
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore and double bond functionalisation of cyclopentadithiophene-phosphaalkenes†‡
Jordann A. L.
Wells§
 ,
Muhammad Anwar
Shameem¶§
,
Arvind Kumar
Gupta
and
Andreas
Orthaber
*
,
Muhammad Anwar
Shameem¶§
,
Arvind Kumar
Gupta
and
Andreas
Orthaber
*
Synthetic Molecular Chemistry. Dept. Chemistry – Ångström laboratories. Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: andreas.orthaber@kemi.uu.se
First published on 8th September 2020
Abstract
The heterofulvenoid cyclopentadithiophene-phosphaalkene is a versatile building block for opto-electronic tuning with donor and acceptor moieties. Both the annulated thienyl rings and the phosphaalkene bond can be functionalised using a variety of chemical transformations, e.g. forming C–C, C–E (E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si, Br) bonds, or oxidation and metal coordination, respectively. Solid-state structures, optical and electronic properties are probed theoretically and experimentally, illustrating the opto-electronic tailoring opportunities at this motif.
Si, Br) bonds, or oxidation and metal coordination, respectively. Solid-state structures, optical and electronic properties are probed theoretically and experimentally, illustrating the opto-electronic tailoring opportunities at this motif.
Introduction
Combining electron donor and electron acceptor units to tailor the HOMO and LUMO of dye molecules has been an extensively used concept in the design of dyes for sensors, solar cells, etc.1 In the cyclopentadithiophene (CPDT) motif two thiophene units are linked using a methylene bridge improving the orbital delocalisation by removing a torsional degree of freedom, i.e. planarising the conjugated system.2 Moreover, functionalisation at the bridgehead positions allowed addressing of the solubility (Fig. 1, I) and electronic nature of this annulated bithiophene framework.2,3 In particular, electron deficient bridgehead substituents, e.g. ketone (II) and dicyanoalkenes (III) have led to interesting opto-electronic materials and building blocks for donor–acceptor polymers, in which the exocyclic fulvenoid repeating unit acts as an outstanding acceptor unit.4,5 Recently, examples of imine functionalised CPDT units have been incorporated into polymers allowing controlled modifications of the HOMO and LUMO levels of the molecular components.6 The diagonal relationship of phosphorus and carbon has led to a variety of conjugated compounds in which carbene fragments of double bonds are replaced by isolobal phosphinidenes giving rise to phosphaalkenes.7–9 The introduction of this group 15 heteroelement as part of conjugated frameworks has noticeable stabilising effects on the LUMO levels, while the HOMO remains almost unaltered. In these building blocks, the low-lying heteroalkene π* orbitals provide an acceptor group resulting in properties applicable to opto-electronic materials.10,11 The marked difference to its lighter homologue, i.e. the imine derivatives, hold promising developments for the heavier pnictaalkene systems. Interesting approaches have already been pursued to translate the molecular phosphaalkene motifs into polymeric materials,12 both by keeping the PC motif intact and using it as polymerizable unit.13–17 Furthermore, the lone pair at the phosphaalkene can easily be functionalised by metal coordination allowing for catalytic applications,18–20 but also altering the opto-electronic properties.21,22 A significantly less explored but equally powerful PC functionalisation is its oxidation with chalcogens23–25 and coordination by NHCs.26
 | ||
| Fig. 1 Literature known CPDT-derivatives (I–III), phospha-/arsa-alkene-CPDT-derivative A and B, and novel derivatives studied in this work. | ||
In view of these properties, we have already described the synthesis and excited state behaviour of phospha- and arsaalkene functionalised cyclopentadithiophene derivatives, where the heteroalkenes lead to pronounced acceptor character by stabilizing the heterofulvenoid-type antibonding orbitals. The elemental substitution (phosphorus vs. arsenic) and metal coordination (AuCl) allowed us to address the energy and dynamics of the lowest energy transitions.27
In this manuscript, we address the further functionalisation of the CPDT-phosphaalkene by metal coordination or oxidation at the heteroalkene and chemical functionalisation at the thiophene rings achieving stabilisation of the LUMO and destabilisation of the HOMO levels, respectively. The impact of these functionalisations is illustrated by comparing electronic spectra and electro-chemistry data.
Results and discussion
Cyclopentadithiophene phosphaalkene A is synthesised according to published procedures from readily available starting materials.27 In order to study the versatility of this building block we initially investigated the derivatisation of the thiophene rings using standard organic transformations (Scheme 1).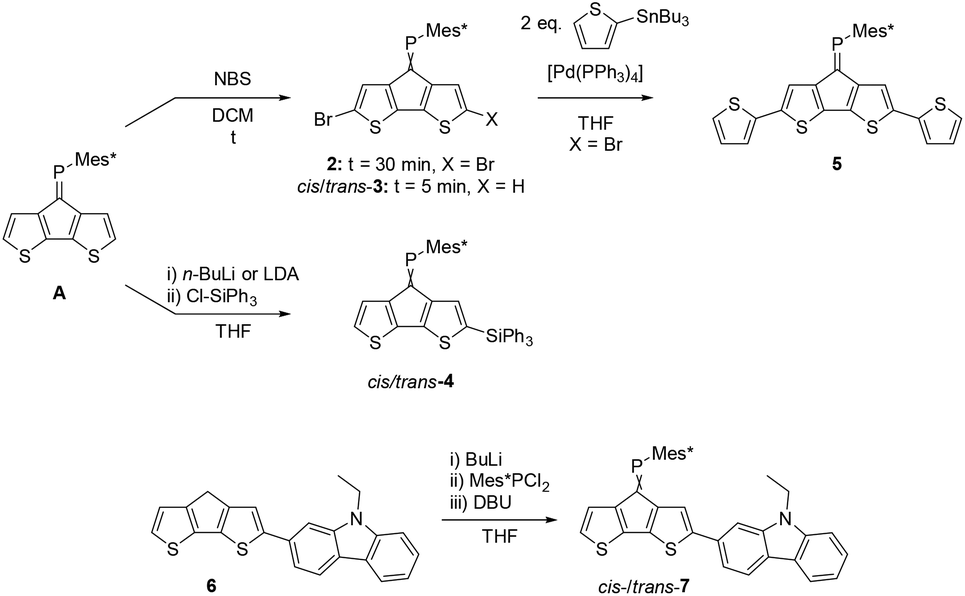 | ||
| Scheme 1 Functionalisation of parent CPDT phosphaalkene A giving brominated (2 and cis-/trans-3), silylated (4), dithienyl (5) and carbazole (7) derivatives. Full experimental details are provided in the ESI.‡ | ||
Expectedly, the bromination with N-bromo succinimide (NBS) occurs selectively at the α,α′-positions under ambient conditions and ultrasonication. Chromatographic workup afforded the dibrominated derivative 2 in approx. 75% isolated yields. Attempts to synthesise the corresponding mono-brominated derivatives cis-3 and trans-3 using strictly one equivalent of NBS and low reaction temperatures suffer from unselective reactivities, affording a statistical mixture of A, 2 and both isomers of 3. The two mono-brominated isomers can be separated chromatographically, however isomerisation within a couple of hours establishes the mixture of cis/trans-3 isomers. The conditions which trigger phosphaalkene E−/Z isomerisation (e.g. light etc.) seem highly dependent on the steric and electronic nature of the substituent and are less explored and understood28 compared to the diphosphene E–Z isomerisation.29 A possible explanation for the isomerisation of 3 could be an increased phosphenium character promoted by the electron withdrawing Br-substituents.
