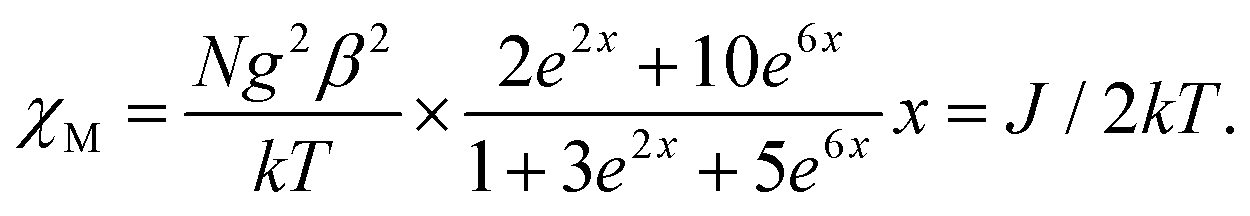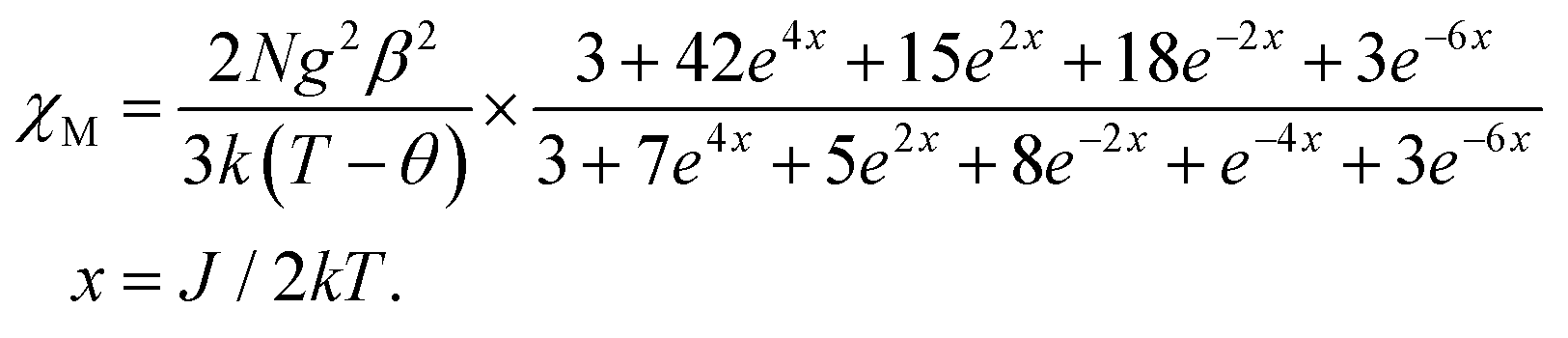DOI:
10.1039/C9QI00975B
(Research Article)
Inorg. Chem. Front., 2020,
7, 247-259
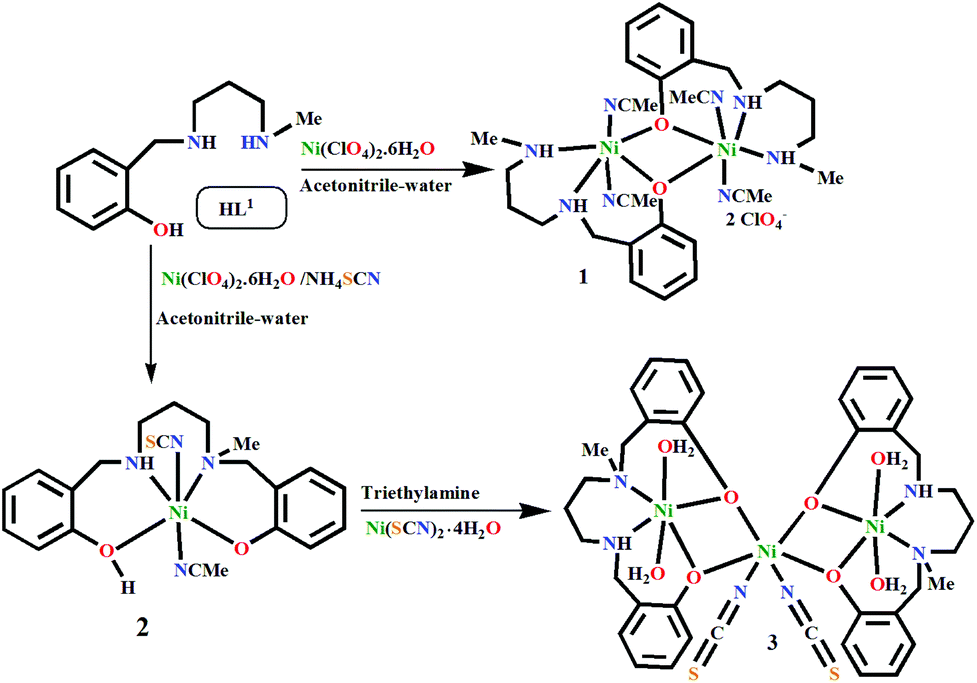
In situ transformation of a tridentate to a tetradentate unsymmetric Schiff base ligand via deaminative coupling in Ni(II) complexes: crystal structures, magnetic properties and catecholase activity study†
Received
2nd August 2019
, Accepted 23rd October 2019
First published on 24th October 2019
Abstract
A new dimeric Ni(II) complex, [Ni2L12(CH3CN)4](ClO4)2·2CH3CN (1), was synthesized using an N2O donor reduced Schiff base [(HL1) = 2-[(3-methylamino-propylamino)-methyl]-4-phenol]. Surprisingly, during an attempt to replace its ClO4− ion with SCN−, the N2O donor ligand in situ converted to a tetradentate N2O2 donor ligand and formed a metal complex, [Ni(HL2)(NCS)(CH3CN)] (2). A probable mechanism via deaminative coupling for this conversion is proposed. Using 2 as a metalloligand under basic conditions, a trinuclear metal complex, [Ni3(L2)2(NCS)2(H2O)4]·H2O (3), was prepared. Single crystal structural characterization revealed that in all three metal complexes, the Ni(II) atoms were in an octahedral environment with coordinated solvent molecules (CH3CN in 1 and 2 and H2O in 3). Among the three metal complexes, 1 and 3 showed catecholase-like biomimicking activity. The calculation of the turnover numbers (Kcat = 7.9 for 1, 14.5 for 3) reveals that 3 is a better catalyst than 1. Mechanistic cycles are proposed for this biomimicking activity on the basis of ESI-MS spectrometry and iodometric measurements. Temperature-dependent magnetic susceptibility measurements suggest that the Ni(II) ions in metal complexes 1 and 3 are antiferromagnetically coupled (J = −32.22 cm−1 for 1, J = −10.4 cm−1 for 3), consistent with their geometries and bridging angles. Theoretically calculated J values (J = −40.15 cm−1 for 1, J = −14.53 cm−1 for 3) by the DFT method corroborate well with the experimental values.
Introduction
N,O-Donor ligands have been widely designed and used for the synthesis of the coordination complexes of predetermined nuclearity and stereochemistry depending upon the type of metal ions.1 However, from a synthetic viewpoint, this synthesis is not free from unexpected outcomes, such as structural serendipity,2in situ ligand transformation,3 and the formation of carbonato-bridged complexes by atmospheric CO2 fixation.4 Structural serendipity is encountered very often during inorganic synthesis; however, the transformation of organic ligand scaffolds has also been observed on few occasions, which opens up new avenues of metal-assisted catalytic transformation to obtain value-added chemicals. These in situ ligand transformations mostly depend on the reactivity of the functional groups towards nucleophilic addition,5 hydro-/solvolysis,5 cycloaddition,5,6 isomerisation,7 elimination,8 oxidation,9 reduction,10 and condensation11 as well as substitution on aromatic side chains.12 The metal ion often plays a very important role in such transformations.13 For example, strongly Lewis acidic redox-inactive metal ions are known to activate the electrophilic center of the imine group of a Schiff base, giving rise to nucleophilic addition.14,15 These transition metal ion-catalyzed coupling reactions have important synthetic applications in single step organic transformation.16
We have been using N2O-donor Schiff bases for quite some time to synthesize polynuclear transition metal complexes with intriguing catalytic and magnetic properties. However, these ligands are prone to undergo rearrangements during complex formation via hydrolytic cleavage of the imine bond. To prevent these rearrangements, the imine bonds are reduced to form reduced Schiff bases, which are usually very stable as ligands; however, in some cases, they can also undergo in situ transformations. As an example, one such conversion was reported where a highly redox active tridentate N2O donor substituted 2-(methylamino)phenol moiety was oxidized by aerial oxygen to form a N3O donor tetradentate Schiff base ligand in the presence of Cu(II) via a oxidative coupling mechanism.17 On the other hand, compounds containing primary and secondary amine groups are known to undergo coupling reactions in the presence of Ni, Ru, Co–Rh and Ir catalysts18via a deaminative pathway.19 However, neither of these coupling reactions have been reported for tridentate N2O donor reduced Schiff bases containing the 2-(aminomethyl)phenol moiety, although these bases have been widely used as ligands for synthesizing complexes with various metal ions and anionic coligands.20 This coupling is of potential interest for single step generation of important unsymmetric salan-type ligands catalysed by non-noble 3d transition metal ions.21
Herein, we synthesize a new dinuclear Ni(II) complex, [Ni2L12(CH3CN)4](ClO4)2·2CH3CN (1) using a tridentate N2O donor reduced Schiff base [(HL1) = 2-[(3-methylamino-propylamino)-methyl]-4-phenol]. However, in our attempt to synthesize its thiocyanate counterpart, two N2O-donor reduced Schiff base ligands (L1)1− undergo in situ coupling to form a new N2O2 donor ligand (HL2)1− [where H2L2 = N,N′-bis(2-hydroxybenzyl)-N′-methyl-1,3-propanediamine], resulting in a mononuclear complex, [Ni(HL2)(NCS)(CH3CN)] (2). 2 is further used as a metalloligand to synthesize a trinuclear metal complex [(NiL2)2Ni(NCS)2(H2O)4]·H2O (3). All three complexes have been characterized by single crystal X-ray analyses. Among the metal complexes, 1 and 3 exhibit catecholase-like biomimicking activities under aerial conditions. ESI-Mass spectra have been used to investigate the detailed mechanism of the coupling as well as the catecholase-like catalytic activity of these complexes. The magnetic properties of 1 and 3 have been investigated experimentally and theoretically. This study shows that both metal complexes are antiferromagnetically coupled, commensurate with wide bridging angles between the Ni(II) centers. The experimentally obtained values are well-matched with the theoretically calculated coupling constants.
Experimental section
Starting materials
Salicylaldehyde, N-methyl-1,3-propanediamine, sodium borohydride and ammonium thiocyanate were purchased from Spectrochem, India and were of reagent grade. They were used without further purification. The other reagents and solvents were of commercially available reagent quality unless otherwise stated.
Caution!
Although this was not encountered during our experiments, perchlorate salts of metal complexes with organic ligands are potentially explosive. Only a small amount of material should be prepared, and it should be handled with care.
Synthesis of 2-[(3-methylamino-propylamino)-methyl]-4-phenol (HL1)
HL1 was synthesized by the usual method:22 5 mmol salicylaldehyde (0.52 mL) was mixed with 5 mmol N-methyl-1,3-propanediamine (0.52 mL) in methanol (30 mL). The resulting solution was refluxed for 1 h and allowed to cool. Solid sodium borohydride (0.222 g, 6 mmol) was added slowly with constant stirring. After completion of the addition, the resulting reaction mixture (HL1) was directly used for further reaction with metal salts.
Synthesis of [Ni2L12(CH3CN)4](ClO4)2·2CH3CN (1)
The methanolic solution (20 mL) of HL1 (5 mmol) was allowed to react with an aqueous solution (10 mL) of Ni(ClO4)2·6H2O (1.828 g, 5 mmol) with constant stirring for 30 minutes. Slow evaporation of the resulting green solution afforded a dark green microcrystalline compound. The green solid was separated by filtration, washed with diethyl ether and dissolved in CH3CN. X-ray quality deep-green single crystals of 1 were obtained by slow evaporation of the acetonitrile solution (yield: 1.9 g; 80%). Anal. Calcd for C34H50Cl2N10Ni2O10: C, 43.02; H, 5.52; N, 14.76. Found: C, 43.14; H, 5.67; N, 14.85. IR (KBr pellet, cm−1): ν(N–H), 3289 cm−1; ν(Cl![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1091 cm−1. λmax (nm) in methanol, 652, 996.
O), 1091 cm−1. λmax (nm) in methanol, 652, 996.
Synthesis of [Ni(HL2)(NCS)(CH3CN)] (2)
Metal complex 2 was synthesized by following the same procedure mentioned above. Here, the only difference was that an aqueous solution of NH4SCN (5 mmol, 0.380 g) was slowly added to the reaction mixture (yield: 0.8 g; 70%). Anal. Calcd for C21H26N4Ni1O2S1: C, 55.17; H, 5.73; N, 12.25. Found: C, 55.28; H, 5.88; N, 12.37. IR (KBr pellet, cm−1): ν(N–H), 3206 cm−1; ν(SCN), 2116 cm−1. λmax (nm) in methanol, 648, 1012. μeff = 3.02μB.
