
A Pd3L6 supramolecular cage incorporating photoactive [2.2]paracyclophane units†
Diego
Rota Martir
a,
Lucie
Delforce
a,
David B.
Cordes
 a,
Alexandra M. Z.
Slawin
a,
Stuart L.
Warriner
b,
Denis
Jacquemin
c and
Eli
Zysman-Colman
*a
a,
Alexandra M. Z.
Slawin
a,
Stuart L.
Warriner
b,
Denis
Jacquemin
c and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife, UK KY16 9ST. E-mail: eli.zysman-colman@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463826
bSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
cCESIAM Laboratory, UMR CNRS 6230, University of Nantes, 2, rue de la Houssinière, 44322 Nantes, Cedex 3, France
First published on 1st November 2019
Abstract
[2.2]Paracyclophane (pCp) scaffolds, unlike many π-conjugated building blocks, have been rarely explored in supramolecular self-assembly. Herein we report the synthesis and characterization of two ligands, pCpd3py and pCpd4py, composed of a pCp core functionalized at its 7- and 15-positions respectively with 3-pyridyl and 4-pyridyl units. The self-assembly of pCpd4py with Pd2+ metal ions afforded a [Pd3(pCpd4py)6]6+ cage structure, pCpd4py-Pd, where the six pCpd4py ligands doubly bridge each edge of the Pd3 triangular core. The ligand pCpd4py and the palladium cage pCpd4py-Pd have been characterized by NMR spectroscopy, ESI-MS spectrometry and X-ray diffraction analyses and their photophysical properties investigated by steady-state and time-resolved emission spectroscopy as well as time-dependent density functional theory calculations.
Introduction
The [2.2]paracyclophane (pCp) is a compact scaffold in which two benzene rings are rigidly held in phase with respect to each other by two para-disposed ethylene bridges.1 Such a skeleton offers the unusual opportunity to study through-space (π–π transannular) and through-bond [σ(bridge)–π(annular)] electronic interactions that strongly perturb the chemical, optical, and electronic properties of the molecule.2,3 In recent years synthetic methodologies to functionalise the pCp core have been developed and a variety of functional groups have been precisely positioned on the rings and bridges of pCp.4,5 For example, Spuling, Sharma and co-workers6 have recently reported the first examples of pCp compounds emitting via a thermally activated delayed fluorescence mechanism7 due to the presence of a diphenylamine donor and a benzophenone acceptor units located on different decks of the pCp. Zafra and co-workers8 reported a pCp skeleton functionalized with stilbenoid compounds possessing terminal donor–donor, acceptor–acceptor or donor–acceptor units that have the ability to promote charge and exciton migration. Park and co-workers9 reported a pCp core functionalized with triarylamine moieties that served as an efficient hole-transporting material in hybrid perovskite solar cells.The use of pCp as a photoactive scaffold in supramolecular self-assembly is, however, rare and almost exclusively limited to 1D-supramolecular architectures formed through transannular π-stacking or H-bonding interactions between pCp rings.10 These 1D assemblies often exhibit a high electron delocalization across the entire supramolecular chains as a result of strong intermolecular π-stacking interactions between adjacent pCp units that produce materials with low-energy optical gaps, infrared emission, and high conductivity.10
There exist examples of 3D-photoactive cage assemblies that have been synthesized through integration of phosphorescent metal complexes,11,12 porphyrins13 or fluorescent aromatic compounds14–16 into the ligand backbone of the metallosupramolecular architectures. These cages generally show red-shifted emission with lower photoluminescence quantum yields, ΦPL, and shorter emission lifetimes, τPL, than the corresponding luminescent ligands12 used in self-assembly. In the majority of the cases this is due to the formation of lower-energy charge-transfer states that involve both the photoactive ligand and the metal ions used as the structural component. To date, there are only three examples of pCp scaffolds used in metallosupramolecular self-assembly.5,17,18 The first is a small PdL2 macrocyclic structure where L represents pCp compounds functionalized with two ethynyl-2-pyridyl groups,17 the second example reported by Lützen and co-workers,5 describes a Pd2L4 cage structures incorporating chiral pCp ligands functionalized with 3-pyridyl units, while in the third most recent example by Lützen and co-workers,19 a Fe4L6 helicate was formed upon mixing iron(II) triflate, 2-formylpyridine and a pCp ligand functionalized with aniline units (Fig. 1). Although these pCp ligands and their cage structures are photoactive, the emission properties were not discussed.