The electron withdrawing effect of the bromide substituent on the remote PC bond is clearly visible in the 31P NMR spectra, which show resonances shifted to higher frequencies. (δ31P for 2: 269.6 ppm; cis-3: 262.9 ppm; trans-3: 263.1 ppm). Compared to A, which has a resonance at 256.6 ppm, the addition of each Br-atom shields the resonance by approx. 6 ppm. The assignment of the two isomers is based on the coupling constants of the shielded β′ protons positioned below the Mes*-ring current; for cis-3 this characteristic proton (4.36 ppm) shows as a doublet with a small 4JPH coupling of 1.7 Hz. In contrast, trans-3 shows a doublet of doublets (dd) splitting pattern with a 3JHH = 5.1 Hz and a 4JPH = 1.5 Hz (4.44 ppm) coupling constants to the neighbouring hydrogen and phosphorus atoms, respectively. Single crystals obtained from slow solvent evaporation of pentane/DCM solutions allowed us to perform single crystal X-ray diffraction experiments (Fig. 2). The solid-state structure of 2 gives a PC bond length of 1.687(2) Å, which is slightly longer than that of A, while the C–PC angle of 102.5(1)° is almost identical. These metrics suggest that the electronic variation has a larger influence on the bond length than the “hybridisation” and steric hindrance, which is more reflected in the angles around the P-centre. The packing is dominated by weak slipped π–π interactions (C-centroid distance 3.711(5) Å) and π⋯Br interactions (3.782(1) Å).
 | ||
Fig. 2 ORTEP plot of the solid-state structure of 2 and cis-4. Selected atoms are drawn at 50% probability levels of thermal ellipsoids, while the Mes* and phenyl groups are depicted as wireframe and capped sticks. Selected bond lengths [Å] and angles [°]: 2: P1–C1A 1.687(2), P1–C10A 1.841(2), C1A–P1–C10A 102.54(11). cis-4: P1–C1 1.678(2), Si1–C9 1.852(2), Si1–Cipso 1.862(2), 1.867(2). 1872(2), C1–P1–C10(Mes*) 103.1(1). Compound 2 shows a positional disorder (0.92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.08); distances are given for the molecule with the higher occupancy. :0.08); distances are given for the molecule with the higher occupancy. | ||
We further studied the possibility to deprotonate the acidic α-positions using a Li-base and reacting the in situ-formed anion with an electrophile. Compound A was thus reacted with two equivalents of LDA in THF at low temperatures and then allowed to reach r.t., followed by the addition of triphenyl chlorosilane as an electron accepting substituent. Surprisingly, two new resonances in the 31P-NMR spectra are observed indicating formation of two major products. Despite being separable by column chromatography, slow isomerisation in solution precluded detailed studies of the isomers. Selective crystallisation from the mixture afforded single crystals suitable for X-ray diffraction analysis. Indeed, the solid-state structure of one monosilylated product was confirmed as cis-4. This isomer (cis-4) crystallises in the monoclinic space group P21/c (no. 14). The P1–C1 double bond is 1.678(2) Å, slightly longer than in the parent system (A: 1.667(5) Å). Despite the cis-orientation of the sterically demanding triphenylsilyl substituent, the C10–P1–C1 angle increases only marginally from 102.6(2)° in A to 103.1(1)° for cis-4. The Si–C9 bond is significantly shorter (1.852(2) Å) compared to the Si–Cphenyl bonds (1.862(2) to 1.872(2) Å) indicative of negative hyper-conjugation of the CPDT-core with the silyl substituent. The second product is suspected to be the corresponding trans-4, based on proton NMR data, indicating that despite the use of two equivalents of LDA or n-BuLi the double-deprotonation was unsuccessful.
The usefulness of 2 as a synthon towards extended conjugated systems was probed using a Stille coupling protocol with 2-tributylstannyl thiophene. The reaction proceeds in moderate yield to give the desired disubstituted product 5 with a 31P-NMR resonance of 265.5 ppm, which is quite similar to those of the brominated derivatives.
Further attempts to use the brominated derivatives 2 and 3 as starting materials in Suzuki type C–C coupling reactions to target extended conjugated systems has proven difficult. In contrast, the carbazole functionalised ketone 5 can be prepared from readily available iodinated precursors and undergoes facile Wolff–Kishner reduction to the methylene derivative 6. In analogy to the synthesis of A, a two-step, one-pot reaction afforded the desired phosphaalkene as an inseparable isomeric mixture of cis-/trans-7 in an approximately 1:1 ratio. Unlike the brominated derivative 3, no cis/trans isomerisation of 7 was observed in solution.
The two isomers crystallised as slightly differently shaped dark purple blocks. Compound trans-7 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) as a hexane solvate (Fig. 3). The disordered solvent molecule is treated by diffuse contribution to the overall scattering using the solvent masking function implemented in OLEX2.30 Notably, the PC double bond in trans-7 is even longer (1.684(2) Å) compared to that of cis-4, while the C–PC angle (102.6(1)°) is more similar to that found in A. The cis-isomer crystallises in the monoclinic space group C2/c (no. 15) as a hexane solvate (removed also by solvent masking). The PC bond lengths in both isomers are comparable (PC: 1.683(3) Å), but the C–PC angle is wider for cis-7 (103.4(1)°) reflecting the steric demand of the carbazole substituent on the Mes* position. The most pronounced effect in the different isomers is observed for the orientation of the carbazole moiety and its torsion with respect to the CPDT core. The smaller torsion angle between the two chromophores’ planes in trans-7 (8.74°) gives rise to efficient delocalisation, while steric interaction of the large Mes* with the carbazole unit leads to an increased torsion angle (21.61°) in cis-7, with the N-ethyl group pointing in the same and opposite direction of the PC bond, respectively.
(no. 2) as a hexane solvate (Fig. 3). The disordered solvent molecule is treated by diffuse contribution to the overall scattering using the solvent masking function implemented in OLEX2.30 Notably, the PC double bond in trans-7 is even longer (1.684(2) Å) compared to that of cis-4, while the C–PC angle (102.6(1)°) is more similar to that found in A. The cis-isomer crystallises in the monoclinic space group C2/c (no. 15) as a hexane solvate (removed also by solvent masking). The PC bond lengths in both isomers are comparable (PC: 1.683(3) Å), but the C–PC angle is wider for cis-7 (103.4(1)°) reflecting the steric demand of the carbazole substituent on the Mes* position. The most pronounced effect in the different isomers is observed for the orientation of the carbazole moiety and its torsion with respect to the CPDT core. The smaller torsion angle between the two chromophores’ planes in trans-7 (8.74°) gives rise to efficient delocalisation, while steric interaction of the large Mes* with the carbazole unit leads to an increased torsion angle (21.61°) in cis-7, with the N-ethyl group pointing in the same and opposite direction of the PC bond, respectively.