Synthesis of [Ni3(L2)2(NCS)2(H2O)4]·H2O (3)
1 mmol (0.457 g) of metal complex 2 dissolved in 10 ml of methanol was added to a methanolic solution of Ni(SCN)2·4H2O (0.123 g, 0.5 mmol) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with slow stirring. This nickel thiocyanate salt was synthesized by a previously reported method.22c After ca. 15 min, triethylamine (0.14 mL, 1 mmol) was added to the mixture. Slow evaporation of the resulting green solution afforded a dark green microcrystalline compound. The green solid was then filtered and washed with diethyl ether and dissolved in an acetonitrile–water mixture. X-ray quality deep-green single crystals of metal complex 3 were obtained by slow evaporation of the acetonitrile–water solution (yield: 0.4 g; 80%). Anal. Calcd for C38H54N6Ni3O9S2: C, 46.62; H, 5.56; N, 8.58 Found: C, 46.74; H, 5.68; N, 8.69. IR (KBr pellet, cm−1): ν(N–H), 3214 cm−1; ν(SCN), 2102 cm−1; ν(OH) in H2O, 3419 cm−1. λmax (nm) in methanol, 650, 1024.
:1 ratio with slow stirring. This nickel thiocyanate salt was synthesized by a previously reported method.22c After ca. 15 min, triethylamine (0.14 mL, 1 mmol) was added to the mixture. Slow evaporation of the resulting green solution afforded a dark green microcrystalline compound. The green solid was then filtered and washed with diethyl ether and dissolved in an acetonitrile–water mixture. X-ray quality deep-green single crystals of metal complex 3 were obtained by slow evaporation of the acetonitrile–water solution (yield: 0.4 g; 80%). Anal. Calcd for C38H54N6Ni3O9S2: C, 46.62; H, 5.56; N, 8.58 Found: C, 46.74; H, 5.68; N, 8.69. IR (KBr pellet, cm−1): ν(N–H), 3214 cm−1; ν(SCN), 2102 cm−1; ν(OH) in H2O, 3419 cm−1. λmax (nm) in methanol, 650, 1024.
Physical measurements
Elemental analyses (C, H and N) were performed using a PerkinElmer 2400 series II elemental analyzer. IR spectra in KBr pellets (4500 to 500 cm−1) were recorded using a PerkinElmer RXI FT-IR spectrophotometer. Electronic spectra (1500 to 250 nm) were recorded in a Hitachi U-3501 spectrophotometer. Electrospray ionization mass spectra (ESI-MS positive) were recorded using a Xevo G2-S QTof (Waters) mass spectrometer equipped with a Z-spray interface over a mass range of 100 to 1200 Da in continuum mode. The temperature-dependent molar susceptibilities of crystalline powdered samples 1 and 3 were measured using a superconducting quantum interference device vibrating sample magnetometer (SQUID-VSM, Quantum Design) with an applied field of 500 Oe throughout the temperature range of 2 to 300 K. The susceptibility data were corrected according to literature values of Pascal's table for diamagnetic contributions.23 Isothermal magnetization measurements were performed at 2 K up to a 5 Tesla magnetic field. The cyclic voltammetry measurements were carried out on an Epsilon Basi-C3 Cell instrument at a scan rate of 100 to 300 mV s−1 within the potential range of 0 to −1.20 V vs. Ag/AgCl in the presence of TBAP as supporting electrolyte.
X-ray crystallographic data collection and refinement
Collected diffractable single crystals of metal complexes 1–3 were mounted on a Bruker-AXS SMART APEX II diffractometer equipped with a graphite monochromator and Mo-Kα (λ = 0.71073 Å) radiation. The crystals were positioned 60 mm from the CCD. 360 frames were measured with a counting time of 5 s. The structures were solved using SHELXL-2016/4 for complexes 1 and 324a,b and SHELXT 2014/4 for 2.24c Non-hydrogen atoms were refined with independent anisotropic displacement parameters. Hydrogen atoms were fixed in their respective geometric positions. The displacement parameters of the H atoms were fixed to be 1.2 (1.5 for methyl hydrogens) times larger than the other atoms to which the H atoms were attached. Absorption corrections were carried out using the SADABS program.25 The structures were refined on F2 using SHELXL-2016/4 for 1 and 3 and SHELXL-2018/324c for 2. The hydrogen atoms bonded to the solvent water molecule in 3 could not be located. Data collection, structure refinement parameters and crystallographic data for the complexes are given in Table 1. CCDC 1944919 (1), 1944920 (2) and 1944921 (3)† contain the supplementary crystallographic data for this paper.
Table 1 Crystal data and structure refinement of 1, 2 and 3
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|.
wR2 (Fo2) = [∑[w(Fo2 − Fc2)2/∑wFo4]1/2.
GOF = [∑[w(Fo2 − Fc2)2/(Nobs − Nparams)]1/2.
|
| Metal complexes |
1
|
2
|
3
|
| Chemical formula |
C34H52Cl2N10Ni2O10 |
C21H26N4Ni1O2S1 |
C38H54N6Ni3O9S2 |
| Formula weight |
949.14 |
457.21 |
979.08 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/n |
C2/c |
|
a (Å) |
13.1525(5) |
10.976(3) |
19.913(5) |
|
b (Å) |
8.4556(4) |
14.442(4) |
11.258(5) |
|
c (Å) |
21.0201(9) |
14.246(4) |
19.410(5) |
|
β (°) |
107.942(2) |
103.399(3) |
92.010(5) |
|
V (Å3) |
2224.01(17) |
2196.7(11) |
4349(2) |
|
Z
|
2 |
4 |
4 |
|
ρ
calc (g cm−3) |
1.417 |
1.383 |
1.492 |
|
μ (Mo Kα) (mm−1) |
1.029 |
1.002 |
1.440 |
|
F (000) |
992 |
960 |
2040 |
|
R (int) |
0.070 |
0.061 |
0.044 |
| Total reflections |
44701 |
9755 |
30552 |
| Unique reflections |
3930 |
3544 |
4642 |
| Reflections with I > 2σ(I) |
3359 |
2288 |
3784 |
|
R
1a, wR2b |
0.0503, 0.1401 |
0.0706, 0.2318 |
0.0487, 0.1456 |
| Temp (K) |
293 |
293 |
293 |
| GOFc |
1.067 |
1.053 |
1.069 |
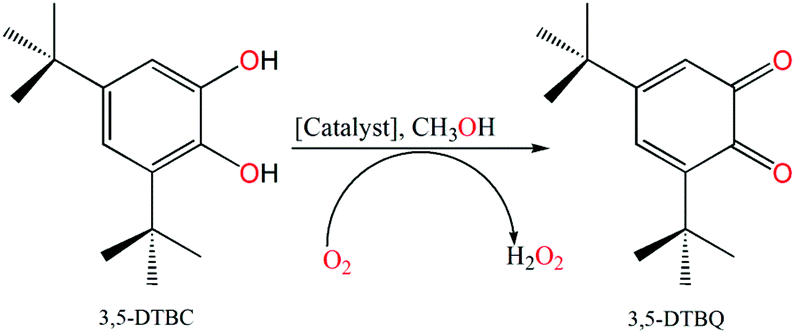
Catalytic oxidation of 3,5-DTBC
The catecholase activity of 1–3 and the detection of hydrogen peroxide during the catalytic reaction via the iodometric method were investigated according to earlier reported methods.26
Computational methodology
Theoretical calculations based on broken symmetry DFT were used to estimate the coupling constants (J) of 1 and 3 as proposed by Ruiz et al.27,28 All the calculations were accomplished with the consideration of the experimental X-ray structures. The hybrid B3LYP functional29–31 and the widely employed TZVP32 basis set were considered in all calculations as implemented in the ORCA package.33 RI approximations with the auxiliary Coulomb fitting basis set TZV/J and tight SCF convergence criteria (Grid4) were also included.34
Results and discussion
Syntheses of the complexes
The monocondensed unreduced Schiff base, 2-[(3-methylamino-propylimino)-methyl]-4-phenol, was synthesized by the reaction between N-methyl-1,3-propanediamine and salicylaldehyde in a 1:1 molar ratio in methanol. On reduction with sodium borohydride, it produced the desired tridentate N2O donor reduced Schiff base (HL1). The phenoxido-bridged dinuclear NiII complex 1 was prepared by reacting HL1 with Ni(ClO4)2·6H2O in a 1:1 ratio (Scheme 1). However, to our surprise, when NH4SCN was added to this reaction mixture, a mononuclear Ni(II) complex (2) of a new N2O2 donor tetradentate ligand (H2L2) formed. Metal complex 2 reacts with a methanolic solution of Ni(SCN)2·4H2O under basic conditions to produce trinuclear metal complex 3. It should be noted that the formation of 2 depicts the first single step synthesis of an unsymmetric singly N-alkylated salan-type ligand, which is otherwise a multistep process.35
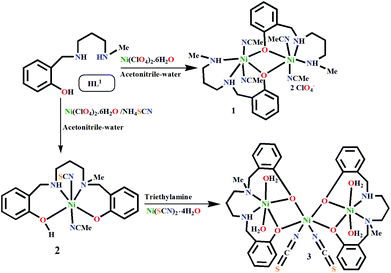 |
| | Scheme 1 Formation of the metal complexes 1–3 (the lattice solvents are not shown here). | |
Mechanistic interpretation
To obtain insight into the mechanism of formation of 2 and the influences of Ni(II) and thiocyanate on this particular in situ transformation of the tridentate ligand HL1 to the tetradentate ligand H2L2, we recorded ESI-MS spectra under various conditions.
The mass spectrum of the dinuclear metal complex 1 in methanol (Fig. S1, ESI†) shows a base peak at 501.13 (calcd 501.13) and an intense peak at 601.09 (calcd 601.08), which can be assigned to the dinuclear species [Ni2(L1)2–H+]+ and [Ni2(L1)2(ClO4)]+, respectively. Along with this, the peak at 251.06 (calcd 251.06) was assigned to the mononuclear [(NiL1)]+ species originating from the fragmentation of 1. A few intense, singly positive sodiated peaks were also found at m/z values of 577.15 (calcd 577.14) and 845.23 (calcd 845.22) and were assigned to [Ni2(L1)2(OH)2(H2O) + Na]+ and [Ni3(L1)3(OH)3(H2O) + Na]+, respectively. The mass spectra of metal complexes 2 and 3 (Fig. S2 and S3, ESI†) are quite similar; they show base peaks at 357.10 and 357.11, respectively (calcd 357.11), for the protonated mononuclear chelate [(NiL2) + H]+. The peak for the dimeric form [(NiL2)2 + H]+ of this mononuclear chelate appears at 713.22 (calcd 713.21) for both 2 and 3. Along with these, another important peak at 301.18 (calcd 301.19) is observed for both 2 and 3, assignable to the free ligand, [H2L2 + H]+. Moreover, an intense peak appears at 503.06 (calcd 503.06) for both metal complexes 2 and 3 due to the dinuclear species, [(NiL2)Ni(OH)2(CH3OH) + Na]+.
We then performed mass spectrometric analysis (Fig. S4, ESI†) of a reaction mixture after one week, prepared by mixing the reactants in the same molar ratios as for the synthesis of complex 2 in methanol, to detect the species which are formed during the conversion of the ligand. A base peak was found at 503.07, which was assigned to the species [(NiL2)Ni(OH)2(CH3OH) + Na]+. Along with this, a peak at a m/z value of 607.17, assignable to the species [(NiL2) + (NiL1)]+ (calcd 607.17), was observed. Two peaks at m/z values of 251.07 and 357.11 confirm the coexistence of [(NiL1)]+ and [(NiL2) + H]+ in the reaction mixture. Another signal at m/z at 415.04 [calcd 415.04], assignable to [(NiL2) + Ni(II) + H−]+, suggests the formation of nickel hydride in situ. Previous studies indicate that the methanolysis of NaBH4 is very rapid at 25 °C, producing hydrogen and NaB(OCH3)4; thus, borohydride cannot be the source of the hydride after one week.36 Thus, we assume that the slow conversion of the reduced ligand of (NiL1)+ to its unreduced imine form in alkaline solution is the source of the hydride. On the other hand, mass analysis of this reaction mixture immediately after mixing the reactants (Fig. S5, ESI†) showed signals for [Ni2(L1)2(ClO4)]+, [Ni2(L1)2–H+]+, and [(NiL1)]+ species at m/z values of 601.09, 501.13 and 251.06, respectively, along with a base peak at a m/z value of 560.12 [calcd 560.11], assignable to the [Ni2(L1)2(NCS)]+ species; this indicates instant formation of metal complex 1 and its thiocyanate adduct. However, no species of unreacted reduced salicylaldehyde or diamine were detected, suggesting that the concentrations of these reactants are negligible compared to that of 1 and its thiocyanate adduct. Thus, we assumed that the contribution of unreacted salicylaldehyde or diamine as a reactant to the transformation of 1 (or its thiocyanate adduct) is minimal; however, their role in the formation of any active catalytic species with nickel in situ cannot be ruled out completely. On the other hand, this [Ni2(L1)2(NCS)]+ species was not observed in the same mixture after one week (Fig. S4, ESI†), indicating that this adduct may be transformed to form metal complex 2.
