
Porous organic polymers: a promising platform for efficient photocatalysis
Ting
Zhang
,
Guolong
Xing
,
Weiben
Chen
and
Long
Chen
 *
*
Department of Chemistry, Institute of Molecular Plus, Tianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University, Tianjin 300072, China. E-mail: long.chen@tju.edu.cn
First published on 19th October 2019
Abstract
Porous organic polymers (POPs) are a class of multi-dimensional porous network materials, which are built via strong covalent linkages between various organic building blocks with different geometries and topologies, and have recently become a rising research field in porous materials. POPs can be generally divided into two categories based on their degree of long-range order, including amorphous (e.g., CMPs, HCPs, PIMs, and PAFs) and crystalline (e.g., COFs). Owning to their advantages of light-weight, superior inherent porosity, excellent stability, pre-designable and tunable structures and functions, POPs have received increasing attention and research interest for their tremendous potential applications in gas storage/separation, heterogeneous catalysis, photoelectric conversion, chemical- and bio-sensing, energy storage and conversion, etc. Their porous structure, pore size, specific surface areas and functions can be directly designed and facilely tuned by introducing specific functional building blocks. In this review article, we summarize the latest representative advances in the field of POPs, focusing on their design and synthetic strategies, with emphasis on their specific applications in photocatalysis, including photocatalytic chemical transformation, photodetoxification of contaminants from water, and water splitting.
1. Introduction
The limited resources and rapid consumption of fossil fuels have caused severe problems for human beings, including energy shortage, greenhouse effects, and environmental pollution. Thus, many efforts have been devoted to pursuing alternative clean and sustainable energy and technology to address these challenges. Among them, visible-light driven photocatalytic technology, which directly converts solar energy to chemical energy, is very promising and can effectively catalyze chemical transformations,1–5 removal of contaminants from water,6–9 water splitting for hydrogen evolution,10–18etc. Therefore, developing efficient photocatalysts with high catalytic activity is of great importance. The past few decades have witnessed burgeoning development in photocatalysts, mainly focusing on inorganic semiconductors (e.g., TiO2,19–21 CdSe,22 WO3,23,24 ZnS,25 and ZnO26–28), metal complexes (e.g., ruthenium and iridium complexes29–31) and organic dyes (e.g., fluorescein32,33 and rose bengal34,35). However, the toxicity and high cost of rare metal components and the intrinsic drawback of not being recyclable limit the practical applications of homogenous photocatalysts. Furthermore, many of them are only active upon UV-light irradiation, and thus are not conducive to the full use of the sunlight. Porous organic polymers (POPs)36–39 feature large pores, high specific surface areas, good stability and robustness in various solvents, which are favorable for mass transfer, providing more active sites, and the recycling process in photocatalysis. Thus, POPs have emerged as a promising platform for efficient photocatalysis.POPs can be classified into either amorphous or crystalline. Amorphous POPs mainly include hyper-crosslinked polymers (HCPs),40–42 polymers of intrinsic microporosity (PIMs),43–45 conjugated microporous polymers (CMPs)46–48 and porous aromatic frameworks (PAFs),49,50 while crystalline POPs are represented by covalent organic frameworks (COFs).51–53 CMPs feature extended π-conjugated skeletons in comparison to other amorphous POPs, endowing them with attractive light-harvesting capability, which is advantageous for photocatalysis. On the other hand, COFs possessing periodic, ordered and extended skeletons with uniform nanopores enable the precise integration of various functional building blocks. Since the seminal work reported by O. M. Yaghi et al.,51 numerous COFs with different topologies have been extensively explored in gas storage54–56 and separation,57 catalysis,58,59 and energy storage.60,61 By integrating organic semiconductor building blocks with photoelectric properties into the COF skeleton, the functions of the organic semiconductor can be retained or even amplified.62,63 Thus, COFs synthesized via the bottom-up strategy are usually photocatalytically active and can be applied for photocatalysis. Another special class of COFs, covalent triazine frameworks (CTFs), feature triazine rings, which are usually formed by trimerization of aromatic nitriles.53 However, compared to COFs with imine,64,65 boroxine or boronate ester,66,67 and azine linkages,68–70 the development of CTFs is hindered by the harsh synthetic conditions71 (e.g. high temperature of over 400 °C) and irreversible formation of aromatic 1,3,5-triazine rings. Thus, most reported CTFs have low crystallinity but excellent chemical stability and a high nitrogen content, making them attractive for photocatalysis applications. Recently, B. Tan et al. had great success in developing a low temperature polycondensation approach to construct CTFs under mild conditions, and further obtained CTFs with high crystallinity either via the in situ oxidation of alcohols to aldehyde monomers or controlling the monomer feeding rate.72–74 In addition, both amorphous CMPs and crystalline COFs exhibit remarkably tunable photocatalytic activity by proper molecular design. In this review, the applications of photocatalytic CMP and COFs in chemical transformations, removal of contaminants from water, and hydrogen evolution via water splitting are briefly summarized.
2. Principle
In general, as displayed in Scheme 1, when POPs absorb photons with energy equal or higher than their band gaps, the electrons at the valence bands (VB) are excited to the conduction bands (CB) and simultaneously leave holes at the VB, resulting in excited electrons and holes in the CB and VB, respectively.14 Subsequently, they transfer to the surface of the photocatalyst for redox reaction.14b A series of reactive species including singlet oxygen (1O2), superoxide radical (O2˙−), hydroxyl radical (˙OH), hydrogen peroxide (H2O2), photogenerated hole (h+) and electron (e−) may participate in these photocatalytic processes. The possible reactive species are usually determined by control experiments with the addition of the corresponding scavenger.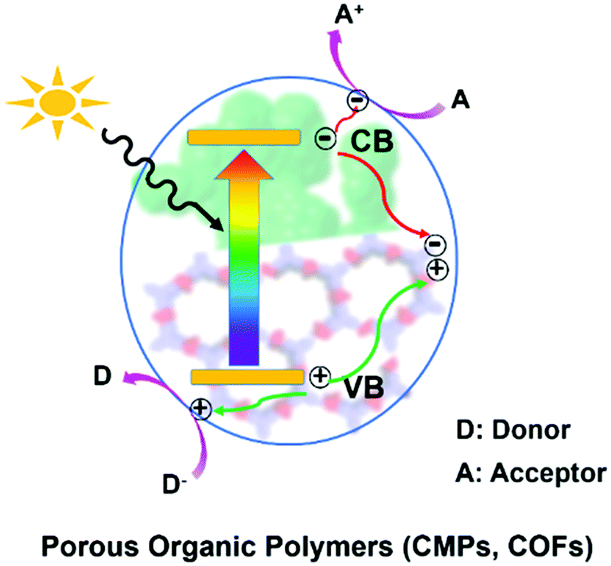 | ||
| Scheme 1 General illustration for the mechanism of photocatalysis with POPs. | ||
3. Visible-light-driven chemical transformation
The photocatalytic systems based on poly(benzothiadiazoles) and their derivatives have been recently reviewed by K. A. I. Zhang et al.75 As a strong electron-withdrawing unit, benzothiadiazole (BT) is often used for the construction of CMPs, and thus BT-based CMPs have become dominant in CMP-photocatalytic chemical transformation. Here, we critically summarize some representative CMPs and COFs, focusing on their recent progress in photocatalytic transformations. For easy understanding, we simply divide the reactions (or chemical transformations) into two types (i.e., oxidation reactions and other reactions) according to whether oxygen is involved.3.1 Photodriven oxidation
Two main reactive species (i.e., 1O2 and O2˙−) may be involved in photodriven oxidation in the presence of oxygen when POPs are used as photocatalysts. They are formed through the energy transfer and charge transfer pathway from the photocatalyst to oxygen.76 Sometimes 1O2 alone acts as an oxidant, while sometimes O2˙− is the main oxidant.F. Vilela et al. reported the first CMPs (CMP_X, where X indicates the amount of SiO2NPs in mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mL−1 used for templating, Fig. 1a) as sensitizers to produce 1O2 and further to photocatalyze the oxidation of α-terpinene to ascaridole.79 The introduction of a template greatly increased the Brunauer–Emmett–Teller (BET) surface areas and pore volumes. CMP_X possessed donor (D)–acceptor (A) built-in structures with benzothiadiazole (BT) as the strong electron acceptor and benzene as the weak electron donor, which were beneficial for preventing the fast recombination of excitons and conducive to the intersystem crossing process, thus favouring photosensitization. Among them, CMP_60 possessed the highest surface area. When it was dispersed in a solution (1 mg mL−1) containing α-terpinene (0.1 M) and the mixture was pumped at a flow rate of 1 mL min−1 through a photoreactor under blue light (420 nm) irradiation in the presence of oxygen (2 equiv.), the conversion of α-terpinene was greater than 90%, much higher than that with the other CMP_X. Even though benzothiadiazole itself can work as a homogeneous photosensitizer for oxidation reactions, the heterogeneous CMPs showed attractive properties of easy separation and recycling.
mL−1 used for templating, Fig. 1a) as sensitizers to produce 1O2 and further to photocatalyze the oxidation of α-terpinene to ascaridole.79 The introduction of a template greatly increased the Brunauer–Emmett–Teller (BET) surface areas and pore volumes. CMP_X possessed donor (D)–acceptor (A) built-in structures with benzothiadiazole (BT) as the strong electron acceptor and benzene as the weak electron donor, which were beneficial for preventing the fast recombination of excitons and conducive to the intersystem crossing process, thus favouring photosensitization. Among them, CMP_60 possessed the highest surface area. When it was dispersed in a solution (1 mg mL−1) containing α-terpinene (0.1 M) and the mixture was pumped at a flow rate of 1 mL min−1 through a photoreactor under blue light (420 nm) irradiation in the presence of oxygen (2 equiv.), the conversion of α-terpinene was greater than 90%, much higher than that with the other CMP_X. Even though benzothiadiazole itself can work as a homogeneous photosensitizer for oxidation reactions, the heterogeneous CMPs showed attractive properties of easy separation and recycling.
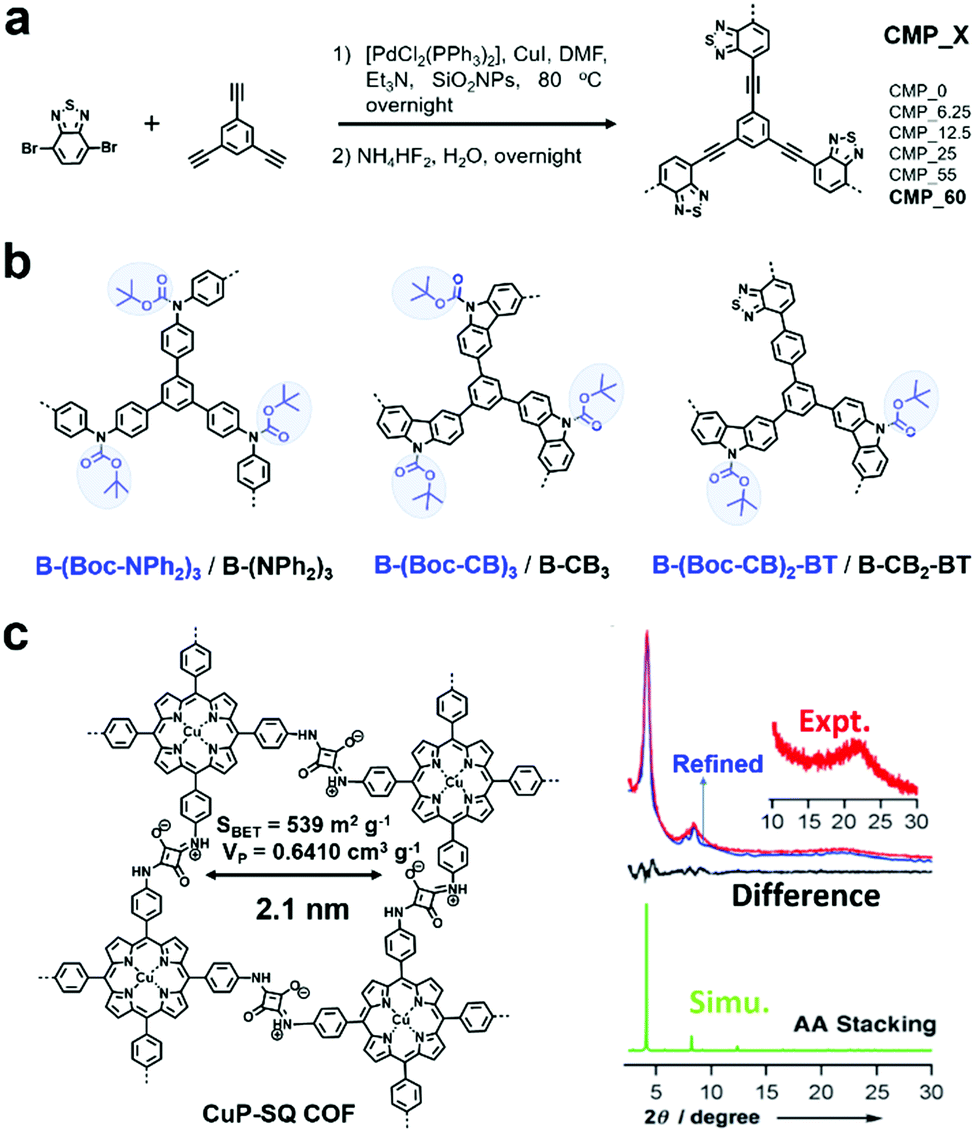 | ||
| Fig. 1 (a) Synthetic route for CMP_X. Modified from ref. 79 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2013. (b) Chemical structures of various poly(HIPE)s. Modified from ref. 82 with permission from the Royal Society of Chemistry, Copyright 2014. (c) Structure of CuP-SQ-COF and its PXRD patterns. Reproduced from ref. 83 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2013. | ||
Furthermore, by combining the high internal phase emulsion polymerization technique and micropore engineering by incorporating and removing a cleavable functional group (Boc) in the polymer network, CMPs (B-(NPh2)3, B-CB3, and B-CB2-BT) with increased surface area and porosity were reported (Fig. 1b).82 The obtained CMPs showed effective photocatalytic activity for the highly selective oxidation of organic sulfides under visible light irradiation (Fig. 2a). However, the CMPs with or without Boc moieties exhibited no obvious differences in photocatalytic activity, while the activities of B-(Boc-NPh2)3 and B-(NPh2)3 were much worse, which can be attributed to their higher HOMO–LUMO band gaps, especially higher LUMO levels. As displayed in Fig. 2b, various phenyl sulfides bearing electron-donating (–CH3, –OCH3) or electron-withdrawing (–F, –Br, –Cl) substituents could be selectively converted to the desired mono-oxidized sulfoxides. The oxidation of sulfides was achieved by the photochemically generated 1O2.
