
Solid-state-stabilization of molecular vanadium oxides for reversible electrochemical charge storage†
Simon
Greiner‡
ab,
Montaha H.
Anjass‡
 *ab,
Maximilian
Fichtner
bc and
Carsten
Streb
*ab
*ab,
Maximilian
Fichtner
bc and
Carsten
Streb
*ab
aInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: montaha.anjass@uni-ulm.de; carsten.streb@uni-ulm.de
bHelmholtz Institute Ulm (HIU), Helmholtzstr. 11, 89081 Ulm, Germany
cKarlsruhe Institute of Technology (KIT), Institute of Nanotechnology, P. O. Box 3640, 76021 Karlsruhe, Germany
First published on 24th October 2019
Abstract
Molecular vanadium oxides are promising active materials for cathodes in lithium and post-lithium batteries due to their high redox activity, low molecular weight and facile tuneability. However, a major challenge for this application is the transformation of the molecular clusters into solid-state oxides under typical electrode fabrication conditions. Here, we report a molecular crystal engineering approach for the stabilization of molecular vanadium oxides in the crystal lattice, enabling initial studies on reversible electron storage in a lithium ion battery test cell.
Introduction
The development of new, high-performance battery electrode materials is critical for advanced lithium-ion batteries and post-lithium systems.1 For cathode active materials, metal oxides are compounds of choice, as they combine high redox-activity with economic viability and long-term stability. For solid-state metal oxides, chemical property tuning on the atomic level can be challenging and often requires detailed empirical knowledge of a given system. For this reason, molecular metal oxides, so-called polyoxometalates (POMs), have been explored as tuneable alternatives for battery electrode materials.2–4 POMs are anionic molecular metal oxides of early, high-valent transition metals (mainly Mo, W, V).5 POMs are ideal electrode material candidates as their redox-properties can be tuned over a wide range, and as they can undergo multiple, reversible redox processes.6,7 Pioneering studies by Awaga and colleagues have shown the reversible uptake of up to 24 electrons for the molybdenum Keggin cluster [PMo12O40].8 This reactivity has been referred to as “electron sponge” behaviour, and highlights the high potential of POMs for electrochemical energy storage.While initial studies focused mainly on molybdenum-based polyoxometalates,9 polyoxovanadates (POV) have recently gained more attention.10–12 In addition to the significantly lower atomic weight, which allows higher gravimetric energy densities, the extraordinary redox-activity of vanadium could enable the storage of multiple electrons per transition metal atom.13 Furthermore, POVs offer easier redox-tuning14,15 and wider structural tunability than molybdates.16–19
To-date, most POV battery research has been focused on the prototype decavanadate cluster Q6−x[HxV10O28](6−x)−.10,12,20,21 For typical battery applications, the cluster is isolated using inorganic counter-cations Q such as Li+ or Na+, as this prevents cluster leaching into the organic electrolyte used in high voltage batteries. However, due to this synthetic approach, the crystal lattice typically contains lattice water5 which is detrimental in lithium-ion batteries, as it could lead to gassing (O2 and H2 evolution) at the typical battery voltages employed.
In most studies, lattice water removal involved thermal treatment at high temperatures (up to 600 °C).20,22,23 However, POVs are known to easily convert into solid-state metal oxides,24–26 and the decavanadate cluster is particularly prone to undergo thermally induced structural rearrangement into solid-state vanadium oxides. As recently reported by some of us, even moderate heating and dehydration of the lithium decavanadate Li6[V10O28]·16H2O at temperatures of ∼120 °C leads to the formation of the solid-state oxides LiVO3 and LiV3O8.27 In consequence, most studies of decavanadates as molecular battery components reported to-date in fact were analysing the performance of nano- or microscale lithium vanadates LiVO3 and LiV3O8.27 Therefore, structural stabilization routes are required which enable the dehydration of hydrated decavanadate materials under typical battery processing conditions to allow mechanistic studies of the performance of truly molecular vanadium oxide molecular cluster batteries. Interestingly, Matson and co-workers recently reported pioneering studies which showed that the introduction of organic ligands and organic cations in polyoxovanadates28–31 enables sustained performance, e.g. in redox-flow batteries.30,31
Building on this work, here, we show how the introduction of hydrogen bonding organo-ammonium cations into the decavanadate lattice can be used to establish a thermal stability window in decavanadates which allows the facile dehydration whilst retaining the molecular structural integrity of the species. Full materials analysis is provided together with thermal stability tests, electrochemical performance analyses and initial battery electrode tests. The study therefore shows how crystal engineering concepts can be used to stabilize important molecular model components for electrochemical energy storage.
