
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStep-growth polymerisation of alkyl acrylates via concomitant oxa-Michael and transesterification reactions†
Karin
Ratzenböck
ac,
David
Pahovnik
 b and
Christian
Slugovc
*ac
b and
Christian
Slugovc
*ac
aChristian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, A 8010 Graz, Austria
bNational Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia
cInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, A 8010 Graz, Austria. E-mail: slugovc@tugraz.at
First published on 23rd November 2020
Abstract
Herein we propose an auto-tandem catalytic approach towards the preparation of poly(ester–ether)s from simple alkyl acrylates and diols. By combining oxa-Michael addition with transesterification the preparation of hydroxy functionalised acrylate monomers can be avoided.
Introduction
Polymerisation of acrylates is typically performed by free radical polymerisation. Formed poly(acrylates) based on C–C main chains are widely used as paintings, surface coatings, adhesives, or fibres.1 However, the oxa-Michael addition reaction2 presents an alternative method for the polymerisation of acrylates leading to potentially more easily degradable poly(ester–ether)s in which functional groups are incorporated in the polymer main chain. Oxa-Michael addition polymerisation of hydroxyalkyl acrylates, also referred to as hydrogen-transfer polymerisation, was first reported in 1975.3 The polymerisation of 2-hydroxyethyl acrylate (HEA) could be initiated with LiH or PPh3. Since then few studies on the polymerisation of hydroxy functionalized acrylates with base- or nucleophile-type catalysts have been reported.4–6 Suzuki et al. presented the homopolymerisation of hydroxy functionalized acrylate monomers, such as HEA, catalysed by N-heterocyclic carbenes (NHC) at room temperature.7 Within the same group, step-growth polymerisation of acrylic acid catalysed by NHC ligands or PPh3 was studied under harsh conditions (100 °C) and oligomers with number average molecular masses (Mn) up to 400 Da were obtained.8 Moreover, diacrylates and diols have been employed to synthesize poly(ester–ether)s catalysed by phosphazene base.9,10 In all mentioned reactions, inert conditions were necessary and oligomers with Mns up to 2000 Da were obtained. Moreover, concomitant transesterification was identified as a side reaction leading to the reconstruction of the repeat units.4–10Further step-growth polymerisations of acrylates comprising two different reaction mechanisms are a combined condensation and oxa-Michael addition reaction of acrylic acid and diols catalysed by a Brønsted acid and a tail-to-tail dimerisation combined with a transesterification of hydroxy-functionalized methacrylates activated by a NHC catalyst.8,11 Further, a combination of an oxa-Michael addition of HEA with a ring-opening polymerisation catalysed by phosphazene base has been disclosed.12
Herein, we report the direct synthesis of poly(ester–ether)s via an auto-tandem catalysis13 of a polyaddition reaction – the oxa-Michael reaction – and a polycondensation reaction, namely transesterification. Starting form readily accessible alkyl acrylate monomers and diols, instead of hydroxy functionalized acrylates, polymerisation via two mechanistically distinct reactions was accessible with a single catalyst under ambient and solvent-free conditions (Scheme 1).