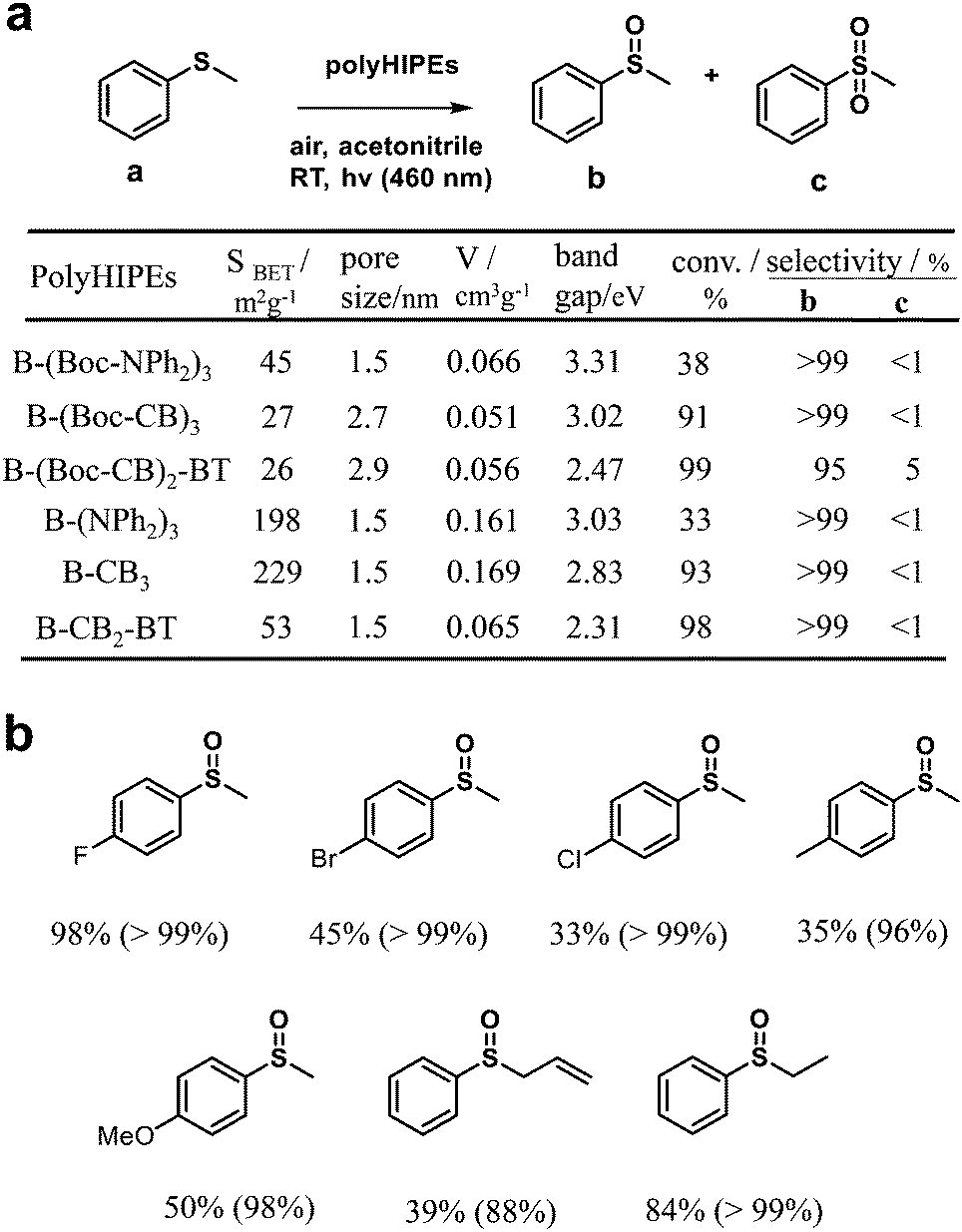 | ||
| Fig. 2 (a) Surface area, pore size characterization and optical and electronic properties of various poly(HIPE)s and their photocatalytic performance for the selective oxidation of thioanisole under visible light. (b) Photocatalytic selective oxidation of a range of sulfides by B-(Boc-CB)2-BT under visible light irradiation. The value out and in parentheses indicates the conversion and selectivity of sulfoxides, respectively. Modified from ref. 82 with permission from the Royal Society of Chemistry, Copyright 2014. | ||
Featuring extended skeletons with high crystallinity and porosity, COFs can also be used as photocatalysts. In 2013, D. Jiang et al. reported a squaraine-linked mesoporous COF (CuP-SQ COF, Fig. 1c) for singlet oxygen generation.83 Benefiting from the zigzagged zwitterionic resonance structure of squaraines, the CuP-SQ COF possessed a zigzag conformation, which protected the extended π-conjugated layers from sideslip, thus rendering it with high stability. Also, it exhibited robust solvent stability with retained crystallinity after harsh treatments. Considering its strong light-harvesting ability, low band gap and ordered structure, CuP-SQ COF was used as a heterogeneous photocatalyst. CuP-SQ COF could effectively harvest visible light and activate molecular oxygen to generate singlet oxygen, and subsequently catalyze the conversion of 1,3-diphenylisobenzofuran (DPBF) to o-dibenzoylbenzene (ODB). Its photocatalytic activity was much higher than that of monomeric CuP, demonstrating that the ordered structure was conducive to improve the catalytic activities.
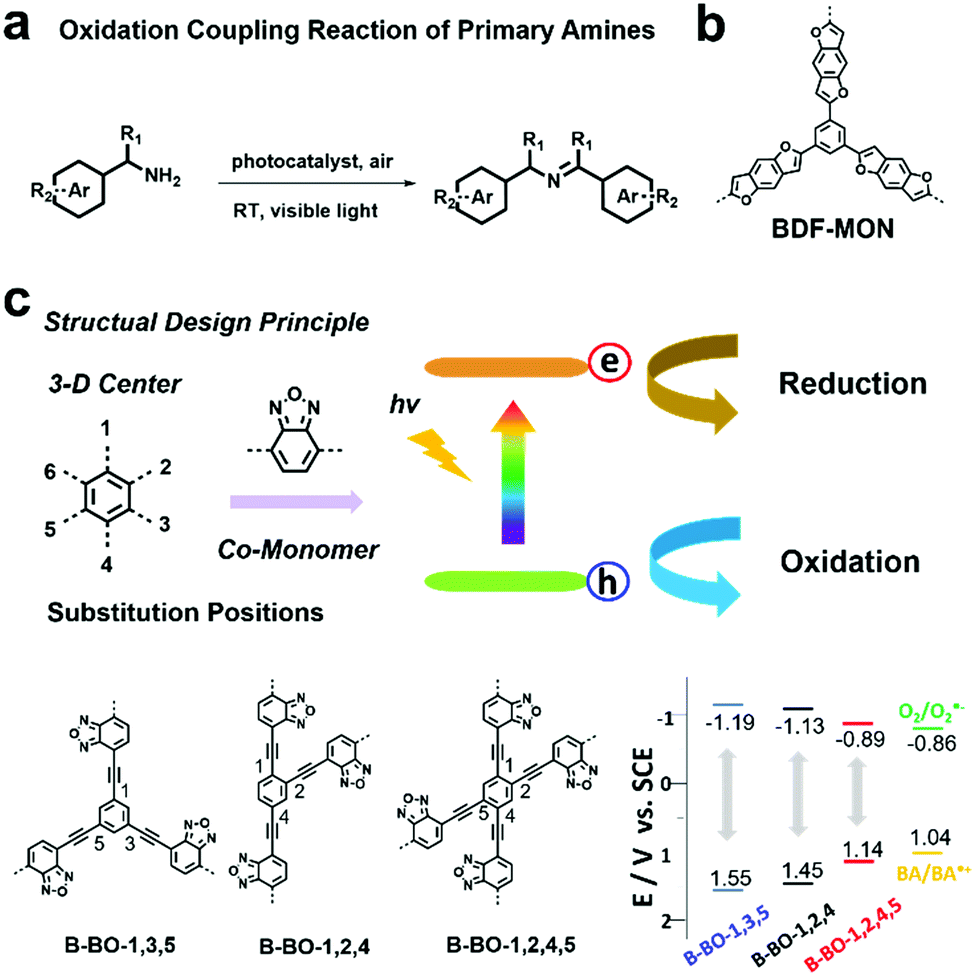 | ||
| Fig. 3 (a) Oxidation coupling reaction of primary amines using a photocatalyst. (b) Chemical structure of BDF-MON. Modified from ref. 93 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2013. (c) Structure design principle, building block structures and the VB and CB positions of B-BO-1,3,5, B-BO-1,2,4, B-BO-1,2,4,5 and redox potentials of benzylamine (BA) and O2 to O2˙−. Reproduced from ref. 94 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2015. | ||
Improving photocatalytic activity is one of the key issues to develop efficient photocatalysts. In addition to variation in the building blocks, the VB and CB position of POPs can also be tuned by modifying the connectivity of the same building blocks, thus leading to modulation of photocatalytic performances. K. A. I. Zhang et al. prepared a series of benzooxadiazole-based CMPs (B-BO-1,3,5 B-BO-1,2,4 and B-BO-1,2,4,5) with the same building blocks; however, the electron-donating benzooxadiazole moieties were connected at different positions on the centered phenyl unit (Fig. 3c).94 Among the three CMPs, B-BO-1,3,5 featuring a larger optical band gap and higher redox potential exhibited better photocatalytic activity than the other two CMPs (B-BO-1,2,4 and B-BO-1,2,4,5) for catalyzing the oxidation coupling reaction of benzylamine and also showed good catalytic efficiency even after 5 cycles.
Besides the oxidation of primary amide, Por-sp2c-COF reported by C. Wang et al. also exhibited excellent photocatalytic capacity towards the oxidation of secondary amine to imine (Fig. 4).62 Por-sp2c-COF featured sp2-carbon linked conjugated 2D skeletons, which are highly stable and exhibit excellent electrochemical and charge-transfer properties. Por-sp2c-COF was synthesized using 5,10,15,20-tetrakis(4-benzaldehyde)porphyrin (p-Por-CHO) and 1,4-phenylenediacetonitrile (PDAN) through Knoevenagel condensation, which was demonstrated before for preparation of COFs95–98 and CMPs.99 Due to the fully sp2-carbon-conjugated structure and high chemical stability, higher photocatalytic efficiency for the oxidation of secondary amine was achieved compared to its imine-based Por-COF counterpart. In addition, both the crystalline framework structure and photocatalytic activities of Por-sp2c-COF could be well retained after several reaction cycles.
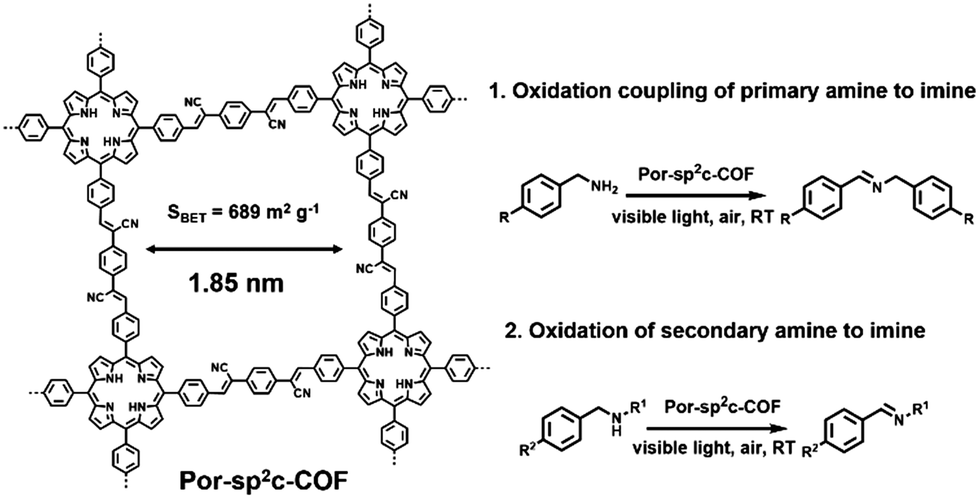 | ||
| Fig. 4 Structure of Por-sp2c-COF and photocatalytic oxidation coupling of primary amine to imine and oxidation of secondary amine to imine. Modified from ref. 62 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2019. | ||
The azo-Henry reaction is a typical oxidation coupling reaction to realize the C–H functionalization and is frequently used as a model reaction to evaluate the photocatalytic performance of CMPs or COFs (Fig. 5a).100–102 By virtue of the effective photocatalytic ability of rose bengal (RB) dye, a bottom-up strategy was adopted by A. I. Cooper et al. to prepare two CMPs (RB-CMP1 and RB-CMP2, Fig. 5b), which were further used for photocatalytic aza-Henry reaction.100 As an example, N-substituted tetrahydroisoquinoline and CH3NO2 were used as substrates. In the presence of air and a 60 W bulb as the light source, the reaction afforded full conversion in 12 h using only 5 mol% CMP catalyst with comparable activity to its homogeneous counterpart (RB). Additionally, the catalyst was effective for the aza-Henry reaction over a wide scope of substrates. The two CMP catalysts also retained good recyclability for at least 10 times.
