialysis (MWCO 3000 Da) in deionized water to remove free HOOC-PEG-COOH and other small molecules. In Fig. 2a, the proton peaks at 1 (δ 3.50) and 2 (δ 4.08) of HOOC-PEG-COOH and characteristic peaks of the DHBP were both present in the 1H NMR spectrum of DHBP-g-PEG, implying that the carboxyl groups of HOOC-PEG-COOH were successfully condensed with the amino groups of the ends of the DHBP by the amidation reaction. The IR absorption bands of DHBP-g-PEG showed both characteristic peaks of HOOC-PEG-COOH at 1114 cm−1 and the amide bond absorption band at 1658 cm−1 from the DHBP, also confirming the above results (Fig. 2b). Moreover, the Mw of DHBP-g-PEG was 317.5 kDa with a PDI of 1.55 (Table 1), further indicating that the PEG chains have been successfully grafted onto the terminals of the DHBP. All these results supported that DHBP-g-PEG has been successfully synthesized.
 | ||
| Fig. 2 Structural characterization of DHBP-g-PEG. (a) 1H NMR spectrum of DHBP-g-PEG in DMSO-d6; (b) FTIR spectra of HOOC-PEG-COOH, DHBP and DHBP-g-PEG. | ||
Furthermore, the grafting ratio of PEG segments in DHBP-g-PEG directly determined the ratio of hydrophilic–hydrophobic components and the proportion of active ingredients of anticancer drugs, which may affect the subsequent amphiphilic self-assembly and its final anticancer activity. To effectively calculate the grafting ratio of PEG in DHBP-g-PEG, we integrated the characteristic peaks of the DHBP at δ 7.00 and HOOC-PEG-COOH at δ 4.09 in the 1H NMR spectrum of DHBP-g-PEG (Fig. 2a). The grafting ratio was 0.47, which not only ensured the necessary ratio of the hydrophilic component, but also guaranteed the certain active ingredients of anticancer drugs in DHBP-g-PEG.
Preparation of HBPMs based on DHBP-g-PEG
Due to the inherent amphiphilic nature, DHBP-g-PEG could further self-assemble into hyperbranched polymer micelles (HBPMs) in aqueous solution. Firstly, the critical aggregation concentration (CAC) of HBPMs was measured to be 4.29 μg ml−1 by using pyrene as a fluorescent probe (Fig. S20†), indicating the formation of HBPMs. Subsequently, transmission electron microscopy (TEM) was directly utilized to study their morphology and size. TEM results showed spherical self-assemblies with an average size of 60.3 ± 3.7 nm (Dav, TEM) (Fig. 3a), which