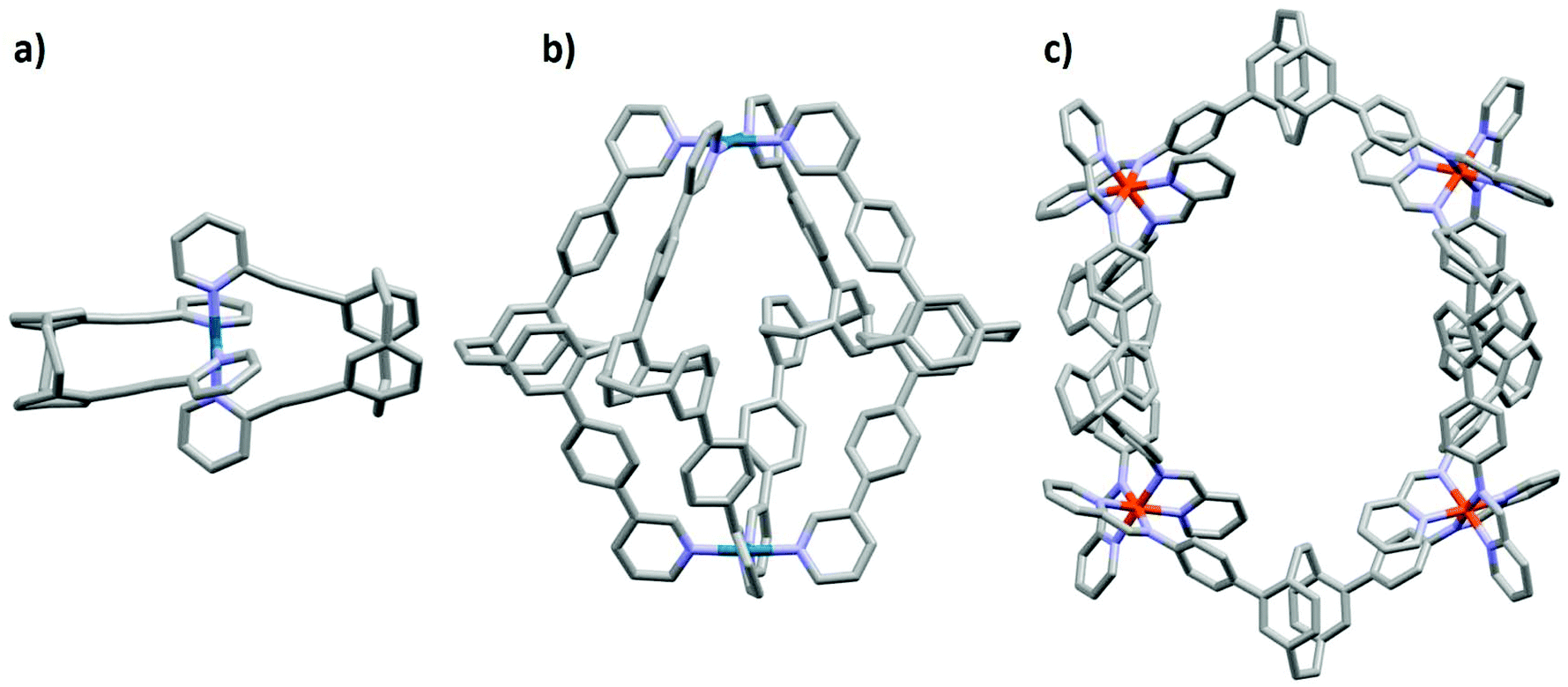 | ||
| Fig. 1 Illustration of the X-ray crystal structures of (a) PdL2 macrocyclic structure,17 (b) Pd2L4 cage structure5 and (c) Fe4L6 helicate19 incorporating pCp compounds. Hydrogen atoms, solvent molecules and counterions have been omitted for clarity. | ||
We report herein two pCp compounds functionalized at the 7- and 15-positions with 3- and 4-pyridyl coordinating units, pCpd3py and pCpd4py (Scheme 1). The self-assembly of pCp4py with Pd2+ ions gives rise to a luminescent cage of composition [Pd3(pCpd4py)6]6+, pCpd4py-Pd while under the same conditions the reaction of pCpd3py with Pd2+ produces multiple products, the structures of which could not be identified by ESI-mass spectrometry. As a result of the six photoactive pCp molecules rigidly assembled in the 3D cage-like structure, pCpd4py-Pd is emissive in both dichloromethane solution, λPL = 455 nm, and as a powder, λPL = 450 nm, with ΦPL of 5% and 3%, respectively in solution and the solid state.
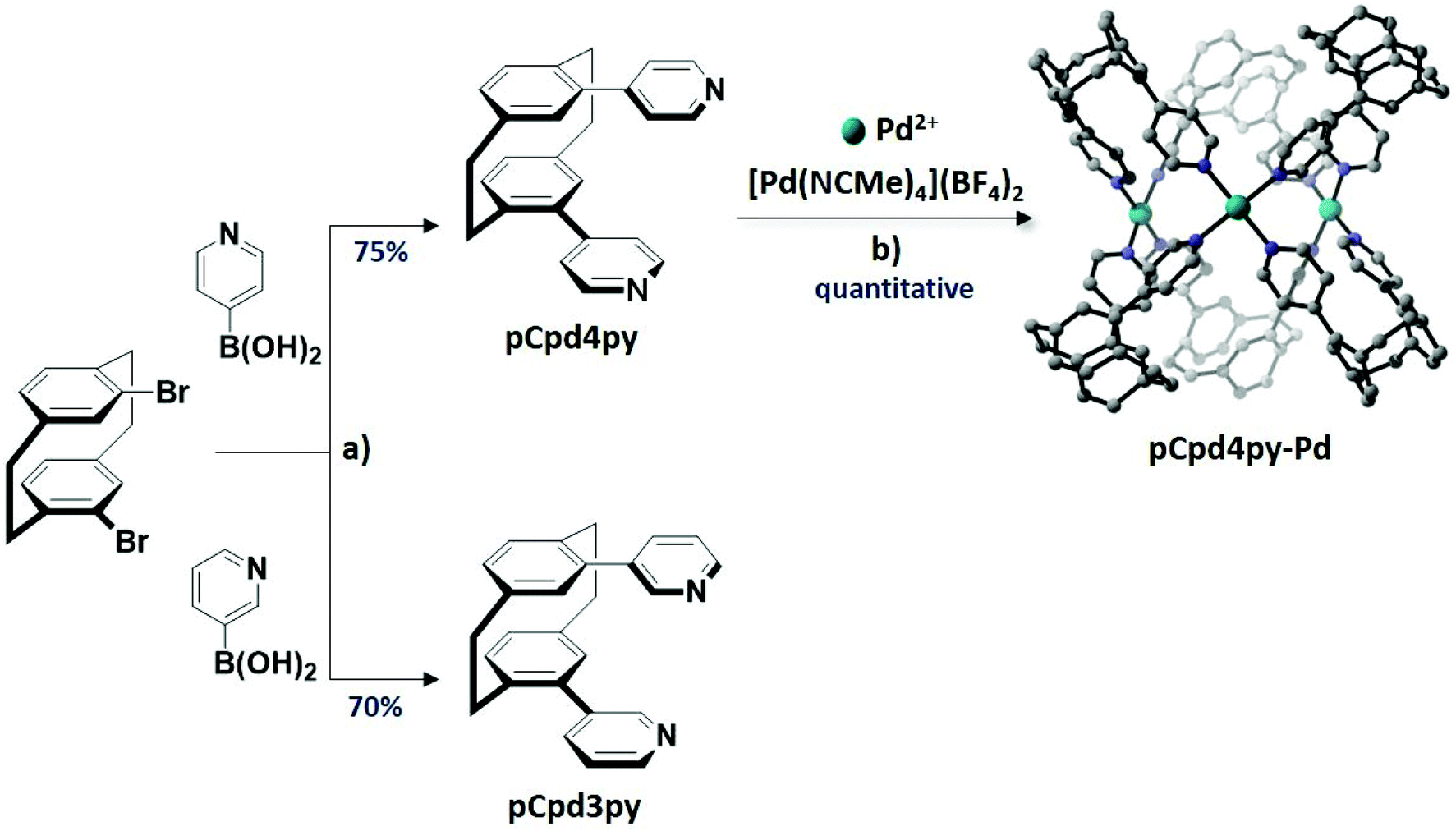 | ||
Scheme 1 Synthesis of ligands pCpd4py and pCpd3py and cage pCpd4py-Pd. Reagents and conditions. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2.0 equiv. CsCO3, 5 mol% Pd(PPh3)4, N2, 1,4-dioxane/H2O (4:1 v/v), 105 °C, 56 h; bDMSO, 85 °C, 12 h. 2.0 equiv. CsCO3, 5 mol% Pd(PPh3)4, N2, 1,4-dioxane/H2O (4:1 v/v), 105 °C, 56 h; bDMSO, 85 °C, 12 h. | ||
Results and discussion