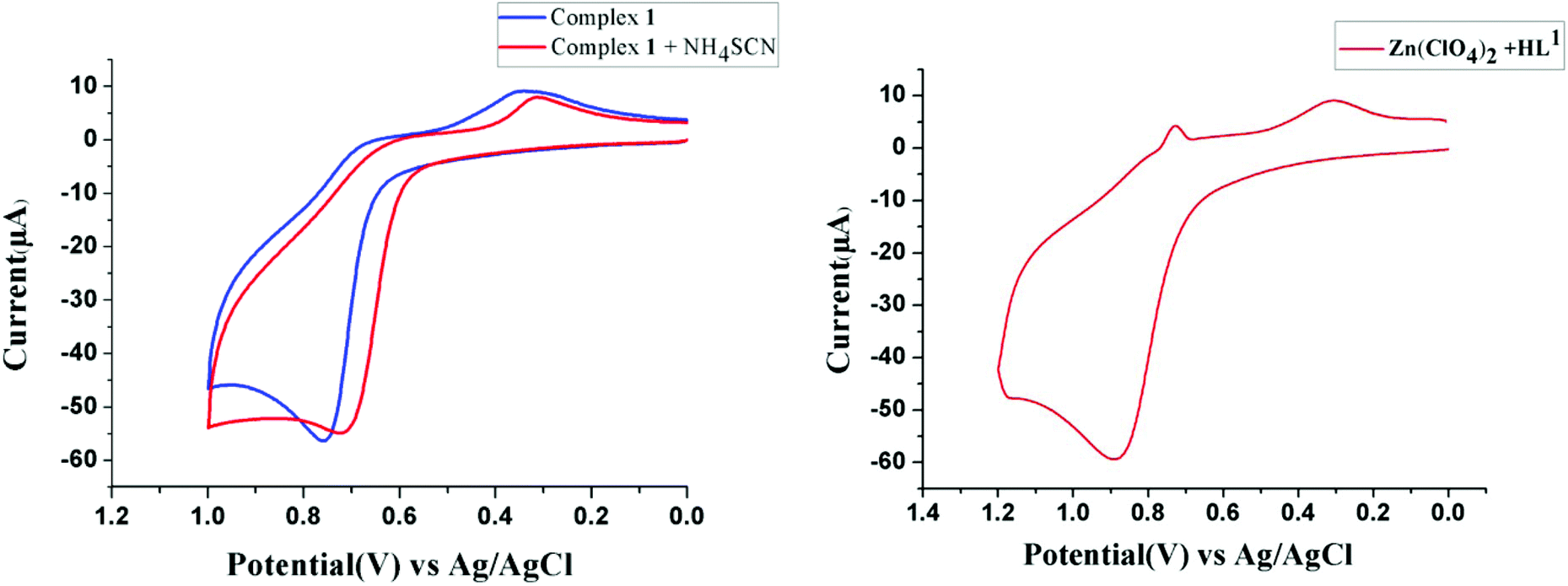
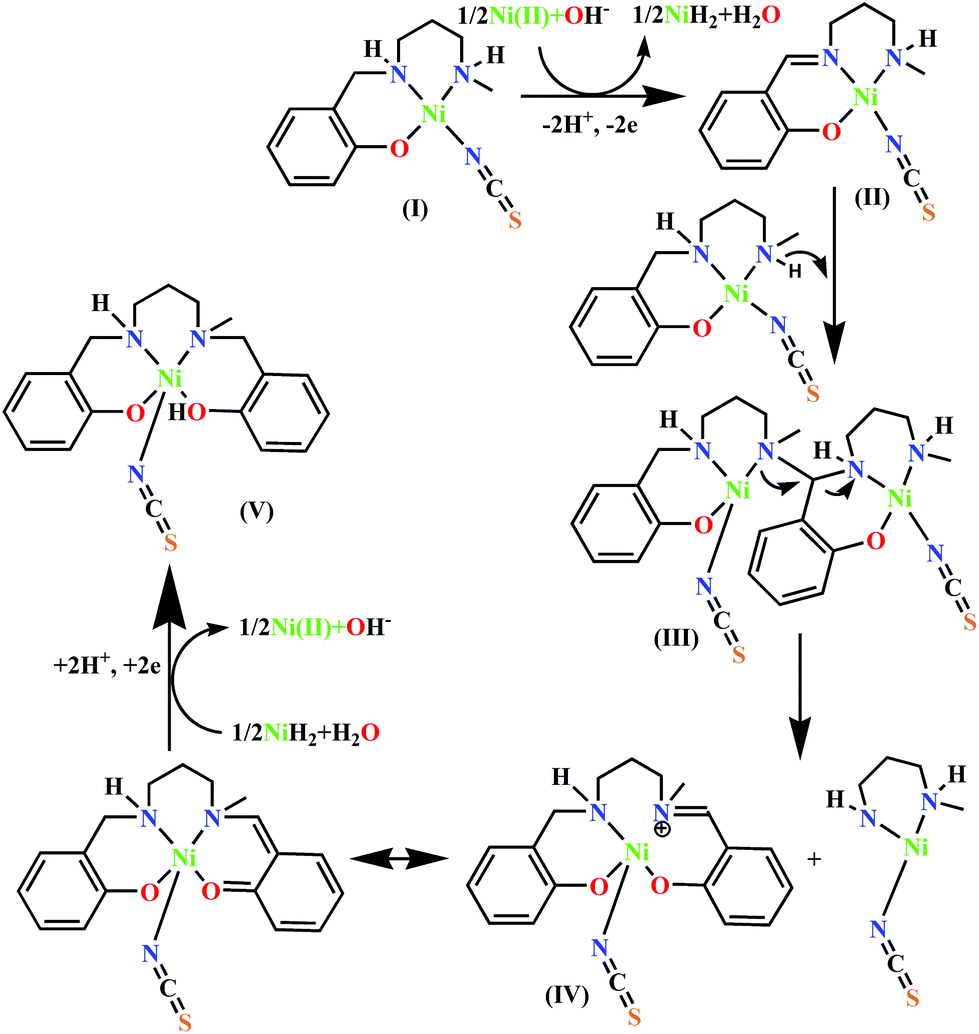
On the other hand, because the reaction takes place in presence of NH4SCN, it may have some influence on the ease of oxidation of (NiL1)+. To explore the influence of SCN− on the oxidation potential of (NiL1)+ with respect to unbound metal complex 1, we also performed a cyclic voltammetric analysis of this mixture. As expected, the anodic peak potential (vs. Ag/AgCl electrode) of 1 in methanol (+0.76 V) also shifted to a lower value (+0.71 V) in the presence of SCN−, indicating a faster rate of electron transfer; this reflects the role of this coligand in increasing the ease of oxidation (Fig. 1). On the other hand, cyclic voltammetric analysis of the compound isolated from the mixture of HL1 and zinc perchlorate in a 1:1 ratio also shows an anodic peak at a similar potential (+0.89 V), suggesting that the oxidation is most likely ligand-based. Based on these observations and analyses, the most probable pathway is depicted in Scheme 2. In the first step, the complex [(NiL1)SCN] (I) undergoes dehydrogenative oxidation to eliminate an H2 molecule and return to its original unreduced form (II), where Ni(II) acts as a hydride transfer agent. In the second step, the Schiff base group undergoes a nucleophilic attack by another [(NiL1)SCN] molecule. The terminal amine group is more likely to be the attacking nucleophile to the imine carbon; thus, the tertiary amine formed (III) has less steric repulsion. The reaction further proceeds to C–N bond cleavage to form the eneamine (IV). Lastly, the eneamine is reduced by the nickel hydride to form the metal complex 2 (V) and regenerates free Ni(II).
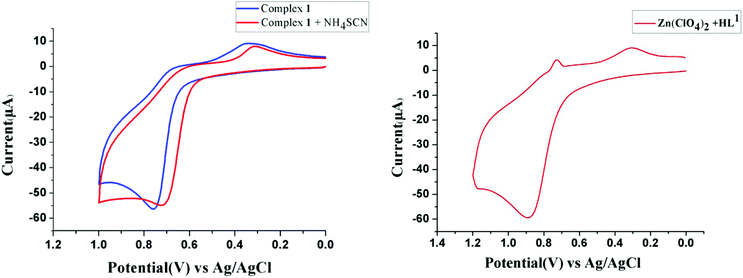 |
| | Fig. 1 Cyclic voltammograms of 1 and a 1:2 mixture of 1 and NH4SCN (left) and of the complex of HL1 and Zn(ClO4)2·6H2O in a 1:1 ratio (right) in methanol. | |
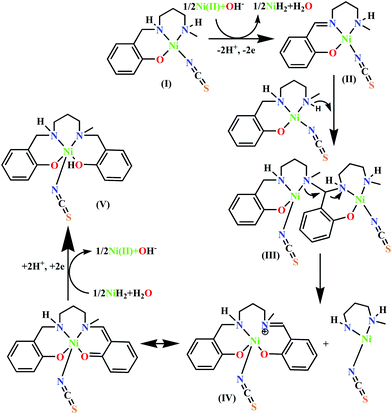 |
| | Scheme 2 Probable mechanism for the transformation of HL1 to H2L2. | |
In order to establish the participation of the imine intermediate in the catalytic reaction, 1 was mixed with the purified nickel perchlorate complex of the unreduced ligand in a 1:1 ratio in methanol without the addition of thiocyanate, and the mixture was analysed by ESI-MS after one week. In the absence of SCN−, a weak signal of [(NiL2) + H]+ was obtained at m/z = 357.11 (Fig. S6, ESI†); this supports the plausible mechanism through a deaminative pathway. Moreover, the signal at m/z = 355.09 [calcd 355.09] indicates the presence of intermediate (IV), excluding SCN−, i.e. [(NiL2)–H−]+. This also suggests that the coordination of SCN− to Ni(II) increases the rate of dehydrogenation of the C–N bond from (NiL1)+, as supported by the electrochemistry studies. In the presence of SCN−, ESI-MS analysis of the mixture of metal complex 1 and the unreduced complex in a 4:1:1 ratio (Fig. S7, ESI†) also depicts weak signals of [(NiL2) + H]+, [(H2L2) + H]+ and [(H2L2)–H−]+ at m/z = 357.11, 301.19 [calcd 301.19] and 299.17 [calcd 299.17], respectively; this supports that the mechanism is also valid in the presence of SCN−. Moreover, mass analysis of a mixture of only 1 and SCN− in a 1:1 ratio after one week (Fig. S8, ESI†) depicts weak signals of [(NiL2) + H]+ at m/z = 357.11 and [(NiL1) + (NiL2)]+ at m/z = 607.17, indicating that the in situ transformation of complex 1 is catalysed by thiocyanate.