was slightly smaller than the hydrodynamic diameter (Dh) (approximately 76.2 nm, PDI = 0.182) determined by DLS. The zeta potential value of HBPMs was −3.1 ± 0.3 mV, indicating weak negative charges on the surfaces of HBPMs. The inset of Fig. 3a presents a typical enlarged TEM image of one HBPM, clearly indicating the solid sphere structure of HBPMs. Additionally, the inner structures of the HBPMs were quantitatively confirmed by SLS/DLS techniques to prove the architecture of these self-assemblies. The Rg/Rh value of these HBPMs was 0.73, which is close to the theoretical value of 0.77, also indicating a solid micellar structure.11 Correspondingly, we also used scanning electron microscopy (SEM) and atomic force microscopy (AFM) to further verify the morphology and size of HBPMs, which were consistent with the TEM results (Fig. 3d and e). Moreover, the 3D AFM height image (Fig. 3f) did not collapse in the central structures of the spherical self-assemblies, demonstrating a solid micellar structure rather than a hollow vesicular structure.12 What's more, sulfur and chlorine elements derived from the disulfide bond of A2 and chlorambucil of B3 could be clearly found in the element mapping (Fig. 3b), indicating the designed structure of HBPMs.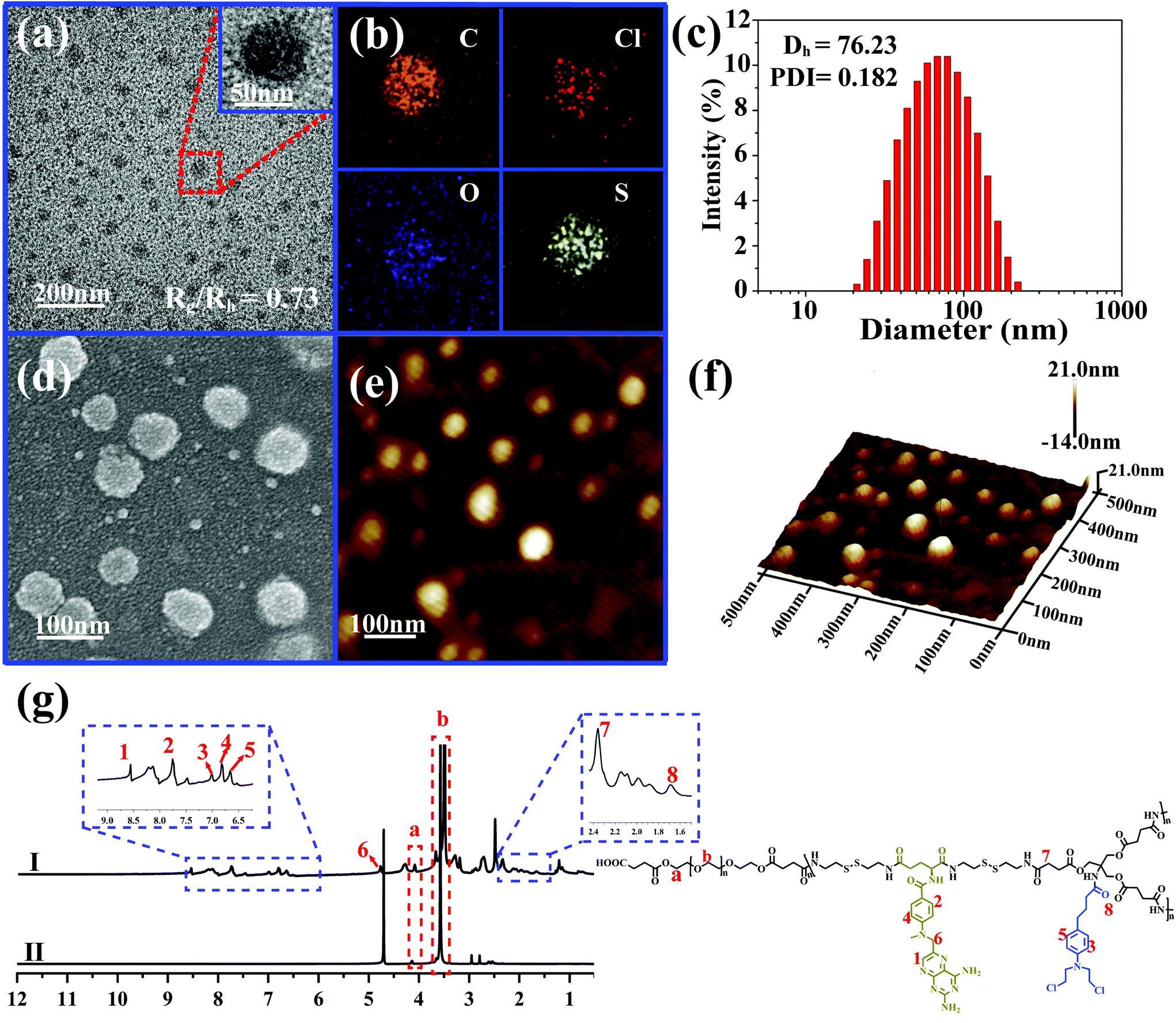 | ||
| Fig. 3 Morphology, size and structure of HBPMs in aqueous solution. (a) TEM image of HBPMs; (b) elemental mapping images of HBPMs; (c) hydrodynamic diameter (Dh) and polydispersity index (PDI) of HBPMs measured by dynamic light scattering (DLS); (d) SEM image and AFM image (e and f) of HBPMs; (g) 1H NMR spectra of DHBP-g-PEG in DMSO-d6 (I) and D2O (II), respectively. | ||
To further study the well-defined structure of self-assemblies, we used 1H NMR analysis to explore the hydrophilic and hydrophobic components of HBPMs. Compared with the 1H NMR spectrum of DHBP-g-PEG in DMSO-d6 (Fig. 3g(I)), the proton signals at 1–8 (δ 8.56, 7.75, 7.01, 6.82, 6.66, 4.80, 2.37, and 1.67) associated with the DHBP disappeared in D2O, while the proton peaks at a (δ 4.10) and b (δ 3.55) in PEG chains could still be found (Fig. 3g(II)). These results clearly showed that the DHBP moieties formed the hydrophobic core layer of HBPMs, while PEG segments consisted of a hydrophilic shell layer in aqueous solution.