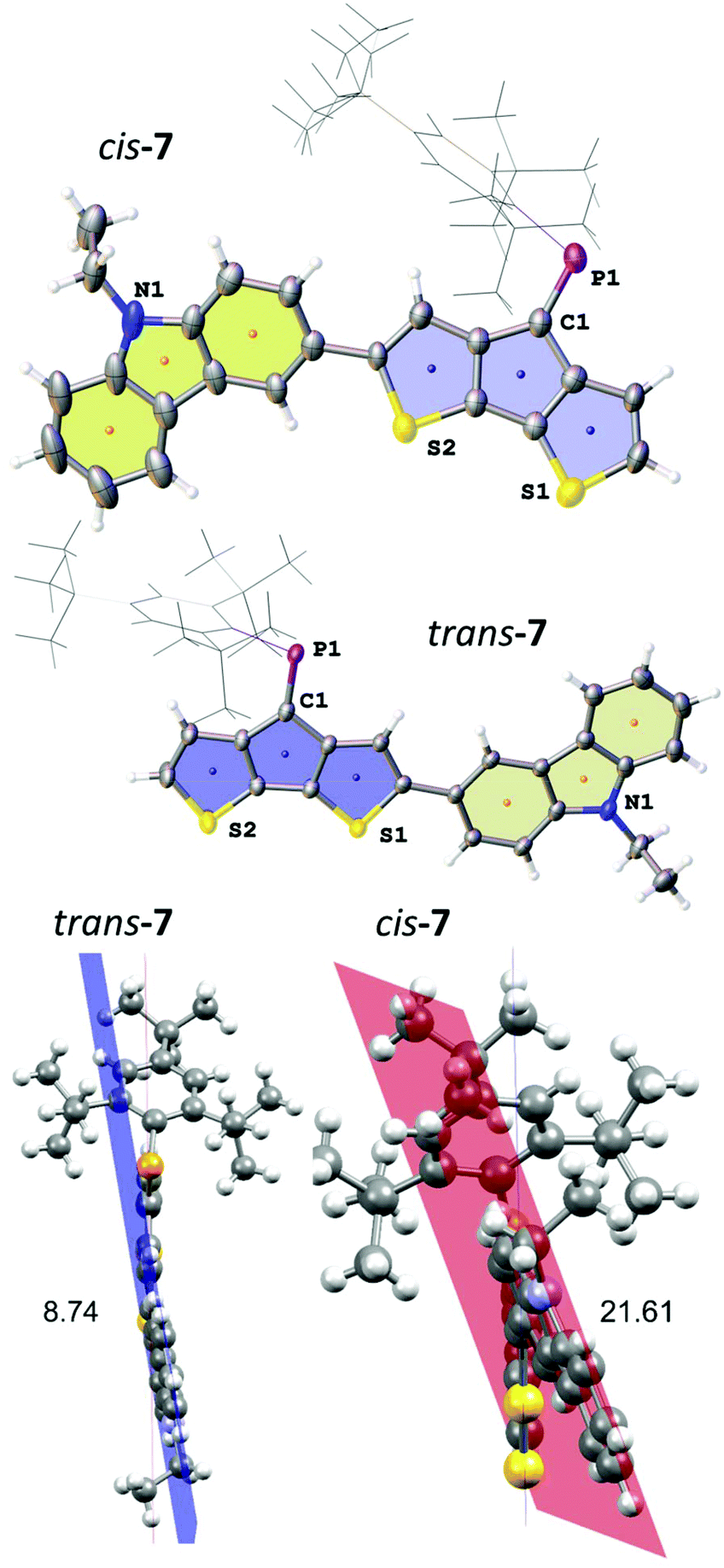 | ||
| Fig. 3 Top: solid-state structures of cis- and trans-7. Bottom: planes through the CPDT and carbazole rings and the torsion angles between both planes (bottom). Selected bond lengths [Å] and angles [°] cis-7: P1–C1 1.683(4), P1–C24 1.835(4), C9–C10 1.478(5) C1–P1–C24 103.38(16). trans-7: P1–C1 1.683(3), P1–C24 1.845(2), C9–10 1.467(3), C1–P1–C24 102.59(11). | ||
The previously reported gold(I) complex [AuCl·A]27 is only moderately stable under ambient conditions and slowly decomposes in the presence of water (Scheme 2). We report the solid-state structure of [AuCl·A] together with one of the decomposition products (8) using single crystal X-ray diffraction analysis (Scheme 2). Complex [AuCl·A] shows fairly short PC and P–Au bonds lengths of 1.668(7) and 2.213(2) Å, respectively. On the other hand, the C10–P1–C1 angle is fairly large indicating an increase of s-character of the coordinating lone pair. Notably, the almost linear P–AuCl fragment (178.16(8)°) is strongly twisted out of the CPDT plane (angle between the CPDT least squares plane (l.s.pl.) and the Cl–P vector of 24.15(3)°). In the solid-state structure of decomposition product 8, the tautomeric hydroxyphosphine and phosphine oxide of AuCl·A form a hydrogen bonded quasi-dimer where the two tautomers act as a hydrogen bond donor and a hydrogen bond acceptor, respectively. The colourless crystals solved in the space group P (no. 2). The gold coordination is barely affected by the hydrolysis of the PC bond with typical P–Au and Au–Cl distances of 2.232(1) and 2.298(1) Å, respectively. The phosphorus oxygen distances are in the typical range of P–O single (1.578(3) Å) and PO double bonds (1.489(3) Å). The hydrogen bond donor⋯acceptor distance is 2.550(4) Å and the O–H⋯O angle 164° indicating strong hydrogen bonding.31,32 These findings are in line with previous observations of changing the tautomeric forms upon coordination33–36 or shifting the equilibrium towards the hydroxyphosphine tautomer using electron deficient P-substituents.37,38
 | ||
| Scheme 2 Left: metal coordination of the CDPT-phosphaalkene. The previously reported complex [AuCl·A] slowly decomposed hydrolytically with partial metal loss giving the hydrogen bonded monometallic complex 8. Formation of unstable cationic complex 9 by adding slight excess of AgPF6 to a DCM solution of A. More stable derivatives are obtained by adding 2,6-dimethylphenyl isocyanide giving 10, or using two equivalents of A giving homoleptic complex 11. Coordination to palladium proceeds less cleanly giving the mononuclear complex 12 as crystallographically identified product. Reactions in THF and different stoichiometries let us assume that alternative products such as 12′ could be present in solution. Right: solid-state-structures of [AuCl·A] (top) and product of its partial hydrolysis (8, bottom) forming the metal coordinated hydroxyphosphine hydrogen bonded to the demetallated phosphine oxide subunit. Selected bond lengths [Å] and angles [°]. [AuCl·A]: P1–C1 1.668(7), P1–C10 1.800(6), P1–Au1 2.2130(15), C1–P1–C1 108.3(3), P1–Au1–Cl1 178.16(8). 8: P1–O1 1.489(3), P2–O2 1.578(3), P2–Au1 2.232(1), Au1–Cl1 2.298(1). O1⋯H–O2 164, O1⋯O2 2.550(4). | ||
Complex [AuCl·A] can also be converted into a cationic gold(I) species 9 upon reaction with AgPF6 characterised by a broad shielded 31P-NMR resonance at ca. 172 ppm. However, the low coordinate gold(I) species decomposes. Overnight, trace amounts of water and/or reactivity with the PF6− counter ion give a cationic phosphenium species (based on the deshielded resonances around 320 ppm in the 31P-NMR spectra), which is followed by further decomposition to untraceable products and formation of a clearly visible gold mirror. Noteworthy, during this process the counter ion decomposes to trifluorophosphine oxide (see ESI‡). The cationic intermediate can be stabilised using 2,6-dimethylphenyl isocyanide39 yielding the heteroleptic complex 10, which is characterised by a very broad 31P-NMR resonance at 203.8 ppm (FWHM = 316 Hz) and a sharp septet for the counter ion at −143.9 ppm. Similarly, the reaction of two equivalents of A with [AuCl(tht)] in the presence of AgPF6 gives the moderately stable homoleptic complex 11 with a broad 31P-NMR resonance at 197.9 ppm (FWHM ca. 125 Hz). The 31P-NMR resonances of the cationic gold complexes 10 and 11 are shifted to higher frequencies compared to the gold(I) chloride complex [AuCl·A] (δ31P = 175.6 ppm) indicating reduced electron density. The broadening of the 31P-NMR resonances can be tentatively attributed to a dynamic behaviour in solution and is commonly observed for phosphaalkene coinage metal complexes.40,41 Interestingly, the deshielded β proton changes from 8.05 ppm in [AuCl·A] to 7.58 and 7.67 ppm in 10 and 11, respectively, illustrating the different electrostatic situations proximal to the Au(I)+ centre, whereas the shielded β′ proton is barely influenced. The low-energy absorption band in the UV/vis spectrum of the gold complexes is red-shifted by ca. 30 nm upon formation of 9. This band is detected as a broad feature after coordination of the isocyanide (10) and barely visible in the homoleptic complex 11. Based on previous calculations this band is assigned to a HOMO (πCPDT) to LUMO (π*heterofulvene+metal) transition of very low probability.27
Addition of 0.5 equivalents of [PdCl2(MeCN)2] to a dichloromethane solution of A immediately results in the formation of a dark green-brown solution. The 31P NMR spectrum of the reaction mixture contains a major peak at 194 ppm which can be ascribed to [PdCl2(A)2] (12). Additionally, a broad peak can be observed at 162 ppm as well as unreacted A. We attribute these observations to the dynamic dissociation of 12 in solution to a monosubstituted palladium complex [PdCl2(A)L] (12′; L = solvent) and free A. The species corresponding to the peak at 162 ppm becomes the major product when excessive amounts of [PdCl2(MeCN)2] are used, supporting the idea that the A:Pd ratio is closer to 1:1 in this species. Formation multi-/di-nuclear complexes with bridging chloride ligands (12′; L = μ-ClPdCl·A) is also possible (ESI‡). The homoleptic complex 12 crystallises as dark green needles from dichloromethane in the centrosymmetric space group P with half a molecule in the asymmetric unit (Fig. 4). The observed Pd–Cl and Pd–P distances are 2.2902(8) and 2.3088(8) Å, respectively which form a regular square planar palladium environment (P–Pd–Cl 91.64(3)°). The resulting trans-complex shows an anti-arrangement of the ligand A minimising the steric interaction of the bulky Mes* substituents. Quite interestingly, the PC bond is only slightly elongated to 1.674(3) Å upon metal coordination, while the C–PC angle is significantly wider (106.8(2)°), quite similar to that of the gold complex [AuCl·A].