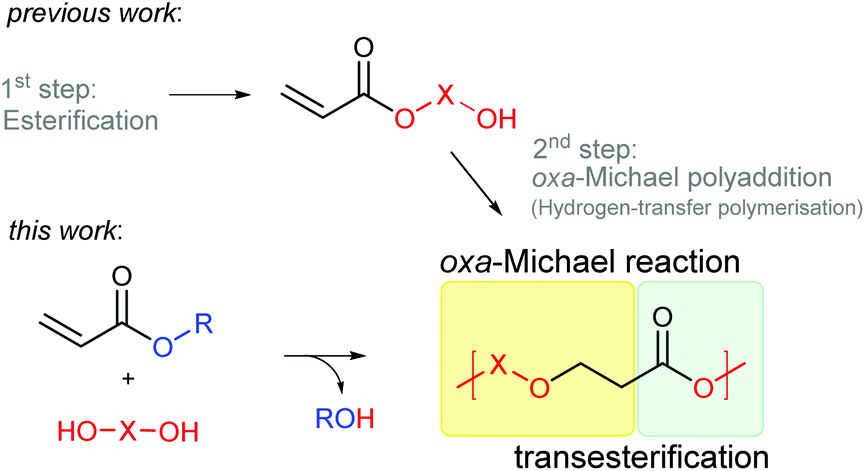 | ||
| Scheme 1 Tandem polymerisation of acrylates with diols. | ||
For the polymerisation a single catalyst that activates both types of reactions was chosen. The nucleophile 4-dimethylaminopyridine (DMAP) and the putatively non-nucleophilic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were tested as catalysts. DMAP has previously been reported to catalyse oxa-Michael reactions such as the addition of alcohols to divinyl sulfone or the phenol-yne click polymerisation of diphenols and dialkynes.14,15 Additionally, DMAP is well known to catalyse esterification reactions.16 DBU has been used as a catalyst which efficiently promotes oxa-Michael reactions of mono-alcohols and activated alkenes under mild conditions.17 It has also been used as catalyst in the oxa-Michael addition of glycerol and acrylic compounds.18 Moreover, transesterification reactions efficiently catalysed by DBU have been reported.19 Recently it has been used as a dual catalyst allowing DBU-pterin salt formation and concomitant ester-amide-transformation.20
Results and discussion
Initially, the polymerisation of 1 equivalent methyl acrylate (1) with 1 equivalent (Z)-2-butene-1,4-diol (4) using 5 mol% DMAP or DBU was tested under solvent-free conditions. In order to avoid the evaporation of the acrylate the reaction was started at 50 °C well below the boiling point of 1 (80 °C) in a closed reaction vessel. Upon full consumption of the acrylate (typically after 2 h, for details see ESI†) the reaction temperature was increased to 80 °C and the reaction vessel was opened in order to facilitate transesterification by removing the formed alcohol by evaporation. After 24 h overall reaction time, the outcome of the reaction was analysed using 1H- and 13C-NMR spectroscopy, infrared spectroscopy (IR), size exclusion chromatography (SEC) and in some cases matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS). In case of DMAP, oligomers with a Mn of 430 Da determined by SEC were observed (Table 1, entry 1). By changing the catalyst to DBU the Mn could be more than doubled (Table 1, entry 2). To rule out any unwanted side reactions e.g. saponification caused by traces of water, the reaction was performed under inert conditions in the presence of 4 Å molecular sieve using dried reagents. As the obtained oligomers showed a similar molecular mass (Table 1, entry 3) and no evidence for the formation of carboxylic acid or carboxylate groups in any experiment could be retrieved from 13C-NMR measurements, the use of inert conditions does not have any benefits. However, in entries 1–3, using 1 as the acrylate, methanol formed during the transesterification. Methanol itself is a reactive Michael donor and thus, is competing with 4 in the oxa-Michael reaction. A 3-methoxypropionate species formed in considerably large amounts (20% of all end groups – determined by 1H-NMR spectroscopy (ESI Table 3†)) as side product during the reaction causing undesired chain termination. Previously, it has been shown in our group that MeOH reacts faster than EtOH in case of oxa-Michael addition and t-BuOH does not show any reactivity.21 Therefore, ethyl acrylate (2) was studied as an alternative Michael