Results and discussion
This study presents a straightforward approach to overcome the decomposition of POVs during thermal dehydration by the introduction of a stabilizing counter-cation, dimethyl ammonium (DMA), which is capable of electrostatic and hydrogen bonding stabilization of the decavanadate clusters. In addition, the low molecular weight of DMA makes it an ideal addition for battery electrode materials. In a straightforward and green one-pot synthesis vanadium pentoxide was first dissolved in water using 4 M aqueous LiOH solution. The decavanadate cluster self-assembled from this solution upon acidification. After addition of a di-methyl ammonium salt, direct precipitation with ethanol yielded the crude product, which was re-crystallized by diffusion of ethanol into an aqueous solution.Structural characterization of the resulting yellow block crystals using single-crystal X-ray diffraction (SC-XRD) together with elemental-analysis (see ESI†) gave the sum formula (DMA)5Li[V10O28]·5H2O (DMA{V10}, DMA = dimethyl ammonium). In the lattice of DMA{V10}, each decavanadate anion is charge-balanced by five DMA and one Li+ cation. Note that the Li+ cation was not detected by SC-XRD but its presence was inferred by charge-balance considerations and verified by Inductively-coupled plasma optical emission spectroscopy (ICP-OES). The decavanadate clusters arrange in hexagonal layers (Fig. 1A), which are stacked in an ABA configuration. Analysis of the DMA cations revealed close proximity to the decavanadate cluster within the hydrogen bonding distance (<3 Å). Both, within and in between these layers, DMA cations form hydrogen bonds to more than one cluster. This linkage between the clusters leads to the formation of a stabilizing 3D network. This network is shown in Fig. 1B, where only the bridging DMA cations are displayed. Note furthermore the low number of crystal water molecules in comparison to related compounds like Li6[V10O28]·16H2O (Li6{V10}).20
 | ||
| Fig. 1 (A) Mixed polyhedral/stick-and-ball representation of the hexagonal layers of DMA{V10} in the crystallographic ac-plane. (B) Mixed polyhedral/stick-and-ball representation of the 3D H-bond network in a unit cell along the a-axis. Only the bridging DMA cations are shown. All lattice water molecules have been removed for clarity in both pictures. Colour scheme: orange: vanadium; red: oxygen; blue: nitrogen; black: carbon; white: hydrogen; grey: lithium; yellow: [VO6] octahedra; purple: N–H–O hydrogen bonds. | ||
Structural stability upon removal of crystal water
The structural stability upon thermal dehydration was investigated by thermogravimetric analysis (TGA) (Fig. 2A). At about 120 °C a plateau is observed after the loss of 5.6 wt% corresponding to four water molecules. This plateau gives a first indication for improved stability after dehydration. In comparison, Li6[V10O28]·16H2O shows a more sloped behaviour and the dehydration is only completed at much higher temperatures. Between 120 °C and 360 °C all five DMA molecules and the remaining water molecule is lost (19.8 wt%). This is in line with earlier observations, where DMA in polyoxovanadate compounds decomposes at similar temperatures.14 | ||
| Fig. 2 (A) Comparison of the thermogravimetric analysis of DMA{V10} (black) and Li6[V10O28]·16H2O (green) showing a plateau at 120 °C after losing 4 water molecules for DMA{V10}. (B) IR spectra and (C) powder XRD pattern of DMA{V10}, DMA{V10}-120 and DMA{V10}-145 showing the structural stability of the compound at 120 °C and the significant changes upon slightly higher temperatures. | ||
To further investigate the stability after dehydration, infrared (IR) (Fig. 2B) and Raman spectroscopy (see Fig. S1†) and powder XRD (Fig. 2C) were performed after the dehydration for 12 h at 120 °C under vacuum (DMA{V10}-120), as usually employed during electrode fabrication, and after heating to 145 °C for 12 h und vacuum (DMA{V10}-145), corresponding to the edge of the described plateau in the TGA curve.