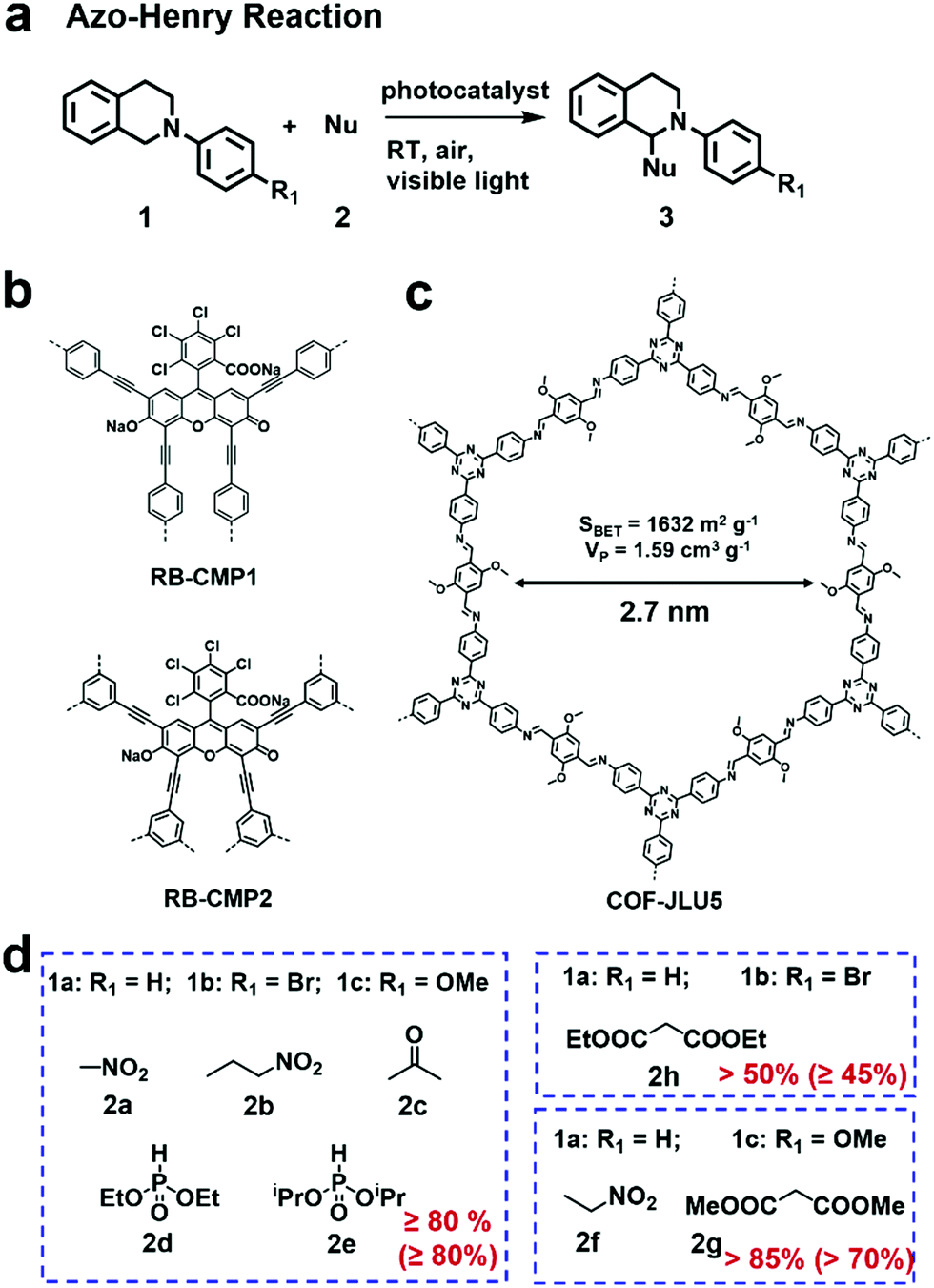 | ||
| Fig. 5 (a) Photocatalytic aza-Henry reaction. (b) Structures of RB-CMP1 and RB-CMP2. Modified from ref. 100 with permission from the American Chemical Society, Copyright 2013. (c) Structure of COF-JLU5. (d) Photocatalytic aza-Henry reaction of different substrates with various nucleophiles in high yields using COF-JLU5 as the photocatalyst. Modified from ref. 102 with permission from the Royal Society of Chemistry, Copyright 2017. | ||
Some 2D COFs featuring excellent porosity, periodic columnar π-arrays and high crystallinity have also exhibited great potential applications for the oxidative azo-Henry reaction.101,102 X. Liu et al. reported a triazine-based COF (COF-JLU5, Fig. 5c) showcasing high photocatalytic activities for the aza-Henry reaction.102 COF-JLU5 exhibited a high BET surface area (1632 m2 g−1) and outstanding chemical stability. The built-in D–A skeleton facilitated the charge separation of photo-generated electron–hole pairs. Notably, various substrates bearing different substituents were subjected to the aza-Henry reaction and gave the desired products in high yields (Fig. 5d). Interestingly, besides the formation of the C–C bond, C–P bond formation could also be catalyzed when phosphonates were used as the nucleophilic reagents. The photocatalytic performances of COF-JLU5 was superior to most heterogeneous catalysts ever reported, and even comparable to the state-of-the-art homogeneous photocatalysts. Furthermore, it could be reused for at least 10 cycles without any obvious deterioration in its catalytic activities.
As mentioned above, the D–A structure is favorable to construct photocatalysts with good performances. However, for a D–A-type CMP, its energy levels can be further fine-tuned by simply regulating the content of electron-withdrawing building blocks during the copolymerization process. In 2016, a series of CMPs (P-BBT-0, P-BBT-10, P-BBT-50 and P-BBT-100, Fig. 6a) incorporating different amounts of the electron-withdrawing moiety benzobisthiadiazole (BBT) was systematically investigated.103 With an increase in the content of BBT, the BET surface area and pore volume of the CMPs increased, while the corresponding reduction and oxidation potentials as well as the band gaps greatly decreased. Among the four POPs, P-BBT-10 exhibited the highest photocatalytic ability to generate O2˙− due to its high redox potentials and suitable band gap for visible light absorption, which was further used for the synthesis of tetrahydroquinoline via the oxidative cyclization of N-methylanilines and maleimide (Fig. 6b). As expected, the oxidative cyclization catalyzed by P-BBT-10 led to the highest yield of 82%. Furthermore, P-BBT-10 exhibited good substrate tolerance and excellent recyclability after 5 cycles.
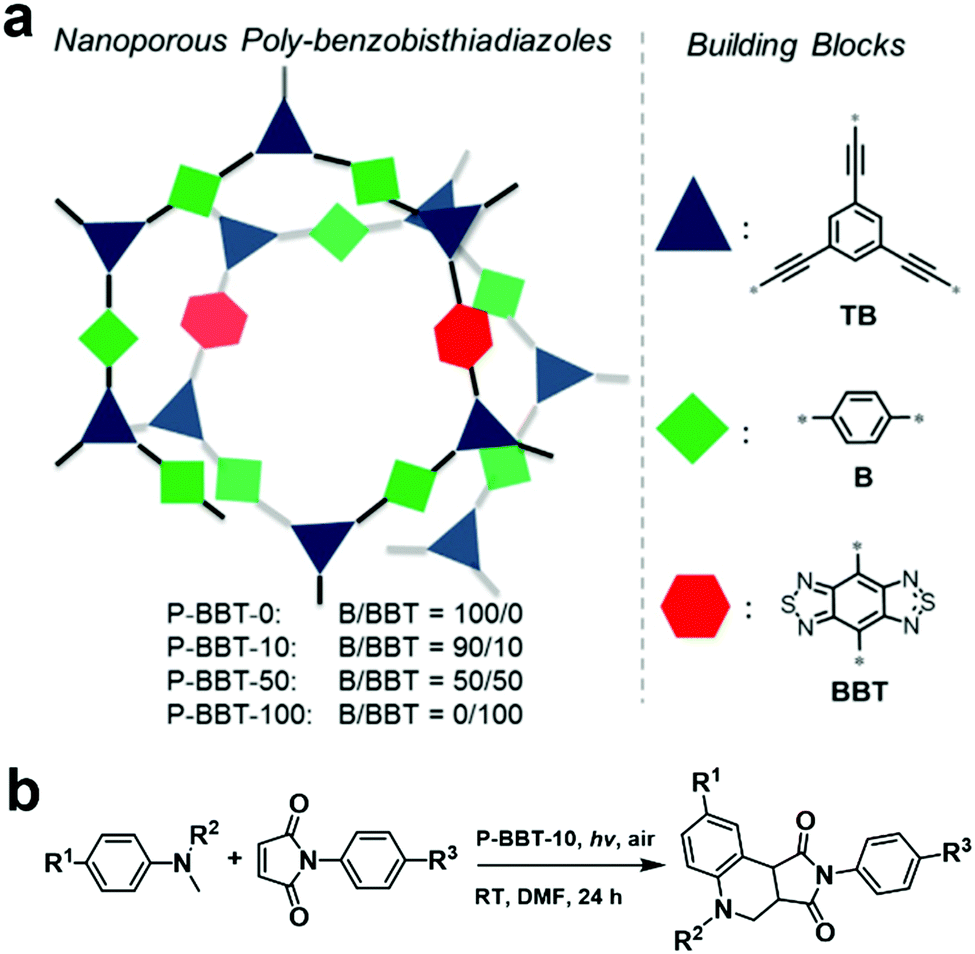 | ||
| Fig. 6 (a) Structure design principle of P-BBT-0, P-BBT-10, P-BBT-50, and P-BBT-100 and (b) photocatalytic oxidative cyclization of N-methylanilines and maleimide using P-BBT-10. Reproduced from ref. 103 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2016. | ||
3-Substituted indoles are important medical intermediates.35,104–107 Formylation and thiocyanation are two typical methods for the C-3 functionalization of indoles. G. Yu et al. reported three carbazole-based CMPs (CMP-CSU5, CMP-CSU6, and CMP-CSU7) and their use as photocatalysts for the C-3 functionalization of indoles (Fig. 7).108 It was noteworthy that the effect of the electronic properties of the cores (i.e., 1,4-dibenzyl, 1,3,5-tribenzyl, or 1,3,5-triazin-2,4,6-triyl) on the photocatalytic activities was investigated, which received little attention in previous works. Their reduction potentials were only slightly negative enough to reduce oxygen to generate O2˙−, while their oxidation potentials were quite different. CMP-CSU6 possessing the highest oxidation potential was the most effective in the photocatalytic reactions. Under the optimized reaction conditions, CMP-CSU6 was able to catalyze the C-3 formylation and thiocyanation of a series of indoles in high yields. The catalytic activity of CMP-CSU6 was comparable or even better than the well-established catalysts such as rose bengal, I2 for the formylation of N-methylindole, and Ru(bpy)3Cl2 and Ir(ppy)3 for the thiocyanation of 1H-indole. In addition, CMP-CSU6 could function at room temperature without the aid of acid, base or strong oxidant and showed good recyclability for at least 5 continuous cycles.
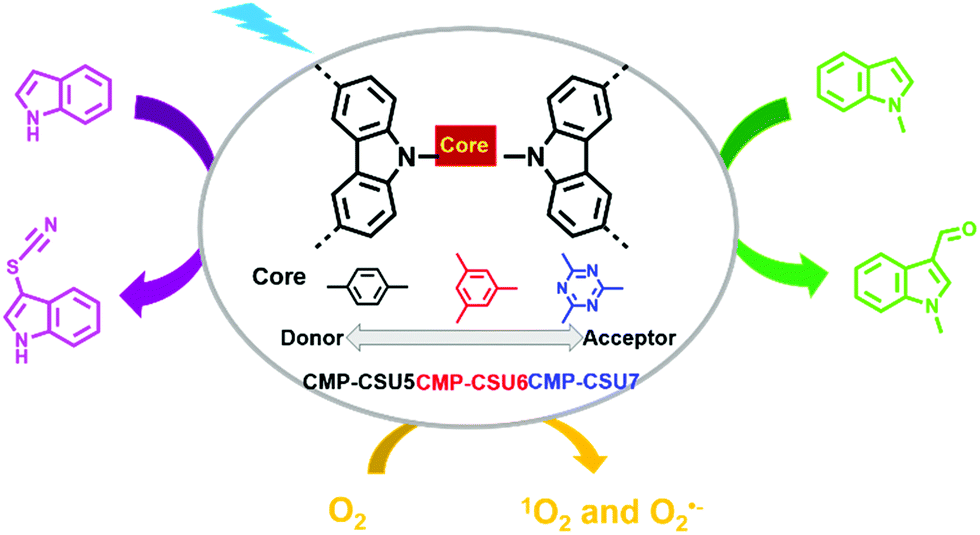 | ||
| Fig. 7 Structures of CMP-CSU5, CMP-CSU6, CMP-CSU7 and photocatalytic C-3 functionalization of indoles. Modified from ref. 108 with permission from the American Chemical Society, Copyright 2018. | ||
Even though some outstanding works have shown great prospects for the application of COFs in visible-light-driven photocatalysis, they still face the severe problem of relatively poor photo-stability and reusability, which limit their practical applications.101 Hence, it is attractive to design robust COFs for photocatalysis. Instead of applying the classical reversible reaction to fabricate COF, three benzoxazole-linked ultrastable COFs (i.e., LZU-190, LZU-191, and LZU-192) were prepared by utilizing reversible/irreversible cascade reactions to form a benzoxazole ring (Fig. 8).63 All three COFs showed great thermal and chemical stability and also exhibited excellent photostability under continuous visible light irradiation (20 W white LED). Among the three COFs, LZU-190 could catalyze the aerobic reaction of 4-carboxyphenylboronic acid to 4-hydroxybenzoic acid with a high yield of 99% when i-Pr2NEt was used as the sacrificial agent (86% in the absence of i-Pr2NEt) and presented no obvious changes in crystallinity, morphology, porosity and photocatalytic activity even after 20 runs. This outstanding work not only presented a prototypical COF with extraordinary performance, but also inspired us to design and synthesize more robust photocatalytic COFs using irreversible condensation reactions.