In vitro stability and controlled drug release of HBPMs
Stability is very important for further practical biomedical applications of nanoscale drug delivery systems.8 Thus, the change of morphology and size of HBPMs under different conditions was measured to study the stability of micelles in aqueous solution.13 As shown in Fig. S21 and 22,† the TEM and SEM images of HBPMs demonstrated that almost no obvious change was observed at different concentrations. DLS results at different time intervals were also studied to further evaluate the stability of HBPMs (Fig. 4a). The Dh values of HBPMs showed no significant change in solution from 0 to 7 d. And the value of the PDI is always about 0.2 in every measurement. Additionally, the Dh of these micelles also showed almost the same size in PBS buffer and cell culture medium for 48 h (Fig. 4b). Therefore, the HBPMs were considered to be stable for further biological evaluation in vitro.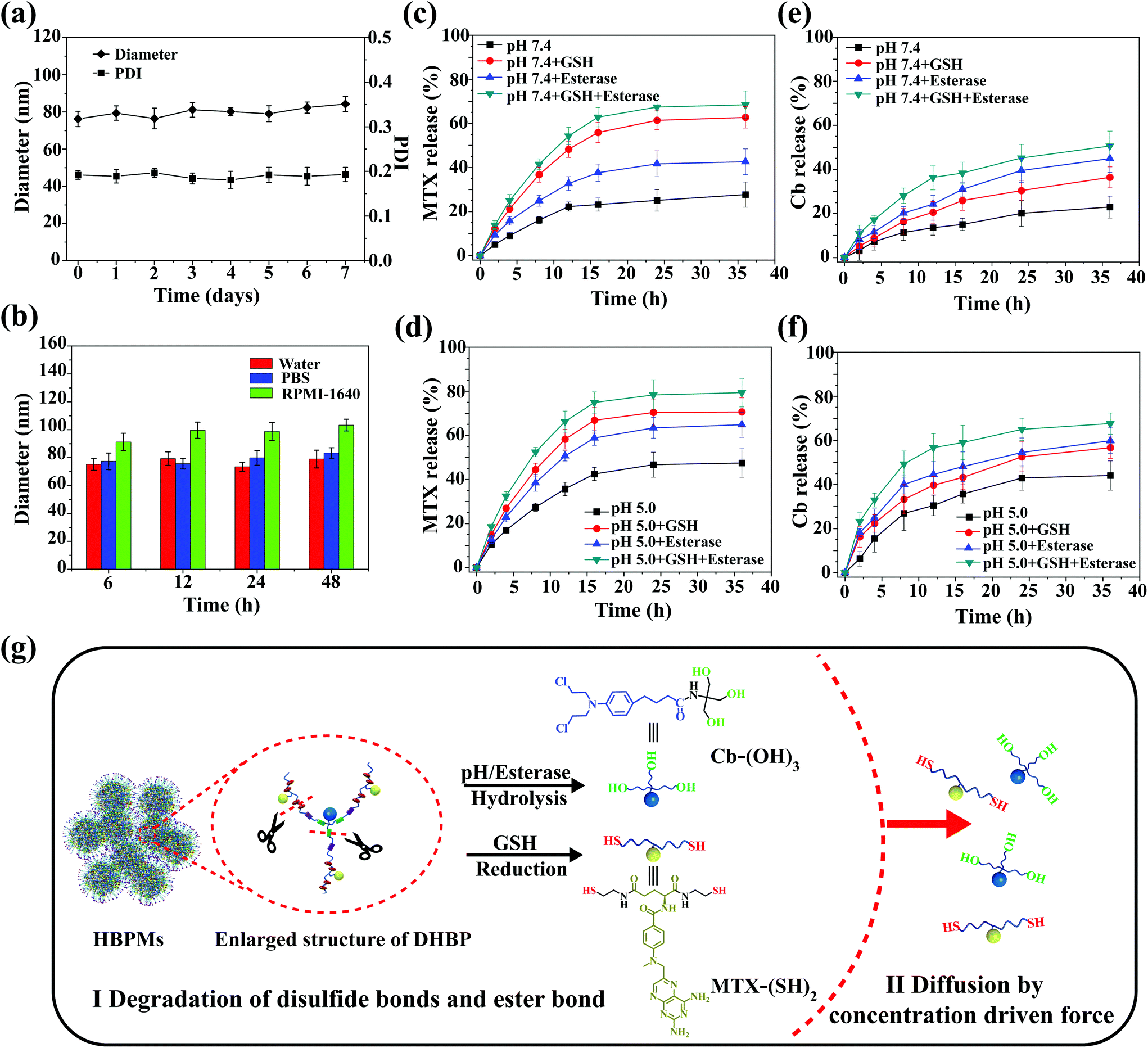 | ||
| Fig. 4 Stability and controlled dug release of HBPMs in vitro. (a) Influence of storage on Dh and PDI after storage for 7 d. (b) Change of Dh in water, PBS and cell culture medium after incubation for 48 h. (c and d) Cumulative release curves of MTX content from HBPMs as a function of time over a period of 36 h. (e and f) Cumulative release curves of Cb content from HBPMs as a function of time over a period of 36 h. (g) Schematic illustration of the release mechanism of active MTX and Cb drugs from HBPMs. | ||
Due to the special physiological microenvironment of tumour cells such as GSH-rich, low pH and enzyme conditions,14 the disulfide bonds and ester bond in the HBPMs could be gradually disintegrated, releasing MTX and Cb. The active MTX content from HBPMs was evaluated using UV/Vis spectrometry through measuring the characteristic absorbance at 385 nm and the Cb content was evaluated by high-performance liquid chromatography to test the Cb characteristic absorbance at 295 nm. In PBS (pH 7.4), HBPMs released only ∼27% MTX and ∼21% Cb over 36 h, respectively (Fig. 4c and e). However, through the addition of GSH or esterase, the concentration of MTX was 62% (pH 7.4, GSH) and 42% (pH 7.4, esterase), and that of Cb was 36% (pH 7.4, GSH) and 45% (pH 7.4, esterase) over 36 h. These results displayed that the disulfide bonds were disintegrated by GSH and the ester bond was cleaved under the enzyme condition. Furthermore, the concentration of MTX and Cb significantly increased to 68% and 51% in the presence of both GSH and esterase, because of the accelerated disintegration of both disulfide bonds and ester bond. On the other hand, all the cumulative amounts of MTX and Cb were released much more rapidly at pH 5.0 than the same samples at pH 7.4 (Fig. 4d and f) because the weak acidic conditions (pH 5.0) accelerated the hydrolysis of the ester bonds, promoting the effective release of drugs.4a,d Therefore, the release of MTX and Cb content from HBPMs could be effectively adjusted by GSH, acid and esterase.