The ligands pCpd4py and pCpd3py (Scheme 1) were prepared in a single step via Suzuki–Miyaura palladium-catalysed cross-coupling of 7,15-dibromocyclophene with the corresponding pyridylboronic acid in yields of 75% and 70%, respectively. When pCpd4py and [Pd(NCMe)4](BF4)2 were heated in a 2:1 ratio in DMSO-d6 at 85 °C for 12 h, the proton resonances associated with pCpd4py broadened and shifted downfield (Fig. 2a). The broad 1H NMR signals are indicative of the formation of a large structure, the tumbling motion of which is very slow on the NMR timescale.16 Evidence for the formation of a single species is confirmed by 1H DOSY NMR spectroscopy where the diffusion coefficient, D, in DMSO-d6 attains 9.2 × 10−11 m2 s−1. The magnitude of D correlates to the presence of a larger structure than the pCpd4py ligand, which shows a diffusion coefficient of 2.5 × 10−10 m2 s−1 (Fig. 2b). The corresponding hydrodynamic radius (rs) of pCpd4py-Pd is estimated to be 12.2 Å (Table S1†), larger than that of the Pd2L4 cage structure reported by Lützen and co-workers (10.4 Å, measured in DMSO-d6, Fig. 1b) but smaller compared to the Fe4L6 helicate illustrated in Fig. 1c (14.0 Å, measured in CD3CN).
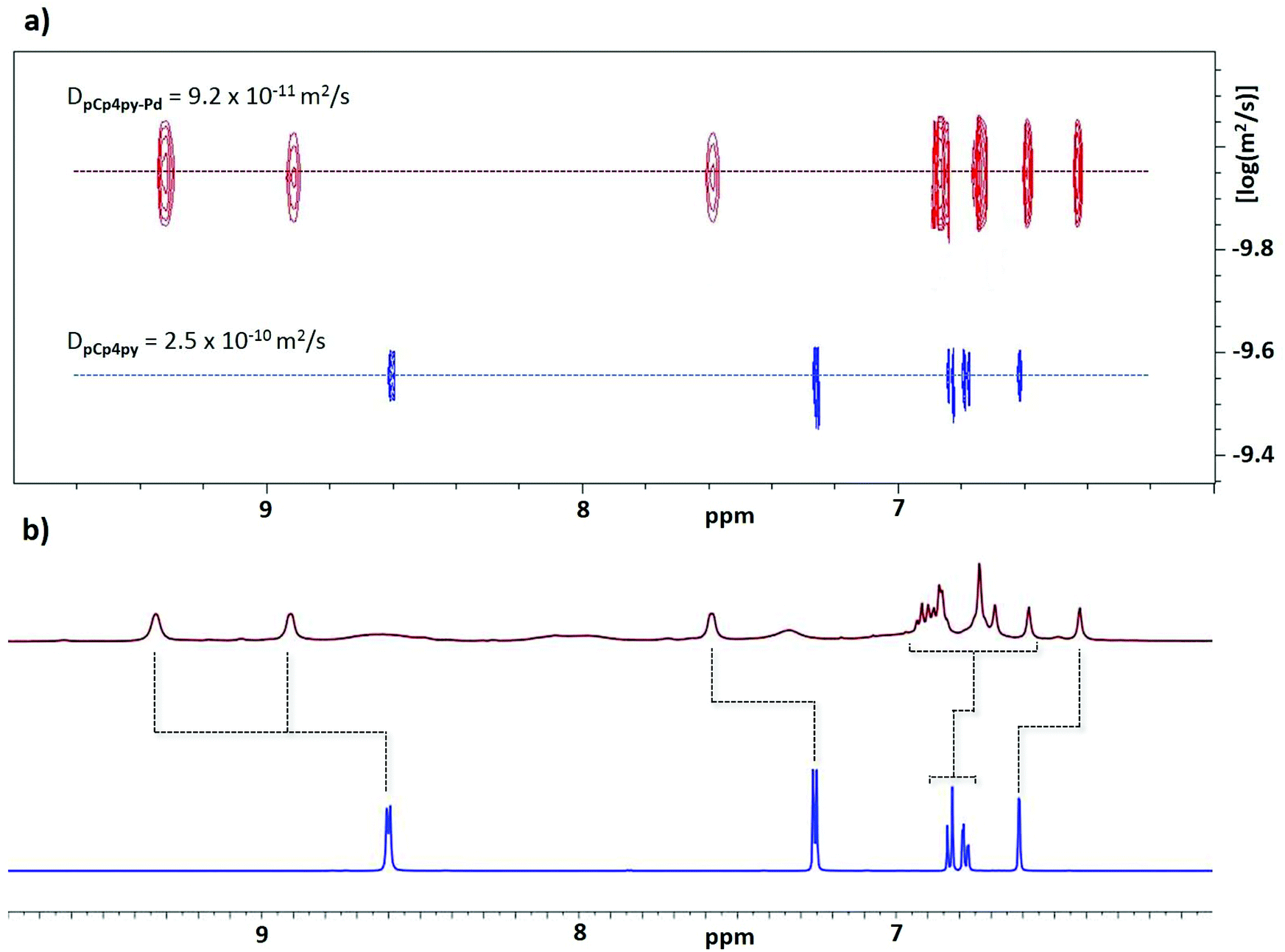 | ||
| Fig. 2 (a) 1H NMR of pCpd4py, in blue and pCpd4py-Pd, in red. (b) 1H DOSY NMR of pCp4py, in blue and pCp4py-Pd, in red. The NMR spectra were collected in DMSO-d6 at 298 K. | ||
The composition of the assembly pCpd4py-Pd has been unequivocally established to be [Pd3(pCpd4py)6](BF4)6 by HR-ESI-mass spectrometry, showing isotopically resolved peaks for [(pCpd4py-Pd)-(BF4)n]n+ (n = 2–5) (Fig. 3 and S10–S13†). The angle between the two 4-pyridyl coordinating motifs in pCpd4py is 50.2°, which is adequate for the formation of a Pd3L6 cage.20 Under similar reaction conditions, the self-assembly of pCpd3py with [Pd(NCMe)4](BF4)2 produced multiple products the structure of which could not be identified by ESI-MS.