acceptor. Even though ethanol can also react as Michael donor, less side product formation (8% of chains terminated with 3-ethoxypropionate) and a similar molecular mass in comparison to methyl acrylate could be observed with DBU as catalyst (Table 1, entry 4). Moreover, t-butyl acrylate (3) was studied, as formed t-BuOH should be inactive in terms of oxa-Michael addition reaction (Table 1, entry 5). However, in this case hardly any transesterification could be observed and cis-di-t-butyl-3,3′-(but-2-ene-1,4-diyloxy)dipropionate was identified as main product (ESI Fig. 34†). These findings are in accordance with Oget et al. who reported that in the oxa-Michael addition reaction of heptanols and t-butyl acrylate hardly any transesterification takes place.18 Subsequently, 2 was used as Michael acceptor in all further experiments. In case of DMAP, only shorter oligomers formed, mainly because transesterification remained a challenge, leaving approx. 30% ethyl ester groups unreacted (Table 1, entry 6, ESI Fig. 32†). Similarly, using a lower DBU loading of 2.5 mol%, the transesterification efficacy suffered, resulting in approx. 18% ethyl ester end groups and leading to a rather low Mn value of 760 Da (Table 1, entry 7). Increasing the DBU loading to 10 mol% did not result in an increase in molecular mass (Table 1, entry 8), but other than in entry 7 no ethyl ester groups were detected and complete transesterification could be achieved (ESI Fig. 28†).|
|
||||
|---|---|---|---|---|
| Entry | R | Catalyst (loading [mol%]) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [Da] b [Da] |
Đ |
| a Reaction conditions: 1 equiv. acrylate, 1 equiv. diol, reaction temperature first 50 °C (until oxa-Michael reaction complete) then 80 °C, total reaction time 24 h, no solvent used. b Number average molecular mass and dispersity (Đ) determined by SEC in THF relative to poly(styrene) standards. c Water-free conditions. d Oxa-Michael reaction was performed at 20 °C. e Reaction temperature first 50 °C for 2.5 h, then 5 h at 80 °C under vacuum. | ||||
| 1 | Me | DMAP (5) | 430 | 1.2 |
| 2 | Me | DBU (5) | 910 | 1.5 |
| 3c | Me | DBU (5) | 950 | 1.6 |
| 4 | Et | DBU (5) | 950 | 1.6 |
| 5 | t Bu | DBU (5) | — | — |
| 6 | Et | DMAP (5) | 530 | 1.2 |
| 7 | Et | DBU (2.5) | 760 | 1.5 |
| 8 | Et | DBU (10) | 810 | 1.5 |
| 9d | Et | DBU (10) | 810 | 1.5 |
| 10e | Me | DBU (5) | 1180 | 1.8 |
The reaction progress was monitored by NMR spectroscopy (ESI Fig. 33†). Even though both reactions, oxa-Michael addition as well as transesterification, can be observed after several minutes, the oxa-Michael reaction proceeds at a faster rate. After 24 h of total reaction time the reaction is in equilibrium, preventing further chain growth. Longer curing at 80 °C (3 d) did not increase the molecular mass. The oxa-Michael reaction itself proceeded also at room temperature, albeit at a slightly slower rate. However, for transesterification elevated temperatures are necessary.22 The polymer's Mn is not influenced by the initial reaction temperature (Table 1, entry 9). Finally, the principal possibility of obtaining higher molecular masses in this reaction was demonstrated by applying vacuum during the second reaction step (Table 1, entry 10).