To further investigate the release mechanism of MTX and Cb from HBPMs, we studied their release kinetics through eqn (1) and (2).14
| Mt/M∞ = ktn | (1) |
| ln r = ln k + n ln t, r = Mt/M∞ | (2) |
In vitro cellular uptake of HBPMs
Cellular uptake efficiency is one of the most important factors for the therapeutic efficacy of HBPMs. To further evaluate their cellular uptake behaviour, HBPMs were first labelled by using the Cy5 fluorescently. The preparation method is described in the ESI.† In order to investigate the cellular uptake behaviour of HBPMs, CLSM was utilized to evaluate their internalization results using human prostate carcinoma cell line (PC-3) cells as the model for 4 h and 12 h before observation. As can be seen from Fig. 5, the red fluorescence generated from Cy5-HBPMs was clearly observed inside the tumour cell and distributed around nuclei, indicating that the HBPMs were quickly taken up. When the incubation time was prolonged from 4 h to 12 h, the red fluorescence intensity was increased significantly. The results indicate that Cy5-HBPMs could be effectively internalized into PC-3 cells.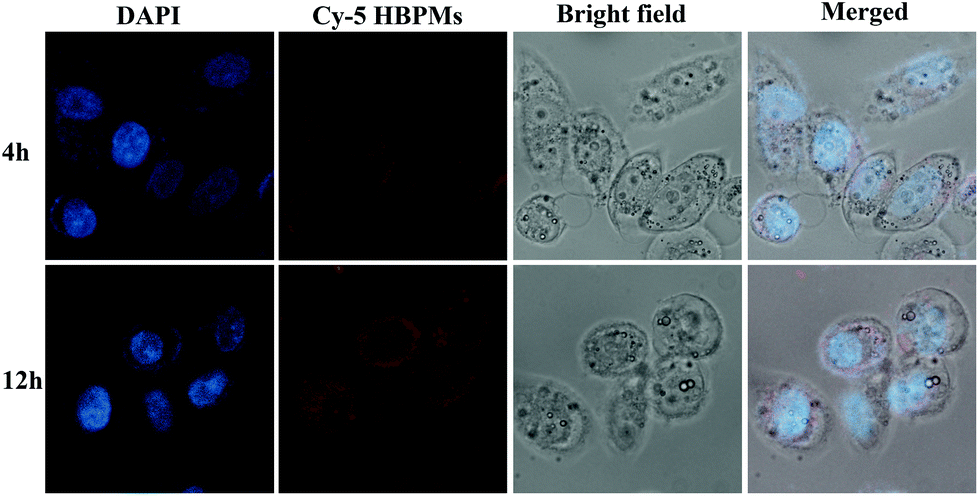 | ||
| Fig. 5 Fluorescence microscopy images of PC-3 cells incubated with the Cy5-HBPMs for 4 h and 12 h at a concentration of 32 μg ml−1. From left to right: DAPI (blue), Cy5-HBPMs (red), bright field and a merge of the two images. | ||
In vitro cytotoxicity and apoptosis of HBPMs
To evaluate the proliferation inhibition effect of HBPMs, MTT assay was used to measure the cellular toxicity of the HBPMs. Firstly, the cytotoxic effect of micelles on normal LO-2 cells was evaluated. The result indicated that HBPMs showed a low cytotoxicity to LO-2 cells, even when the effective drug concentration increased to 64 mg ml−1 (Fig. S26†).15 Then, the cell viability against PC-3 and MCF-7 cells was assessed. The cells were incubated with free MTX, free Cb, MTX/Cb mixture, and HBPMs at a series of concentrations for 48 h. As shown in Fig. 6a, both cancer cells showed drug-concentration dependent cytotoxicity and the group treated with HBPMs displayed a remarkably higher cytotoxicity than the other groups incubated with free MTX, free Cb, and MTX/Cb mixture. However, the lack of synergism at lower drug concentration may be explained that