IR and UV-Vis spectra of the complexes
Metal complexes 1–3 show moderately strong and sharp peaks at 3289, 3206 and 3214 cm−1, respectively, due to the symmetric stretching vibration of the amine (N–H) group. Therefore, it is confirmed that the Schiff base ligand has been reduced. The reduction of the (>CN–) moiety is also clearly indicated by the absence of a typically strong and sharp peak due to the imine bond vibration found in the corresponding complexes of the unreduced Schiff bases in the range of 1620 to 1650 cm−1.22,26 Absorption bands at 2116 for 2 and 2102 cm−1 for 3 characterize the stretching frequency of the thiocyanate (SCN−) co-ligand. The strong and single band at 1091 cm−1 for 1 indicates the presence of an anionic perchlorate group. The electronic spectra of complexes 1–3 were acquired in methanolic solution. The spectra show broad absorption bands at 652 and 996 nm for 1, 648 and 1012 nm for 2, and 650 and 1024 nm for 3; these can be assigned to the spin-allowed d–d transitions 3T1g(F) ← 3A2g and 3T2g(F) ← 3A2g, respectively, for octahedral d8 systems. These values are in agreement with the literature values for octahedral Ni(II) compounds.26b,37
Description of the crystal structures
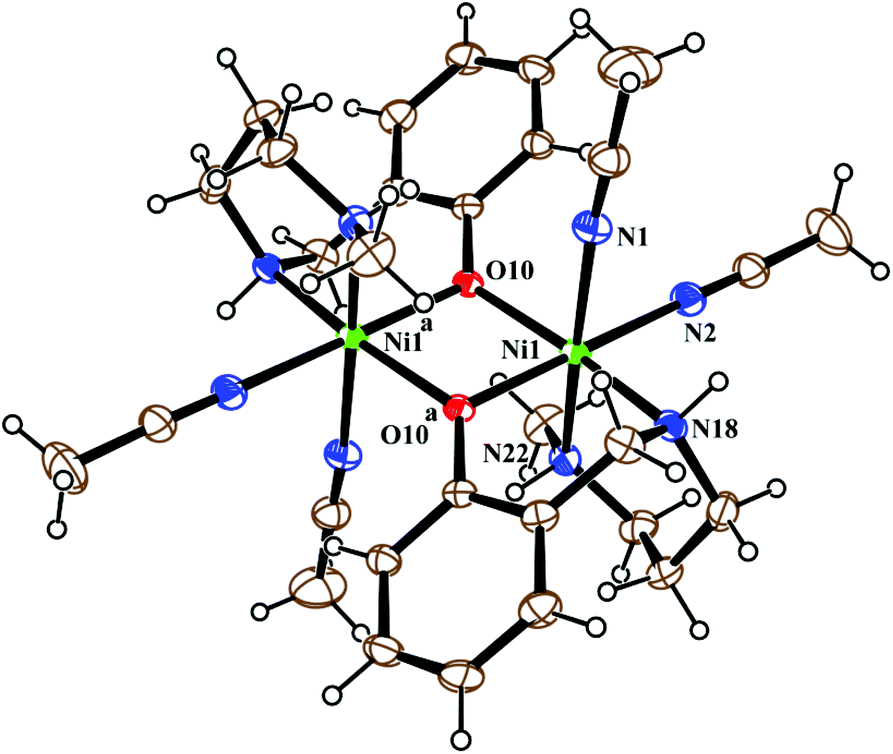
Metal complex 1 consists of a discrete centro-symmetric diphenoxido bridged dinuclear unit with the formula [Ni2L12(CH3CN)4](ClO4)2·2CH3CN (Fig. 2). The dinuclear core consists of two Ni(II) atoms, labeled Ni(1) and Ni(1)a (a = 1 − x, 1 − y, 1 − z), bridged by phenoxido oxygen atoms O(10), O(10)a with a Ni(1)–Ni(1)a distance of 3.207 Å and a Ni(1)–O(10)–Ni(1)a angle of 102.44(8)°. Each nickel atom has a slightly distorted octahedral environment and is coordinated by three donor atoms, N(18), N(22) and O(10), of the deprotonated tridentate reduced Schiff base ligand (L1)− in a facial configuration along with two solvent acetonitrile molecules [N(1) and N(2)] and a phenoxido oxygen atom, O(10)a (a = 1 − x, 1 − y, 1 − z), from the symmetry-related reduced Schiff base ligand, with the usual bond distances.22a,38 The basal plane around the nickel center, Ni(1), is composed of four donor atoms: N(18), O(10), O(10a) (a = 1 − x, 1 − y, 1 − z) and N(2). The distances in the basal plane are in the range of 2.055(2) to 2.097(4) Å. The deviations of the coordinating atoms O(10), N(2), N(18), and O(10a) from the least-square mean plane through them are 0.097(2), −0.043(3), −0.047(3), and −0.006(2) Å, respectively, and that of the Ni(1) atom from the same plane is −0.007(1) Å towards the axially coordinated N(22) atom. The axial positions are occupied by one acetonitrile molecule, N(1), at a distance of 2.208(3) Å, and N(22) of the tridentate Schiff base at a distance of 2.097(3) Å, with a N(1)–Ni(1)–N(22) bond angle of 176.07(11)°. Two noncoordinated perchlorate anions are present to balance the charge of the complex. Selected distances and angles are summarized in Tables S1 and S2, ESI.†
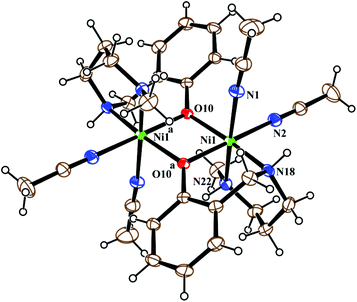 |
| | Fig. 2 ORTEP view of the structure of 1 with ellipsoids at 30% probability. The ClO4− anions and noncoordinated acetonitrile molecules are omitted for the sake of clarity. | |
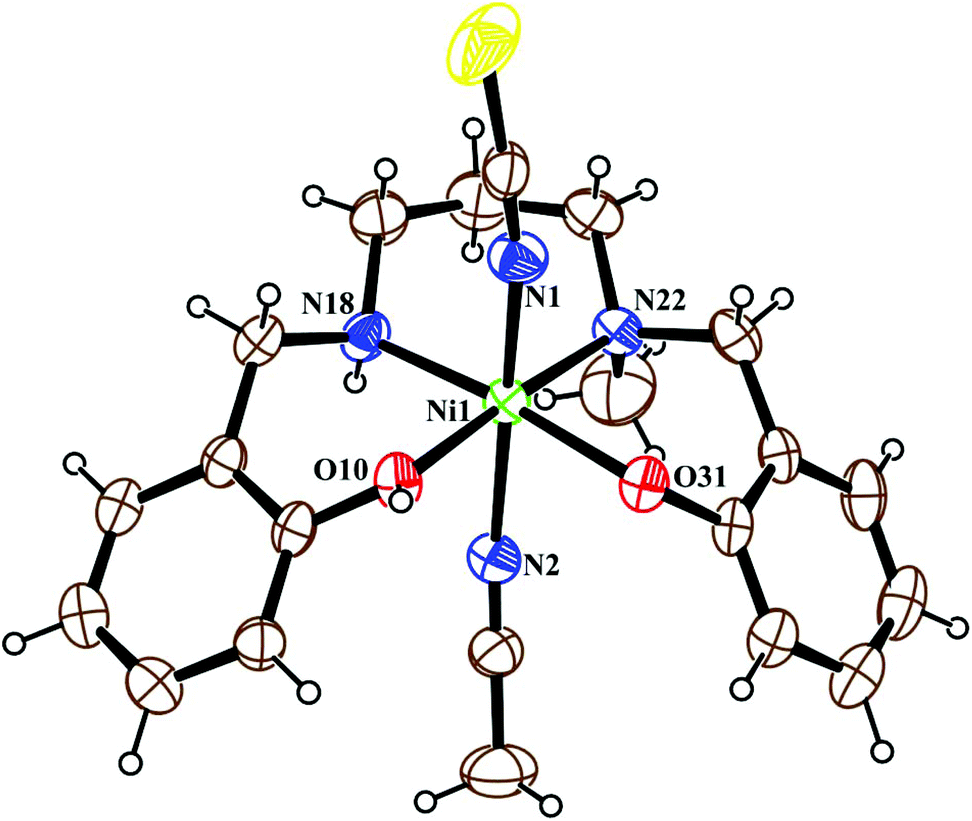
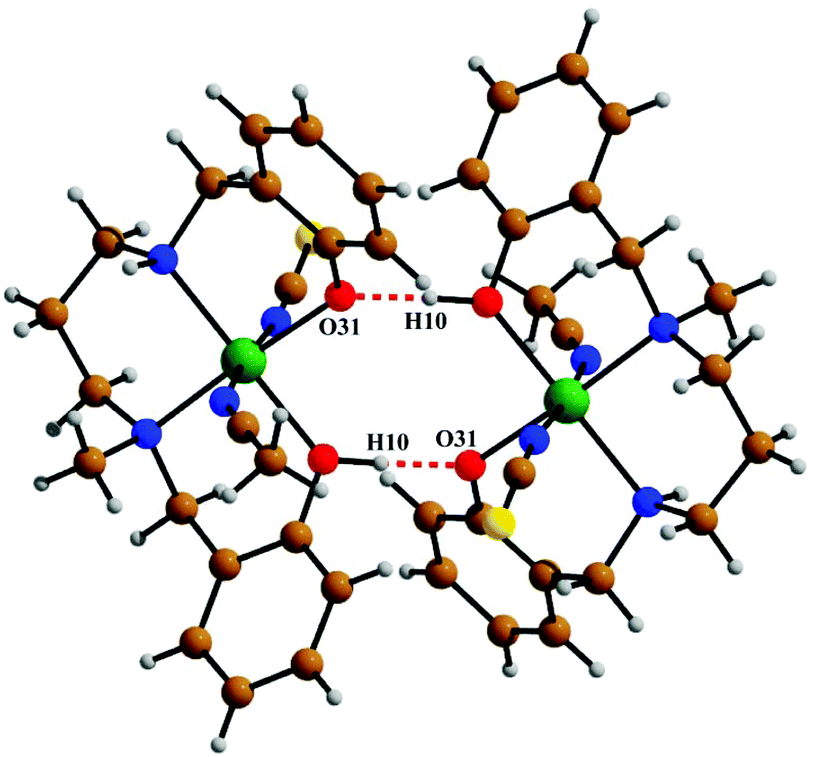
The crystal structure of metal complex 2 consists of a monomeric unit of the neutral complex [Ni(HL2)(NCS)(CH3CN)] (Fig. 3). The metal atom Ni(1) has a distorted octahedral environment, being coordinated by the mono-negative chelating tetradentate ligand (HL2)− through the secondary amine nitrogen atom N(18), the tertiary amine nitrogen atom N(22) and two phenoxido oxygen atoms [O(10), O(31)]. The nitrogen atom of isothiocyanate, N(1), and of acetonitrile, N(2), complete the hexacoordination around the nickel atom. Four donor atoms, N(18), N(22), O(10), and O(31), constitute the basal plane around the nickel center Ni(1). The distances in the basal plane are in the range of 2.044(4) to 2.128(6) Å. The deviations of the coordinating atoms O(10), O(31), N(18) and N(22) from the least-square mean plane through them are −0.027(4), 0.027(4), 0.025(6), and −0.025(6) Å, respectively, and that of the Ni(1) atom from the same plane is 0.065(1) Å towards the axially coordinated N(1) atom. The two axial distances, Ni(1)–N(1) and Ni(1)–N(2), are 2.029(6) Å and 2.155(6) Å, respectively, and the N(1)–Ni(1)–N(2) bond angle is 173.2(2)°. The hydrogen atom H(10) of the tetradentate ligand participates in strong intermolecular hydrogen bonds with O(31) of another molecule (H(10)⋯O(31); 1.47(6) Å, O(31)–H(31)⋯O(10); 172(8)° and O(31)⋯O(10); 2.428(6) Å) to form a H-bonded dimer (Fig. 5). Selected distances and angles are summarized in Tables S1–S3, ESI.†
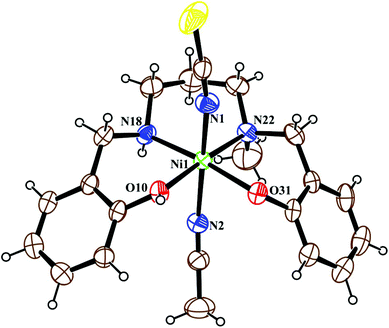 |
| | Fig. 3 ORTEP view of the structure of 2 with ellipsoids at 30% probability. | |
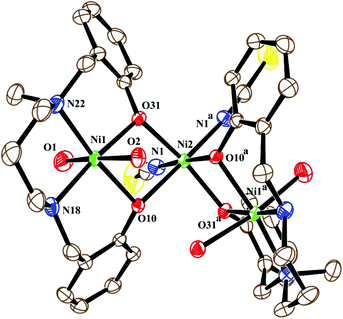 |
| | Fig. 4 ORTEP view of the structure of 3 with ellipsoids at 30% probability. H-Atoms and non-coordinated water molecules are omitted for clarity. | |
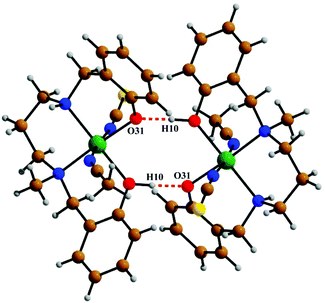 |
| | Fig. 5 The intermolecular hydrogen bonds shown in 2. | |
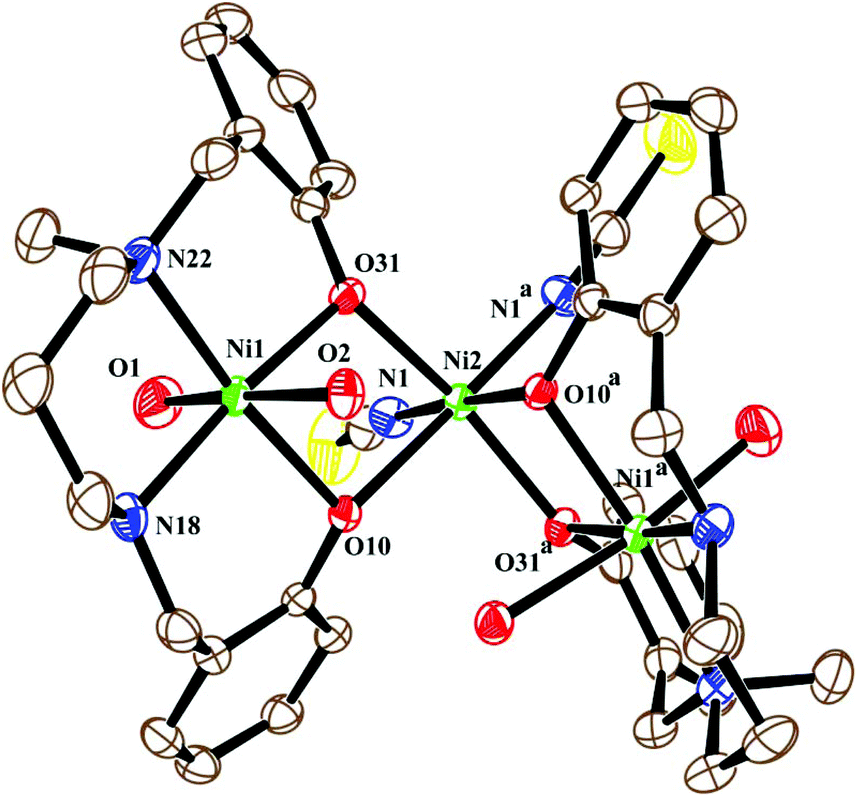
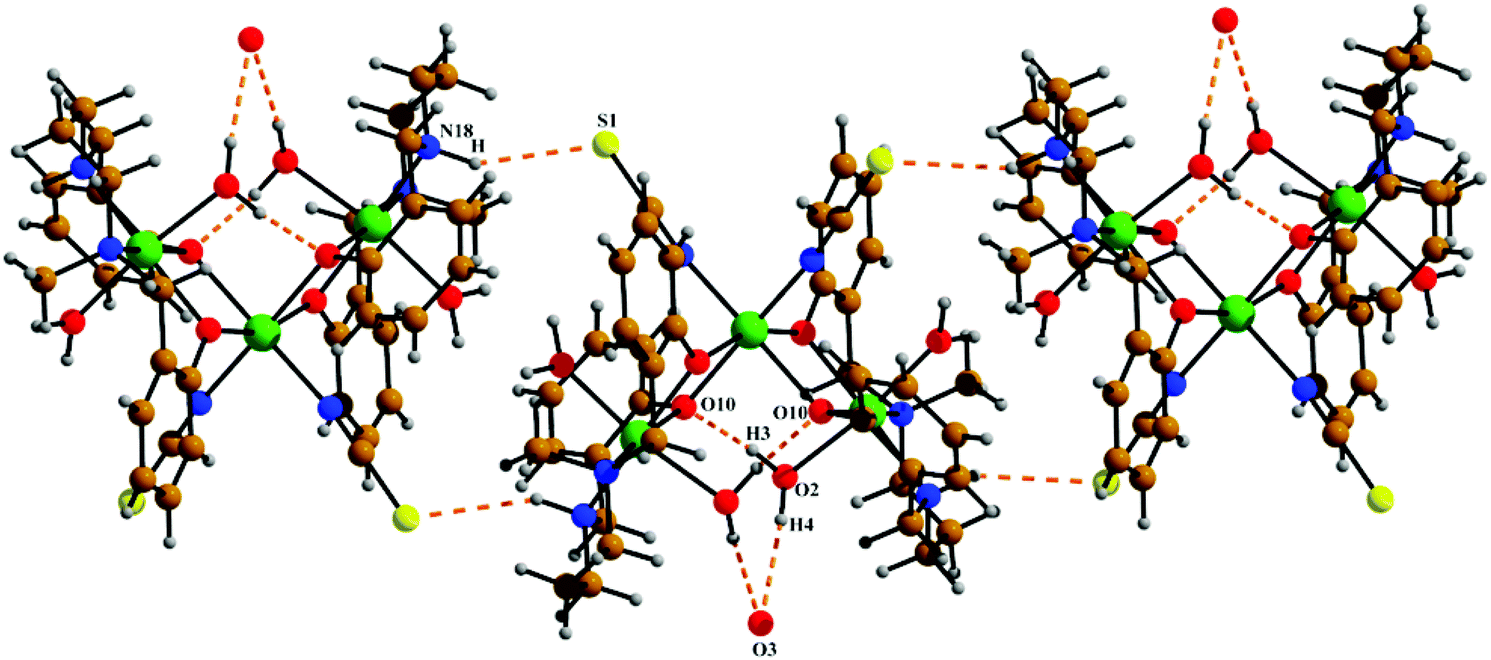