The reaction product of 2 and 4 using 5 mol% DBU at 50 °C under bulk conditions (Table 1, entry 4) was investigated in detail. The 1H-NMR spectrum is dominated by the signals representing the repeat units of the oligomer giving rise to five nearly equally intense signals at 5.70, 4.68, 4.10, 3.69 and 2.58 ppm corresponding to the olefinic hydrogens, the methylene groups of the esterified and etherified diol, and the methylene groups resulting from the oxa-Michael addition respectively (Fig. 1, top). Four different end groups were identified. With 72% end group A, featuring a free OH-group stemming from a 2-butene-1,4-diol unit, is the most abundant one, exhibiting a signal at 4.21 ppm. It cannot be differentiated whether A is located on the oxa-Michael addition side (EA), the transesterification side (EE), or on both ends. Signals at 6.41 and 6.11 ppm are attributed to residual vinyl end groups B (8%). Side product formation by ethanol led to a 3-ethoxypropionate end group C (8%) with signals at 3.48 and 1.17 ppm. About 12% of the ethyl ester did not undergo transesterification giving rise to signals at 4.18 and 1.25 ppm (end group D). Based on this evaluation, an average degree of polymerisation (n) of 5.5 was calculated (for details see ESI†). Accordingly, the Mn is about 770 Da, a value which compares fairly well with the Mn of 950 Da determined by SEC. MALDI-TOF MS confirmed the presence of the end groups identified by NMR spectroscopy (Fig. 1). Additionally, species bearing [DBU]+ moieties, most likely as end groups, were identified. Moreover, series containing decomposition products of DBU were observed. As recently described, traces of H2O can cause hydrolysis of DBU resulting in primary amines23 which can then react in an aza-Michael addition manner. Further evidence of such species is provided by IR spectroscopy as the formed lactam derivative gives rise to bands at 1648 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1575 cm−1 (N–H) (ESI Fig. 7†). However, species of such decomposition adducts were only observed in minor amounts. According to NMR spectroscopy and MALDI-TOF MS the most abundant species bears alcohol end groups.
O) and 1575 cm−1 (N–H) (ESI Fig. 7†). However, species of such decomposition adducts were only observed in minor amounts. According to NMR spectroscopy and MALDI-TOF MS the most abundant species bears alcohol end groups.
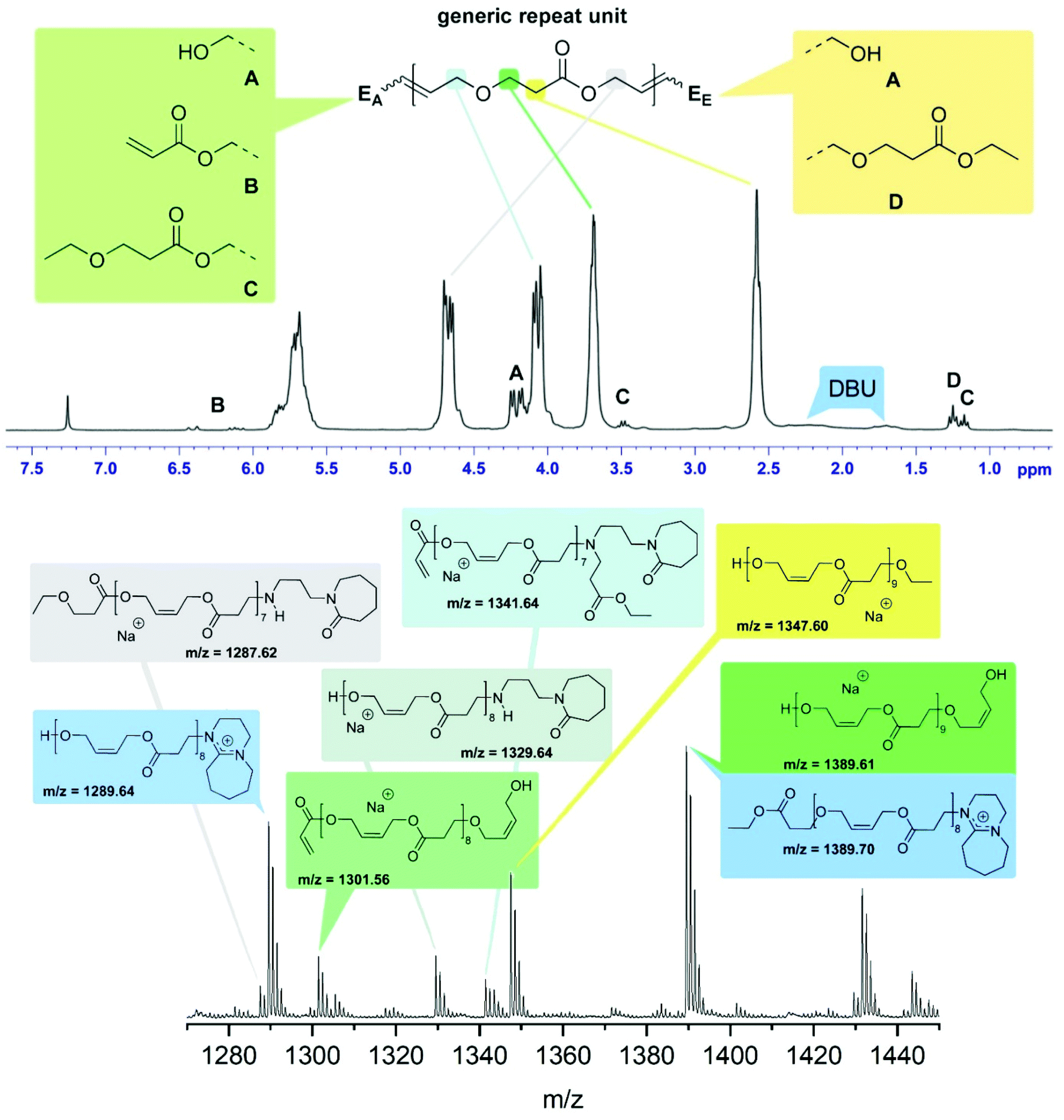 | ||
| Fig. 1 Top: 1H-NMR spectrum of poly2/4 in CDCl3 including a peak assignment of the repeat unit (coloured) and the end-groups (representative peaks marked with capital letters); bottom: detail of the MALDI-TOF MS with peak assignment; the repeat unit is 142.06 Da; note that the sketched structures are representative for the many isomeric species possible. | ||