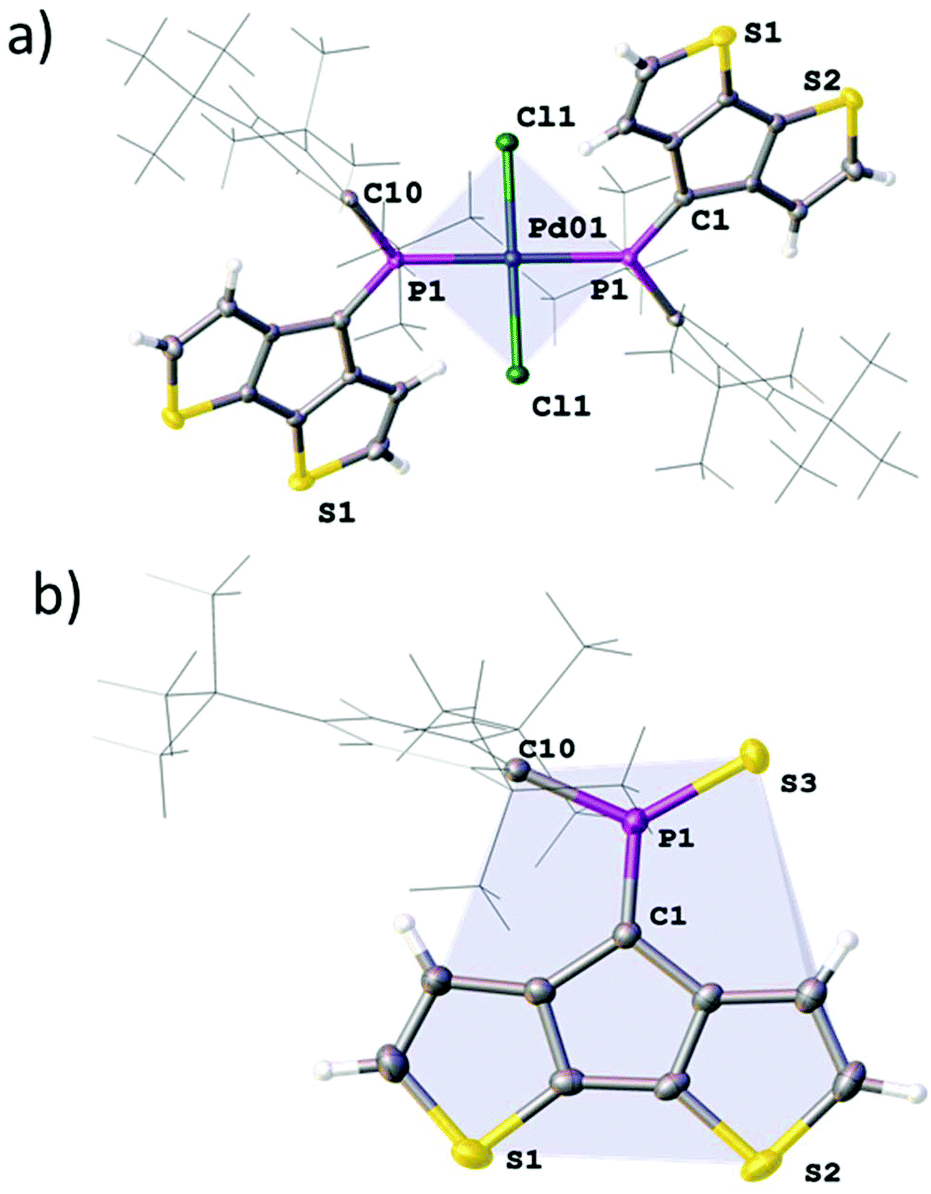 | ||
| Fig. 4 ORTEP plot (50% probability ellipsoids) of the solid-state structure of palladium complex 12 (a) and thiaphosphorane 13 (b). (a) Light blue: least squares plane of the square planar Pd-coordination environment including (Pd1, P1, Cl1). Selected bond lengths [Å] and angles [°] 13: Cl1–Pd01 2.2902(8), P1–Pd01 2.3088(8) P1–Pd01–Cl01 91.64(3). P1–C1 1.674(3), C1–P1–C(Mes*) 106.8(2). Light blue: least squares plane of the CPDT-PCS subunit. Selected bond lengths [Å] and angles [°] 15: P1–C1 1.669(4), P1–S3 1.917(1), C1–P1–C10 111.0(2), C1–P1–S3 124.2(1). | ||
A significantly less explored route to functionalise the phosphaalkene bonds is its oxidation with elemental chalcogens (e.g. O3, S8, Segrey), the reactivity of which decreases down the group.42–44 While methyleneoxophosphoranes are reported to be very unstable, methylenethia- and seleno-phosphoranes have been spectroscopically characterised.24 The oxidation products of Mes*PC(TMS)2 are all isolable and partially stable in air.42 Importantly, the oxidation of the PC moiety extends the acceptor orbitals including the chalcogen atoms, effectively lowering the LUMO levels (vide infra), resulting in a dramatic change of the electronic situation. The reaction of A with excess S8 in benzene or chloroform did not afford the anticipated methylenethiaphosphorane even after refluxing for 12 hours (Scheme 3). Similarly, A does not react with grey selenium in refluxing chloroform or dichloromethane. Interestingly, some reports describe the reaction in neat triethylamine or use small amounts of a non-nucleophilic base to promote the desired oxidation. Addition of diazabicycloundecene (DBU, 2% in the respective solvent), results in immediate consumption of the starting material in cases of sulfur, and initiated the reaction of grey selenium. The latter gives a new species (15), with a 31P-NMR resonance of 144.2 ppm and the typical 77Se satellites. The coupling constant of 915.7 Hz is indicative of very high s-character in the P–Se bond compared to e.g. phosphine selenides, which typically have coupling constants of around 750 Hz. Previous work by Appel and co-workers already hinted at this situation with a large 1JPSe of 890 Hz. Some examples of selenoiminophosphirenes are currently still the representatives with the largest P–Se coupling constants (960 Hz).42
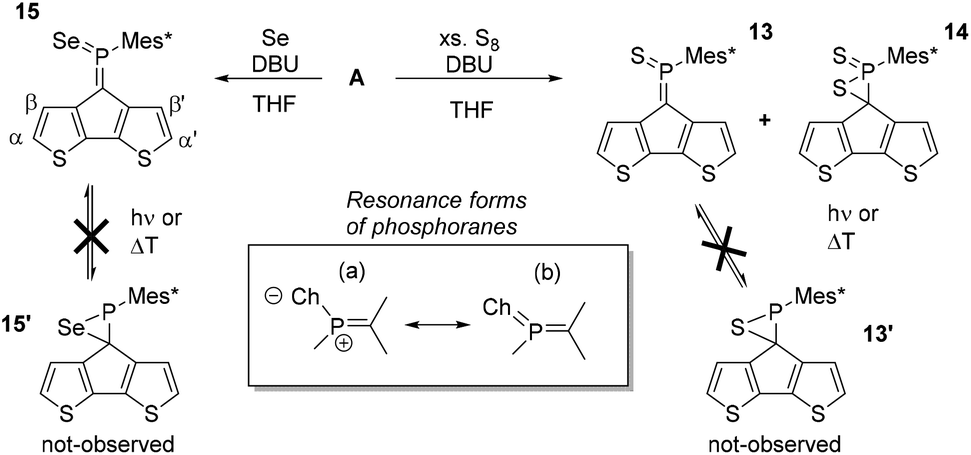 | ||
| Scheme 3 Top: functionalisation of the CPDT phosphaalkene using S8 and Segrey as oxidants. Thiaphosphorane 13 and selenophosphorane 15 could be isolated using column chromatography as ambiently stable compounds. Rearrangement of the thiaphosphorane into the thiaphosphirane was not observed under various conditions. Bottom: relevant resonance structures of the phosphoranes. The compound with a 31P NMR resonance of −1.26 ppm was isolated by column chromatographic work-up. 2D NMR analysis indicates a P–C(CPDT) single bond, and the 31P-NMR resonance agrees with the formation of 14. | ||
In case of the sulfur reaction, the crude reaction mixture presents three new resonances in the 31P-NMR spectra at around 160, 80, and 0 ppm. The latter signal is tentatively assigned to the di-oxidised species 14 based on 2D-NMR data (see ESI‡) and literature references.45,46 Chromatographic workup of the crude mixture on silica gel (DCM/pentane) allowed isolation of the methylenethiaphosphorane (13) with a characteristic 31P-NMR resonance at 157.2 ppm. The more deshielded 31P-NMR signal for 13 compared to 15 agrees with the slightly higher electronegativity of sulfur, favouring an ionic resonance structure (a, Scheme 3). The β′ protons are again strongly shielded (13: 4.48 and 7: 4.53 ppm) suggesting similar orientation of the Mes* group. The close proximity to the chalcogen atoms has also a strong impact on the β-proton resonances shifting them to higher frequencies (13: 7.88 and 15: 8.09 ppm). Yoshifuji and co-workers have described the thermal conversion of the methylenethiaphosphorane into the thiaphosphirane, however we were unable to trigger this conversion to give 13′.45,46
The formation of 13 was structurally confirmed by SC-XRD; 13 crystallises in the hexagonal space group P65 as dark red blocks. The CPDT core displays the expected co-planarity with the exocyclic unsaturated CPS fragment. The PC (1.669(4) Å) and PS (1.917(1) Å) bonds are in the typical range of strong double bonds. The angle of the PC unit towards the Mes* and S are 111.0(2) and 124.2(1); the former being much wider compared to the parent compound as well as the metallated derivatives 12 and [AuCl·A]. The thiaphosphorane unit is coplanar with the CPDT core, efficiently extending the π-conjugated framework. The NBO analysis carried out on these thia- and selenophosphoranes support the strong contribution of the ionic resonance forms in Scheme 3(a) having strong P(δ+)Ch(δ−) character and no d-orbital contributions from phosphorus in agreement with the common views of hypervalency in p-block compounds. The significantly larger polarisation in the thiaphosphorane compared to the selenophosphorane is evidenced by the relatively higher negative NBO charge at sulfur (−0.52) compared to Se (−0.45). (vide infra and ESI‡).