While hardly any changes can be observed in the IR spectra after heating the sample to 120 °C, significant changes are visible after heating to 145 °C. The characteristic V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bond at 944 cm−1 (green line) is shifted to 970 cm−1, while the V–O–V vibration bonds at 835 cm−1 and 805 cm−1 vanish. This indicates a structural transformation upon heating to 145 °C and is in line with the thermal behaviour and presence of a flat plateau at 120 °C observed by TGA.
O stretching bond at 944 cm−1 (green line) is shifted to 970 cm−1, while the V–O–V vibration bonds at 835 cm−1 and 805 cm−1 vanish. This indicates a structural transformation upon heating to 145 °C and is in line with the thermal behaviour and presence of a flat plateau at 120 °C observed by TGA.
Further evidence is supplied by powder XRD patterns of the respective compounds. While the pattern of DMA{V10}-120 is missing some peaks in comparison to the pristine compound, the remaining signals are matching the initial pattern regarding diffraction angle and intensity. This indicates the loss of molecules, which form a reflection layer, without changes to the remaining structure and is in line with the loss of crystal water without transformation of the {V10} framework. No new signals, which would indicate the formation of a new phase, are observed.27 However, after heating the sample to 145 °C nearly all crystallinity is lost. The loss of nearly all diffractions indicates the formation of a new amorphous phase. This transformation into a new amorphous phase with new, broad signals arising at higher diffraction angles has also been observed during the thermal decomposition of Li6{V10} (Fig. S2†).
Virtually the same results were obtained by Raman spectroscopy (see Fig. S1†).
Based on this data, the stabilization of decavanadate by rational design can be reported. DMA{V10} remains as molecular POV cluster under much harsher conditions than comparable compounds like Li6[V10O26]·16H2O. The material remains stable under standard electrode fabrication conditions and does not undergo transformation into a solid-state oxide. However, only small increase of the temperature leads to the decomposition of the molecular cluster, emphasizing the easy conversion of decavanadate species into solid-state oxides upon dehydration at moderate temperatures.
Electrochemical investigation on DMA{V10}
In order to test the electrochemical properties of DMA{V10}, electrodes containing 70 wt% POV, 20 wt% carbon black and 10 wt.–% PVDF (1) were fabricated. During the slurry preparation using N-methyl-2-pyrrolidone (NMP) a partial dissolution of DMA{V10} was observed. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) of the coating showed a good dispersion of vanadium oxide species in the carbon matrix (see Fig. S3†). Although pXRD measurements of the coated electrode (see Fig. S4†) show only weak diffractions, suggesting the loss of some crystallinity, these diffractions match with the ones observed for DMA{V10}-120. No new diffractions, which would indicate the formation of a new crystalline phase, were observed. In isolated cases larger particles (ca. 3 μm) were found (see Fig. S5†). These are expected to be disadvantageous due to the expected low electrical conductivity of DMA{V10} and the resulting loss of active material. Post-mortem analysis furthermore showed similar sized holes indicating the disconnection of these microparticles during cycling (see Fig. S5†).The cyclic voltammetry data (Fig. 3) shows an open circuit voltage of about 3.2 V vs. Li+/Li. In the first cycle, two broad reductive signals can be observed around 2.0 V and 1.5 V vs. Li+/Li. During oxidation, a broad peak around 2.2 V vs. Li+/Li can be distinguished. The cyclic voltammogram further shows a large irreversible oxidation peak at 3.7 V, which is not visible in consecutive cycles. While the origin of this peak could not be identified, it is hypothesized, that it correlates to the oxidation of traces of crystal water, which might remain in the material. Evolution of O2 from trace water would be expected at this potential (>3.3 V vs. Li+/Li).32 During the second cycle a third reductive peak can be distinguished around 3.0 V. This could be explained by a partial reduction of the pristine material, as has been observed for precipitation of decavanadates with ethanol in the literature.10 A slight shift of the other two signals to lower potentials indicates an increasing overpotential of the reduction caused by increasing resistance. This is supported by the shift of the oxidation peak to a higher potential.