The scope of the reaction was further tested by employing 2-butyne-1,4-diol (5), ethylene glycol (6), 1,4-butanediol (7), and 1,2-propanediol (8) as diols and 2 as acrylate under optimized conditions. Results are gathered in Table 2. All introduced alcohols allowed polymerisation resulting in oligomers with similar molecular masses between 560 and 700 Da. Again, DBU performed better than DMAP (Table 2, entries 2 and 3). The results of the reaction of ethylene glycol (6) with 2 and the oxa-Michael addition polymerisation of 2-hydroxyethyl acrylate (HEA) using the same reaction conditions were compared. The homopolymerisation of HEA resulted in a polymer with a Mn of 880 Da while reacting 2 and 6 gave a lower Mn of 600 Da (ESI Fig. 31†). This observation can be rationalised by the importance of adjusting the right stoichiometry of the reactants in the auto-tandem polymerisation. The relatively low molecular masses obtained here are attributed to the reaction conditions (catalyst choice, reaction time and temperature), as in literature similar oligomers from hydroxy-functionalized acrylates are described with Mn-values of 155 to 2400 Da.4–9 Thus, it can be assumed that by tuning the reaction conditions or changing the catalyst not only for the homopolymerisation of HEA but also for the herein presented auto-tandem polymerisation, higher molecular masses might be accessible. The principle feasibility of such an optimization was demonstrated by applying vacuum during the second reaction step (Table 1, entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Diol | Yielda [%] | n |
M
nc [Da] |
Đ |
| a Crude yield after 24 h reaction time, calculated according to: massreaction mixture/(massmonomers + masscatalyst − massEtOH) × 100. b Repeat units calculated from 1H-NMR spectra. c Number average molecular mass and dispersity determined by SEC in THF relative to poly(styrene) standards. d Reaction performed with 5 mol% DMAP. e Not determined due to overlapping signals. | |||||
| 1 | 4 | 98 | 5.5 | 950 | 1.6 |
| 2d | 5 | 89 | 1.9 | 420 | 1.1 |
| 3 | 5 | 99 | 2.8 | 560 | 1.3 |
| 4 | 6 | 93 | n.d.e | 600 | 1.3 |
| 5 | 7 | 95 | 3.1 | 700 | 1.4 |
| 6 | 8 | 87 | n.d.e | 590 | 1.3 |
Mechanistically, the reaction is initiated either by deprotonation of the alcohol or by nucleophilic attack of the base/nucleophile on the Michael acceptor in a 1,4- or 1,2-fashion, generating a strongly basic zwitterion, which can subsequently deprotonate an alcohol. During propagation, the formed alkoxide undergoes either an oxa-Michael addition or a transesterification with the acrylate. In the later stage, additional transesterification of newly formed esters is feasible, resulting in structural diverse oligomers. Thus, not only alternating oxa-Michael addition and transesterification fragments, but also di-ester and di-ether structure elements as well as cyclic species are likely to be present (ESI Fig. 6†).4–10