In order to elucidate the impact of the modifications of the parent CPDT phosphaalkene A we performed a detailed theoretical study including the newly synthesised and further in silico derivatives. For all calculated systems the Mes* substituent has been replaced by the electronically similar, but computationally simpler mesityl (Mes) substituent (Fig. 5). We have focused on the electronic impact of the derivatisations in the ground state and its consequences for the excited states. The structures are optimised in their ground state using a continuum solvation model for DCM at the DFT cam-B3LYP/6-311G** level of theory. At least 30 singlet and triplet transitions are calculated using time-dependent DFT (TD-DFT). As previously noted27 the charge transfer character complicates computation of the transition energies, thus we scaled the low-energy transitions to the experimentally observed absorption maximum of A with respect to the calculated value of (I).
 | ||
| Fig. 5 Left: depiction of all theoretically studied systems using the simplified mesityl (1,3,5-trimethylphenyl) phosphorus protecting group at the TD-DFT B3LYP/6-311G** level of theory using the continuum solvation model for DCM. Right: summarised results of the optical and electronic data of the studied systems. The calculated lowest energy absorption energy/wavelength (grey number) is scaled with respect to the experimental wavelength for A (red numbers). The theoretical Eg (cross) is depicted on the secondary axis (right). HOMO and LUMO energy levels are provided in eV. | ||
Substitution of the core with electron withdrawing halogen substituents (II, III, trans-/cis-IV, and XVIII) has a notable stabilising influence on the LUMO energies compared to the parent model compound (I). No significant effects on the HOMO levels have been noted, with the exception of fluoride which also results in the lowest energy absorption. Introducing a silyl group (cis- and trans-V) leads to a destabilisation of both HOMO and LUMO levels, however with no net effect on the absorption energies (or Eg = HOMO–LUMO gap). The dibromo derivative 2 shows a slightly milder reduction potential compared to the parent compound, which is in line with the calculated LUMO levels of I and XVIII. Noteworthy, the introduction of these substituents has almost no influence on the extinction coefficients of the low energy transitions. Both, alkene (VI) and imine (VII) derivatives lead to an increased transition probability (by a factor of three to five), however only the imine (VII) substituent also causes a pronounced red-shift. Similar effects are obtained using diphenyl (VIII), and triphenylamine (IX–XI) substitutions. The electron richer ditolyl(phenyl)amine derivative (X) is most red-shifted in this series. Separating the donor and acceptor moieties by another phenyl linker has no further effects on the orbital shapes and transition energies.
Interestingly, the N-linked carbazole derivative (XV) displays a blue shifted low-probability absorption (0.04), while the 2-linked N-methyl carbazole (XVI) shows red-shifted absorption and increased transition probabilities (0.08). This is explained by the different donor sites within the seemingly similar derivatives. The former has a CPDT localised HOMO, while the latter has a significantly extended HOMO stretching over the carbazole and the CPDT core leading to a pronounced charge-transfer character (Fig. 6). In order to further support these findings, electrochemical analyses of isomeric 7 were carried out. The CPDT-phosphaalkene core gives rise to a reversible reduction at E1/2 = −1.806 V (vs. Fc/Fc+), which is shifted to more negative potentials compared to the parent compound A (E1/2 = −1.62 V). In contrast to A, we observe a reversible oxidation at 0.277 V and a quasi-reversible oxidation 0.650 V, followed by an irreversible oxidation around 1.08 V. The former two are assigned to the (quasi-)reversible oxidation of the carbazole moiety, while the latter is attributed to the irreversible oxidation of the CPDT-phosphaalkene core, similar to that observed in A (+0.84 V). These experimental values are corroborated by the 0.4 eV lower LUMO levels of XVI compared to parent phosphaalkene I. The different isomers have almost identical optical properties, and their free energies are within less than 0.5 kcal mol−1 substantiating that different isomers can be present in solution.