The neutral trinuclear entity of the metal complex (Fig. 4) [Ni3(L2)2(NCS)2(H2O)4]·H2O (3) can be assumed to be composed of two terminal mononuclear units of [NiL2(H2O)2] connected to a central [Ni(SCN)2] unit through the phenoxido oxygen atoms of the Schiff base ligand. The trinuclear structure possesses crystallographic C2 symmetry running through the central nickel atom Ni(2). Each of the two equivalent terminal Ni(1) atoms possesses an octahedral environment, where four donor atoms of the di-negative tetradentate Schiff base ligand (L2)−, with dimensions of Ni(1)–N(18) 2.092(3) Å, Ni(1)–N(22) 2.133(3) Å, Ni(1)–O(10) 2.060(2) Å and Ni(1)–O(31) 2.051(2) Å, constitute the basal plane. Two axial positions are occupied by oxygen atoms (O1, O2) of two water molecules with bond distances of Ni(1)–O(1) 2.134(4) Å and Ni(1)–O(2) 2.132(3) Å. The deviations of the coordinating atoms N(18), N(22), O(10), and O(31) from the mean plane passing through them are −0.044(3) Å, 0.043(3) Å, 0.049(2) Å, and −0.049(2) Å, respectively, and that of Ni(1) from the same plane is −0.060(1) Å. The six-coordinated central Ni(2) has also a distorted octahedral environment, constructed by the coordination of four phenoxido oxygen atoms O(10), O(10)a, O(31), and O(31)a [a = −x, y, ½ − z] from the two terminal [NiL2(H2O)2] moieties and two mutually cis nitrogen atoms of the isothiocyanate ligands at N(1) and N(1)a. Three phenoxido-bridged oxygen atoms O(10), O(31), and O(31)a [Ni(2)–O(10) 2.181(2) Å, Ni(2)–O(31) 2.053(2), Ni(2)–O(31)a Å] and one N atom (N(1)a) [Ni(2)–N(1)a, 2.063(3) Å] of the ligating isothiocyanate form the basal plane. The deviations of the coordinating atoms O(10), O(31), O(31)a, and N(1)a from the mean plane passing through them are −0.114(2) Å, 0.107(2) Å, 0.102(2) Å, and −0.095(3) Å, respectively, and that of Ni(2) from the same plane is −0.135(1) Å. The N(1) of isothiocyanate and the phenoxido O(10)a are in the axial positions, where the bond angle of O(10)a–Ni(2)–N(1) is 177.60(11)°. Both hydrogens of the water molecule O(2) are involved in hydrogen bonding (Fig. 6). One of them, H(3), forms a hydrogen bond with the phenoxido oxygen, O(10), of the ligand (H(3)⋯O(10); 1.76 Å, O(2)–H(3)⋯O(10); 174° and O(3)⋯O(10); 2.681 Å). The other hydrogen atom, H(4), forms an intermolecular hydrogen bond with a solvent water molecule, O(3); (H(4)⋯O(3); 2.24 Å, O(2)–H(4)⋯O(3); 174° and O(3)⋯O(2); 3.156 Å). The amine N(18) hydrogen atom also forms a weak H-bond interaction with a sulphur atom, S(1), of a coordinated isothiocyanate molecule (H⋯S(1); 2.61 Å, N(18)–H⋯S(1); 152° and N(18)⋯S(1); 3.159 Å). Selected distances and angles are summarized in Tables S1–S3, ESI.†
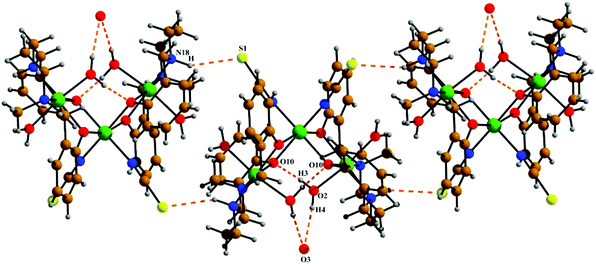 |
| | Fig. 6 The intra and intermolecular hydrogen bonds shown in 3. Symmetry transformations for the intermolecular H-bonds are shown in Table 2. | |
Table 2 Hydrogen bond parameters in 2 and 3 (in Å and °)
| |
D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
<(D–H⋯A) |
Symmetry transformations |
| D = donor atom, A = acceptor atom, H = hydrogen atom. |
|
2
|
O(10)–H(10)⋯O(31) |
0.96(5) |
1.47(6) |
2.428(6) |
172(8) |
|
|
3
|
N(18)–H⋯S(1)a |
0.99(3) |
2.61(3) |
3.519(4) |
152(3) |
a½ + x, ½ + y, z |
| O(2)–H(3)⋯O(10)b |
0.92(3) |
1.76(3) |
2.681(4) |
174(3) |
b−x, y, ½ − z |
| O(2)–H(4)⋯O(3)c |
0.92(4) |
2.24(4) |
3.156(9) |
174(5) |
c−1 + x, y, z |
Catecholase-like activity studies
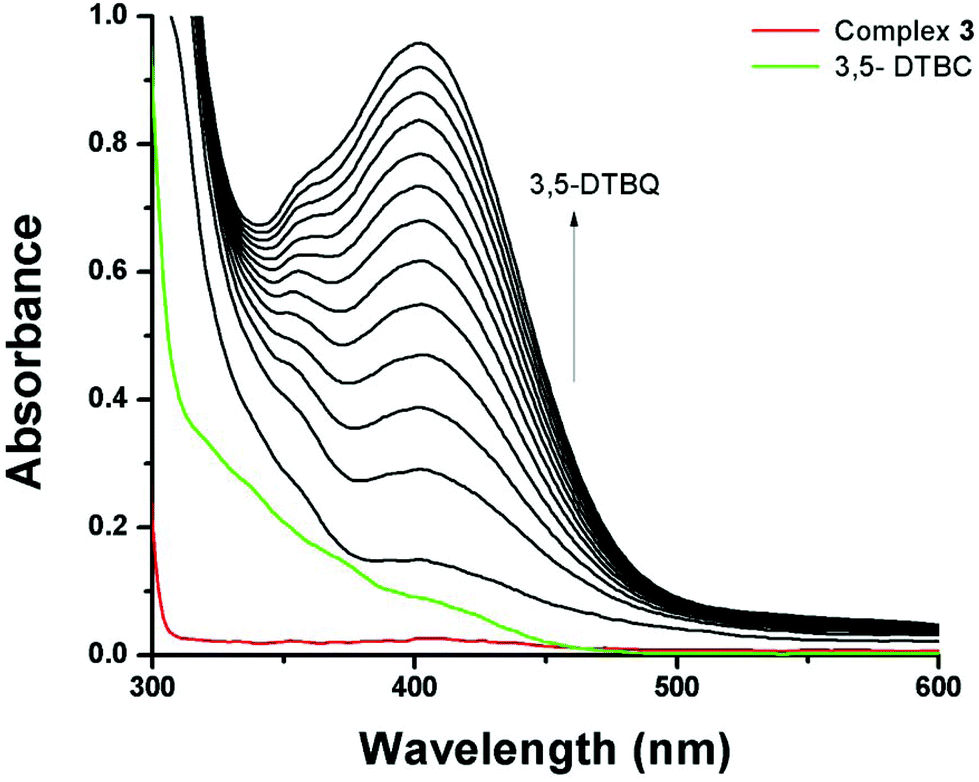
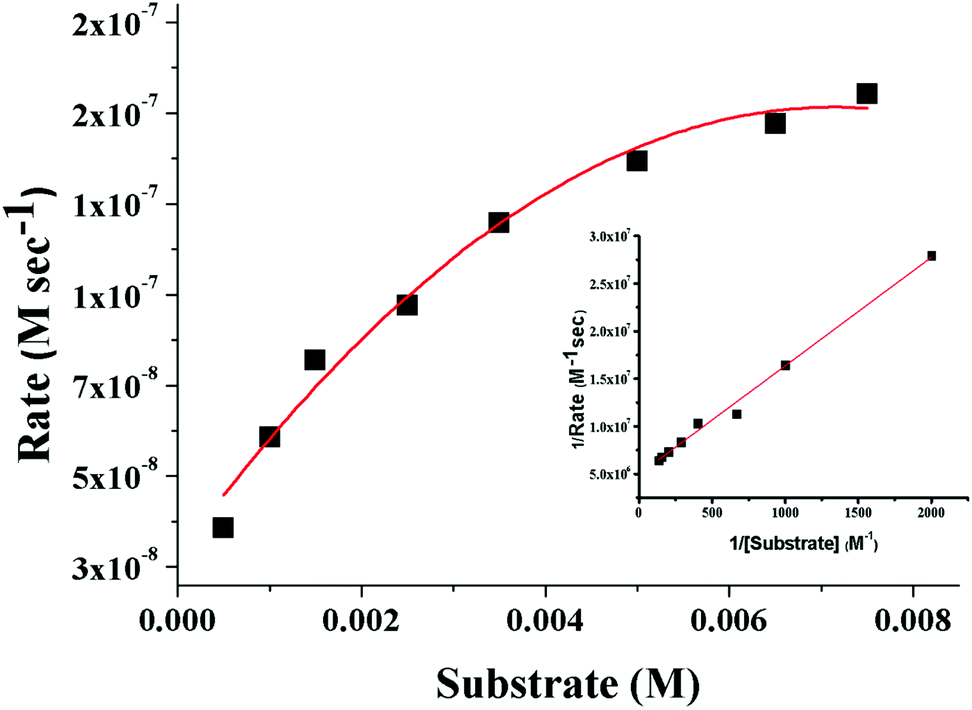
3,5-Di-tert-butylcatechol (3,5-DTBC) was used as a substrate for the measurements of the catecholase-like activity of the metal complexes 1–3. First, we examined the catalytic activity of all three metal complexes for the oxidation of 3,5-DTBC to 3,5-DTBQ in air-saturated methanol (Fig. 7). For this purpose, we mixed equal volumes of 1 × 10−4 M solutions of each of 1–3 and 1 × 10−2 M substrate solution (3,5-DTBC) in methanol at room temperature, and UV–vis spectra of the mixtures were recorded at 5 min time intervals. While 1 and 3 showed gradual increases of an absorption band around 401 nm due to the formation of 3,5-DTBQ, this gradual increase was absent for 2. Hence, detailed kinetic studies of this oxidation reaction were performed on 1 and 3. Fig. 8 presents the gradual increase of the absorption bands at 401 nm for 3 in the presence of 3,5-DTBC, and similar plots for complex 1 are given in Fig. S9, ESI.† The kinetic parameters of both complexes were measured by the initial rate method.39 The substrate concentration dependence of the oxidation rates and various kinetic parameters were determined by mixing equal volumes of 1 × 10−4 M solutions of 1 and 3 with different concentrations (Table S4, ESI†) of 3,5-DTBC under aerobic conditions. In all cases, a first-order kinetic dependence was followed at low concentrations of 3,5-DTBC, whereas saturation kinetics was observed at higher concentrations. The observed rate vs. [substrate] data of the enzymatic kinetics were then analyzed by the Michaelis–Menten model to obtain Lineweaver–Burk plots and values of different kinetic parameters. Both the observed rate vs. [substrate] and Lineweaver–Burk plot for 3 are shown in Fig. 9, whereas similar plots for 1 are shown in the ESI.† The kinetic parameters for 1 and 3 are listed in Table S5, ESI.† The turnover numbers (Kcat) were calculated by dividing the Vmax values by the concentration of the corresponding complexes (Table S5, ESI†).