Conclusions
This work demonstrates the accessibility of poly(ester–ether)s from alkyl acrylates and diols by an auto-tandem catalysis. Concomitant oxa-Michael addition and transesterification reactions induced by the same base or nucleophile result in the desired macromolecule. Adjusting the stoichiometry of the acrylate and the diol has been identified as the critical step of this approach. Moreover, the reaction can be performed without exclusion of humidity and pre-preparation of hydroxy functionalised acrylate monomers, traditionally employed in the oxa-Michael (or the hydrogen-transfer) polymerisation, is no longer required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Christian Doppler Research Association (CDL-Organocatalysis in Polymerization) is gratefully acknowledged.Notes and references
- E. Penzel, N. Ballard and J. M. Asua, Polyacrylates, in Ullmann's encyclopedia of industrial chemistry, ed. W. Gerhartz and Y. S. Yamamoto, Wiley-VCH, Weinheim, 1985–1996, pp. 1–20. DOI:10.1002/14356007.a21_157.pub2.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Michael addition reactions in macromolecular design for emerging technologies, Prog. Polym. Sci., 2006, 31, 487–531, DOI:10.1016/j.progpolymsci.2006.03.001.
- T. Saegusa, S. Kobayashi and Y. Kimura, Hydrogen-Transfer Polymerization of Hydroxyalkyl Acrylates, Macromolecules, 1975, 8, 950–952, DOI:10.1021/ma60048a051.
- B. A. Rozenberg, Macromolecular design with the use of acrylic monomers containing groups with mobile hydrogen atoms, Polym. Sci., Ser. C, 2007, 49, 355–385, DOI:10.1134/S1811238207040042.
- J.-I. Kadokawa, Y. Kaneko, S. Yamada, K. Ikuma, H. Tagaya and K. Chiba, Synthesis of hyperbranched polymers via proton–transfer polymerization of acrylate monomer containing two hydroxy groups, Macromol. Rapid. Commun., 2000, 21, 362–368, DOI:10.1002/(SICI)1521-3927(20000401)21:7<362::AID-MARC362>3.0.CO;2-F.
- Y. Lin, J.-W. Gao, H.-W. Liu and Y.-S. Li, Synthesis and Characterization of Hyperbranched Poly(ether amide)s with Thermoresponsive Property and Unexpected Strong Blue Photoluminescence, Macromolecules, 2009, 42, 3237–3246, DOI:10.1021/ma802353f.
- S. Matsuoka, S. Namera and M. Suzuki, Oxa-Michael addition polymerization of acrylates catalyzed by N-heterocyclic carbenes, Polym. Chem., 2015, 6, 294–301, 10.1039/C4PY01184H.
- T. Murase, S. Matsuoka and M. Suzuki, Hydrogen-transfer and condensation–addition polymerizations of acrylic acid, Polym. Chem., 2018, 9, 2984–2990, 10.1039/C8PY00271A.
- H. Yang, Y. Zuo, J. Zhang, Y. Song, W. Huang, X. Xue, Q. Jiang, A. Sun and B. Jiang, Phosphazene-catalyzed oxa-Michael addition click polymerization, Polym. Chem., 2018, 9, 4716–4723, 10.1039/C8PY01089G.