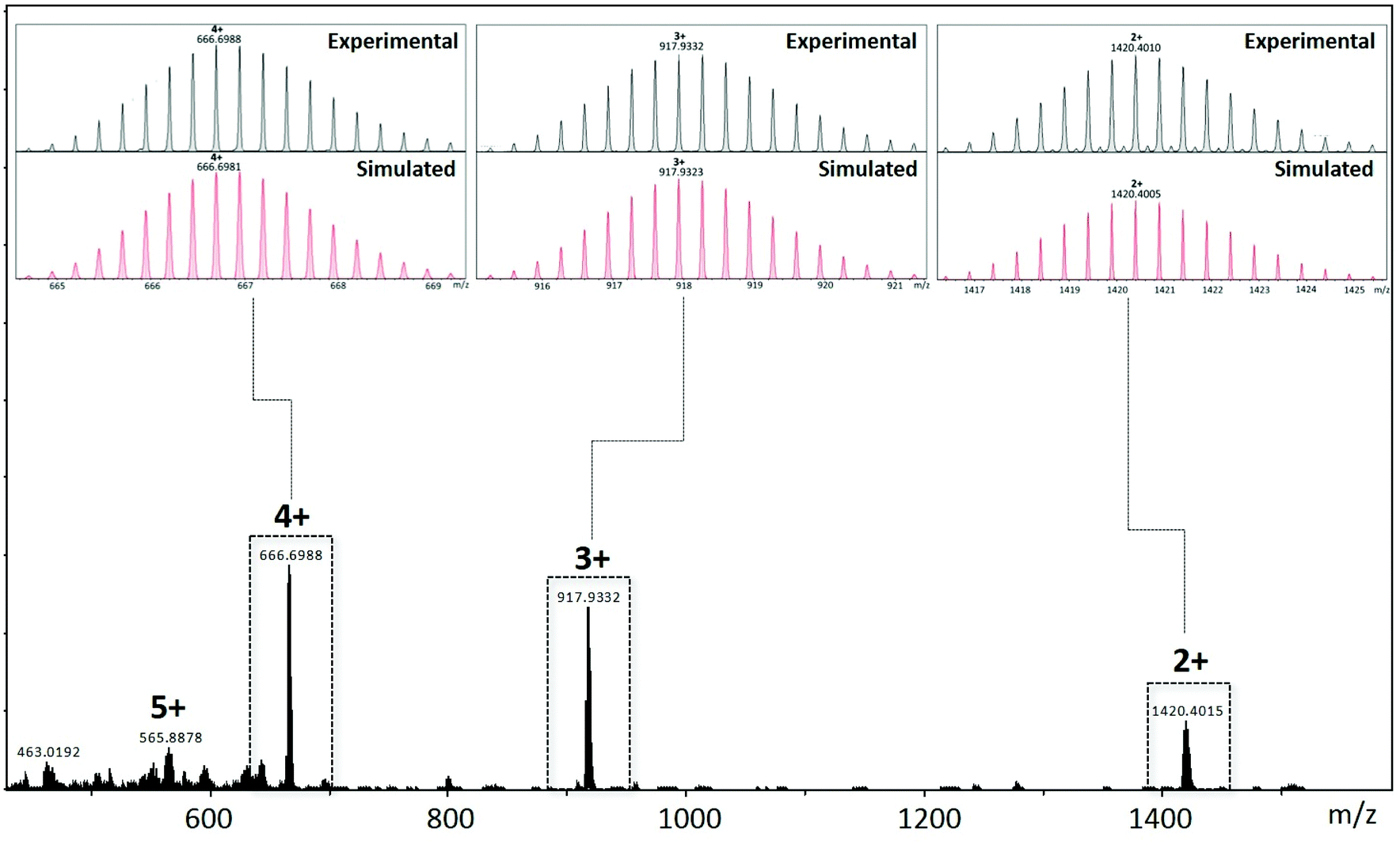 | ||
| Fig. 3 ESI-MS of a DMSO-d6 solution containing cage pCpd4py-Pd. Inserts show the measured (in black) and simulated (in red) isotope patterns for the 4+ (left), 3+ (center) and 2+ (right) charge states. Additional data is provided in the ESI.† | ||
The X-ray structures of ligands pCpd3py and pCpd4py are shown in Fig. S14 and S15 in the ESI.† The structure of pCpd4py-Pd was also unambiguously determined by X-ray crystallography (Fig. 4). Crystals suitable for X-ray analysis were grown by the slow diffusion of a 1:1 mixture of acetone–hexane into a DMSO-d6 solution of pCpd4py-Pd (30 mM) over 5 days. The topology of pCpd4py-Pd resembles that reported by Fujita et al.20 in which the cage is constructed such that two 4-pyridyl units of ligand pCpd4py doubly bridge adjacent Pd(II) centres, which are arranged in a regular triangle. Among Pd cages, this specific Pd3L6 arrangement is rare with only two other examples reported to date.20,21 More generally, only a handful of metallocages of stoichiometry M3L6 have been reported.22–24 The Pd⋯Pd distance around the cavity is 7.7 Å, and the inner cavity diameter of the metallocage, determined from the distance between the centroid of the three palladiums, and a palladium, attains 8.8 Å. The longest external dimension of the cage is 18.0 Å (Fig. S7†). The inner diameter of cage pCpd4py-Pd is smaller than that of the Pd3L6 cage of Fujita et al.,20 which showed a Pd⋯Pd distance of 8.7 Å.