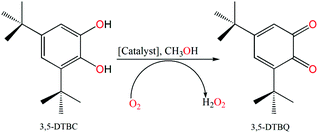 |
| | Fig. 7 Catalytic oxidation of 3,5-DTBC to 3,5-DTBQ in air-saturated methanol. | |
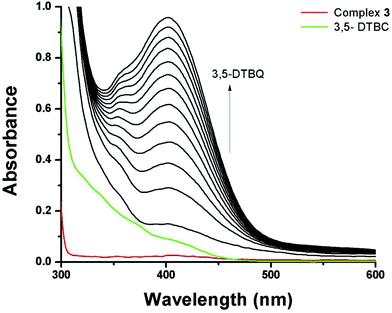 |
| | Fig. 8 Increase in absorbance around 401 nm after mixing equal volumes of methanolic solutions of 3,5-DTBC (1 × 10−2 M) and 3 (1 × 10−4 M). The spectra were recorded at 5 min intervals. | |
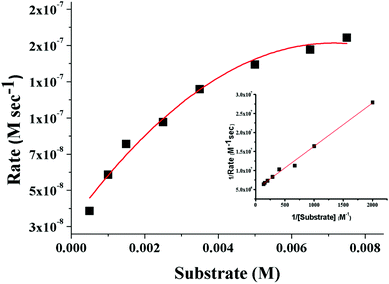 |
| | Fig. 9 Plot of the initial rate versus the substrate concentration for the oxidation of 3,5-DTBC catalyzed by 3. The inset shows the Lineweaver–Burk plot. | |
The binding of 3,5-DTBC to the complexes was investigated by mass spectrometry (ESI; positive mode) to obtain an idea regarding the possible complex–substrate intermediates and mechanistic inference of catecholase activity during the oxidation reaction. We recorded ESI-MS spectra for complexes 1 and 3 of 1:100 mixtures (v/v) of the complexes and 3,5-DTBC in methanol solution within 5 min of mixing. The detailed analysis of the mass spectra of complexes 1 and 3 was previously mentioned in the mechanistic interpretation section. Now, after addition of 3,5-DTBC to a methanolic solution of complex 1, a considerable change was observed in the mass spectrum (Fig. S12, ESI†). The base peak was observed at m/z = 472.23 (calcd 472.22), which can be assigned to the semiquinonate form of the intermediate species [Ni(L1)(3,5-DTBSQ) + H]+ of complex 1. In addition, low intensity peaks at 494.20 (calcd 494.20) and 251.06 (calcd 251.06) were observed for [Ni(L1)(3,5-DTBSQ) + Na]+ and [Ni(L1)]+, respectively. Another important peak appears at m/z = 722.30 (calcd 722.29), which is due to the formation of [Ni2(L1)2(3,5-HDTBC) + H]+. This proves the formation of the catalyst–substrate intermediate species and suggests that 3,5-DTBC binds with the binuclear unit at first. In the case of complex 3, again, a considerable change was observed after the addition of 3,5-DTBC (Fig. S13, ESI†). In addition to some common assigned peaks of complex 3, three new peaks at m/z = 243.12 (calcd 243.14), m/z = 634.18 (calcd 634.18), and m/z = 578.26 (calcd 578.26) were observed due to the quinine-sodium aggregate ([3,5-DTBQ-Na]+) and semiquinonate intermediate species ([Ni2(L2)(3,5-DTBSQ)]+ and [Ni(L2)(3,5-DTBSQ) + H]+), respectively. Another peak at m/z = 445.05 (calcd 445.05) was assigned to ([Ni2(L2)(CH3O)]+. The semiquinonate species clearly establish the complex–substrate bonding as well as the formation of intermediates.
Mechanistic insight
Various detailed mechanistic approaches have been reported for the conversion of substituted catechols to their corresponding quinines.40 In most homonuclear Ni(II) systems, metal-semiquinoate intermediates are formed via radical pathways.41 This intermediate reacts with oxygen and also produces hydrogen peroxide as a byproduct. It has been found that for both complexes, H2O2 is formed during the reaction with 3,5-DTBC. The quantitative estimation shows that 91% H2O2 is generated after 1 h of reaction for 1, while 74.67% is generated in the case of 3.
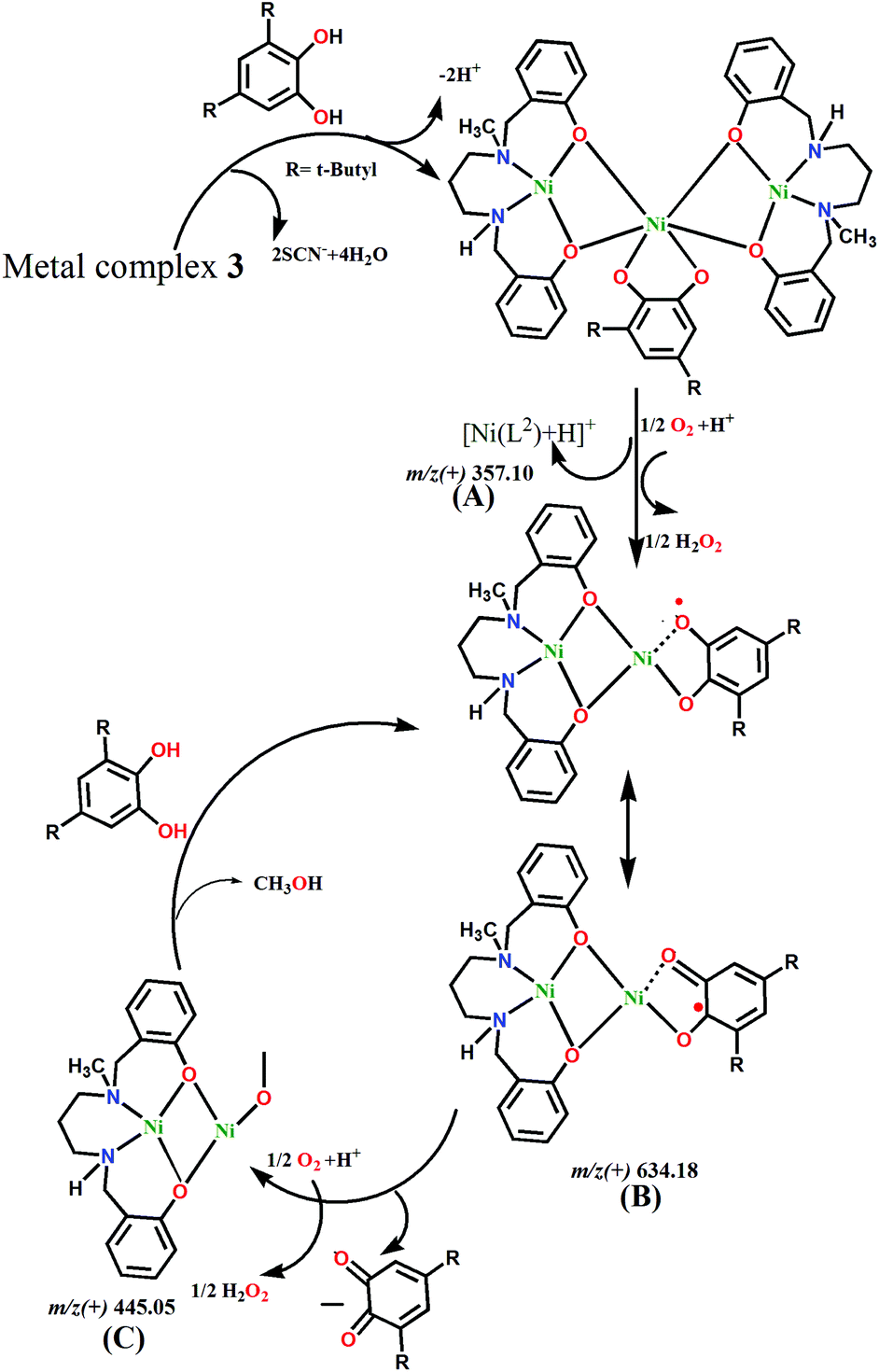
Based on the ESI-MS spectra, we propose mechanism cycles for 1 (Fig. S11, ESI†) and 3 (Fig. 10). In the case of 1, the dinuclear species [Ni2(L1)2–H+]+ reacts with 3,5-DTBC to generate the intermediate [Ni2(L1)2(3,5-HDTBC) + H+]+. However, this less stable dinuclear entity dissociates into mononuclear [Ni(L1)]+, which reacts with aerial O2 and forms a radical intermediate, [Ni(L1)(3,5-DTBQ)]+. H2O2 is produced as a byproduct from the reduction of oxygen molecule. The semiquinoate intermediate itself converts to 3,5-DTBQ by leaving the reactive species [Ni(L1)]+ and H2O2. The regenerated mononuclear species then further reacts with 3,5-DTBC, and the cycle continues. However, in 3, the central Ni(II) can bind with 3,5-DTBC by replacing two thiocyanate co-anions. However, this large molecule dissociates and forms two small species, [Ni2(L2)]2+ and [Ni(L2)]+. Both these reactive species are capable of binding with catechol to form radical semiquinoates. The reaction cycle goes the same way in 1, and 3,5-DTBQ and H2O2 are produced as the product and byproduct, respectively.
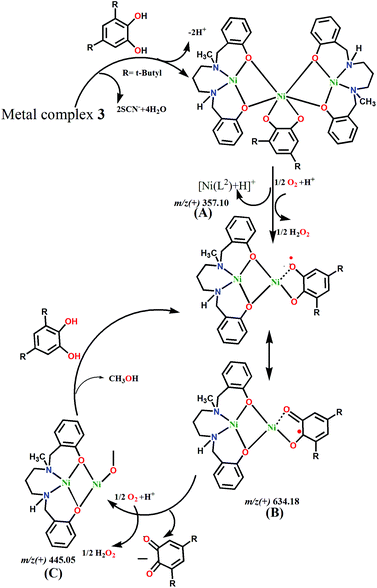 |
| | Fig. 10 Proposed mechanism for the catalytic cycle of the oxidation of 3,5-di-tert-butylcatechol with 3. | |
The activity order of these complexes for this oxidation of 3,5-DTBC to 3,5-DTBQ is 3 > 1. From X-ray analysis, it was observed that coordinated solvent molecules, e.g. H2O and CH3CN, are present in the structures. The central Ni(II) ion of 3 also binds with labile thiocyanate co-ligands. Thus 3,5-DTBC can coordinate easily to Ni(II) ions by replacing these labile groups. ESI-MS analysis indicates the presence of two possible reactive intermediates ([Ni2(L2)(3,5-DTBSQ)]+ and [Ni(L2)(3,5-DTBSQ) + H]+) in solution. However, the latter intermediate does not appear to be the active species because it is also detected in the mass spectra of the solution of 2 + 3,5-DTBC, but no catalytic activity is detected for 2. Therefore, it can be assumed that the oxidation reaction proceeds through the binuclear reactive intermediate species. The central Ni(II) ion of 3 can easily bind with 3,5-DTBC to for the dinuclear intermediate on dissociation, whereas the coordination of 3,5-DTBC to the dinuclear metal complex 1 is somewhat hindered due to steric repulsion resulting from the tridentate ligand. Hence, the catalytic activity of 3 is considerably higher than that of 1. A table (Table S7†) comparing the catecholase activities of reported Ni(II) Schiff base complexes is included in the ESI.† However, the Kcat (h−1) values in the table are inconsistent because different methods (initial or integrated rate) were used to calculate the kinetic parameters.