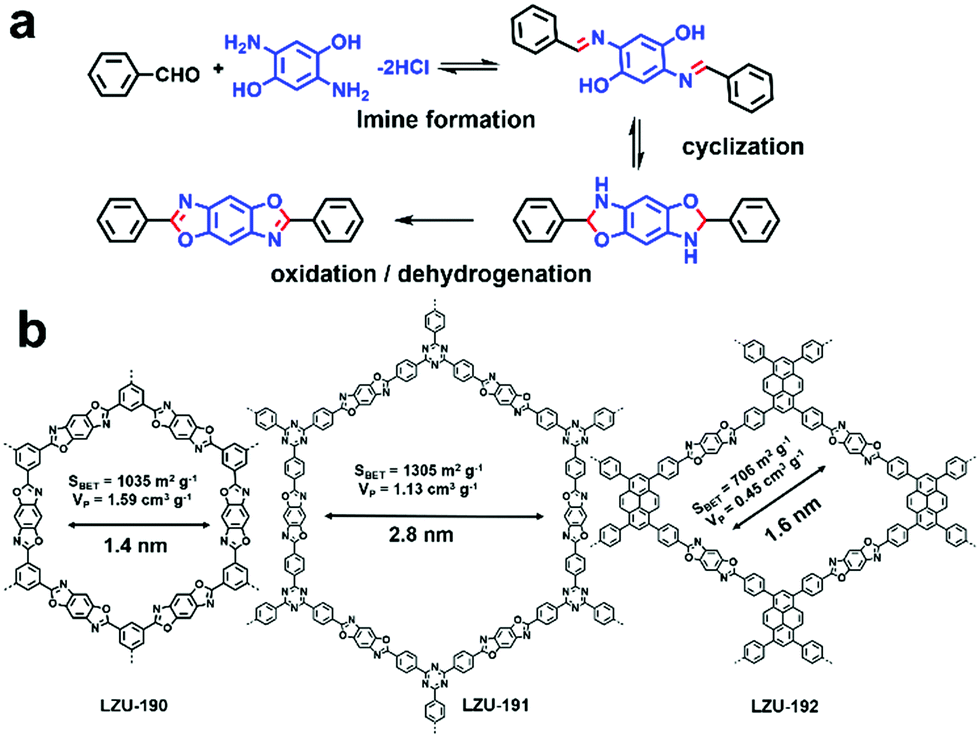 | ||
| Fig. 8 (a) Design concept to construct ultra-stable COFs: LZU-190, LZU-191, and LZU-192. (b) Chemical structures of LZU-190, LZU-191, LZU-192. Modified from ref. 63 with permission from the American Chemical Society, Copyright 2018. | ||
3.2 Other reactions photocatalyzed by POPs
Some chemical transformations can also be photocatalyzed by POPs without the aid of oxygen, which are mostly reduction reactions. For example, organic bromides represent a type of important intermediate, but may cause serious pollution and are harmful to humans.109 Reductive dehalogenation of organic bromides is an effective way to detoxify organic bromides.110 Using the high internal phase polymerization strategy, two conjugated porous polymers, B2-FL3 and B2-FL2-BBT (Fig. 9a) were prepared and further used for the photoreductive dehalogenation of various α-bromoacetophenone derivatives bearing substituents with varying electronic properties in high conversion efficiencies.111 With the electron-withdrawing benzobisthiadiazole (BBT), B2-FL2-BBT exhibited higher photocatalytic activity. The proposed reaction mechanism is shown in Fig. 9b. The photogenerated hole was firstly captured by amine (DIPEA) to form a radical cation. Then the photogenerated electron reacted with α-bromoacetophenone to form phenone radicals with cleaving of the Br leaving group. Later, the Hantzsch ester donated a hydrogen radical to the phenone radical to afford the final product. Meanwhile, pyridine ester and ammonium bromide were obtained as side products, which originated from the reaction of the dehydrogenated Hantzsch ester radical and the DIPEA radical cation. Another imine-based D–A type COF (COF-JLU22) reported by X. Liu et al. was also utilized for the photoreductive dehalogenation of phenacyl bromide.112 The D–A structure rendered the COF-JLU22 with red-shifted absorption and also enhanced photocatalytic activity. Meanwhile, its high crystallinity was well-retained after continuous green light (30 W) irradiation, indicating its high photochemical stability. In view of the above advantages, COF-JLU22 efficiently photocatalyzed the reductive dehalogenation of phenacyl bromide in a high yield of 85% in DMF and many α-bromoacetophenones were able to convert to the corresponding acetophenone in high yields by utilizing the optimized conditions (Fig. 9c). In addition, COF-JLU22 could also effectively photocatalyze the enantioselective α-alkylation of aldehydes (Fig. 9c). Also, in both reactions, COF-JLU22 exhibited high tolerance to a large number of substrates, excellent stabilities and recycling performances.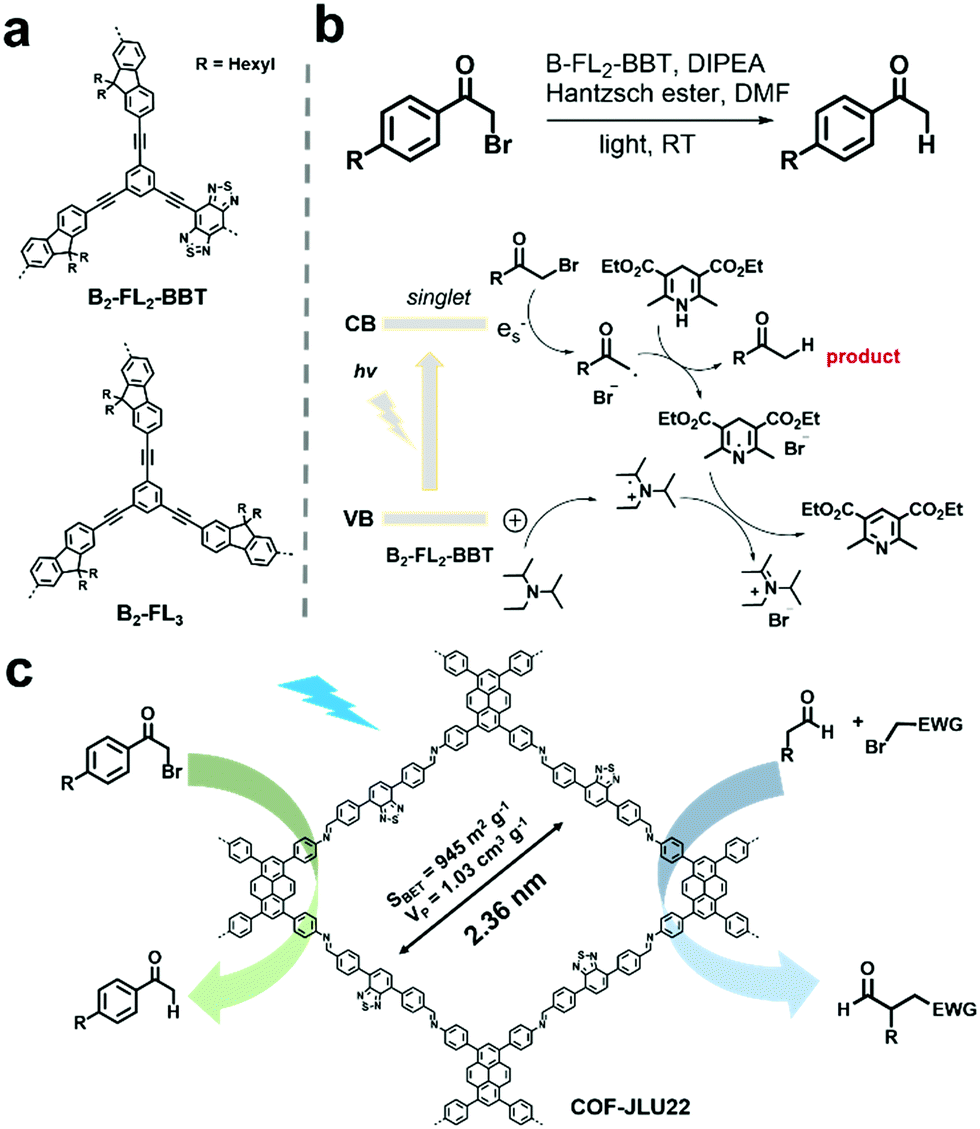 | ||
| Fig. 9 (a) Chemical structures of B2-FL2-BBT and B2-FL3. (b) Proposed mechanism of the dehalogenation reaction under visible light irradiation using B2-FL2-BBT as the photocatalyst. Modified from ref. 111 with permission from the Royal Society of Chemistry, Copyright 2014. (c) Structure of COF-JLU22 and photocatalytic photoreductive dehalogenation reaction of phenacyl bromide and the enantioselective α-alkylation of aldehydes. Modified from ref. 112 with permission from Elsevier, Copyright 2019. | ||
Photopolymerization is an efficient way to convert small monomers to high molecular weight polymers, which are widely used as adhesives, coatings, etc.113 Numerous photoinitiators have been developed for photopolymerization, but most of them are UV-light-triggered photoinitiators.114,115 In 2014, a porous polymer (B-(Boc-CB)2-BO) obtained via high internal phase emulsion (HIPE) polymerization, which was used as a visible light photoinitiator for the free radical polymerization of methyl methacrylate (MMA).116 Benefiting from the π-conjugated structure as well as the strong electron acceptor BO, which is beneficial for the efficient separation of excitons, the obtained B-(Boc-CB)2-BO exhibited high photocatalytic activity under visible light. The free polymerization of MMA reached as high as 82% conversion when B-(BOC-CB)2-BO was employed as the photoinitiator.
A fully conjugated 2D sp2-carbon COF (TP-COF) was recently reported by Z. Li et al., which served as an artificial photosystem I for the synthesis of L-glutamate via the photocatalysis of NADH regeneration (Fig. 10).117a TP-COF features a band gap of 2.36 eV and a high reduction potential of 3.23 eV. Upon irradiation with sunlight, TP-COF is excited to generate excited electron/hole pairs. Subsequently, NAD+ is reduced to NADH with the help of an electron mediator. Also, NADH could further initiate the chemical transformation of α-ketoglutarate to L-glutamate with a yield up to 97% within 12 min, which is among the best performances to date in this field.
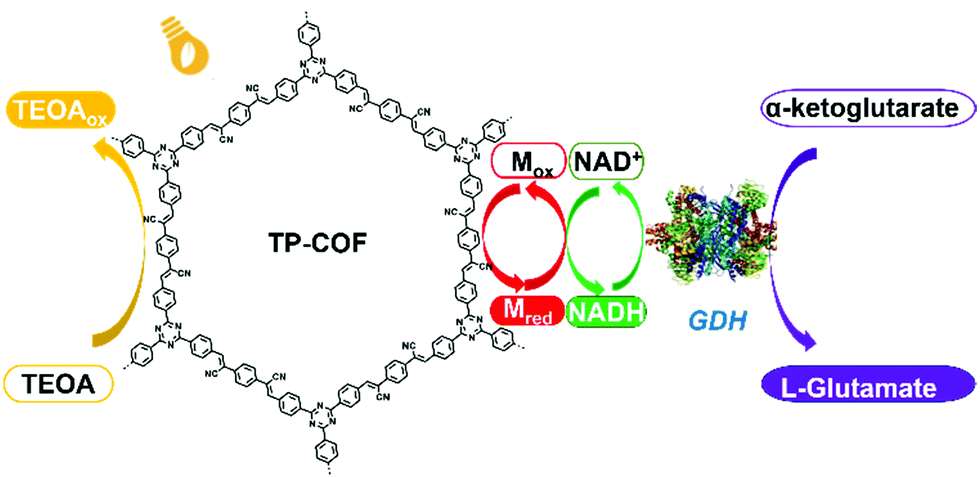 | ||
| Fig. 10 Schematic illustration of the artificial photosystem I using TP-COF. Reproduced from ref. 117a with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2019. | ||
The reduction of CO2 with H2O to value-added chemicals and renewable fuels utilizing sunlight is an appealing approach to solve the energy shortage and global warming problems caused by the emitted CO2 from daily life.117b Ideal photocatalysts to promote this conversion should possess strong CO2 adsorption capacity together with high catalytic activity.117c Upon light irradiation, the photogenerated holes of photocatalysts are captured by H2O to form O2 and H+, and the photogenerated electrons in the CB reduce CO2via a sequence of complex reactions to produce valuable chemicals, including HCHO, HCOOH, CH3OH and CH4, depending on the energy level correlation between the photocatalysts and the redox agents.117d With large surface areas and abundant nitrogen content, the two azine-based COFs (N3-COF and ACOF-1) reported by W. Zhu et al. showed superior performances towards the adsorption and subsequent conversion of CO2 into CH3OH.117b Due to their extended structures, they are highly visible light-active compared to the commonly used inorganic photocatalysts. Besides, they are thermally and chemically stable, enabling the use of water as the reductant and avoiding the use of sacrificial agents. Moreover, for practical light-harvesting application, a triazine-based COF (2D CTF) film was developed by J.-O. Baeg et al. for converting CO2 to HCOOH.117e Thus, these pioneering works will trigger wider studies on solar energy conversion.
In addition, some POPs can efficiently photocatalyze both oxidation and reduction reactions. J. Zhang et al. reported a carbazole-based conjugated porous polymer (Cz-POF-1).118 Cz-POF-1 was prepared via the FeCl3-assisted oxidative polymerization of carbazole-based monomers. Cz-POF-1 exhibited a high BET surface area of 2065 m2 g−1 and large pore volume of 1.57 mL g−1 (72% mesopores), which are beneficial for mass transfer. As shown in Fig. 11a, the calculated E1/2 of the Cz-POF-1+/*Cz-POF-1 couple (Ered1/2 = −1.53 V) was sufficiently negative, thus phenacyl bromide (Ered1/2 = −0.49 V) and oxygen (Ered1/2 = −0.86 V) could be reduced through singlet electron transfer, while the POF-1+ intermediate (Ered1/2 = +1.38 V) was a good oxidant for oxidizing the Hantzsch ester (Ered1/2 = +0.72 V). Thus, Cz-POF-1 was capable of photocatalyzing three prototypic organic transformations in mild conditions, including net reductive dehalogenation of phenacyl bromide, net oxidative hydroxylation of arylboronic acids, and redox-neutral α-alkylation of aldehydes (Fig. 11b). In addition, the above three reactions photocatalyzed by Cz-POF-1 exhibited a good tolerance to various substrates and the photocatalyst could be easily recovered for reuse. Benefiting from its large conjugated structure and porous structure, Cz-POF-1 exhibited better catalytic activity compared to the linear and non-porous carbazole-based polymer.