HBPMs couldn't be formed when the concentration of DHBP-g-PEG was lower than the CAC value. Thus, the endocytosis of micelles into tumor cells was greatly inhibited. According to the previous literature,16 the degree of synergy between two drugs can be quantified by calculating the combination index (CI). Generally, CI value < 1 indicates synergism, CI value = 1 indicates an additive effect, and CI value > 1 indicates antagonism. Herein, the CI values of HBPMs calculated by using the IC50 values were 0.746 and 0.912 for PC-3 and MCF-7, respectively (Table S3†). The results clearly indicated that the synergistic effect of dual drugs was utilized to inhibit cancer cells.16 In addition, HBPMs did not show obvious cytotoxicity on red blood cells in a hemolysis assay, thus indicating good biocompatibility (Fig. S27†).17 All these results indicated that the group treated with HBPMs showed more significant anticancer efficiency than the other groups, suggesting the significantly enhanced combination chemotherapy of the HBPMs.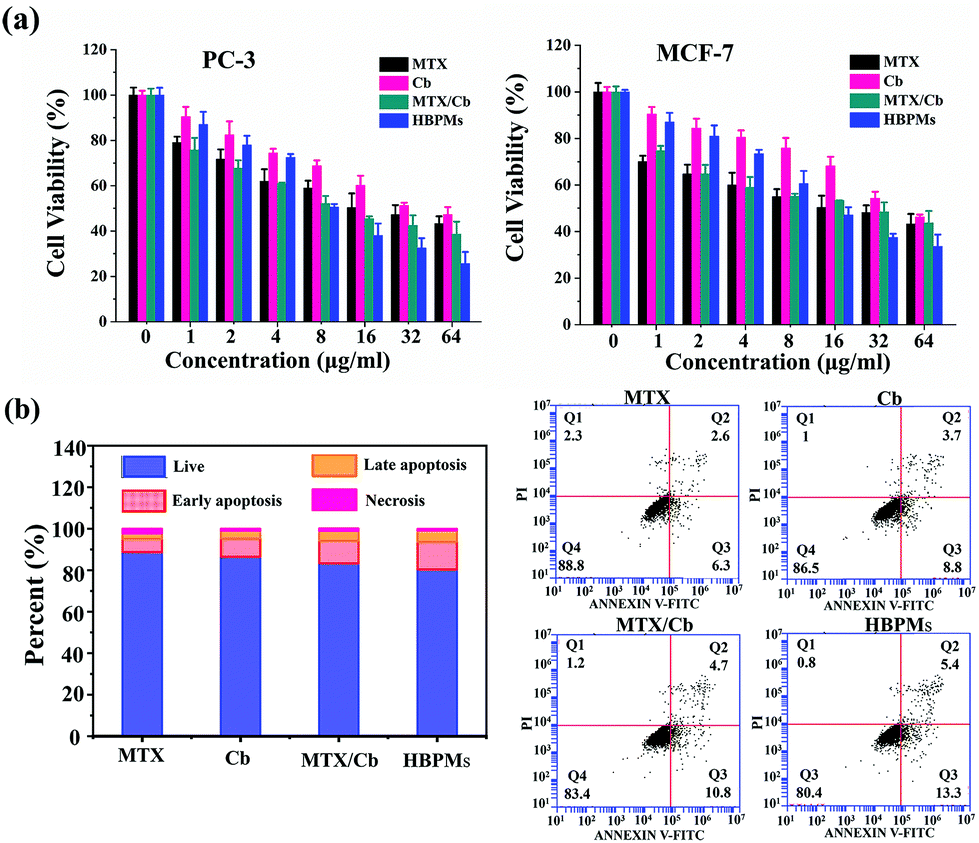 | ||
| Fig. 6 Effect of HBPMs on cancer cell growth and cell apoptosis. (a) Cytotoxicity of MTX, Cb, MTX/Cb mixture, and HBPMs at different concentrations to PC-3 and MCF-7. (b) Flow cytometry analysis for the apoptosis of PC-3 cells induced by MTX, Cb, MTX/Cb mixture, and HBPMs at a drug concentration of 32 μg ml−1 for 24 h. | ||
Generally, most of the free anticancer drugs could induce mitochondrial mediated apoptosis in tumour cells.9 To study the apoptosis-inducing capability of HBPMs, PC-3 cells were utilized as the model cells. Basically, PC-3 cells were treated with HBPMs at an equivalent drug concentration of 32 μg ml−1 and Annexin V-FITC/PI apoptosis staining was used to detect the percentage of apoptotic cells by flow cytometry analysis (Fig. 6b). After treatment with the free MTX, Cb, MTX/Cb mixture, and HBPMs for 24 h of incubation at the equivalent drug concentration, the total apoptotic ratio (sum of early and late apoptotic ratios of PC-3 cells) was ∼8.9%, ∼12.5%, ∼15.5% and ∼18.7%, respectively. The result demonstrated that the HBPMs induced a significantly higher apoptotic ratio than individual free drug groups and the mixture of two drug group which was also consistent with the in vitro cytotoxicity.