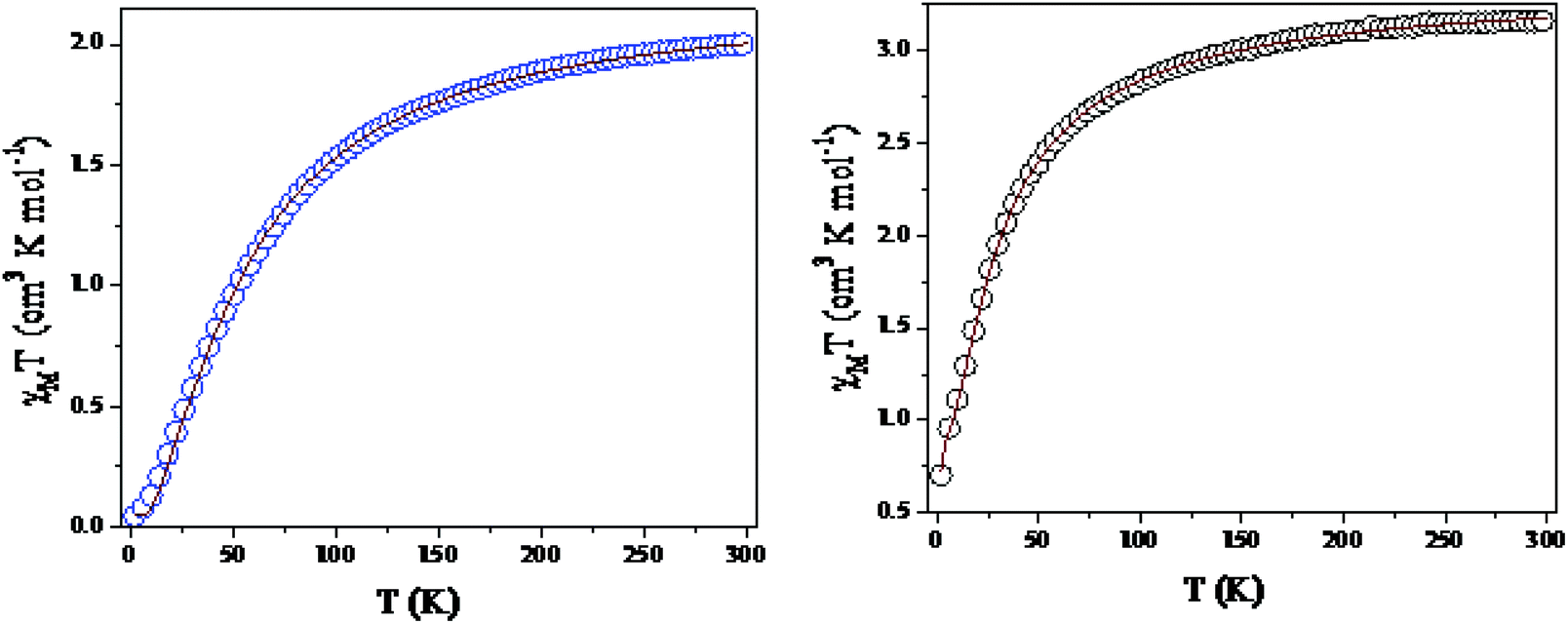
Magnetic properties
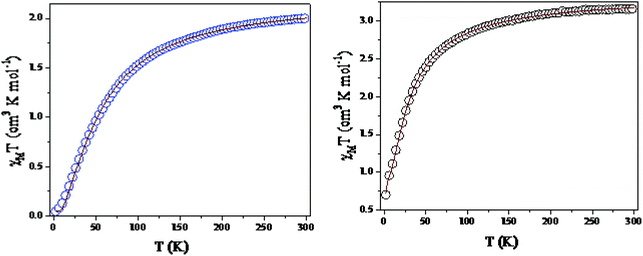
Temperature-dependent molar magnetic susceptibility measurements of microcrystalline powdered samples of 1 and 3 were carried out in the temperature range of 2 to 300 K. The results are shown in the form of χMT vs. T plots in Fig. 11. For 1, the χMT value is 2.11 cm3 K mol−1 at room temperature (300 K), which is expected for a magnetically non-interacting NiII dimer (the spin only χMT value is 2.0 cm3 K mol−1 for Ni(II) with g = 2). Upon cooling, χMT shows a smooth decrease and reaches a value of 0.01 cm3 K mol−1 at 2 K. This behavior indicates the presence of predominant intramolecular antiferromagnetic interactions between the Ni(II) ions. The magnetization measurements of 1 at 2 K also confirm that a diamagnetic ground state arises from strong antiferromagnetic exchange interactions (Fig. S14, ESI†).
 |
| | Fig. 11 Variations of χmT as a function of temperature for 1 (left) and 3 (right). | |
Because the structure of 1 shows the presence of isolated Ni(II) dimers connected through two phenoxido bridges, we fitted the experimental magnetic data of this compound by following the Van-Vleck equation (eqn (1)). The Hamiltonian for this equation is considered to be H = −JS1S2, where S1 = S2 = SNi = 1. This simple model gives a very satisfactory fit of the magnetic properties of 1 in the whole temperature range with the following set of parameters: J = −32.22 ± 0.12 cm−1, g = 2.11 ± 0.07, θ = 0 K, R = 7.1 × 10−5.
| | 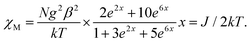 | (1) |
The thermal variation of the χmT product of 3, Ni(II) trimer, shows a room-temperature value of 3.52 cm3 K mol−1; this is close to the expected spin only non-interacting Ni(II) trimer (3.0 cm3 K mol−1). Upon cooling, the χmT product shows a steady decrease from 300 K to 100 K. This decrease becomes more pronounced below 100 K and reaches a value of ca. 0.75 cm3 K mol−1 at 2 K. This behavior indicates that 3 also presents antiferromagnetic exchange interactions within the trinuclear unit.
Although 3 is a bent trinuclear unit, we consider it as a linear trinuclear unit from the magnetic point of view because there is no direct linkage between the two terminal Ni atoms. Because the central Ni(2) atom is also connected to two terminal Ni(II) via the same double phenoxido bridges, we consider metal complex 3 as a symmetric linear trinuclear unit with only one intra-trinuclear coupling constant (J). We fitted the magnetic properties of this complex to the simple model derived for an S1 = S2 = S3 = SNi = 1 linear trimer with the Hamiltonian H = −J(S1S2 + S2S3), where S2 is the spin state of the central Ni(II) ion. The best fits lead to the following data: J = −10.4 ± 0.06 cm−1, g = 2.12 ± 0.04, θ = −0.36 ± 0.01 K, R = 1.1 × 10−5.
| | 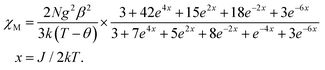 | (2) |
Magneto-structural correlations
In the last two decades, several studies on magnetostructural correlations on diphenoxido-bridged metal complexes have been reported. All these studies conclude that the exchange interaction (J) has strong dependence on the bridging angle. It is shown that diphenoxido-bridged Ni(II) dimers are ferromagnetically coupled if the Ni–O–Ni angle is less than ∼97° due to orthogonality of the magnetic orbitals, and antiferromagnetic coupling is expected at larger angles.42 The relationship between the J values and the Ni–O–Ni bridging angles is almost linear.43 The out-of-plane shift of the phenoxido rings, the hinge distortion of the M2O2 core frameworks and the metal⋯Ophenoxido distances are also important factors that affect the J values.44 It is very difficult to synthesize complexes whose structures have the required variations of all these parameters. However, theoretical calculations can provide an idea because one can control the variation of each parameter, leaving the rest unaffected. To better understand the magnetic exchange properties, we performed DFT calculations using the broken symmetry approach to calculate the J values. By calculating, we obtained values of J = −40.15 cm−1 for 1 and −14.53 cm−1 for 3; these are comparable with the experimentally observed values of J = −32.22 cm−1 and −10.4 cm−1, respectively (Table 3). The diphenoxido bridged Ni(II) dimers are usually antiferromagnetic in nature, and the coupling constant becomes more negative with increasing bridging angle. To visualize this fact, we plotted the J values45 with respect to the Ni–O–Ni angles of earlier reported diphenoxido-bridged Ni(II) dimers (Fig. S15, ESI†). The results showed sign alterations of the J values around ∼97°. The phenoxido bond angle (Ni–O–Ni, 102.44(8)°) in 1 indicates that it should show strong antiferromagnetic interactions. Metal complex 3, a trinuclear unit, also shows antiferromagnetic coupling at room temperature, which results in the S = 1 ground state. In 3, the Ni–O–Ni angles are 96.81° and 102.59°, with an average value of 99.70°. Therefore, the magnetic coupling is expected to be weak and antiferromagnetic, as observed experimentally.
Table 3 Selected structural parameters and magnetic coupling constants (J) for 1 and 3
| Metal complex |
Ni–O bond length (Å) |
Ni–Ni distance (Å) |
<Ni–O–Ni (°) |
Coupling constant (J) by DFT (cm−1) |
Coupling constant (J) experimental (cm−1) |
|
1
|
2.060(2) |
3.207 |
102.44(8) |
−40.15 |
−32.22 ± 0.12 |
| 2.055(2) |
|
|
|
|
|
3
|
2.100(2) |
3.202 |
96.81(9) |
−14.53 |
−10.40 ± 0.06 |
| 2.051(2) |
|
102.59(10) |
|
|
Conclusions
The N2O donor reduced Schiff base ligand (HL1) on reaction with Ni(ClO4)2·6H2O produces the usual phenoxido bridged dinuclear metal complex 1. However, addition of NH4SCN to the reaction mixture results in an unexpected mononuclear Ni(II) complex (2) of an N2O2 donor ligand (H2L2). This unprecedented in situ transformation of HL1 to H2L2 appears to occur via deaminative coupling, as is evident from mass spectral analysis. The thiocyanate ion appears to play a catalytic role in this transformation. Metal complex 2 on reaction with Ni(SCN)2·4H2O produces a phenoxido-bridged trinuclear metal complex, 3. Variable temperature magnetic measurements of 1 and 3 show antiferromagnetic coupling between the Ni(II) centers, consistent with their high bridging angles. The theoretically calculated coupling constants (J) are in conformity with those obtained experimentally. Due to their loosely bonded solvent molecules (H2O, CH3CN), dinuclear metal complex 1 and trinuclear metal complex 3 are useful catalysts for biomimicking catecholase-like activity; however, the mononuclear metal complex 2 is nearly inert for this oxidation reaction. ESI-MS spectra reveal that the binuclear entity [Ni2(L2)]2+ is responsible for the catalytic activity of 3. In 1, the catalyst–substrate binding appears to be hindered by the tridentate ligand bonded to Ni(II) ion; hence, its catalytic activity is less than that of 3.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. G. thanks the DST-FIST funded single crystal X-ray diffractometer facility at the Department of Chemistry, University of Calcutta for providing the single crystal X-ray diffractometer facility. We also thank the CRNN, University of Calcutta, Kolkata, India, for magnetic data measurements. A. G. also thanks the University Grants Commission (UGC), New Delhi for funding the CAS-V, Department of Chemistry, University of Calcutta. S. M. thanks the University Grants Commission (UGC), New Delhi, for the Senior Research Fellowship [Sr. No. 2061510240, Ref No. –21/06/2015 (i) EU-V].
References
-
(a) M. Suzuki, H. Furutachi and H. Ōkawa, Coord. Chem. Rev., 2000, 200–202, 105–129 CrossRef CAS
 ;
(b) P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS ;
(c) C. T. Lyons and T. D. P. Stack, Coord. Chem. Rev., 2013, 257, 528–540 CrossRef CAS .
;
(b) P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS ;
(c) C. T. Lyons and T. D. P. Stack, Coord. Chem. Rev., 2013, 257, 528–540 CrossRef CAS .
-
(a)
R. W. Saalfrank, E. Uller, B. Demleitner and I. Bernt, in Molecular Self-Assembly Organic Versus Inorganic Approaches, Springer, Berlin, Heidelberg, 2000, pp. 149–175 Search PubMed ;
(b) R. E. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC ;
(c) R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794–8824 CrossRef CAS .