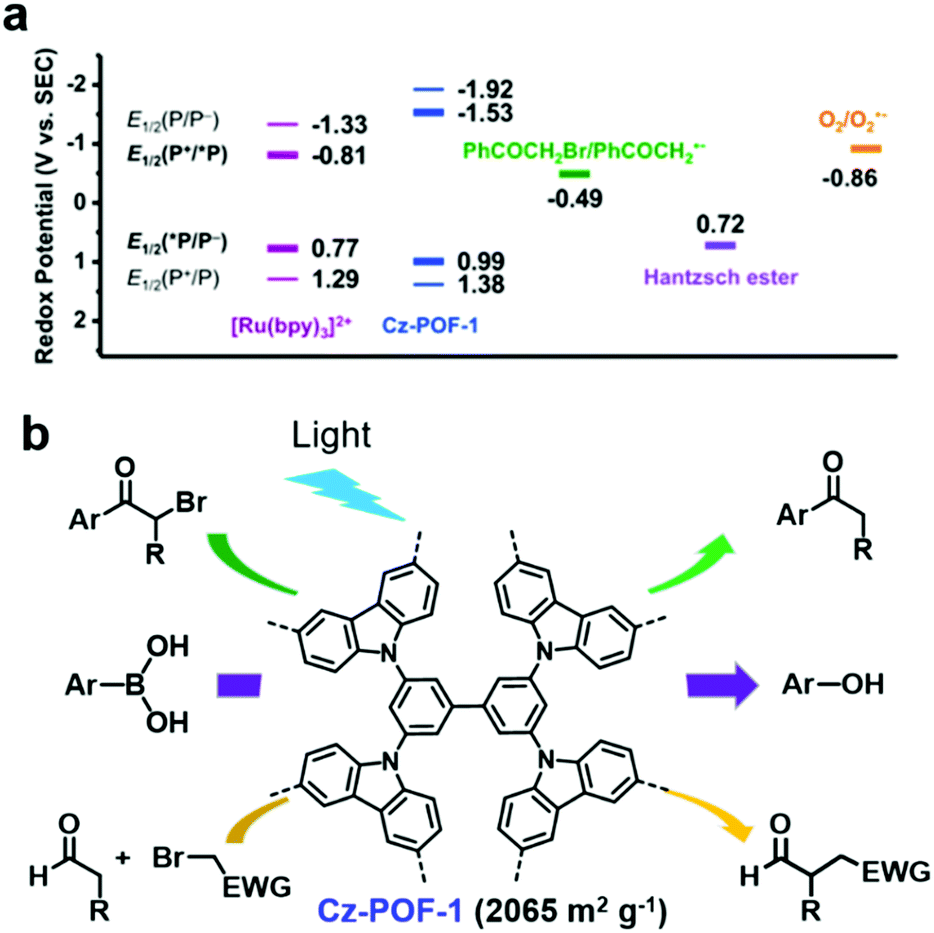 | ||
| Fig. 11 (a) Redox potentials of Cz-POF-1 and several related substrates. (b) Three prototypic organic transformations using POF-1 as the photocatalyst: net reductive dehalogenation of phenacyl bromide, net oxidative hydroxylation of arylboronic acids, and redox-neutral α-alkylation of aldehydes. Reproduced from ref. 118 with permission from the American Chemical Society, Copyright 2015. | ||
4. Treatment of pollutants in water
Water pollution not only affects the growth of aquatic organisms but also poses a threat to the health of human beings. Organic dyes and heavy metal ions with nonbiodegradability and chemical stability are the two major types of water pollutants.119 To date, various methods such as sorption,119 ion exchange,120 and photocatalysis121 have been employed to remove or degrade organic dyes in aqueous solution. Adsorption is one of the most costless, effective and eco-friendly methods. Many porous materials including zeolites122,123 and activated carbon124,125 have been widely used as sorbents, while CMPs together with COFs as burgeoning new POPs have recently been explored for removing pollutants from water.126–131 Our group also reported several CMPs with strong adsorption capacity towards pollutants in water.129,132 However, these sorbents often suffer from limited adsorption capacities. Benefiting from the large electron delocalized network structure, efficient light absorption and charge carrier transportation, many CMPs and COFs can act as both sorbents and photocatalysts for the removal of dyes or photodetoxification of heavy metal ions for water remediation, which make them attractive.4.1 Photodegradation of organic dyes
The release of common organic dyes such as methyl orange (MO), rhodamine B (RhB), methylene blue (MB), and Congo Red (CR) from the paper, leather and food industries into the ecosystem is highly toxic to aquatic life and causes serious threats to the environment. When CMPs or COFs are used as photocatalysts, one or several active species (i.e., superoxide radical (O2˙−), singlet oxygen (1O2), photogenerated hole (h+), and ˙OH) generated by light irradiation may be involved to photodegrade dyes in water. The general process can be summarized as follows: (1) upon light irradiation, the electron in the VB is excited to the CB, leaving holes in the VB; (2) two active oxygen species, O2˙− and 1O2, are generated through singlet electron transfer (SET) and energy transfer (ET) from the excited photocatalyst to molecular oxygen; (3) O2˙− reacts with water to form ˙OH, another active species, while the holes spread over the whole polymer and further catch pollutants, facilitating the separation of excitons; and (4) the dyes are degraded by one or more of the generated reactive oxidative species.Our group reported two polyoxometalate built-in CMPs (POM-CMPs, i.e., Bn-Anderson-CMP and Th-Anderson-CMP, Fig. 12a) for the photodegradation of organic dyes.133 Both CMPs showcased broad UV/vis absorption, which is crucial for photocatalysis. Due to the fascinating photochemical properties of polyoxometalates (POMs), high porosity and good reusability of CMPs, the hybrid materials showed higher photocatalytic activity than both building blocks. Additionally, under similar reaction conditions, due to the strong electrostatic interactions between the positively charged Anderson-POMs and the negatively charged dyes (RhB and MB), the photocatalytic activities of these POM-CMPs were comparable or even higher than that of benzothiadiazole-based CMP nanoparticles and porous carbon nitride (C3N4). Furthermore, Bn-Anderson-CMP exhibited higher photocatalytic activity than that of Th-Anderson-CMP with less time needed to reach the same degradation efficiency of RhB upon irradiation, which can be attributed to its higher BET surface area, providing more reactive sites during the photocatalytic process (Fig. 12b). 1O2 and H2O2 were determined to be the main active species participating in the photodegradation of RhB and POM-CMPs maintained high catalytic activity during this process (Fig. 12c).
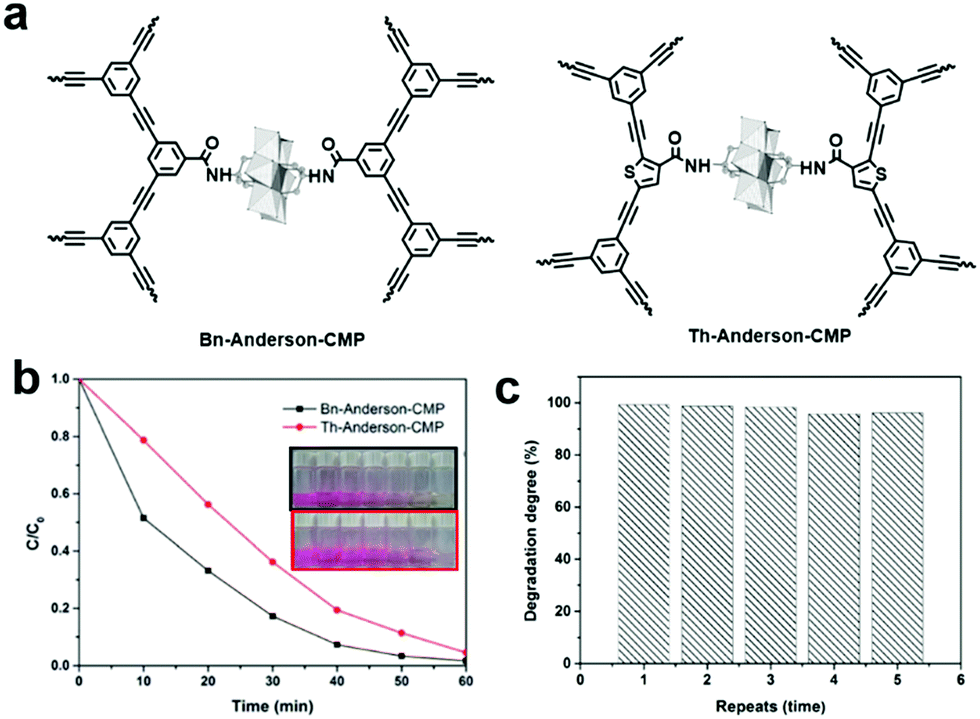 | ||
| Fig. 12 (a) Chemical structures of Bn-Anderson-CMP and Th-Anderson-CMP. (b) Photocatalytic degradation of RhB in the presence of POM-CMPs in water and the insert shows the corresponding color changes. (c) Reusability of Bn-Anderson-CMP for the photocatalytic degradation of MB in water under visible light irradiation. Reproduced from ref. 133 with permission from the Royal Society of Chemistry, Copyright 2017. | ||
The presence of metal in organic/inorganic hybrid porous materials may not only affect the photocatalytic activities of materials, but also induce secondary pollution. Thus, a series of metal-free porous polymers BF-CMPs (Py-BF-CMP, TPE-BF-CMP and TPA-BF-CMP, Fig. 13a) with the D–A structure was also reported for the photodegradation organic dyes in water.134 The main difference among these CMPs is the electronic nature of the electron donor units. Due to the strong electron-donating property and extended conjugation between the pyrene (Py) and bifluorenylidene (BF) units, Py-BF-CMP possessed the lowest band gap, together with the highest surface area and largest pore volume. Thus, Py-BF-CMPs are not only a sorbent but also serve as a photocatalyst (Fig. 13b). For example, the maximum adsorption capacity of the three BF-CMPs for RhB was calculated to be 1905, 1024, and 926 mg g−1 for Py-BF-CMP, TPA-BF-CMP and TPE-BF-CMP, respectively. Then, the photocatalytic activity of these BF-CMPs for the degradation RhB was explored, and the contribution of adsorption was evaluated with the same mixture in the dark. Py-BF-CMP showed the highest photocatalytic activity among the three BF-CMPs under continuous visible light (>450 nm) irradiation for 30 min, where more than 81% of RhB was degraded, which is comparable or even better than that of some reported porous polymers (C3N4, and POM-based CMPs (Bn-Anderson-CMP)). O2˙− was the primary active species during the photodegradation process and it was noteworthy that Py-BF-CMP exhibited high stability and recyclability both as an adsorbent and photocatalyst.
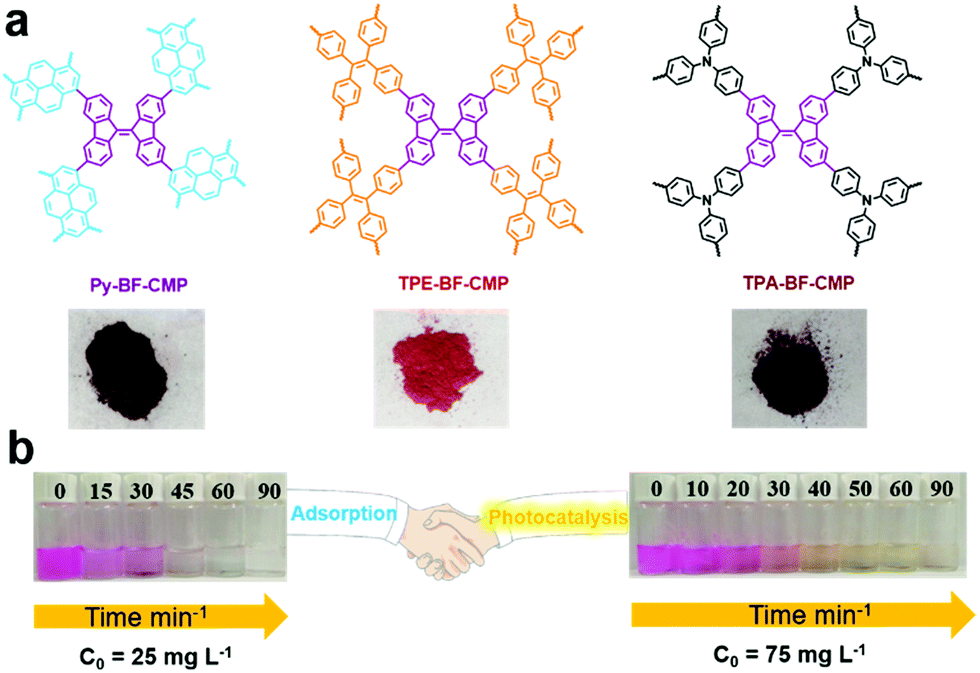 | ||
| Fig. 13 (a) Chemical structures of BF-CMPs and pictures of the BF-CMPs powder. (b) Adsorption and photocatalytic degradation capacities of Py-BF-CMP towards RhB. Reproduced from ref. 134 with permission from the American Chemical Society, Copyright 2018. | ||
Besides CMPs, a series of imine-linked COFs (COFA+C, COFA+B and COFA+D, Fig. 14) reported by Y. Cai et al. have been used for the photodegradation of water pollutants and the structure–property relationship was investigated in detail.135 The COFs were synthesized via Schiff base chemistry with the same aldehyde (A) but different NH2-terminated building blocks (B, C, and D). All the COFs were thermally stable up to 300 °C. Dark adsorption tests for the three COFs towards MO dye were firstly conducted. Generally, a higher BET surface area and larger pore volume result in better adsorption capacity. In this work, the author demonstrated that the existence of noncovalent interactions such as π–π stacking and hydrogen bonding between the porous polymer skeleton and dye molecules promoted the adsorption process. It was estimated that π–π stacking and H-bonding interactions may exist between MO and the polymers, while the former interaction played a decisive role. Thus, the existence of the largest amount of benzene rings in COFA+B rendered it with the highest adsorption ability toward MO. On the other hand, since the –NH– group in COFA+D can form H-bonds with the azo groups (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) of MO, COFA+D exhibited a higher adsorption capacity than COFA+B. Accordingly, the adsorption capacity of these COFs towards MO followed the order of COFA+B > COFA+D > COFA+C. Then the COFs were further evaluated for degrading MO under visible light. After irradiation with visible light for 30 min, MO was completely degraded by COFA+C and the performance of COFA+C was superior to the other COFs. Under the same conditions, only 29.6% MO was removed by COFA+B while almost no MO was degraded in the presence of COFA+D. Based on the control experiments, it is deduced that the photodegradation of organic pollutants was mainly driven by the superoxide radicals (O2˙−) with the assistance of hydroxyl radicals (˙OH).
N–) of MO, COFA+D exhibited a higher adsorption capacity than COFA+B. Accordingly, the adsorption capacity of these COFs towards MO followed the order of COFA+B > COFA+D > COFA+C. Then the COFs were further evaluated for degrading MO under visible light. After irradiation with visible light for 30 min, MO was completely degraded by COFA+C and the performance of COFA+C was superior to the other COFs. Under the same conditions, only 29.6% MO was removed by COFA+B while almost no MO was degraded in the presence of COFA+D. Based on the control experiments, it is deduced that the photodegradation of organic pollutants was mainly driven by the superoxide radicals (O2˙−) with the assistance of hydroxyl radicals (˙OH).
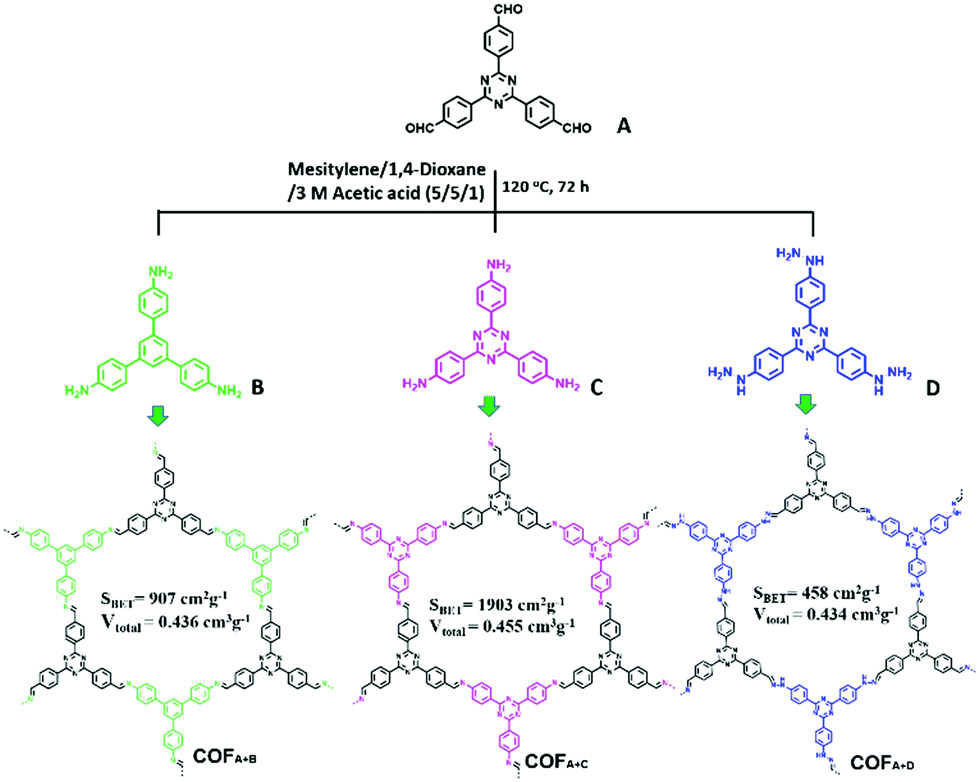 | ||
| Fig. 14 Synthesis and structures of COFA+B, COFA+C and COFA+D. Modified from ref. 135 with permission from Elsevier, Copyright 2018. | ||
4.2 Photodegradation of heavy metal ions
Hexavalent chromium (Cr(VI)), which is known as a toxic heavy metal due to its mutagenic and carcinogenic effect, is widely used in the leather tanning, printing, and pigment industries.136,137 Thus, groundwater pollutants containing Cr(VI) may cause great damage to human health. However, when Cr(VI) is reduced to Cr(III), the toxicity is reduced by 500–1000 times.138 Therefore, photocatalytic reduction is an effective way to detoxify wastewater that contains Cr(VI) heavy metal ions.Benefiting from the photocatalytic activity of Bi2MoO6 for environmental decontamination and the high photocatalytic activity and stability of CMPs, a porous polymer (BBT) was synthesized in situ on the surface Bi2MoO6 nanosheets, resulting in a Z-scheme heterojunction (BBT–BMO, Fig. 15a).139 The construction of the Z-scheme heterojunction facilitated the separation of charge carriers, thus improving the photocatalytic ability (Fig. 15b). BBT–BMO-10% with a mass ratio of 10% (BBT to BMO) exhibited the maximum photocatalytic rate. It had a relatively high BET surface area of 35.9 m2 g−1 and a broader absorption spectrum, providing more active sites. In addition, the weaker fluorescence intensity, lower photocurrent and structure-resulting efficient exciton separation indicated that more photogenerated holes and electrons were available, further enhancing the photocatalytic performance. During the photocatalytic process, both photogenerated electrons and O2˙− contributed to the reduction of Cr(VI) and the photocatalyst could be reused after at least 3 runs.