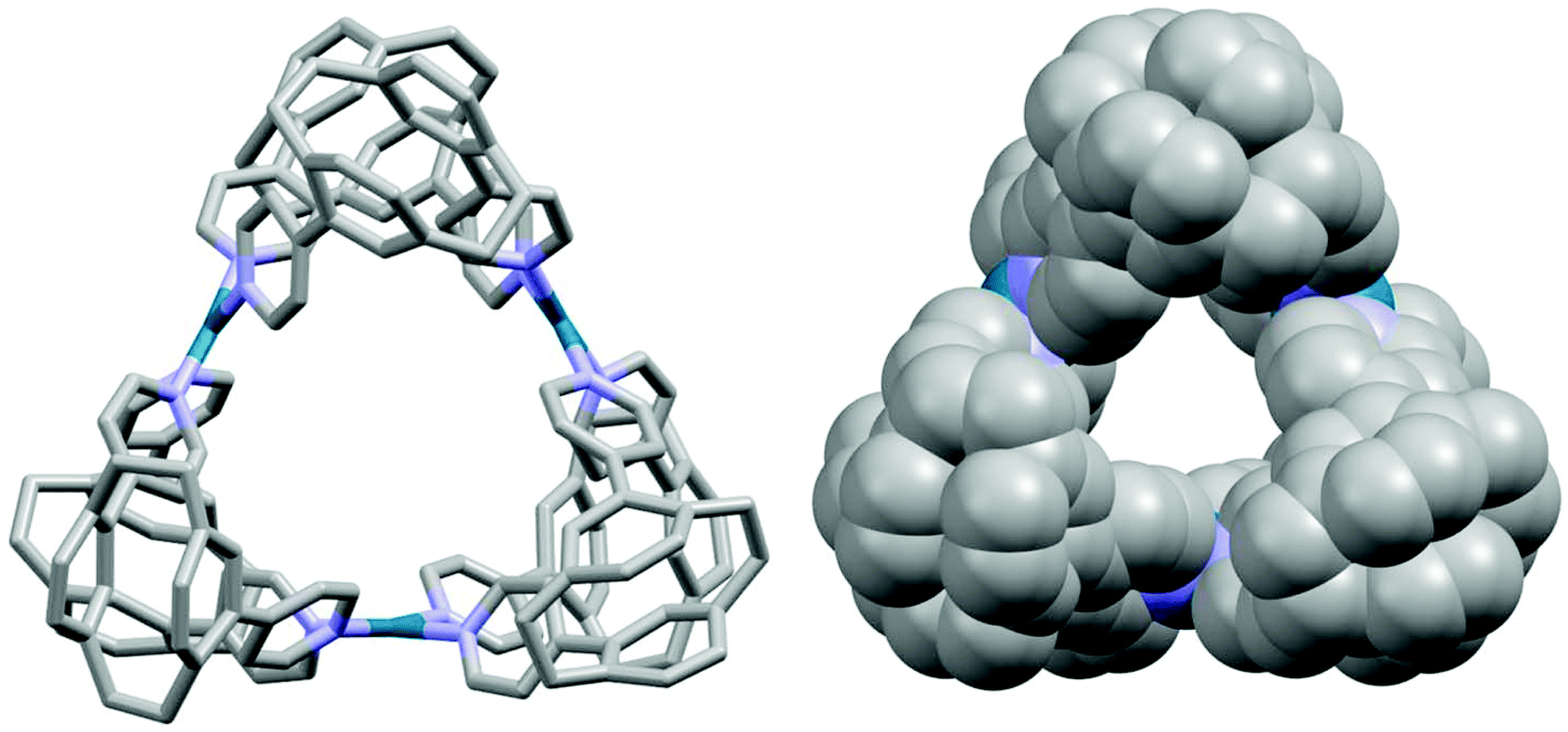 | ||
| Fig. 4 Illustration of the X-ray crystal structure of pCpd4py-Pd. Capped sticks view (left) and space-filling view (right). Hydrogen atoms, solvent molecules and counterions have been omitted for clarity. | ||
A racemic mixture of ligand pCpd4py containing both enantiomers (Sp)-pCpd4py and (Rp)-pCpd4py was used to self-assemble the cage pCpd4py-Pd. Circular Dichroism (CD) experiments reveal that cage pCpd4py-Pd is also formed as expected as a racemic mixture during the self-assembly process (Fig. S17†). However, this does not enable us to determine if pCpd4py-Pd is a racemic cage incorporating both (Sp)-pCpd4py and (Rp)-pCpd4py ligands in a statistical mixture, or whether there exists a racemic mixture of enantiopure cages. The X-ray structure of pCp4py-Pd reveals that each individual cage is enantiopure, but with a racemic mixture of cages occurring in individual crystals.