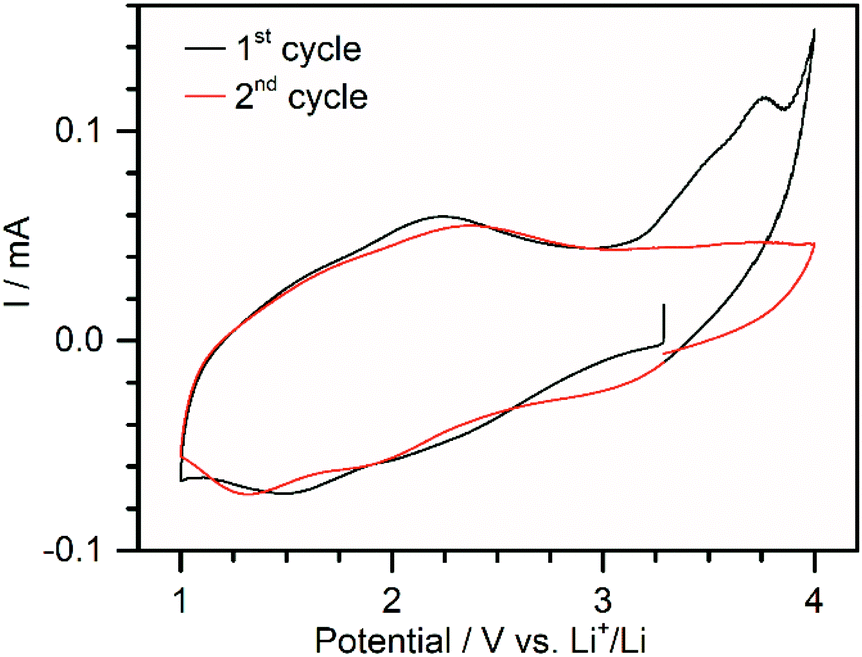 | ||
| Fig. 3 Cyclic voltammogram of 1 in a potential range between 1.0 and 4.0 V vs. Li+/Li at a sweep rate of 0.05 mV s−1. | ||
The electrochemical performance of 1 as cathode material in lithium-ion batteries was investigated. Initial galvanostatic cycling of 1 in a lithium half-cell was measured in the voltage range between 1.2 and 3.4 V applying a current density of 50 mA g−1 (C/5 rate) at 25 °C. Fig. 4 presents the voltage profile in the first 3 cycles and the 10th cycle. Assuming the V(V)/V(IV) redox couple for all 10 vanadium centres, dehydrated DMA{V10} has a theoretical capacity of 224 mA h g−1. The discharge capacity in the first cycle is 290 mA h g−1, which could be due to reduction of three vanadium atoms per cluster to V(III) within this voltage window. However, no evidence for the reversible reduction to V(III) in the decavanadate cluster could be reported and requires further investigation. The sloped curve of the voltage profiles indicates a solid solution system in contrast to the phase transition in classical intercalation materials. The described voltage profile as well as the broad signals in the CV matches with other decavanadate compounds published in very recent literature, where careful attention has been paid to the gentle processing of the POV and similar strategies for the thermal stabilization have been employed. Yoshikawa and colleagues reported similar charge–discharge behaviour using [H2Metf]3[V10O28] (H2Metf = di-protonated metformin).11 The H2Metf counterion also enables the formation of hydrogen bonds, although the molecular weight is higher. They achieved a discharge capacity of 156 mA h g−1 in a voltage range between 1.5 and 3.8 V vs. Li+/Li, which remained above 100 mA h g−1 for 20 cycles. Similarly, Liu and colleagues21 very recently leveraged the stabilizing effect of a 3D structure in Mg2(NH4)2[V10O28]. While the charge–discharge curve shows similar behaviour to the results reported here, the group achieved an initial discharge capacity of ca. 200 mA h g−1 between 1.0 and 3.8 V. In contrast to the present study, the discharge capacity remained above 180 mA h g−1 for up to 60 cycles. These recent studies are all in contrast to earlier reports, where several plateaus could be observed.22,23 In these studies the decavanadate compounds were dehydrated at elevated temperatures, which is expected to decompose the POVs into solid-state oxides.27
 | ||
| Fig. 4 Galvanostatic charge/discharge profile of 1 cycled between 1.2 and 3.4 V vs. Li+/Li at a current density of 50 mA g−1 at 25 °C. | ||
After a few cycles, the capacity of 1 has dropped significantly to ca. 20 mA h g−1. Dissolution of vanadium is expected to contribute strongly to the capacity fading as has been observed in literature.13,33 Post-mortem ICP-OES confirmed the dissolution of the cluster by high vanadium content at the lithium anode. Addition of pristine DMA{V10} to electrolyte solution shows a partial dissolution and yellow coloration in a similar extent as Li6{V10}.