Conclusions
In summary, we successfully constructed a dual drug-based hyperbranched polymer (DHBP) simultaneously with redox-responsive disulfide bonds and a pH/esterase-responsive ester bond. After a graft modification with functionalized PEG segments, the resulting DHBP-g-PEG could self-assemble into hyperbranched polymer micelles (HBPMs) in water, serving as both carries and cargos. The obtained HBPMs exhibited excellent controlled drug release due to the sensitive responsiveness in physiological solution. Most importantly, HBPMs presented enhanced synergistic cancer chemotherapy efficiency compared with either individual free drugs or a mixture of the two free drugs and in vitro anticancer activity. These HBPMs will contribute to remarkable advances in synergistic chemotherapy for cancer.Experimental section
Materials
Chlorambucil, 4-dimethyl-aminopyridine (DMAP) and 2-bromoisobutyryl bromide were purchased from J&K Scientific Ltd, China. Tris(hydroxymethyl)methyl aminomethane (TRIS) and succinic anhydride were purchased from Energy Chemical, China. PEG (Mw = 2000 g mol−1, PDI = 1.04) was purchased from Shanghai Yare Biotech, Inc. EDC·HCl, methotrexate and 1-hydroxybenzotriazole (HOBt) were purchased form Adamas, China. Organic solvents and inorganic salts were purchased from Tianjin Kermel Chemical Reagents Development Center. All reagents were used as received unless special statement.Structural characterization methods
IR spectra were measured using a TENSOR27 spectrometer and are reported in cm−1. UV-Vis spectra were recorded on an Agilent Cary-100 spectrophotometer. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III 400 MHz spectrometer in CDCl3, DMSO-d6 or D2O. Electrospray ionization (ESI) mass spectra were acquired with a Bruker micrOTOF-Q II electrospray instrument. DLS was performed on a Zetasizer Nano ZS90 before the sample was filtered. The molecular weight was measured with a Gel Permeation Chromatograph waters 1515, and the standard reagent was poly(methyl methacrylate) (PMMA). SEM was performed by using a Verios G4 microscope. AFM was performed by using a Dimension Fast Scan and Dimension Icon instrument. The image for TEM measurements was obtained using an FEI Talos F200X instrument.Determination of the critical aggregation concentration (CAC)
To measure the CAC value of DHBP-g-PEG aggregates, we used pyrene as the fluorescent probe. 3 μL of pyrene ethanol solution (6 × 10−4 mol L−1) was added to 3 mL of DHBP-g-PEG aggregate aqueous solution with different concentrations to maintain the theoretical concentration of pyrene, 6 × 10−7 mol L−1. The I1/I3 values of all solutions were recorded on a Horiba Fluorolog-3 spectrometer at 332 nm excitation wavelength and 5 nm slit width.Preparation of HBPMs
Briefly, 1 mg of DHBP-g-PEG was dissolved in 20 μL of dimethyl sulfoxide. Then, the solution was slowly added to 5 mL of deionized water and stirred for 1 hour. After that, the solution was dialyzed against deionized water for 24 h (MWCO = 1000 Da), during which the water was renewed every 6 h. The volume of the solution was increased to 5 mL to produce a solution with a concentration of 0.5 mg mL−1.In vitro drug release from HBPMs
1 mg HBPMs were dispersed in eight PBS buffer solutions (30 ml), (1) pH 7.4; (2) pH 7.4 + esterase (30 U mL−1); (3) pH 7.4 + GSH (1 mg ml−1); (4) pH 7.4 + esterase (30 U mL−1) + GSH (1 mg ml−1); (5) pH 5.0; (6) pH 5.0 + esterase (30 U mL−1); (7) pH 5.0 + GSH (1 mg ml−1); (8) pH 5.0 + esterase (30 U mL−1) + GSH (1 mg ml−1) at 37 °C, and stirred at 100 rpm. At determined time intervals, 4 ml of solution was collected and replaced by fresh PBS (4 mL). The amount of MTX content was determined by UV/Vis spectrometry at λ = 385 nm. The amount of Cb content was analyzed using HPLC with an eluent composed of acetonitrile and water (6:4, v:v) at a flow rate of 1 ml min−1. The detection wavelength of Cb was set at 294 nm.
Cell culture
The LO-2, PC-3 and MCF-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM). The culture media contain 10% fetal bovine serum (FBS) and antibiotics (100 units per mL penicillin and 100 units per mL streptomycin) at 37 °C under a humidified atmosphere containing 5% CO2.Hemocompatibility assay