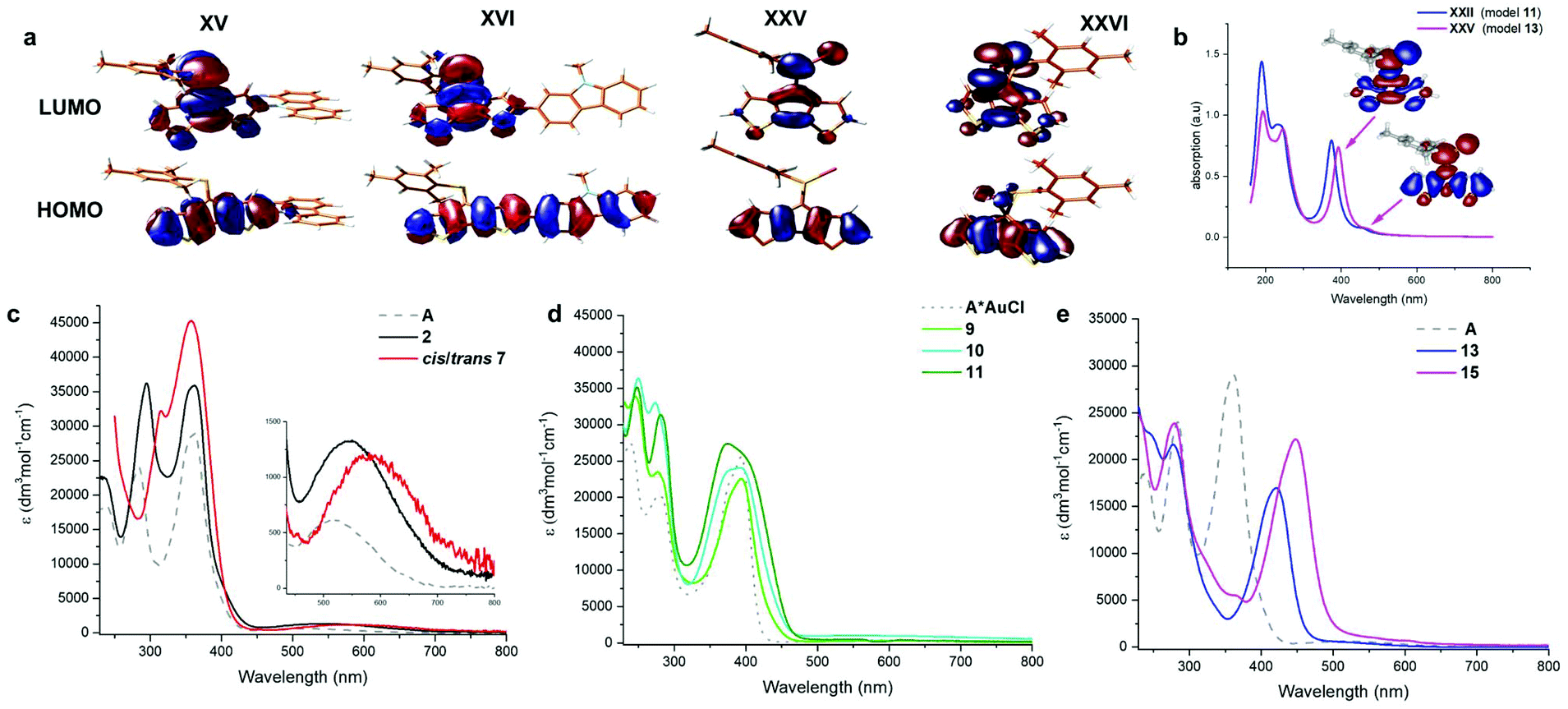 | ||
| Fig. 6 (a) Calculated Frontier molecular orbitals of selected derivatives. (b) calculated spectra of XXII and XXV (models of derivatives 13 and 15), including the EDDMs of the two lowest energy transitions of XXV. (C, d & e) optical spectra of compounds 2, 7, 9, 10, 11, 13, 15, and the literature reported A, [A*AuCl]. Solvent: DCM. Due to the facile decomposition of these cationic complexes the extinction coefficients are only approximated. | ||
Quite surprising is the small impact of replacing the thiophene for selenophene or furan rings, i.e. modifying the core of the donor unit, only slightly shifting the absorption energies and not affecting the transition probabilities. A moderate impact could be achieved combining core modification with fluorine-substitution at the α-position (XXvs.XXI).
We also studied the chemical functionalisation at the P-centre (and the double bond) using sulfur and selenium oxidations.
The thiaphosphirane (XXIII) is less stable (ΔG = +1.4 kcal mol−1), than the methylenethiaphosphorane (XXII); similarly, the selenophosphirane (XXV) is less table (ΔG = +2.9 kcal mol−1) than the isomeric methyleneselenophosphorane (XXVI). The frontier molecular orbitals of the methylenephosphoranes are quite similar to those of unsubstituted A (and e.g. Au(I) coordinated derivatives) with CPDT localised bonding π orbitals and fulvenoid based anti-bonding orbitals that involve the PS (or Se) moiety. The electronic properties of these two systems (13 and 15) have also been probed by means of electrochemical analysis showing clear differences but also a number of similarities. The oxidative side shows two irreversible processes at slightly milder potentials for the sulfur-derivative 13 compared to the selenophosphorane. (13: 0.671 and 0.905 V; 15: 0.707 and 1.012 V). The reductive side is however markedly different. In the case of 13 a mild reversible reduction (E1/2 = −1.585 V) is followed by an irreversible reduction (−1.944 V). In contrast, 15 shows an irreversible reduction at −1.658 V, followed by a chemical step (reoxidation at −0.640 V). This initial EC process is followed by a quasi-reversible reduction at (E1/2 = −1.956 V). The small difference in the oxidative process is in line with the HOMO, being mainly localised on the “organic core”, while the different reductive behaviour agrees with the localisation of the LUMO on the heterofulvenoid + chalcogen framework. Model compounds of the radical anions (for S: XXII˙− and Se: XXV˙−) have been investigated showing only small differences in spin and charge localisation. The negative charges on the chalcogen atoms are very similar (S: −0.82, Se: −0.78), however the significantly larger radius of Se implies less nucleophilic character. On the other hand, the spin density at the P atom is higher for XXII˙− (0.30) compared to XXV˙− (0.27). In the latter case, spin density is increased at the chalcogen and phosphaalkene carbon centres (XXV˙−: Se 0.14 and CP 0.31 vs.XXII˙− S: 0.13 and CP: 0.28), which might indicate a more facile rearrangement to the three-membered ring structure of the Se-derivative compared to its lighter S-analogue.
The FMOs of the phosphirane have significant bonding and antibonding contributions from the three-membered ring as well as the CPDT core. The calculated S0 → S1 transitions are slightly blue-shifted for the phosphoranes (XXII: 457 (496) nm and XXV: 470 (510) nm) compared to the parent motif (I: 476 (517) nm), and dramatically blue shifted for the corresponding phosphiranes (XXIII: 332(360) nm and XXVI: 361(392) nm). These calculations also support the observed inability to trigger conversion of phosphoranes 13 and 15 in the isomeric phosphiranes 13′ and 15′ by illumination or at elevated temperatures.
Re-investigation of the parent phosphaalkenes A's electro-chemistry revealed that oxidative cycling gives rise to two new reversible redox features. These are accompanied with a gradual shift of the main oxidation peak to milder potential. Based on our previous work we assign this behaviour to an oxidative electro-polymerisation based on the CPDT core (Fig. 7). Detailed investigations of the films for organic electronics applications are currently under way.