In DCM, ligands pCpd4py and pCpd3py exhibit absorption maxima at 289 nm and 328 nm for pCpd4py and at 280 nm with a shoulder at 328 nm for pCpd3py. The blue-shifted absorption bands of pCpd3py reflect the greater electron withdrawing character of the 3-pyridyl moiety compared to the 4-pyridyl moiety.25 TD-PBE0 calculations (see the ESI† for details) return the three lowest vertical absorptions at 300 nm (f = 0.004), 287 nm (f = 0.140), and 276 nm (f = 0.046) for pCpd3py and 306 nm (f = 0.003), 287 nm (f = 0.100), and 277 nm (f = 0.098) for pCpd4py. These values are in reasonable agreement with experiment (Fig. 5) with first a weak band and next stronger absorptions. In both ligands, the two lowest transitions can be mainly ascribed to HOMO to LUMO and HOMO to LUMO+1 electronic promotions, respectively (Table S3†). Both transitions do present a CT character from the phenyl rings to the pyridyl moieties (Fig. S24†).
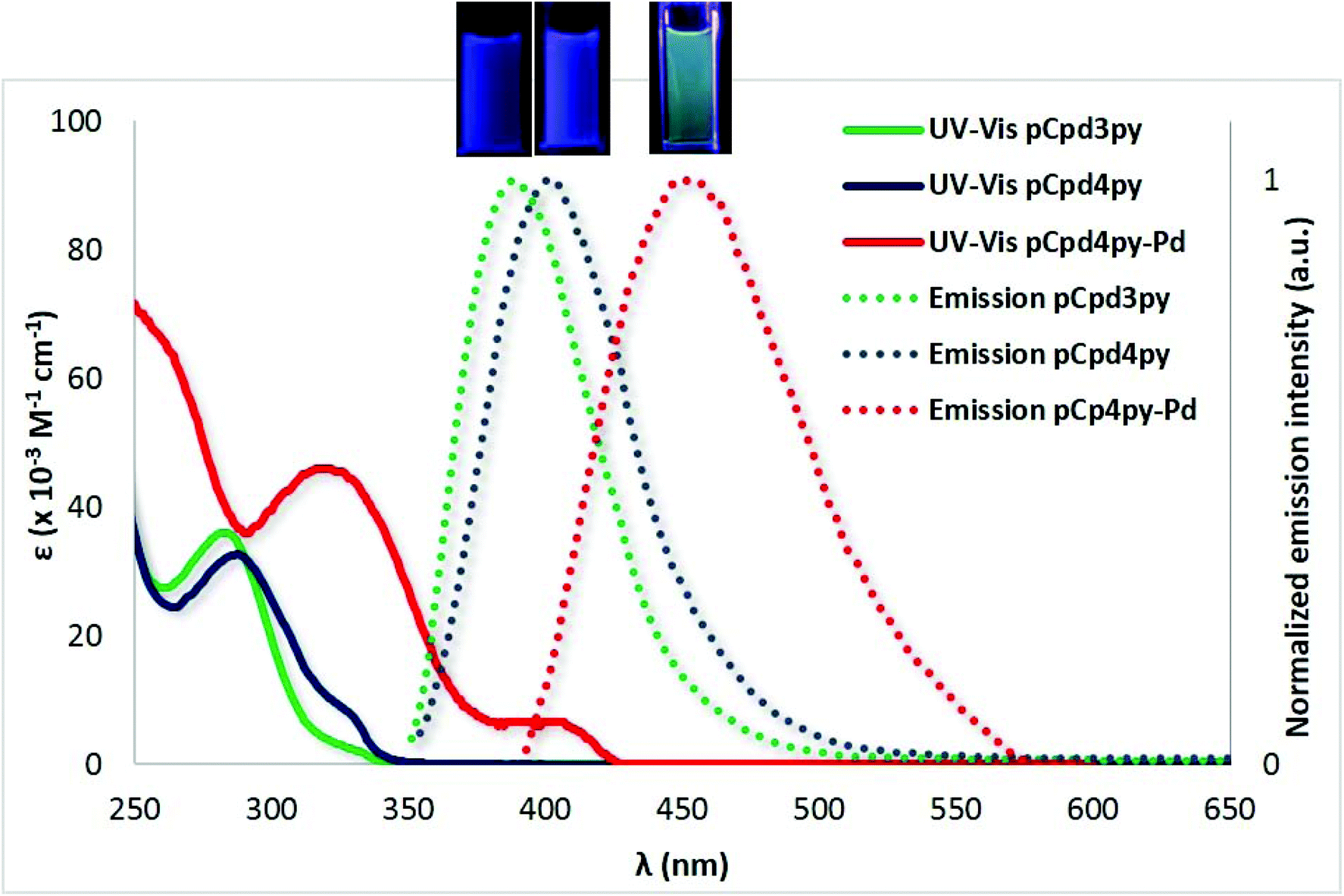 | ||
| Fig. 5 UV-Vis absorption spectra of pCp3dpy (solid green line), pCpd4py (solid blue line) and pCpd4py-Pd (solid red line) collected in DCM at 298 K and normalized emission spectra of pCpd3py (dashed green line), pCpd4py (dashed blue line) and pCpd4py-Pd (dashed red line) collected in degassed DCM at 298 K (λexc = 340 nm). Photographs of DCM solutions of pCpd3py, pCpd4py and pCpd4py-Pd are shown in the inset. | ||
The measured trends in their emission are similar with both showing broad and unstructured spectra characteristic of an intramolecular charge transfer state at λPL = 400 nm and λPL = 395 nm, respectively, for pCpd4py and pCpd3py and with ΦPL of 12% and 14%, and τPL of 5 ns and 4 ns (Fig. 5). The powder emission of pCpd4py and pCpd3py is similar to those collected in DCM (for pCpd4py: λPL = 394 nm, ΦPL = 9%, τPL = 6 ns; for pCpd3py: λPL = 390 nm, ΦPL = 11%, τPL = 4 ns) (Table 1). At the TD-PBE0 level, the computed emission wavelengths in DCM are 371 nm (f = 0.006) and 375 nm (f = 0.006) for pCpd3py and pCpd4py, respectively. The small oscillator strengths determined for these two emissions are consistent with the rather small quantum yield measured in both solution and powder.