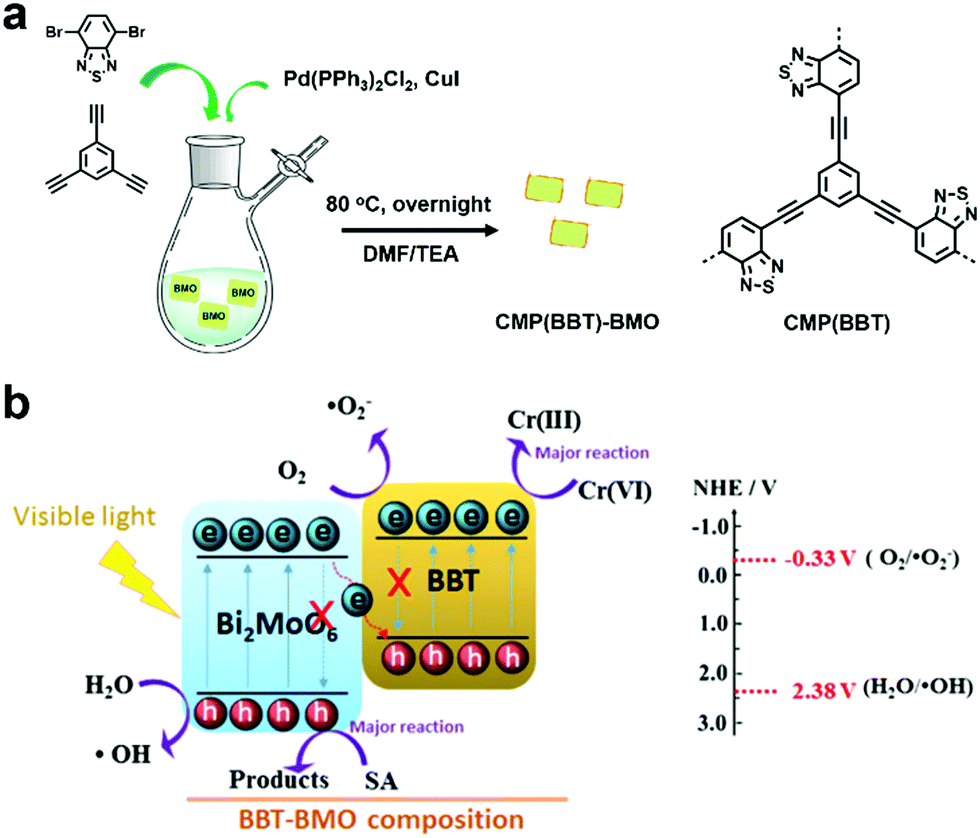 | ||
| Fig. 15 (a) Preparation of BBT–BMO. (b) Proposed photocatalytic mechanism for the photoreduction of Cr(VI) using BBT–BMO. Reproduced from ref. 139 with permission from Elsevier, Copyright 2017. | ||
Our group developed two D–A-type BT-based COFs (BT-COFs, i.e., TPB-BT-COF and TAPT-BT-COF, Fig. 16), which were further applied for the photoreduction of Cr(VI).140 Both BT-COFs were not soluble in common solvents and displayed good chemical stability in various harsh conditions. Fortunately, the conduction bands of these two BT-COFs were negative enough for the reduction of Cr(VI). The concentration of Cr(VI) in solution was detected with the assistance of DPC and sulfuric acid. With a more negative conduction band as well as a narrower band gap, TPB-BT-COF exhibited a faster photocatalytic rate. Cr(VI) was totally reduced upon visible light irradiation for 75 min without any hole scavenger and no additional acid or base was needed to adjust the pH during the photocatalytic process.
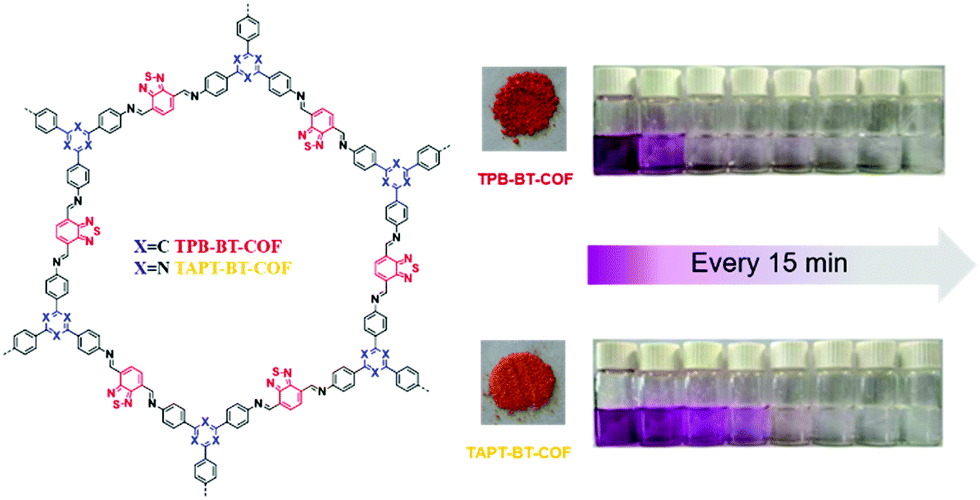 | ||
| Fig. 16 Structures of BT-COFs and pictures of the corresponding powder and photographs of the photocatalytic reduction using TPB-BT-COF and TAPT-BT-COF. Reproduced from ref. 140 with permission from the Royal Society of Chemistry, Copyright 2019. | ||
Notably, the influence of photocatalyst structures on photocatalytic activity for the treatment of pollutants in water can be concluded from the following aspects:
1. Generation of reactive oxygen species
Suitable energy levels are needed for generating reactive oxygen species, particularly the CB edge potential of COFs should be lower than the standard redox potential E(O2/O2˙−) (−0.33 vs. NHE). Besides, the hydrophilicity of porous polymers also affects the production of reactive oxygen species (mainly ˙OH).
2. Available active sites
More active sites tend to generate more excitons, thus affecting the photocatalytic ability, which can be deduced from the transient photocurrent intensity. A higher transient photocurrent intensity indicates more visible-light active centres, which is beneficial for the generation of photoexcited charger carriers.
3. Effective exciton separation
Effective separation of photoinduced electron–hole pairs is a prerequisite to initiate photocatalysis. First, it can be inferred from the theoretical calculation whether there is a clear separation between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). In addition, fully quenched photoluminescence (PL) emissions originating from the recombination of free charge carriers corresponds to a high separation efficiency of photogenerated electron–hole pairs. Furthermore, a small arc radius obtained from the electrochemical impedance spectroscopy (EIS) Nyquist plot also reflects the good separation of electron–hole pairs.
Besides the above aspects, the generation of stable reaction intermediates is necessary for efficient photocatalysis.
5. Hydrogen evolution
Hydrogen fuel is a type of clean energy because it only produces safe and pollution-free water and a tremendous amount of heat upon combustion, which is expected to be a potential alternative to fossil fuels. The abundant water resources on earth provide a wide range of sources for hydrogen production. However, the hydrogen obtained from water undergoes an energy-raising (uphill) reaction that requires an energy of 1.23 eV and a high activation barrier to cross, and thus catalysts are needed to lower the activation barrier of the reaction.12,141,142 Most of the reported photocatalysts for hydrogen evolution are inorganic semiconductors such as TiO219 and noble metal catalysts;143 however, they suffer from the disadvantages of expensive metal components and low catalytic activity under visible light. Thus, organic semiconductors with diverse synthetic strategies, tunable electronic structures, wide visible-light absorption and high carrier mobility have attracted great research interest. Typically, C3N4144,145 has been widely used as a photocatalyst for hydrogen production. However, its structure and energy levels are not easy to adjust and its synthetic temperature is usually high. In contrast, CMPs and COFs are structurally tunable and can be easily prepared, and thus, an increasing number of CMPs and COFs have recently been reported as efficient photocatalysts for hydrogen evolution, which is critically summarized below.5.1 General mechanism
As shown in Scheme 2, the general process of photocatalytic hydrogen evolution can be roughly divided into three steps:142 (1) firstly, generation of excited electrons and holes (excitons) under light irradiation; (2) excitons are separated and migrate to the active sites on the catalyst surface; and (3) finally, water undergoes a redox reaction on the surface of the catalyst releasing hydrogen as a product.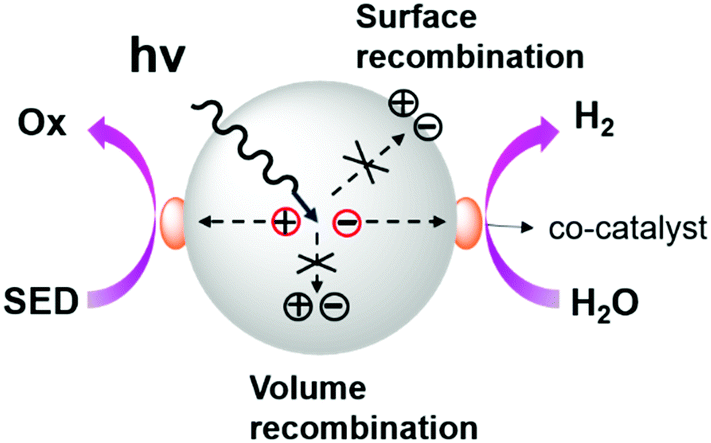 | ||
| Scheme 2 General photocatalytic hydrogen evolution process. Reproduced from ref. 142 with permission from the American Chemical Society, Copyright 2013. | ||
Usually, the hydrogen evolution rate (HER) is an important parameter to evaluate the photocatalytic activity towards the evolution of hydrogen from water. Furthermore, to investigate the mechanism, an isotope-labelling experiment is additionally conducted using D2O instead of H2O to confirm that the produced hydrogen comes from water.
It is well accepted that whether a photocatalyst can be applied for hydrogen evolution depends on the compatibility of the energy levels of its VB and CB with the oxidation potential of H2O/H2. A promising photocatalyst should possess the following elements: (1) strong visible light absorption for the utilization of sunlight; (2) high photocatalytic activity; (3) good chemical stability; and (4) excellent reusability. The band gap can be narrowed to fit the visible region via proper structural design. Effective exciton separation is an efficient way to improve the photocatalytic activity. In addition to optimizing the structure and morphology (e.g., surface morphology and structural defects) of the catalyst to promote exciton separation, sacrificial electron donors (SEDs) are often added to the catalytic system to inhibit the recombination of electrons and holes. Besides, co-catalysts (such as Pt nanoparticles) are usually deposited, and the transfer of electrons from the photocatalyst to co-catalyst is greatly promoted due to the existence of heterojunctions between Pt and the photocatalyst.142
In the following section, we will introduce the recent research progress of CMPs and COFs (representative structures are listed in Scheme 3) for photocatalytic hydrogen evolution.