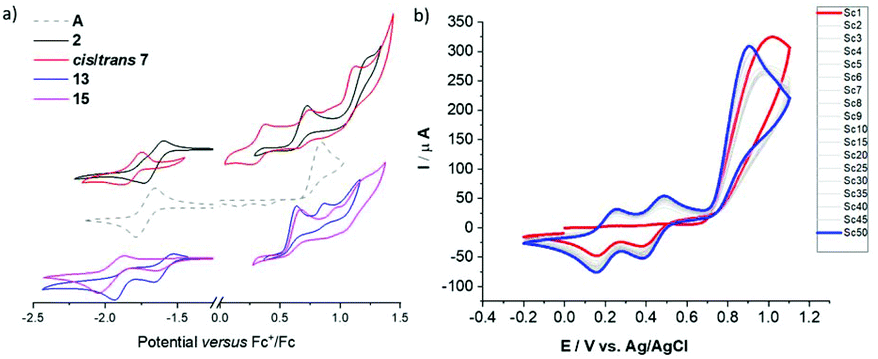 | ||
| Fig. 7 (a) Electrochemical analysis of 2, 7, 13, and 15 in comparison with parent compound A. Oxidative and reductive scans at a scan rate of 100 mV s−1. Three electrode setup with glassy carbon working electrode (WE), Pt coil counter electrode (CE), Ag wire reference electrode; DCM/Bu4NPF6 (0.1 M) solution; referenced to the Fc+/Fc couple. The voltammograms are offset and scaled. (b) Electropolymerisation of A (1 mM) by repetitive oxidative cycling (−0.2 V to +1.1 V) at a scan rate of 100 mV s−1. First (red trace) and 50th (blue trace) are highlighted indicating the increased current due to material deposition at the WE. | ||
Conclusions
In this manuscript, we have experimentally and theoretically illustrated that the cyclopentadithiophene-phosphaalkene core (A) is an interesting building block for optically and electronically tuneable materials. This scaffold offers a large variety of possible functionalisation including the organic core as well as the exocyclic phosphaalkene unit. Oxidation of the P-centre with chalcogens and introduction of a donor substituent at the α,α′-position are most promising. Theoretical studies support these findings and provide further examples of suitable functionalisations. Our current efforts are directed towards using these systems in opto-electronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Swedish research council (Vetenskapsrådet), the Carl-Trygger foundation, Lars-Hiertas minne, and the Olle-Engkvist foundation for their continuous support. The authors would also like to extend their gratitude to Dr Faye L. Cruikshank (University of Edinburgh) for collecting mass spectrometry data and analysis support during the ongoing COVID-19 pandemic.Notes and references
- A. Arjona-Esteban, M. R. Lenze, K. Meerholz and F. Würthner, in Elementary Processes in Organic Photovoltaics, ed. K. Leo, Springer International Publishing, Cham, 2017, pp. 193–214 Search PubMed.
- P. Coppo and M. L. Turner, Cyclopentadithiophene based electroactive materials, J. Mater. Chem., 2005, 15, 1123–1133 RSC.
- M. Horie, L. A. Majewski, M. J. Fearn, C.-Y. Yu, Y. Luo, A. Song, B. R. Saunders and M. L. Turner, Cyclopentadithiophene based polymers-a comparison of optical, electrochemical and organic field-effect transistor characteristics, J. Mater. Chem., 2010, 20, 4347–4355 RSC.
- J. S. Reddy, T. Kale, G. Balaji, A. Chandrasekaran and S. Thayumanavan, Cyclopentadithiophene-Based Organic Semiconductors: Effect of Fluorinated Substituents on Electrochemical and Charge Transport Properties, J. Phys. Chem. Lett., 2011, 2, 648–654 CrossRef CAS.
- S. Wanwong, A. Poe, G. Balaji and S. Thayumanavan, The effect of heteroatom conformation on optoelectronic properties of cyclopentadithiophene derivatives, Org. Biomol. Chem., 2014, 12, 2474–2478 RSC.
- J. D. Azoulay, Z. A. Koretz, B. M. Wong and G. C. Bazan, Bridgehead Imine Substituted Cyclopentadithiophene Derivatives: An Effective Strategy for Band Gap Control in Donor–Acceptor Polymers, Macromolecules, 2013, 46, 1337–1342 CrossRef CAS.
- R. Hoffmann, Building Bridges Between Inorganic and Organic Chemistry (Nobel Lecture), Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
- R. Appel, F. Knoll and I. Ruppert, Phospha–alkenes and Phospha–alkynes, Genesis and Properties of the (p–p)π–Multiple Bond, Angew. Chem., Int. Ed. Engl., 1981, 20, 731–744 CrossRef.
- R. Pietschnig and A. Orthaber, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2016 Search PubMed.
- M. A. Shameem and A. Orthaber, Organophosphorus Compounds in Organic Electronics, Chem. – Eur. J., 2016, 22, 10718–10735 CrossRef CAS.
- F. Vidal and F. Jäkle, Functional Polymeric Materials Based on Main-Group Elements, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CAS.
- J. P. Green and A. Orthaber, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, 2019, DOI:10.1002/9781119951438.eibc2716.
- L. Chen, B. W. Rawe, K. Adachi and D. P. Gates, Phosphorus-Containing Block Copolymers from the Sequential Living Anionic Copolymerization of a Phosphaalkene with Methyl Methacrylate, Chem. – Eur. J., 2018, 24, 18012–18019 CrossRef CAS.
- K. Dück, B. W. Rawe, M. R. Scott and D. P. Gates, Polymerization of 1-Phosphaisoprene: Synthesis and Characterization of a Chemically Functional Phosphorus Version of Natural Rubber, Angew. Chem., Int. Ed., 2017, 56, 9507–9511 CrossRef.
- V. A. Wright and D. P. Gates, Poly(p-phenylenephosphaalkene): A π-conjugated macromolecule containing P=C bonds in the main chain, Angew. Chem., Int. Ed., 2002, 41, 2389–2392 CrossRef CAS.
- R. C. Smith and J. D. Protasiewicz, Conjugated Polymers Featuring Heavier Main Group Element Multiple Bonds: A Diphosphene-PPV, J. Am. Chem. Soc., 2004, 126, 2268–2269 CrossRef CAS.
- D. Morales Salazar, E. Mijangos, S. Pullen, M. Gao and A. Orthaber, Functional small-molecules & polymers containing P=C and As=C bonds as hybrid π-conjugated materials, Chem. Commun., 2017, 53, 1120–1123 RSC.
- S. Ikeda, F. Ohhata, M. Miyoshi, R. Tanaka, T. Minami, F. Ozawa and M. Yoshifuji, Synthesis and Reactions of Palladium and Platinum Complexes Bearing Diphosphinidenecyclobutene Ligands: A Thermally Stable Catalyst for Ethylene Polymerization, Angew. Chem., Int. Ed., 2000, 39, 4512–4513 CrossRef CAS.
- M. Freytag, S. Ito and M. Yoshifuji, Coordination Behavior of Sterically Protected Phosphaalkenes on the AuCl Moiety Leading to Catalytic 1,6-Enyne Cycloisomerization, Chem. – Asian J., 2006, 1, 693–700 CrossRef CAS.
- J. Dugal-Tessier, G. R. Dake and D. P. Gates, P,N-Chelate Complexes of Pd(II) and Pt(II) Based on a Phosphaalkene Motif: A Catalyst for the Overman−Claisen Rearrangement, Organometallics, 2007, 26, 6481–6486 CrossRef CAS.
- S. Ito, M. Freytag and M. Yoshifuji, Structural and coordination properties of 1,2-bis(cyclopropyl)-3,4-bis(2,4,6-tri-tert-butylphenyl)-3,4-diphosphinidenecyclobutene prepared by dehydrogenative homocoupling of 3-cyclopropyl-1-(2,4,6-tri-tert-butylphenyl)-1-phosphaallene, Dalton Trans., 2006, 710–713 CAS.