Such a high discharge capacity of about 290 mA h g−1 using a homo-POV based electrode has to our knowledge only be reported by Wang and colleagues.22 However, high temperatures during dehydration (>400 °C) and powder diffraction pattern strongly indicate the formation of solid-state oxides as active material in their work.
In order to get further insights about the enhanced capacities of 1, electrochemical impedance spectroscopy was performed between 10 mHz and 200 kHz. The Nyquist plot is shown in Fig. 5. Moreover, the equivalent circuit model (ECM) used for the fit is also presented in the inset.34,35 The Inductance I represents external wiring of the cell and measuring device, Re corresponds to the electrolyte resistance, the finite Warburg element Ws and the capacitor C represent the solid-state diffusion and accumulation of Li ions in the electrode, RCT is the charge-transfer resistance and CPEDL represents the formation of a double-layer on the electrode–electrolyte interface. CPE1 could so far not be assigned to a physical process and is assumed as a corrective element. The very low charge-transfer resistance RCT of 1 (49 Ω) indicate a good electronic conductivity and an efficient charge transfer.34–36 This suggests a good electronic contact between the DMA{V10} clusters and the current collector and is in line the morphological observation, where most of the material seems to be well dispersed in the carbon black matrix.
 | ||
| Fig. 5 Nyquist plot of 1 before cycling at OCV of 3.2 V and the corresponding fits according to the equivalent circuit model of the cell (inlet). | ||
Conclusions
In conclusion, we demonstrated how crystal engineering concepts can be leveraged to tune polyoxovanadates for the application in electrochemical energy storage. Introduction of di-methyl ammonium cations into the decavanadate lattice was used to stabilize the material against thermal transformation into solid-state oxides by the formation of a hydrogen bonding network. This enables the thermal dehydration within the stability window of the cluster as well as the fabrication of coated electrodes for the applications of molecular vanadium oxides in battery electrodes. Initial tests as cathodes in lithium-ion batteries show very promising performance with high capacity.Experimental
All reagent-grade chemicals were purchased from Sigma Aldrich, Alfa Aesar or VWR and were used without further purification.Synthesis and heat treatment of DMA{V10}
V2O5 (5 g, 27.5 mmol, 1 eq.) was suspended in water (50 ml) and basified with 4 M aqueous LiOH until pH 11 was reached. The solution was stirred for 1 h at 50 °C, which yielded a colorless solution. After cooling the solution to room temperature, the pH was decreased to 4.5 using 4 M HCl. To this solution, a solution of DMA·HCl (4.2 g, 51.5 mmol, 2 eq.) in water (10 ml) was added dropwise while stirring. Diffusion of ethanol into the reaction mixture gave orange plate crystals of DMA{V10} suitable for X-ray diffraction.DMA{V 10 }–120 and DMA{V10}-145 were obtained by heating the sample under vacuum for 12 h at 120 °C and 145 °C, respectively.
Structural characterization
Single-crystal X-ray diffraction (SC-XRD) was performed on a Bruker D8 Quest single-crystal diffractometer with a Photon II detector using Mo Kα radiation (λ = 0.71073 Å).§ Powder X-ray Diffraction (pXRD) data were collected on a STOE Stadi P diffractometer with Cu Kα radiation (λ = 1.5405 Å) using Debye-Scherrer geometry. ATR-FT-IR spectra were recorded on a PerkinElmer Spectrum Two. Thermogravimetric analysis (TGA) was carried out on a NETZSCH TG 209F1 analyzer at a heating rate of 10 K min−1 under air up to 700 °C in an Al2O3 crucible.Electrochemical characterization
Electrochemical tests were carried out in Swagelok-type cells versus lithium metal. Electrode slurries for 1 were prepared inside an argon-filled glovebox with recirculation using 70 wt% POM, 20 wt% carbon black and 15 wt% poly (vinylidene difluoride) (PVDF) binder with N-methyl-2-pyrrolidine (NMP). The mixed slurry was coated on an aluminium foil by the doctor blade technique. The solvent was evaporated at 40 °C under vacuum for several hours and subsequently dried at 120 °C under vacuum for 12 h. Each working electrode (12 mm diameter) contained approximately 1–2 mg of active material and Li foil was used as counter electrode. The electrolyte was LiPF6 (1 M) in a mixture of ethylene carbonate and dimethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio). Cyclic voltammetry (CV) experiments and Electrochemical Impedance Spectroscopy were carried out using a Bio-Logic VMP-3 potentiostat. Temperature controlled galvanostatic charge–discharge experiments were conducted using an Arbin electrochemical workstation at 25 °C.