| λ PL/nm | Φ PL/% | τ PL/ns | ||||
|---|---|---|---|---|---|---|
| DCMa | Powderb | DCMc | Powderb,d | DCMa | Powderb,e | |
| a Measurements in degassed DCM at 298 K (λexc = 340 nm). b Measured as powder. c Φ PL measurements were carried out in degassed DCM under nitrogen (λexc = 360 nm) using quinine sulfate as the external reference (ΦPL = 54.6% in 0.5 M H2SO4 at 298 K).26 d Values obtained using an integrating sphere. e λ exc = 378 nm. | ||||||
| pCpd3py | 395 | 390 | 14 | 11 | 4 | 4 |
| pCpd4py | 400 | 394 | 12 | 9 | 5 | 6 |
| pCpd4py-Pd | 455 | 450 | 5 | 3 | 3 | 4 |
The absorption spectrum of pCpd4py-Pd in DCM was red-shifted at 327 nm and 408 nm compare to that of pCpd4py. The higher energy band of pCpd4py-Pd at 327 nm can be attributed to the 1π → π* ligand-centered transition localized on pCpd4py, while the broad, lower intensity band at 408 nm can be assigned to a charge transfer transition from the ligands pCpd4py to Pd centers (ligand-to-metal charge transfer). This evolution of the absorption spectra is consistent with TD-PBE0 results that also indicate the emergence of new transitions, e.g., a series of absorptions in the 365–377 nm domain, red-shifted by ca. +70 nm compared to the ligand, a result coherent with the experimental shift of ca. +80 nm (Table S4†). These transitions involve a CT from occupied orbitals localized on the phenyl rings of the ligands to unoccupied orbitals of the metallic cores (Fig. S25†). The emission profiles of cage pCpd4py-Pd in both DCM solution and as a powder are broader and red-shifted, respectively, at λPL = 455 nm and 450 nm, compared to those of pCpd4py (Table 1). The photoluminescence quantum yields of pCpd4py-Pd are lower compared to those of pCpd4py at 5% in degassed DCM and 3% as a powder. The broad and red-shifted emission of cage pCpd4py-Pd as compared to pCpd4py results from the presence of the Pd(II) ions that effectively enhance the CT character of the lowest transitions, giving rise to smaller optical gaps.11 However, similarly to that observed for other phosphorescent palladium cages incorporating emissive Ir(III) and Ru(II) metalloligands,12 the Pd2+ ions do not completely quench the emission of pCpd4py. The lower ΦPL values observed for pCpd4py-Pd compared to pCpd4py is likely due to the presence of several low-lying nearly perfectly dark states (see Table S4 in the ESI†).
In conclusion, two deep-blue emissive [2.2]paracyclophanes molecules functionalized with 3-pyridyl and 4-pyridyl coordinating units, namely pCpd3py and pCpd4py, respectively, have been synthesized and characterized. The self-assembly of pCpd4py with Pd2+ ions gives rise to a fluorescent cage of the composition of [Pd3(pCpd4py)6](BF4)6, pCpd4py-Pd. The cage has been comprehensively characterized by 1H and 1H-DOSY NMR spectroscopy and HR-ESI-MS and the structure of the cage was further elucidated by single crystal X-ray diffraction. The pCpd4py-Pd cage is emissive at 450 nm with ΦPL of 5%. To the best of our knowledge, this is the first report that shows that supramolecular cages based on the rigid [2.2]paracyclophanes core scaffold are photoactive and can be potentially explored for optoelectronic applications. We therefore believe that the assembly of suitably functionalised pCp structures with metal ions as structural components opens up the possibility to prepare a wide range of supramolecular architectures such as coordination polymers, networks and macrocycles, that retain the optoelectronic properties of photoactive [2.2]paracyclophane molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
EZ-C acknowledges the University of St Andrews and EPSRC (EP/M02105X/1) for financial support. We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for analytical services. We thank Umicore AG for the gift of materials.References
- L. G. Bahrin, L. G. Sarbu, P. G. Jones, L. M. Birsa and H. Hopf, [2.2]Paracyclophane-Bis(triazole) Systems: Synthesis and Photochemical Behavior, Chem. – Eur. J., 2017, 23, 12338–12345 CrossRef CAS.
- A. Kahnt, D. M. Guldi, A. de la Escosura, M. V. Martínez-Díaz and T. Torres, [2.2]Paracyclophane: a pseudoconjugated spacer for long-lived electron transfer in phthalocyanine–C60 dyads, J. Mater. Chem., 2008, 18, 77–82 RSC.
- E. Elacqua and L. R. MacGillivray, From the Decks to the Bridges: Optoelectronics in [2.2]Paracyclophane Chemistry, Eur. J. Org. Chem., 2010, 6883–6894 CrossRef CAS.
- C. Braun, M. Nieger and S. Bräse, Unprecedented One-Pot Reaction towards Chiral, Non-Racemic Copper(I) Complexes of [2.2]Paracyclophane-Based P,N-Ligands, Chem. – Eur. J., 2017, 23, 16452–16455 CrossRef CAS.
- J. Anhäuser, R. Puttreddy, Y. Lorenz, A. Schneider, M. Engeser, K. Rissanen and A. Lützen, Chiral self-sorting behaviour of [2.2]paracyclophane-based bis(pyridine) ligands, Org. Chem. Front., 2019, 6, 1226–1235 RSC.