- Q. Jiang, Y. L. Zhang, Y. Du, M. Tang, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, Preparation of hyperbranched polymers by oxa-Michael addition polymerization, Polym. Chem., 2020, 11, 1298–1306, 10.1039/C9PY01686D.
- A. Fukumoto, M. Arimoto, S.-I. Matsuoka and M. Suzuki, Polycondensation of methacrylates: auto-tandem organocatalysis using N-heterocyclic carbenes, Polym. Chem., 2018, 9, 5295–5302, 10.1039/C8PY01027G.
- H.-J. Yang, C.-Q. Chai, Y.-K. Zuo, J.-F. Huang, Y.-Y. Song, L. Jiang, W.-Y. Huang, Q.-M. Jiang, X.-Q. Xue and B.-B. Jiang, Hybrid Copolymerization via the Combination of Proton Transfer and Ring-opening Polymerization, Chin. J. Polym. Sci., 2020, 38, 231–239, DOI:10.1007/s10118-020-2341-x.
- C. Robert and C. M. Thomas, Tandem catalysis: a new approach to polymers, Chem. Soc. Rev., 2013, 42, 9392–9402, 10.1039/C3CS60287G.
- S. Strasser, C. Wappl and C. Slugovc, Solvent-free macrocyclisation by nucleophile-mediated oxa-Michael addition polymerisation of divinyl sulfone and alcohols, Polym. Chem., 2017, 8, 1797–1804, 10.1039/C7PY00152E.
- Y. Shi, T. Bai, W. Bai, Z. Wang, M. Chen, B. Yao, J. Z. Sun, A. Qin, J. Ling and B. Z. Tang, Phenol–yne Click Polymerization: An Efficient Technique to Facilely Access Regio– and Stereoregular Poly(vinylene ether ketone)s, Chem. – Eur. J., 2017, 23, 10725–10731, DOI:10.1002/chem.201702966.
- B. Neises and W. Steglich, Simple Method for the Esterification of Carboxylic Acids, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524, DOI:10.1002/anie.197805221.
- J. E. Murtagh, S. H. McCooey and S. J. Connon, Novel amine-catalysed hydroalkoxylation reactions of activated alkenes and alkynes, Chem. Commun., 2005, 227–229, 10.1039/B414895A.
- F. Nadeau, M. Sindt and N. Oget, Free-solvent Michael addition of glycerol to acrylic compounds, New J. Chem., 2015, 39, 9155–9161, 10.1039/C5NJ02223A.
- S. R. Samanta, R. Cai and V. Percec, A rational approach to activated polyacrylates and polymethacrylates by using a combination of model reactions and SET-LRP of hexafluoroisopropyl acrylate and methacrylate, Polym. Chem., 2015, 6, 3259–3270, 10.1039/C5PY00082C.
- A. R. Bockman and J. M. Pruet, Exploring the scope of DBU-promoted amidations of 7-methoxycarbonylpterin, Beilstein J. Org. Chem., 2020, 16, 509–514, DOI:10.3762/bjoc.16.46.
- S. Strasser and C. Slugovc, Nucleophile-mediated oxa-Michael addition reactions of divinyl sulfone - a thiol-free option for step-growth polymerisations, Catal. Sci. Technol., 2015, 5, 5091–5094, 10.1039/C5CY01527H.
- J. C. A. Flanagan, E. J. Kang, N. I. Strong and R. M. Waymouth, Catalytic Dimerization of Crotonates, ACS Catal., 2015, 5, 5328–5332, DOI:10.1021/acscatal.5b00930.
- A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Investigating the Underappreciated Hydrolytic Instability of 1,8-Diazabicyclo[5.4.0]undec-7-ene and Related Unsaturated Nitrogenous Bases, Org. Process Res. Dev., 2019, 23, 1860–1871, DOI:10.1021/acs.oprd.9b00187.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and detailed characterisation of all products. See DOI: 10.1039/d0py01271h |
| This journal is © The Royal Society of Chemistry 2020 |