- E. Öberg, A. Orthaber, C. Lescop, R. Réau, M. Hissler and S. Ott, Phosphorus Centers of Different Hybridization in Phosphaalkene-Substituted Phospholes, Chem. – Eur. J., 2014, 20, 8421–8432 CrossRef.
- P. M. Petrar, G. Nemes, I. Silaghi-Dumitrescu, H. Ranaivonjatovo, H. Gornitzka and J. Escudié, 1,3-Digermacyclobutanes with exocyclic C=P and C=P=S double bonds, Chem. Commun., 2007, 40, 4149–4151 RSC.
- T. A. van der Knaap, M. Vos and F. Bickelhaupt, A new stable tricoordinate phosphorus(V) compound: P-2,6-dimethylphenyl-C,C-diphenylselenophosphene, J. Organomet. Chem., 1983, 244, 363–367 CrossRef CAS.
- H. Miyake, T. Sasamori and N. Tokitoh, Sulfurization of 4,5,6-Triphospha[3]Radialene, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1247–1250 CrossRef CAS.
- P. K. Majhi, K. C. F. Chow, T. H. H. Hsieh, E. G. Bowes, G. Schnakenburg, P. Kennepohl, R. Streubel and D. P. Gates, Even the normal is abnormal: N-heterocyclic carbene C2 binding to a phosphaalkene without breaking the P=C π-bond, Chem. Commun., 2016, 52, 998–1001 RSC.
- A. El Nahhas, M. A. Shameem, P. Chabera, J. Uhlig and A. Orthaber, Synthesis and Characterization of Cyclopentadithiophene Heterofulvenes: Design Tools for Light-Activated Processes, Chem. – Eur. J., 2017, 23, 5673–5677 CrossRef CAS.
- V. B. Gudimetla, A. L. Rheingold, J. L. Payton, H.-L. Peng, M. C. Simpson and J. D. Protasiewicz, Photochemical E- Z Isomerization of meta-Terphenyl-Protected Phosphaalkenes and Structural Characterizations, Inorg. Chem., 2006, 45, 4895–4901 CrossRef CAS.
- H.-L. Peng, J. L. Payton, J. D. Protasiewicz and M. C. Simpson, Twisting the Phenyls in Aryl Diphosphenes (Ar-P=P-Ar). Significant Impact upon Lowest Energy Excited States, J. Phys. Chem. A, 2009, 113, 7054–7063 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CAS.
- A. Orthaber, J. H. Albering, F. Belaj and R. Pietschnig, P-C bond formation via P-H addition of a fluoroaryl phosphinic acid to ketones, J. Fluorine Chem., 2010, 131, 1025–1031 CrossRef CAS.
- A. S. Ionkin, W. J. Marshall, B. M. Fish, M. F. Schiffhauer and C. N. McEwen, A stable enol in small ring systems: clear differentiation between penta- and tri-valency of phosphorus atoms, Chem. Commun., 2008, 5432–5434 RSC.
- S. B. Clendenning, P. B. Hitchcock and J. F. Nixon, First η1-ligated 2,4,6-tri-tert-butyl-1,3,5-triphosphabenzene complexes and the remarkable trihydration reaction of trans-[PtCl2(PMe3)(P3C3But3)] to cis-[PtCl(PMe3)(P3O3C3H5But3)], containing the novel CH(But)PH(O)C(But)PH(O)CH(But)P(O) ring system, Chem. Commun., 1999, 1377–1378 RSC.
- A. G. Ginzburg, V. V. Bashilov, F. M. Dolgushin, A. F. Smol'yakov, P. V. Petrovskii and V. I. Sokolov, Change of the coordination type for the phospholyl ligand under nucleophilic attack of H2O on phosphorus atom in 2,5-diphenylphosphacymantrene promoted by aliphatic amines, Inorg. Chim. Acta, 2011, 370, 292–296 CrossRef CAS.
- M. Doux, N. Mézailles, L. Ricard and P. Le Floch, Group 10 Metal Complexes of SPS-Based Pincer Ligands: Syntheses, X-ray Structures, and DFT Calculations, Eur. J. Inorg. Chem., 2003, 2003, 3878–3894 CrossRef.
- J. Francos, D. Elorriaga, P. Crochet and V. Cadierno, The chemistry of Group 8 metal complexes with phosphinous acids and related POH ligands, Coord. Chem. Rev., 2019, 387, 199–234 CrossRef CAS.
- J. Bader, R. J. F. Berger, H.-G. Stammler, N. W. Mitzel and B. Hoge, First Solid State Structures of Real Diorganyl Phosphinous Acids R2POH (R=CF3, C2F5), Chem. – Eur. J., 2011, 17, 13420–13423 CrossRef CAS.
- H. Tavakol and F. Keshavarzipour, The Substituent Effect on the Energy Profiles of Inter and Intramolecular Proton Transfers between Phosphine Oxide-Phosphinous Acid Tautomers, Heteroat. Chem., 2016, 27, 210–220 CrossRef CAS.
- V. P. Dyadchenko, N. M. Belov, M. A. Dyadchenko, Y. L. Slovokhotov, A. M. Banaru and D. A. Lemenovskii, A complex of gold(I) benzenethiolate with isocyanide: synthesis and crystal and molecular structures, Russ. Chem. Bull., 2010, 59, 539–543 CrossRef CAS.
- A. Orthaber, H. Löfås, E. Öberg, A. Grigoriev, A. Wallner, S. H. M. Jafri, M.-P. Santoni, R. Ahuja, K. Leifer, H. Ottosson and S. Ott, Cooperative Gold Nanoparticle Stabilization by Acetylenic Phosphaalkenes, Angew. Chem., Int. Ed., 2015, 54, 10634–10638 CrossRef CAS.
- S. T. Clausing, D. Morales Salazar and A. Orthaber, Preparation, Photo- and Electrochemical Studies of a Homoleptic Imine-Phosphaalkene Cu(I) Complex, Inorg. Chim. Acta, 2020 DOI:10.1016/j.ica.2020.119958.
- R. Appel and C. Casser, Reaktionen des 2,4,6-tri-tert.-butylphenyl[bis(trimethylsilyl)methylen]phosphans, Tetrahedron Lett., 1984, 25, 4109–4112 CrossRef CAS.
- K. V. Turcheniuk, A. B. Rozhenko and I. V. Shevchenko, Synthesis and Some Chemical Properties of a 1,2λ3σ3-Thiaphosphirane, Eur. J. Inorg. Chem., 2011, 1762–1767 CAS.
- B. Hoge, J. Bader, H. Beckers, Y. S. Kim, R. Eujen, H. Willner and N. Ignatiev, The Bis(pentafluoroethyl)phosphinous Acid (C2F5)2POH, Chem. – Eur. J., 2009, 15, 3567–3576 CrossRef CAS.
- K. Toyota, K. Shimura, H. Takahashi and M. Yoshifuji, Preparation and X-Ray Structure Analysis of 3,3-Diphenyl-2-(2,4,6-tri-t-butylphenyl)-1,2-thiaphosphirane, Chem. Lett., 1994, 23, 1927–1930 CrossRef.
- K. Toyota, H. Takahashi, K. Shimura and M. Yoshifuji, Valence Isomerism between Sterically Protected Methylenephosphine P-Sulfide and 1,2-Thiaphosphirane, Bull. Chem. Soc. Jpn., 1996, 69, 141–145 CAS.
Footnotes |
| † This paper is dedicated to all people whose background, ethnicity, sexuality, or gender identity are underrepresented STEM subjects, and who suffer from the systemic flaws preventing them from flourishing. |
| ‡ Electronic supplementary information (ESI) available: Experimental, crystallographic, and theoretical details. CCDC 2003575–2003582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi00714e |
| § These authors contributed equally |
| ¶ Present address: Chemical Biology and Therapeutics, Lund University, Sölveg 19, BMC D10, 221 84, Lund, Sweden |
| This journal is © the Partner Organisations 2020 |