:1 volume ratio). Cyclic voltammetry (CV) experiments and Electrochemical Impedance Spectroscopy were carried out using a Bio-Logic VMP-3 potentiostat. Temperature controlled galvanostatic charge–discharge experiments were conducted using an Arbin electrochemical workstation at 25 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Ulm University, the Helmholtz-Gemeinschaft (HGF) and the Deutsche Forschungsgemeinschaft (DFG) (STR 1164/12, STR1164/14 and Germany's Excellence Strategy, EXC-2154/1) is gratefully acknowledged. S. G. gratefully acknowledges financial support through a PhD fellowship by the Fonds der Chemischen Industrie (FCI). This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe).References
- D. Andre, S. J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- M. Genovese and K. Lian, Curr. Opin. Solid State Mater. Sci., 2015, 19, 126–137 CrossRef CAS.
- Y. Ji, L. Huang, J. Hu, C. Streb and Y. Song, Energy Environ. Sci., 2015, 8, 776–789 RSC.
- S. Herrmann, C. Ritchie and C. Streb, Dalton Trans., 2015, 44, 7092–7104 RSC.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS.
- J. W. Jordan, G. A. Lowe, R. L. McSweeney, C. T. Stoppiello, R. W. Lodge, S. T. Skowron, J. Biskupek, G. A. Rance, U. Kaiser, D. A. Walsh, G. N. Newton and A. N. Khlobystov, Adv. Mater., 2019, 1904182 CrossRef CAS.
- H. Wang, S. Hamanaka, Y. Nishimoto, S. Irle, T. Yokoyama, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2012, 134, 4918–4924 CrossRef CAS.
- M. Lira-Cantú and P. Gómez-Romero, Chem. Mater., 1998, 10, 698–704 CrossRef.
- S. Hartung, N. Bucher, H.-Y. Chen, R. Al-Oweini, S. Sreejith, P. Borah, Z. Yanli, U. Kortz, U. Stimming, H. E. Hoster and M. Srinivasan, J. Power Sources, 2015, 288, 270–277 CrossRef CAS.
- H. Wang, J. Isobe, D. Matsumura and H. Yoshikawa, J. Solid State Electrochem., 2018, 22, 2067–2071 CrossRef CAS.
- H. Y. Chen, J. Friedl, C. J. Pan, A. Haider, R. Al-Oweini, Y. L. Cheah, M. H. Lin, U. Kortz, B. J. Hwang, M. Srinivasan and U. Stimming, Phys. Chem. Chem. Phys., 2017, 19, 3358–3365 RSC.
- H. T. Tan, X. Rui, W. Sun, Q. Yan and T. M. Lim, Nanoscale, 2015, 7, 14595–14607 RSC.
- K. Kastner, J. T. Margraf, T. Clark and C. Streb, Chem. – Eur. J., 2014, 20, 12269–12273 CrossRef CAS.
- J. Tucher, K. Peuntinger, J. T. Margraf, T. Clark, D. M. Guldi and C. Streb, Chem. – Eur. J., 2015, 21, 8716–8719 CrossRef CAS.
- C. Streb, in Structure and Bonding, ed. Y.-F. Song, 2017, vol. 176 Search PubMed.
- T. McGlone, J. Thiel, C. Streb, D.-L. L. Long and L. Cronin, Chem. Commun., 2012, 48, 359–361 RSC.
- A. Seliverstov and C. Streb, Chem. – Eur. J., 2014, 20, 9733–9738 CrossRef CAS.