-
(a) X. J. Kong, T. He, Y. Z. Zhang, X. Q. Wu, S. N. Wang, M. M. Xu, G. R. Si and J. R. Li, Chem. Sci., 2019, 10, 3949–3955 RSC ;
(b) X. Zhang, J. Sun, G. Wei, Z. Liu, H. Yang, K. Wang and H. Fei, Angew. Chem., 2019, 131, 2870–2875 CrossRef ;
(c) B. Dutta, R. Jana, C. Sinha, P. P. Ray and M. H. Mir, Inorg. Chem. Front., 2018, 5, 1998–2005 RSC .
-
(a) T. N. Hooper, R. Inglis, M. A. Palacios, G. S. Nichol, M. B. Pitak, S. J. Coles, G. Lorusso, M. Evangelisti and E. K. Brechin, Chem. Commun., 2014, 50, 3498–3500 RSC ;
(b) L. Jiang, Y. Liu, X. Liu, J. Tian and S. Yan, Dalton Trans., 2017, 46, 12558–12573 RSC ;
(c) M. Mondal, J. Mayans and A. Ghosh, Inorg. Chim. Acta, 2019, 498, 119175 CrossRef CAS .
- M. U. Anwar, Y. Lan, L. M. C. Beltran, R. Clérac, S. Pfirrmann, C. E. Anson and A. K. Powell, Inorg. Chem., 2009, 48, 5177–5186 CrossRef CAS .
-
(a) M. Das, K. Harms and S. Chattopadhyay, Dalton Trans., 2014, 43, 5643–5647 RSC ;
(b) K. Ghosh, A. Banerjee, A. Bauzá, A. Frontera and S. Chattopadhyay, RSC Adv., 2018, 8, 28216–28237 RSC .
- K. T. Mahmudov, M. N. Kopylovich, A. Sabbatini, M. G. B. Drew, L. M. Martins, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2014, 53, 9946–9958 CrossRef CAS PubMed .
- M. H. Sadhu, A. Solanki, T. Kundu, V. Hingu, B. Ganguly and S. B. Kumar, Polyhedron, 2017, 133, 8–15 CrossRef CAS .
- A. K. Kostopoulos, A. D. Katsenis, J. M. Frost, V. G. Kessler, E. K. Brechin and G. S. Papaefstathiou, Chem. Commun., 2014, 50, 15002–15005 RSC .
- C. Camp, J. Andrez, J. Pécaut and M. Mazzanti, Inorg. Chem., 2013, 52, 7078–7086 CrossRef CAS .
- O. A. Adebayo, K. A. Abboud and G. Christou, Polyhedron, 2017, 122, 71–78 CrossRef CAS .
-
(a) Q. Chen, F. Jiang, L. Chen, M. Yang and M. Hong, Chem. – Eur. J., 2012, 18, 9117–9124 CrossRef CAS ;
(b) S. Khan, S. Jana, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, RSC Adv., 2016, 6, 61214–61220 RSC .
- X.-M. Chen and M.-L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS .
-
(a) N. Nimitsiriwat, E. L. Marshall, V. C. Gibson, M. R. J. Elsegood and S. H. Dale, J. Am. Chem. Soc., 2004, 126, 13598–13599 CrossRef CAS ;
(b) D. Qin, F. Han, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2009, 28, 5535–5541 RSC .
-
(a) A. K. Ghosh, T. S. Mahapatra, R. Clérac, C. Mathonière, V. Bertolasi and D. Ray, Inorg. Chem., 2015, 54, 5136–5138 CrossRef CAS ;
(b) S. K. Kurapati and S. Pal, Dalton Trans., 2015, 44, 2401–2408 RSC ;
(c) S. Aime, M. Botta, U. Casellato, S. Tamburini and P. A. Vigato, Inorg. Chem., 1995, 34, 5825–5831 CrossRef CAS .
-
(a) Z. G. Lada, A. S. Beobide, A. Savvidou, C. P. Raptopoulou, V. Psycharis, G. A. Voyiatzis, M. M. Turnbull and S. P. Perlepes, Dalton Trans., 2017, 46, 260–274 RSC ;
(b) J. P. Tong, X. J. Sun, J. Tao, R. B. Huang and L. S. Zheng, Inorg. Chem., 2010, 49, 1289–1291 CrossRef CAS ;
(c) G. L. Liu, S. F. He, S. Zhang and H. Li, Dalton Trans., 2012, 41, 6256–6262 RSC ;
(d) P. Krumholz, Inorg. Chem., 1965, 4, 757–758 CrossRef CAS .
- S. Ghorai and C. Mukherjee, Chem. – Asian J., 2014, 9, 3518–3524 CrossRef CAS .
-
(a) C. F. Winans and H. Adkins, J. Am. Chem. Soc., 1932, 54, 306–312 CrossRef CAS ;
(b) S. Kostera, B. Wyrzykiewicz, P. Pawluć and B. Marciniec, Dalton Trans., 2017, 46, 11552–11555 RSC ;
(c) P. T. K. Arachchige, H. Lee and C. S. Yi, J. Org. Chem., 2018, 83, 4932–4947 CrossRef CAS ;
(d) H. Chung, S. Han, Y. K. Chunga and J. H. Park, Adv. Synth. Catal., 2018, 360, 1267–1272 CrossRef CAS ;
(e) D. Deng, B. Hu, Z. Zhang, S. Mo, M. Yang and D. Chen, Organometallics, 2019, 38, 2218–2226 CrossRef CAS .
-
(a) J. A. Ballantine, H. Purnell, M. Rayanakorn, J. M. Thomas and K. J. Williams, J. Chem. Soc., Chem. Commun., 1981, 1, 9–10 RSC ;
(b) V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS ;
(c) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS .
-
(a) A. Biswas, M. G. B. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2010, 49, 8155–8163 CrossRef CAS ;
(b) C. T. Yang, M. Vetrichelvan, X. Yang, B. Moubaraki, K. S. Murray and J. J. Vittal, Dalton Trans., 2004, 113–121 RSC ;
(c) B. Sreenivasulu, F. Zhao, S. Gao and J. J. Vittal, Eur. J. Inorg. Chem., 2006, 13, 2656–2670 CrossRef .
-
(a) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS ;
(b) M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS .
-
(a) M. Mondal, M. Chakraborty, M. G. B. Drew and A. Ghosh, Magnetochemistry, 2018, 4, 51 CrossRef CAS ;
(b) A. Biswas, M. G. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2010, 49, 8155–8163 CrossRef CAS ;
(c) K. P. Sarma and R. K. Poddar, Transition Met. Chem., 1984, 9, 135–138 CrossRef CAS .
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS .
-
(a) G. M. Sheldrick, Shelxs97, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS ;
(b) G. M. Sheldrick, Shelxl 2014, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed ;
(c)
G. M. Sheldrick, ShelXL 2013, 2013 Search PubMed .
-
SAINT, version 6.02, SADABS, version 2.03, Bruker AXS Inc., Madison, WI, 2002 Search PubMed .
-
(a) A. Biswas, L. K. Das, M. G. B. Drew, G. Aromí, P. Gamez and A. Ghosh, Inorg. Chem., 2012, 51, 7993–8001 CrossRef CAS ;
(b) M. Mondal, P. M. Guha, S. Giri and A. Ghosh, J. Mol. Catal. A: Chem., 2016, 424, 54–64 CrossRef CAS .
- E. Ruiz, A. Rodriguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982–989 CrossRef CAS .
- E. Ruiz, P. Alemany, S. Alvarez and J. Cano, J. Am. Chem. Soc., 1997, 119, 1297–1303 CrossRef CAS .
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS .
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS .
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS .
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef .
-
F. Neese, ORCA, An ab initio, Density Functional and Semiempirical Program Package, Version 3.03, Universität Bonn, Bonn, Germany, 2015 Search PubMed .
- F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS .
- M. Kannan and T. Punniyamurthy, Tetrahedron: Asymmetry, 2014, 25, 1331–1339 CrossRef CAS .
-
(a) H. C. Brown, E. J. Mead and B. C. Subba Rao, J. Am. Chem. Soc., 1955, 77, 6209–6213 CrossRef CAS ;
(b) R. E. Davis and J. A. Gottbrath, J. Am. Chem. Soc., 1962, 84, 895–898 CrossRef CAS ;
(c) C.-T. F. Lo, K. Karan and B. R. Davis, Ind. Eng. Chem. Res., 2007, 46, 5478–5484 CrossRef CAS .
-
(a) P. Mukherjee, M. G. Drew and A. Ghosh, Eur. J. Inorg. Chem., 2008, 2008, 3372–3381 CrossRef ;
(b) R. Biswas, C. Diaz, A. Bauzá, M. Barceló-Oliver, A. Frontera and A. Ghosh, Dalton Trans., 2014, 43, 6455–6467 RSC .
- P. Mukherjee, M. G. B. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2009, 48, 5848–5860 CrossRef CAS .
-
(a) B. Sreenivasulu, F. Zhao, S. Gao and J. J. Vittal, Eur. J. Inorg. Chem., 2006, 2656–2670 CrossRef CAS ;
(b) C. T. Yang, M. Vetrichelvan, X. Yang, B. Moubaraki, K. S. Murray and J. J. Vittal, Dalton Trans., 2004, 113–121 RSC ;
(c) J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429–438 CrossRef CAS ;
(d) M. Pait, M. Shatruk and D. Ray, Dalton Trans., 2015, 44, 11741–11754 RSC ;
(e) T. Chakraborty, S. Mukherjee, S. Dasgupta, B. Biswas and D. Das, Dalton Trans., 2019, 48, 2772–2784 RSC ;
(f) A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki, E. Schwingel, W. Haase and S. Ostrovsky, Inorg. Chem., 2002, 41, 1788–1794 CrossRef CAS .
-
(a) S. K. Dey and A. Mukherjee, Coord. Chem. Rev., 2016, 310, 80–115 CrossRef CAS ;
(b) P. Mahapatra, M. G. B. Drew and A. Ghosh, Inorg. Chem., 2018, 57, 8338–8353 CrossRef CAS ;
(c) Y. Thio, X. Yang and J. J. Vittal, Dalton Trans., 2014, 43, 3545–3556 RSC .
- T. Ghosh, J. Adhikary, P. Chakraborty, P. K. Sukul, M. S. Jana, T. K. Mondal, E. Zangrando and D. Das, Dalton Trans., 2014, 43, 841–852 RSC .
-
(a) R. Biswas, S. Giri, S. K. Saha and A. Ghosh, Eur. J. Inorg. Chem., 2012, 17, 2916–2927 CrossRef ;
(b) R. Biswas, P. Kar, Y. Song and A. Ghosh, Dalton Trans., 2011, 40, 5324–5331 RSC ;
(c) M. Mondal, S. Giri, P. M. Guha and A. Ghosh, Dalton Trans., 2017, 46, 697–708 RSC ;
(d) E. Ruiz, P. Alemany, S. Alvarez and J. Cano, Inorg. Chem., 1997, 36, 3683–3688 CrossRef CAS PubMed .
- E. Berti, A. Caneschi, C. Daiguebonne, P. Dapporto, M. Formica, V. Fusi, L. Giorgi, A. Guerri, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2003, 42, 348–357 CrossRef CAS .
-
(a) D. Venegas-Yazigi, D. Aravena, E. Spodine, E. Ruiz and S. Alvarez, Coord. Chem. Rev., 2010, 254, 2086–2095 CrossRef CAS ;
(b) M. Stylianou, C. Drouza, Z. Viskadourakis, J. Giapintzakis and A. D. Keramidas, Dalton Trans., 2008, 6188–6204 RSC .
-
(a) S. Naiya, H. S. Wang, M. G. Drew, Y. Song and A. Ghosh, Dalton Trans., 2011, 40, 2744–2756 RSC ;
(b) P. Mukherjee, M. G. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2009, 48, 4817–4827 CrossRef CAS ;
(c) A. Burkhardt, A. Buchholz, H. Görls and W. Z. Plass, Anorg. Allg. Chem., 2013, 14, 2516–2520 CrossRef ;
(d) K. K. Nanda, R. Das, L. K. Thompson, K. Venkatsubramanian, P. Paul and K. Nag, Inorg. Chem., 1994, 33, 1188–1193 CrossRef CAS ;
(e) P. Mahapatra, S. Ghosh, S. Giri and A. Ghosh, Polyhedron, 2016, 117, 427–436 CrossRef CAS ;
(f) S. Dasgupta, J. Adhikary, S. Giri, A. Bauza, A. Frontera and D. Das, Dalton Trans., 2017, 46, 5888–5900 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS spectra of metal complexes 1–3; different reaction mixtures as well as spectra of complexes 1 and 3 with 3,5-DTBC; table of the bond parameters of complexes 1–3; different plots and mechanistic cycle of 1 for catecholase activity; magnetization and correlation plots of 1 and 3; table of kinetic parameters for the catecholase activity of 1 and 3. CCDC 1944919–1944921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi00975b |
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Souvik
Maity
a,
Sanjib
Giri
a,
Souvik
Maity
a,
Sanjib
Giri