- E. Spuling, N. Sharma, I. D. W. Samuel, E. Zysman-Colman and S. Brase, (Deep) blue through-space conjugated TADF emitters based on [2.2]paracyclophanes, Chem. Commun., 2018, 54, 9278–9281 RSC.
- M. Y. Wong and E. Zysman-Colman, Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- J. L. Zafra, A. Molina Ontoria, P. Mayorga Burrezo, M. Peña-Alvarez, M. Samoc, J. Szeremeta, F. J. Ramírez, M. D. Lovander, C. J. Droske, T. M. Pappenfus, L. Echegoyen, J. T. López Navarrete, N. Martín and J. Casado, Fingerprints of Through-Bond and Through-Space Exciton and Charge π-Electron Delocalization in Linearly Extended [2.2]Paracyclophanes, J. Am. Chem. Soc., 2017, 139, 3095–3105 CrossRef CAS.
- S. Park, J. H. Heo, C. H. Cheon, H. Kim, S. H. Im and H. J. Son, A [2,2]paracyclophane triarylamine-based hole-transporting material for high performance perovskite solar cells, J. Mater. Chem. A, 2015, 3, 24215–24220 RSC.
- D. E. Fagnani, M. J. Meese Jr., K. A. Abboud and R. K. Castellano, Homochiral [2.2]Paracyclophane Self-Assembly Promoted by Transannular Hydrogen Bonding, Angew. Chem., Int. Ed., 2016, 55, 10726–10731 CrossRef CAS PubMed.
- D. Rota Martir and E. Zysman-Colman, Supramolecular iridium(III) assemblies, Coord. Chem. Rev., 2018, 364, 86–117 CrossRef CAS.
- D. Rota Martir and E. Zysman-Colman, Photoactive supramolecular cages incorporating Ru(ii) and Ir(iii) metal complexes, Chem. Commun., 2019, 55, 139–158 RSC.
- S. Durot, J. Taesch and V. Heitz, Multiporphyrinic cages: architectures and functions, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS.
- N. Kishi, M. Akita and M. Yoshizawa, Selective host-guest interactions of a transformable coordination capsule/tube with fullerenes, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS.
- P. D. Frischmann, V. Kunz and F. Wurthner, Bright Fluorescence and Host-Guest Sensing with a Nanoscale M(4)L(6) Tetrahedron Accessed by Self-Assembly of Zinc-Imine Chelate Vertices and Perylene Bisimide Edges, Angew. Chem., Int. Ed., 2015, 54, 7285–7289 CrossRef CAS.
- D. R. Martir, A. Pizzolante, D. Escudero, D. Jacquemin, S. L. Warriner and E. Zysman-Colman, Photoinduced Energy and Electron Transfer Between a Photoactive Cage Based on a Thermally Activate Delayed Fluorescence Ligand and Encapsulated Fluorescent Dyes, ACS Appl. Energy Mater., 2018, 1, 2971–2978 CrossRef CAS.
- G. Meyer-Eppler, F. Topić, G. Schnakenburg, K. Rissanen and A. Lützen, Chiral Self-Sorting of trans-Chelating Chiral Ligands upon Formation of PdII Complexes, Eur. J. Inorg. Chem., 2014, 2014, 2495–2501 CrossRef CAS.
- M. Gon, Y. Morisaki and Y. Chujo, A silver(I)-induced higher-ordered structure based on planar chiral tetrasubstituted [2.2]paracyclophane, Chem. Commun., 2017, 53, 8304–8307 RSC.
- J. Anhäuser, R. Puttreddy, L. Glanz, A. Schneider, M. Engeser, K. Rissanen and A. Lützen, Subcomponent self-assembly of a cyclic tetranuclear Fe(II) helicate in a highly diastereoselective self-sorting manner, Chem. – Eur. J., 2019, 25, 12294–12297 CrossRef.
- K. Suzuki, M. Kawano and M. Fujita, Solvato-controlled assembly of Pd3L6 and Pd4L8 coordination “boxes”, Angew. Chem., Int. Ed., 2007, 46, 2819–2822 CrossRef CAS.
- M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Light-Controlled Interconversion between a Self-Assembled Triangle and a Rhombicuboctahedral Sphere, Angew. Chem., Int. Ed., 2016, 55, 445–449 CrossRef CAS.
- T. Zhang, L.-P. Zhou, X.-Q. Guo, L.-X. Cai and Q.-F. Sun, Adaptive self-assembly and induced-fit transformations of anion-binding metal-organic macrocycles, Nat. Commun., 2017, 8, 15898 CrossRef CAS.
- Y. Liu, R. Zhang, C. He, D. Dang and C. Duan, A palladium(II) triangle as building blocks of microporous molecular materials: structures and catalytic performance, Chem. Commun., 2010, 46, 746–748 RSC.
- O. Jurček, P. Bonakdarzadeh, E. Kalenius, J. M. Linnanto, M. Groessl, R. Knochenmuss, J. A. Ihalainen and K. Rissanen, Superchiral Pd3L6 Coordination Complex and Its Reversible Structural Conversion into Pd3L3Cl6 Metallocycles, Angew. Chem., Int. Ed., 2015, 54, 15462–15467 CrossRef.
- J. H. Blanch, Determination of the Hammett substituent constants for the 2-, 3-, and 4-pyridyl and -pyridinium groups, J. Chem. Soc. B, 1966, 937–939 RSC.
- W. H. Melhuish, Quantum Efficiences Of Fluorescence Of Organic Substances: Effect Of Solvent And Concentration Of The Fluorescent Solute 1, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of ligands pCpd4py, pCpd3py and cage pCpd4py-Pd, crystal structures, supplementary optoelectronic data and DFT and TD-DFT calculation details. CCDC 1951959–1951961. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01147a. The research data underpinning this publication can be accessed at https://doi.org/10.17630/7b0cfca2-b3b9-4a2a-a557-eb983e5ba132 |
| This journal is © the Partner Organisations 2020 |