- A. Seliverstov and C. Streb, Chem. Commun., 2014, 50, 1827–1829 RSC.
- A. Xie, C.-A. Ma, L. Wang and Y. Chu, Electrochim. Acta, 2007, 52, 2945–2949 CrossRef CAS.
- S. Lu, Y. Lv, W. Ma, X. Lei, R. Zhang, H. Liu and X. Liu, Inorg. Chem. Front., 2017, 4, 2012–2016 RSC.
- H. Liu and J. Wang, Ionics, 2010, 16, 379–383 CrossRef CAS.
- A. L. Xie, L. B. Wang, Y. Q. Chu and C. A. Ma, Adv. Mater. Res., 2015, 1088, 337–342 Search PubMed.
- A. Seliverstov, M. Rangus, M. Hartmann, S. G. G. Mitchell and C. Streb, Inorg. Chem. Front., 2017, 4, 160–164 RSC.
- Y. Ji, J. Hu, J. Biskupek, U. Kaiser, Y.-F. Song and C. Streb, Chem. – Eur. J., 2017, 23, 16637–16643 CrossRef CAS.
- J. Livage, Coord. Chem. Rev., 1998, 180, 999–1018 CrossRef.
- M. H. Anjass, M. Deisböck, S. Greiner, M. Fichtner and C. Streb, ChemElectroChem, 2018, 6, 398–403 CrossRef.
- F. Li, S. H. Carpenter, R. F. Higgins, M. G. Hitt, W. W. Brennessel, M. G. Ferrier, S. K. Cary, J. S. Lezama-Pacheco, J. T. Wright, B. W. Stein, M. P. Shores, M. L. Neidig, S. A. Kozimor and E. M. Matson, Inorg. Chem., 2017, 56, 7065–7080 CrossRef CAS.
- F. Li, L. E. VanGelder, W. W. Brennessel and E. M. Matson, Inorg. Chem., 2016, 55, 7332–7334 CrossRef CAS.
- L. E. Vangelder, A. M. Kosswattaarachchi, P. L. Forrestel, T. R. Cook and E. M. Matson, Chem. Sci., 2018, 9, 1692–1699 RSC.
- L. E. VanGelder and E. M. Matson, J. Mater. Chem. A, 2018, 6, 13874–13882 RSC.
- J. Luo, W. Cui, P. He and Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS.
- C. Wu and Y. Xie, Energy Environ. Sci., 2010, 3, 1191–1206 RSC.
- J. Liu, Z. Chen, S. Chen, B. Zhang, J. Wang, H. Wang, B. Tian, M. Chen, X. Fan, Y. Huang, T. C. Sum, J. Lin and Z. X. Shen, ACS Nano, 2017, 11, 6911–6920 CrossRef CAS.
- J. Hu, Y. Ji, W. Chen, C. Streb and Y.-F. Song, Energy Environ. Sci., 2016, 9, 1095–1101 RSC.
- J.-J. Chen, M. D. Symes, S.-C. Fan, M.-S. Zheng, H. N. Miras, Q.-F. Dong and L. Cronin, Adv. Mater., 2015, 27, 4649–4654 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1955646. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01229j |
| ‡ These authors contributed equally. |
§ Crystallographic data for: C10H37N5O31V10, Mw = 1233.85 g mol−1, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.228 Å, b = 16.754 Å, c = 20.784 Å, α = 81.27, β = 83.96, γ = 82.44 V = 3816.5 Å3, Z = 4, μ(Mo-Kα) = 2.436 mm−1, 163183 reflections collected, 14440 unique which were used in all calculations; structure solution and refinement as done using OLEX2.37 Final R1 = 0.0664 and wR2 = 0.1613 (all data). CCDC 1955646. , a = 11.228 Å, b = 16.754 Å, c = 20.784 Å, α = 81.27, β = 83.96, γ = 82.44 V = 3816.5 Å3, Z = 4, μ(Mo-Kα) = 2.436 mm−1, 163183 reflections collected, 14440 unique which were used in all calculations; structure solution and refinement as done using OLEX2.37 Final R1 = 0.0664 and wR2 = 0.1613 (all data). CCDC 1955646. |
| This journal is © the Partner Organisations 2020 |