Hemolysis tests were performed using a mouse red blood cell (RBC) suspension in vitro.17 All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Northwestern Polytechnical University and approved by the Animal Ethics Committee of Northwestern Polytechnical University. Firstly, the RBCs were collected form fresh mouse blood by centrifugation (2000 rpm, 10 min), and washed with normal PBS three times. Then, the purified RBCs were diluted with PBS to obtain a RBC stock suspension (5%, v/v). The diluted RBC suspension (300 μl) was incubated with (1) 1000 μl of PBS as a negative control; (2) 1000 μL of ultrapure H2O as a positive control; (3) 1000 μl of HBPMs with different concentrations. The mixtures were incubated at 37 °C for 2 h. The hemolysis of RBCs after different treatments was observed by using a digital camera. The absorbance of the supernatant was hemolysis (%) = [(A1 − A2)/(A3 − A2)] × 100%, where, A1, A2, and A3 are the absorbance of samples treated with different types of materials, normal PBS, and ultrapure H2O, respectively.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the funding of the National Natural Science Foundation of China (Grant No. 21674086 and 21801162), Natural Science Basic Research Plan in Shaanxi Province of China (2020JC-20, 2018JZ2003), and the Fundamental Research Funds for the Central Universities (3102019PY003). We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University.Notes and references
- (a) A. M. Caminade, D. Y. Yan and D. K. Smithd, Chem. Soc. Rev., 2015, 44, 3870 RSC; (b) D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857 CrossRef CAS PubMed; (c) Q. B. Mou, Y. Ma, X. Jin, D. Y. Yan and X. Y. Zhu, Chem. Commun., 2016, 52, 11728 RSC; (d) Y. C. Zheng, S. P. Li, Z. L. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091 RSC; (e) G. Qiu, X. Liu, B. R. Wang, H. B. Gu and W. X. Wang, Polym. Chem., 2019, 10, 2527 RSC; (f) X. Liu, W. Lin, D. Astruc and H. B. Gu, Prog. Polym. Sci., 2019, 96, 43 CrossRef CAS; (g) X. Z. Zhu, Z. C. Wang, J. Liu, X. Min, T. Wang and X. D. Fan, Macromol. Rapid Commun., 2019, 40, 1900135 CrossRef PubMed; (h) D. Yang, Y. L. Hou, Q. Zhuang, P. Liu and J. Kong, Macromol. Rapid Commun., 2019, 40, 1900100 CrossRef PubMed; (i) S. E. Stiriba, M. Q. Slagt, H. Kautz, R. J. M. Klein Gebbink, R. Thomann, H. Frey and G. Koten, Chem. – Eur. J., 2004, 10, 1267 CrossRef CAS PubMed; (j) W. F. Jiang, Y. Liu, C. Y. Yu, S. L. Li, Y. J. Li and Y. F. Zhou, Chem. Commun., 2016, 52, 8223 RSC; (k) Y. L. Wang, B. Li, Y. F. Zhou, Z. Y. Lu and D. Y. Yan, Soft Matter, 2013, 9, 3293–3304 RSC; (l) X. D. Xiao, H. B. Chen, X. X. Dong, D. Z. Ren, Q. Deng, D. P. Wang and W. Tian, Angew. Chem., Int. Ed., 2020, 59, 9534–9541 CrossRef CAS PubMed; (m) S. Qiu, Z. Gao, F. Yan, H. X. Yuan, J. X. Wang and W. Tian, Chem. Commun., 2020, 56, 383–386 RSC; (n) L. Petho, G. Kasza, E. Lajkó, O. Láng, L. Kohidai, B. Iván and G. Mezo, Soft Matter, 2020, 16, 5759–5769 RSC; (o) N. Zs. Nagy, Z. Varga, J. Mihály, Gy. Kasza, B. Iván and É. Kiss, eXPRESS Polym. Lett., 2020, 14, 90–101 CrossRef.
- (a) I. N. Kurniasih, J. Keilitz and R. Haag, Chem. Soc. Rev., 2015, 44, 4145 RSC; (b) W. B. Wu, R. L. Tang, Q. Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997 RSC; (c) R. J. Dong, Y. Y. Zhou and X. Y. Zhu, Acc. Chem. Res., 2014, 47, 2006 CrossRef CAS PubMed; (d) D. L. Wang, T. Y. Zhao, X. Y. Zhu, D. Y. Yan and W. X. Wang, Chem. Soc. Rev., 2015, 44, 4023 RSC; (e) S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2012, 64, 102 CrossRef; (f) W. Z. Chen, A. Kretzschmann, W. Tian and S. Wu, Adv. Ther., 2018, 2, 1800103 CrossRef; (g) J. Zhang, H. J. Liu, Y. Yuan, S. Z. Jiang, Y. F. Yao and Y. Chen, ACS Macro Lett., 2013, 2, 67 CrossRef CAS; (h) Y. Chen, Z. Shen, L. Gehringer, H. Frey and S. E. Stiriba, Macromol. Rapid Commun., 2006, 27, 69 CrossRef; (i) Y. Bai, C. P. Liu, F. Y. Xie, R. Ma, L. L. Zhuo, N. Lia and W. Tian, Carbohydr. Polym., 2019, 213, 411–418 CrossRef CAS PubMed; (j) H. Li, Z. Z. Duan, Y. Yang, F. F. Xu, M. F. Chen, T. X. Liang, Y. Bai and R. Q. Li, Macromolecules, 2020, 53, 4255–4263 CrossRef CAS; (k) H. Li, Y. Yang, F. F. Xu, T. X. Liang, H. R. Wen and W. Tian, Chem. Commun., 2019, 55, 271–285 RSC.
- (a) A. N. Kharat and M. Adeli, J. Mater. Chem. B, 2015, 3, 3896 RSC; (b) Z. X. Zhou, X. P. Ma, C. J. Murphy, E. Jin, Q. H. Sun, Y. Q. Shen, E. A. Van Kirk and W. J. Murdoch, Angew. Chem., Int. Ed., 2014, 53, 10949 CrossRef CAS PubMed; (c) X. M. Ma, J. B. Tang, Y. Q. Shen, M. H. Fan, H. D. Tang and M. Radosz, J. Am. Chem. Soc., 2009, 131, 14795 CrossRef CAS PubMed; (d) S. Mignani, J. Rodrigues, H. Tomas, M. Zablocka, X. Y. Shi, A. M. Caminade and J. P. Majoral, Chem. Soc. Rev., 2018, 47, 514 RSC; (e) J. B. Wolinsky and M. W. Grinstaff, Adv. Drug Delivery Rev., 2008, 60, 1037 CrossRef CAS PubMed; (f) N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840 CrossRef CAS PubMed; (g) R. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef.