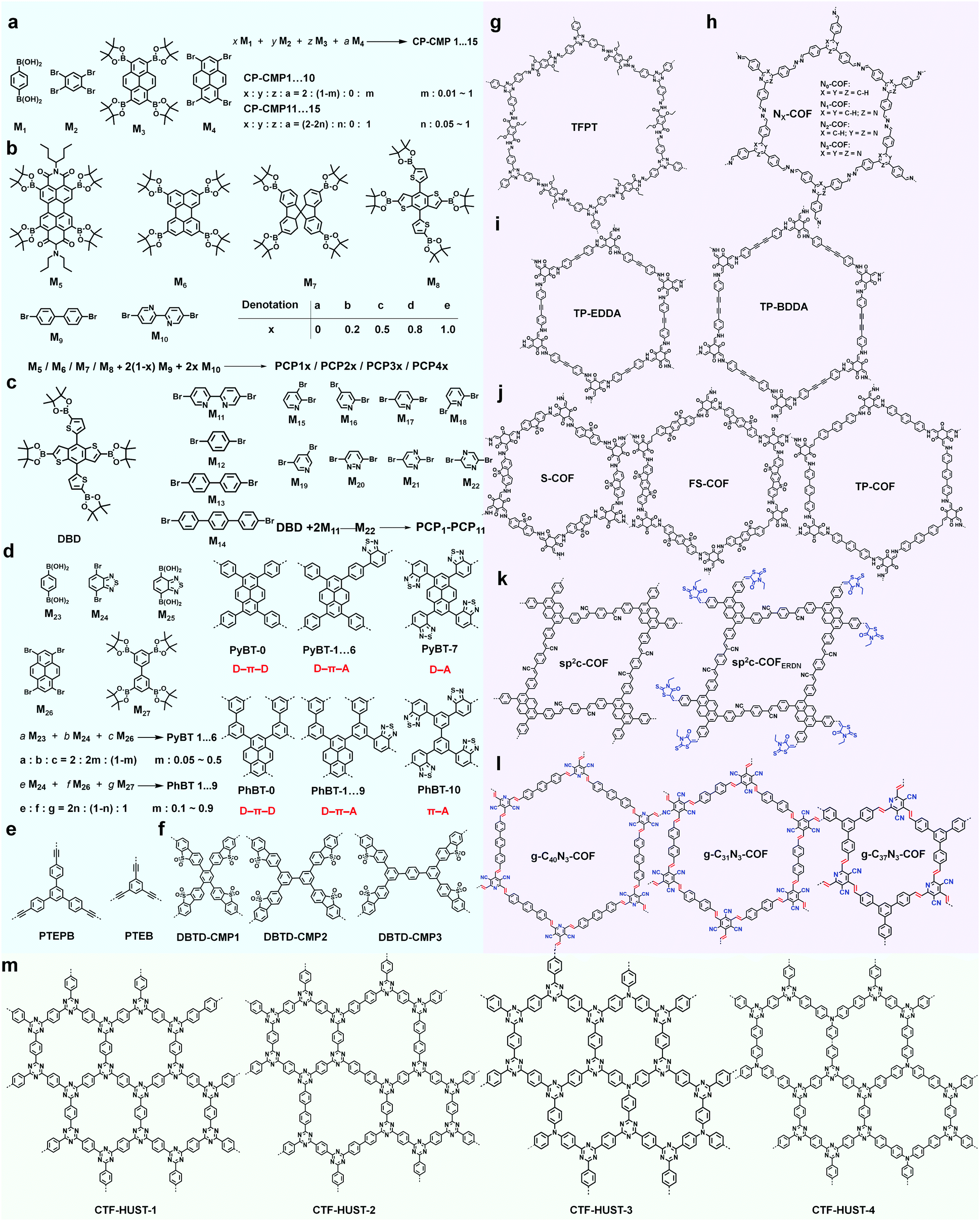 | ||
| Scheme 3 Representative (a–f) CMPs and (g–m) COFs as photocatalysts for hydrogen evolution. | ||
5.2 Representative examples
CMPs, due to their high stability, tunable chemical structures and electron properties, have been recognized as promising candidates for hydrogen evolution. The influence of modulating building blocks on the hydrogen evolution rate (HER) has been systematically investigated, as shown in Scheme 3a–f.With two or three of four monomers (M1, M2, M3, and M4) as building blocks, A. I. Cooper et al. documented a library of pyrene-based CMPs (CP-CMP1–15, Scheme 3a) with varying ratios of building blocks, which were further used for photocatalytic hydrogen production.146 The difference in compositions led to great modulations in their absorption (420 to 640 nm, Fig. 17a), and emission spectra (445 to 588 nm, from blue to red emission, as shown in Fig. 17b), BET surface areas (600 to 1700 m2 g−1) and band gaps (1.94–2.95 eV). In the presence of diethylamine as a sacrificial electron donor, the photocatalytic hydrogen production rate of CP-CMP1–15 increased first and then decreased with an increase in optical gap under visible light (>420 nm) irradiation. This can be attributed to the fact that with an increase in pyrene content, the energy level for charge transfer became lower, and meanwhile the recombination of electrons-holes became easier and became the dominant factor limiting the photocatalytic activity of the CMPs. Therefore, CP-CMP10 with a medium optical gap (2.33 eV) and moderate pyrene content exhibited the highest HER of 174 ± 9 μmol g−1 h−1. Notably, the obtained CMPs were mainly active in the visible range (Fig. 17a) and could achieve effective photocatalytic hydrogen evolution without the addition of co-catalysts. The strategy of composition variation was an effective way to regulate the optical bands of CMPs, providing a new idea for further improving the photocatalytic hydrogen evolution capacity of CMPs.
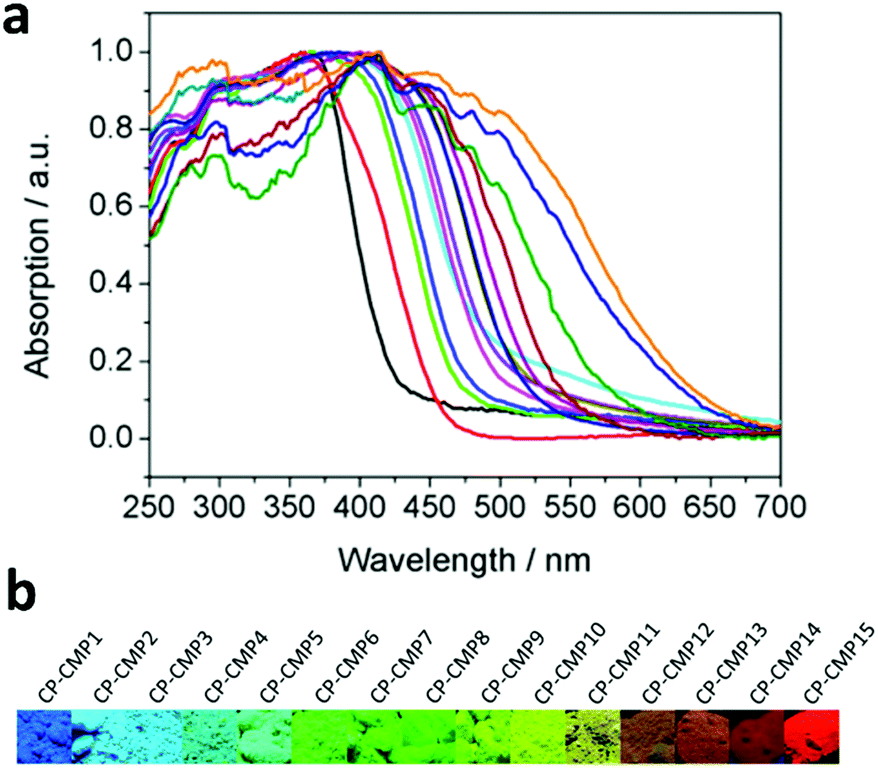 | ||
| Fig. 17 (a) UV-vis absorption spectra of CP-CMP1-15 powder and (b) corresponding images under irradiation with UV light (λ = 365 nm). Reproduced from ref. 146 with permission from the American Chemical Society, Copyright 2015. | ||
Although modulating the bandgap is a very effective way to improve photocatalytic activity, to further improve the efficiency, the photogenerated electrons and holes should be effectively separated.13 Inspired by the fact that intrachain charge transfer is beneficial for exciton dissociation and charge generation, a series of CMPs (PCP1x, PCP2x, PCP3x, and PCP4x, Scheme 3b) with alternating building blocks with different electronic properties were synthesized.153 PCP4e constructed using the strong electron-donating M8 and the weak electron-withdrawing bipyridyl (bpy) monomer featured a D–A structure and exhibited higher photocatalytic ability with an HER of 9.43 mmol g−1h−1. Besides, the D–A structure induced an improvement in electron-hole pair separation, and the higher wettability originating from the hydrogen bonding between the N atoms on bpy and H atoms of water led to a higher concentration of H2O molecules around the catalytic sites for reduction. Finally, it was observed that the residual Pd after the preparation of CMPs may also contribute to their photocatalytic activity (Fig. 18).
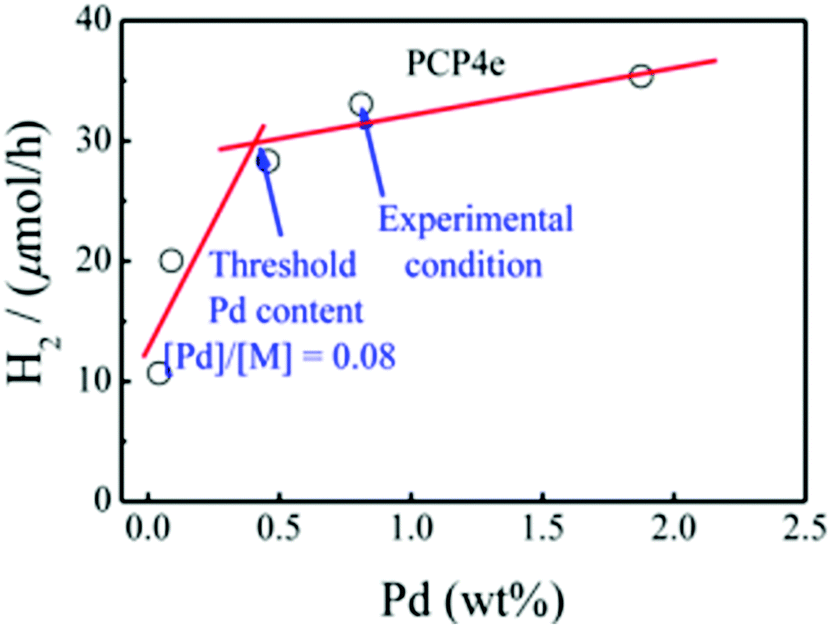 | ||
| Fig. 18 Effect of residual of Pd content on the hydrogen production rate using PCP4e as a photocatalyst. Reproduced from ref. 147 with permission from the American Chemical Society, Copyright 2016. | ||
Internal polarization is also crucial for effective charge separation, which is highly related to hydrogen evolution.148 L. Yu et al. synthesized a broad scope of CMPs (PCP0–11, Scheme 3c) in D–D-type or D–A-type to illustrate how the internal polarization affects the photocatalytic hydrogen evolution performance.149 Furthermore, benzene or biphenyl was introduced as π crosslinkers to construct a series of D–π–A-type CMPs (Scheme 3d) to further promote the charge separation for better photocatalytic performance.150a Besides, highly conjugated 1,3-diyne-linked CMPs (i.e., TEPB and TEB, Scheme 3e) possess high charge carrier mobilities and can effectively narrow the band gaps for visible photocatalysis.150b
Taking the advantages of the strong electron-withdrawing property and rigid, planar structure of benzothiophene dioxide (DBTD), three CMPs containing DBTD with cross-linkers in different lengths (i.e., DBTD-CMP1, DBTD-CMP2, and DBTD-CMP3, Scheme 3f) were reported by J.-X. Jiang et al. and were further used for photocatalytic hydrogen evolution.151 Surprisingly, even the linear conjugated polymer counterparts, P7 and P10, with DBTD units in their backbone (Fig. 19a) exhibited a high HER of 1.49 mmol g−1h−1 and 3.26 mmol g−1h−1, respectively. The photocatalytic performance of the CMPs increased with a decrease in cross-linker length, as explained by the spectra (UV-vis spectra and fluorescent spectra) and the DFT calculation. DBTD-CMP1 with the shortest benzene cross-linker exhibited a greater redshifted absorption, lower fluorescence emission intensity, shorter excitation lifetime and greater LUMO orbital distribution on DBTD, thus resulting in a higher conjugation degree, more efficient charge carrier separation and more reactive sites. All these characters led to a higher photocatalytic performance. Remarkably, DBTD-CMP1 exhibited an attractively high HER of 2460 μmol g−1h−1 (λ > 420 nm) without Pt co-catalyst and an even higher HER of 4600 μmol g−1h−1 when Pt was loaded, which is superior to most reported POP-based photocatalysts. Notably, the Pt-loaded DBTD-CMP1 showed an HER of 9200 μmol g−1h−1 under UV-vis light illumination (Fig. 19b). Furthermore, DBTD-CMP1 exhibited long-term stability and excellent reproducibility.
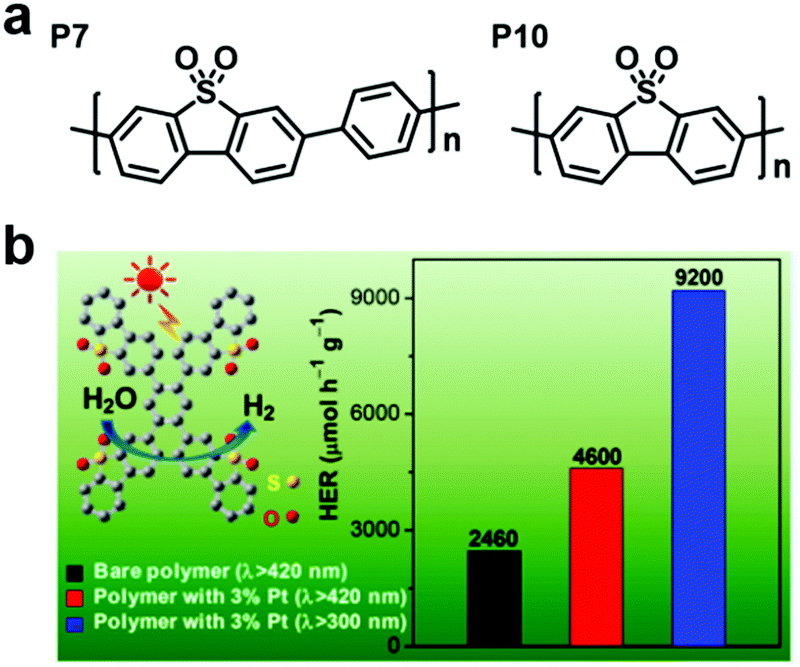 | ||
| Fig. 19 (a) Structures of the liner polymers P7 and P10. (b) HER of the DBTD-CMP1 photocatalyst under different conditions. Reproduced from ref. 151 with permission from the American Chemical Society, Copyright 2018. | ||
Similarly, COFs with an ordered structure, excellent photoelectric properties and tunable energy levels, have also been used as photocatalysts for hydrogen evolution. Although with limited examples, the use of COFs in photocatalytic hydrogen evolution has shown promising results. Triazines with a high electron mobility and strong electron affinity are widely used in photoelectric devices.152 The dihedral angle between the benzene and triazine rings in 2,4,6-triphenyl-1,3,5-triazine (TPT) is much smaller than that with benzene as the centre core, which is conducive to the construction of COFs with π-extended structures and tighter interlayer stacking, endowing them with high crystallinity and strong light absorption.153 B.V. Lotsch et al. reported a hydrazone-linked COF (TFPT-COF, Scheme 3g) containing TPT blocks and systematically investigated its performance in photocatalytic hydrogen evolution.154 As expected, TFPT-COF was highly crystalline and possessed large mesopores of 3.8 nm in diameter together with a high BET surface area of 1603 m−2 g−1. Due to its suitable energy levels, TFPT-COF was able to photocatalytically split water into hydrogen under visible light irradiation. In the presence of 10 vol% aqueous triethanolamine (TEOA) solution as the sacrificial electron donor and with Pt as the co-photocatalyst, the photocatalytic system showcased continuous and stable hydrogen production as high as 1970 μmol g−1h−1 during the first 5 h under standard basic conditions, even higher than that of Pt-modified amorphous melon, and g-C3N4. Although TFPT-COF lost its long-term order after ultrasonic treatment in water, it still remained stable photocatalytic activity, and its HER performance did not exhibit any obvious change after three cycles. It was hypothesized that the triazine moieties primarily acted as photoactive sites and thus played important roles in the catalytic hydrogen evolution.
To better understand the effect of the 1,3,5-triazine moiety on the HER performance, a series of hydrazine-linked COF counterparts (N0, N1, N2 and N3-COF, Scheme 3h) containing different triphenylaryl units with 0–3 nitrogen atoms on the central aromatic ring was prepared by the same group, realizing the regulation of photoelectric properties and photocatalytic hydrogen production performance at the molecular level.68 The integration of nitrogen atoms in the aromatic central aryl rings led to a significant change in the dihedral angle between the peripheral phenyl and central aryl ring (Fig. 20a), and thereby a variation in the planarity of the skeleton. In addition, when the number of nitrogen atoms increased from 0 to 3, the electron density in the central aromatic ring of the COF gradually decreased. While, the electron-deficient properties of the azine moiety could effectively stabilize the photogenerated electrons, facilitating the separation of electron–hole pairs and the subsequent migration to the active sites, thus promoting the hydrogen generation process. The average HERs photocatalyzed by N0, N1, N2 and N3-COF were 23, 90, 438 and 1703 μmol g−1 h−1, respectively. The observed trend was explained by theoretical calculations, and meanwhile two possible pathways after photoexcitation on Nx-COFs were proposed (Fig. 20b). It is estimated that it was more likely to go through the radical anion pathway and the stabilization of the radical anion contributed to the photocatalytic process. The increase in nitrogen numbers resulted in the electron-poor character of the core, which effectively stabilized the radical anion and transferred it to the nearest Pt nanoparticles. Therefore, N3-COF exhibited the highest photocatalytic activity, which was comparable to that of carbonitride photocatalysts and even better than that of Pt-modified amorphous melon (720 μmol g−1 h−1), g-C3N4 (840 μmol g−1 h−1) and crystalline poly (triazine imide) (864 μmol g−1 h−1).