- (a) X. L. Liang, C. Gao, L. G. Cui, S. M. Wang, J. R. Wang and Z. F. Dai, Adv. Mater., 2017, 29, 1703135 CrossRef PubMed; (b) S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234 CrossRef CAS PubMed; H. Su, J. M. Koo and H. G. Cui, J. Controlled Release, 2015, 219, 383 Search PubMed; (c) Y. Bai, N. An, D. Chen, Y. Z. Liu, C. P. Liu, H. Yao, C. Wang, X. Song and W. Tian, Carbohydr. Polym., 2020, 231, 115714 CrossRef PubMed; (d) P. Huang, D. L. Wang, Y. Su, W. Huang, Y. F. Zhou, D. X. Cui, X. Y. Zhu and D. Y. Yan, J. Am. Chem. Soc., 2014, 136, 11748 CrossRef CAS PubMed.
- (a) C. Hu, X. H. Xu, X. Zhang, Y. C. Li, Y. K. Li and Z. W. Gu, Chem. Mater., 2017, 29, 7658 CrossRef CAS; (b) X. J. Xi, S. Q. Hu, Z. X. Zhou, X. R. Liu, J. B. Tang and Y. Q. Shen, J. Mater. Chem. B, 2016, 4, 5236 RSC; (c) Y. J. Huang, X. K. Ding, Y. Qi, B. R. Yu and F. J. Xu, Biomaterials, 2016, 106, 134 CrossRef CAS PubMed.
- (a) K. C. R. Bahadur and P. Xu, Adv. Mater., 2012, 24, 6479–6483 CrossRef CAS PubMed; (b) M. J. Zhou, X. J. Zhang, Y. L. Yang, Z. Liu, B. S. Tian, J. S. Jie and X. H. Zhang, Biomaterials, 2013, 34, 8960–8967 CrossRef CAS PubMed.
- L. Fan, S. S. Zhang, C. Y. Zhang, C. Yin, Z. Q. Chu, C. J. Song, G. Lin and Q. Li, Adv. Sci., 2018, 5, 1700289 CrossRef PubMed.
- Y. Li, J. Y. Lin, J. Y. Ma, L. Song, H. R. Lin, B. Tang, D. Y. Chen, G. H. Su, S. F. Ye, X. Zhu, F. H. Luo and Z. Q. Hou, ACS Appl. Mater. Interfaces, 2017, 9, 34650 CrossRef CAS PubMed.
- (a) X. Duan, H. Chen, L. Fan and J. Kong, ACS Biomater. Sci. Eng., 2016, 2, 2347 CrossRef CAS; (b) C. F. Liu, C. P. Li, C. Pang, M. Q. Li, H. X. Li, P. X. Li, L. Fan, H. Liu and W. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 27940–27950 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, X. B. Zhang, X. Wei, Z. M. Tang and S. B. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 5833 CrossRef CAS PubMed.
- (a) C. Wu and S. Q. Zhou, Chem. Commun., 2013, 49, 5633 RSC; (b) H. J. Dou, M. Jiang, H. S. Peng, D. Y. Chen and Y. Hong, Angew. Chem., Int. Ed., 2003, 42, 1516 CrossRef CAS PubMed.
- (a) J. F. Reuther, D. A. Siriwardane, R. Campos and B. M. Novak, Macromolecules, 2015, 48, 6890 CrossRef CAS; (b) X. Song, X. D. Deng, Q. H. Wang, J. J. Tian, F. L. He, H. Y. Hu and W. Tian, Polym. Chem., 2020, 11, 102 RSC.
- T. Bai, J. J. Du, J. X. Chen, X. Duan, Q. Zhuang, H. Chen and J. Kong, Polym. Chem., 2017, 8, 7160 RSC.
- (a) J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS; (b) H. Yao, M. Qi, Y. Liu and W. Tian, Chem. – Eur. J., 2016, 22, 8508–8519 CrossRef CAS PubMed.
- Y. Bai, C. P. Liu, D. Chen, C. F. Liu, L. H. Zhuo, H. Li, C. Wang, H. T. Bu and W. Tian, Carbohydr. Polym., 2020, 246, 116654 CrossRef CAS PubMed.
- Y. B. Cai, H. S. Shen, J. Zhan, M. L. Lin, L. H. Dai, C. H. Ren, Y. Shi, J. F. Liu, J. Gao and Z. M. Yang, J. Am. Chem. Soc., 2017, 139, 2876–2879 CrossRef CAS PubMed.
- S. Q. Wang, H. Zheng, L. Zhou, F. Cheng, Z. Liu, H. P. Zhang, L. L. Wang and Q. Y. Zhang, Nano Lett., 2020, 20, 5149–5158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00862a |
| This journal is © The Royal Society of Chemistry 2020 |