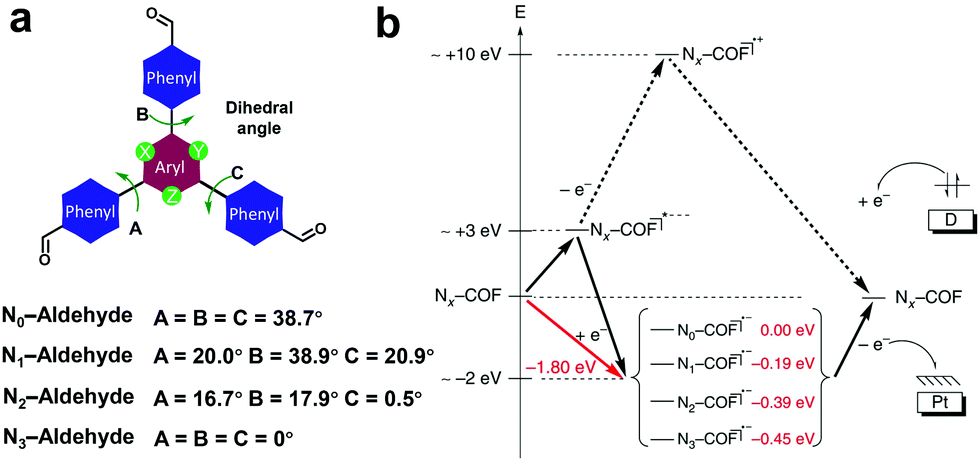 | ||
| Fig. 20 (a) Dihedral angles of Nx-aldehyde. (b) Proposed possible pathways after the photoexcitation of Nx-COFs. Reproduced from ref. 68 with permission from Nature Publishing Group, Copyright 2015. | ||
Diacetylene-bridged COFs, which possess highly conjugated structures and strong electron migration ability, are expected to be excellent photocatalysts.155 However, owing to the decreased formation rate of COFs originating from their larger pore size and weaker interlayer attraction, their synthesis seems to be very challenging.156 A. Thomas et al. recently developed a diacetylene-bridged β-ketoenamine COF (TP-BDDA, Scheme 3i) together with an acetylene-bridged COF counterpart (TP-EDDA in Scheme 3i) for comparison.157 Theoretically, the optical band gaps for both COFs were sufficient for photocatalytic hydrogen evolution. However, as shown in Fig. 21a, using a home-made reactor set-up (Fig. 21b) and in the presence of TEOA and the co-catalyst, only TP-BDDA could efficiently catalyze hydrogen evolution with an average HER of 324 ± 10 μmol g−1 h−1 under visible light irradiation (≥395 nm). In contrast, only negligible H2 evolution (30 ± 5 μmol g−1 h−1) was detected when TP-EDDA was used as the photocatalyst, illustrating that the conjugated diacetylene moiety played a crucial role in improving the photochemical water reduction activity. Interestingly, the quantum efficiency for the TP-BDDA catalyzed hydrogen evolution even reached up to 1.8% upon 520 nm light irradiation. Notably, TP-BDDA was the first example of COF without any heteronuclear atoms in its structure for photocatalytic hydrogen evolution.
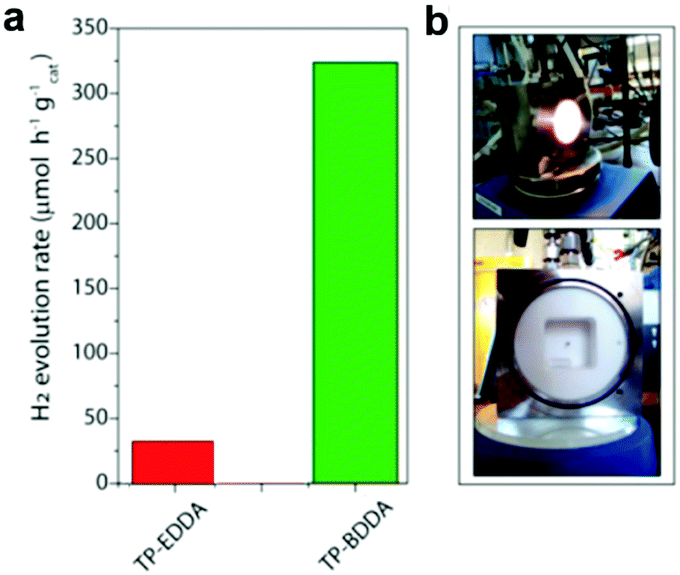 | ||
| Fig. 21 (a) Photocatalytic hydrogen evolution rates of TP-EDDA and TP-BDDA. (b) Reactor setup for hydrogen evolution experiments. Reproduced from ref. 157 with permission from American Chemical Society, Copyright 2018. | ||
The integration of DBTD monomer into COFs (FS-COF and S-COF, Scheme 3j) also greatly improved their photocatalytic activity towards hydrogen production (compared to TP-COF, Scheme 3j).158 FS-COF features a long-range ordering structure, a high BET surface area (1288 m2 g−1), wettable mesopores (3.2 nm, a contact angle of ∼23.6°) and strong visible light absorption, which were favorable for visible light catalytic hydrogen evolution. The HER of FS-COF for visible light catalysis (>420 nm) was up to 10.1 mmol g−1 h−1 with ascorbic acid as the sacrificial electron donor and Pt as the co-catalyst, and it could still reach as high as 1.32 mmol g−1 h−1 without the addition of Pt. Moreover, FS-COF showed a high external quantum yield of 3.2% at 420 nm, excellent optical stability, and a good photostability and steady photoactivity under continuous visible light irradiation during the photocatalytic process. The much improved photocatalytic hydrogen evolution of FS-COF compared to the other analogues was illustrated through electronic structure calculations (Fig. 22a and b). Among the four COF counterparts, FS-COF exhibited the smallest optical gap, which facilitated the visible photon absorption and exciton generation. Notably, the HER could be further increased to 16.3 mmol g−1 h−1 when FS-COF was dye-sensitized by loading a near-infrared absorbing dye.
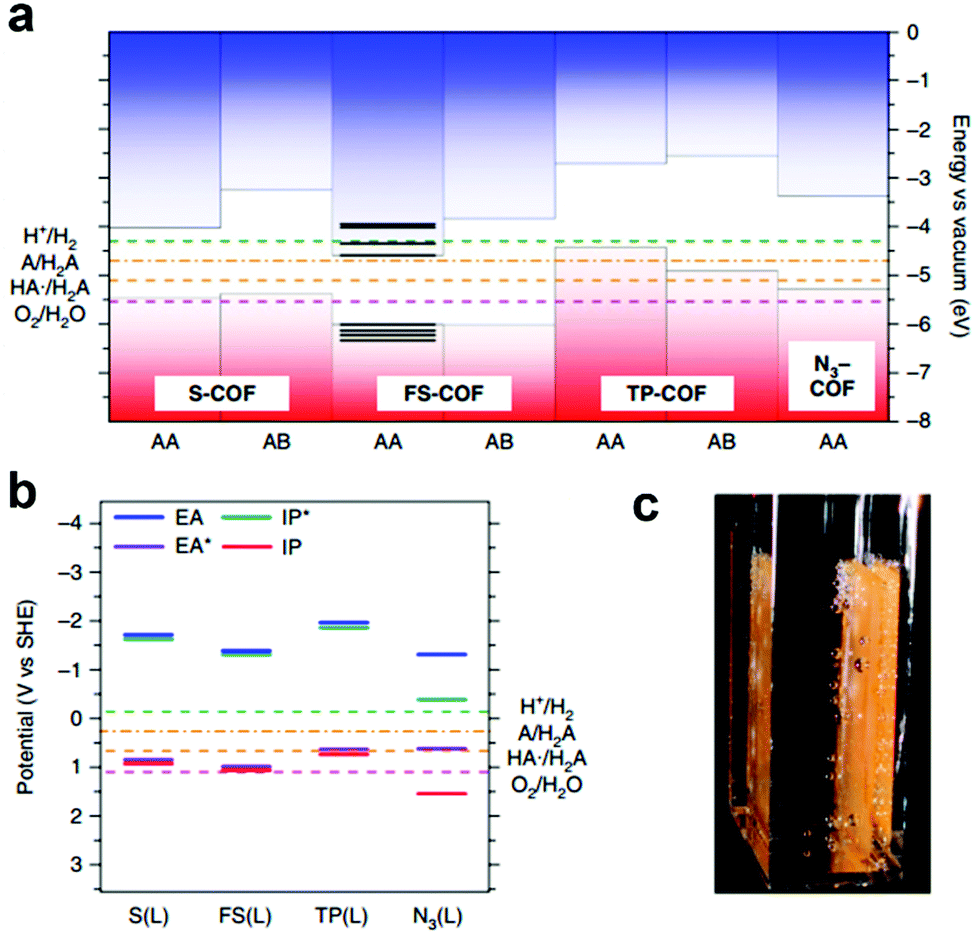 | ||
| Fig. 22 Electronic structure calculations using (a) cluster calculation and (b) periodic calculation approaches to predict the valence band maximum (VBM, red in a), conduction band minimum (CBM, blue in a), electron affinity (EA, blue line in b), exciton ionization potential (IP*, green line in b), ionization potential (IP, red line in b) and exciton electron affinity (EA*, purple line in b) of the COFs, providing insights into their photocatalytic water splitting activities. (c) Photograph of FS-COF film on glass-producing hydrogen. Reproduced from ref. 158 with permission from Nature Publishing Group, Copyright 2018. | ||
Considering the excellent photocatalytic hydrogen evolution performance of FS-COF, thin films of FS-COF by deposition of its colloidal solution onto glass substrate were further fabricated (Fig. 22c). The photocatalytic HER (λ > 420 nm, 0.1 M ascorbic acid, 5 h irradiation) was highly dependent on the film thickness and reached up to 24.9 mmol h−1 m−2, which is a step further for real application. To date, FS-COF exhibited the highest photocatalytic HER performance compared to other reported COFs.
sp2-carbon-linked COFs featured with π-delocalization over 2D networks exhibit unique photophysical properties and have attracted considerable attention for photocatalytic hydrogen evolution. sp2c-COF was prepared as shown in Scheme 3k.159 To further modulate its absorbance and band-gap, the electron-withdrawing group 3-ethylrhodanine (ERDN) was used as an end-capping group to introduce a push–pull effect to the 2D networks (sp2c-COFERDN in Scheme 3k). Both COFs exhibited high stability, crystallinity, porosity, π-conjugation and suitable range of energy levels for hydrogen evolution via water splitting. sp2c-COF provided an HER of 1360 μmol g−1 h−1 in the presence of 10 vol% triethanolamine (TEOA) sacrificial donor and Pt nanoparticles under irradiation (≥420 nm). Specially, it could quickly respond to a broad range of visible light from 400 nm to 590 nm. Remarkably, sp2c-COFERDN exhibited greatly enhanced activity with HERs of 2120 (≥420 nm) and 1240 μmol g−1 h−1 (≥498 nm) which originated from its improved light-harvesting capability (exceptionally broad absorbance centred at 530 nm and even extended to 800 nm), band-gap structure, and built-in heterojunction interface.
To date, most of the reported sp2-COFs have been obtained based on the Knoevenagel reaction between benzaldehyde and phenylacetonitrile (Fig. 23a). Very recently, F. Zhang et al. developed a scalable solution-processing approach to fabricate sp2-COFs.160 Three COFs (g-C40N3-COF, g-C31N3-COF and g-C37N3-COF, Scheme 3l) were synthesized with the key building block 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) and linear/trigonal aldehyde (4,4′′-diformyl-p-terphenyl, 4,4′-diformyl-1,1′-biphenyl or 1,3,5-tris(4-formylphenyl)benzene) utilizing the model reaction of the Knoevenagel condensation reaction between benzaldehyde and 2-methylnicotinonitrile (Fig. 23b). These COFs possessed high crystallinity, especially g-C40N3-COF. The conjugated structure and the incorporated pyridine rendered them with excellent light-harvesting characteristics. Additionally, with its suitable band gap of 2.36 eV and a high reduction potential of 5.83 eV (vs. vacuum level) together with a high oxidation potential of 3.47 eV (vs. vacuum level), g-C40N3-COF is an ideal photocatalyst for water splitting with two half-reactions in the presence of sacrificial reagents. The HER (50 mg) was 4.12 mmol g−1 h−1, with an apparent quantum yield (AQY) of 4.84% at λ = 420 nm, which is even superior most promising POPs sch as N3-COF, FS-COF.
 | ||
| Fig. 23 (a) Model Knoevenagel condensation reaction to construct sp2c-COF in previous works and (b) new strategy used for sp2c-COF by F. Zhang et al. Modified from ref. 160 with permission from Nature Publishing Group, Copyright 2019. | ||
CTFs have shown great potential in photocatalytic hydrogen production due to their high physicochemical stability and high nitrogen content. However, the conventional ionothermal conditions used to synthesize CTFs may cause partial carbonization of the structures and with a large energy penalty. B. Tan et al. provided an alternative strategy to construct CTFs via the low temperature condensation of aldehydes and amidines involving Schiff base formation followed by Michael addition (Fig. 24a).74 Four CTFs named CTF-HUST-1, CTF-HUST-2, CTF-HUST-3 and CTF-HUST-4 (Scheme 3m) were obtained and further explored as photocatalysts for hydrogen evolution. With layered structures, CTF-HUSTs showed a higher HER compared to the CTF-1 prepared by the ionothermal method and among which, CTF-HUST-2 exhibited the maximum HER of 2647 μmol g−1 h−1 in sacrificial photocatalytic hydrogen evolution under visible light irradiation (>420 nm). Nevertheless, the frameworks lacked long-range order, although their enhanced crystallinity can improve charge transport in the framework, which is beneficial for photoelectric properties. In 2018, the same group adopted a strategy of in situ oxidation of alcohols to aldehyde monomers (Fig. 24b).72 The method was effective to construct a series of crystalline CTFs (CTF-HUST-C1, CTF-HUST-C5, and CTF-HUST-C6) due in the fact that a decrease in nucleation rate and generation of a lower concentration of nuclei are the key to improving the crystallization. Under the same conditions, the three CTFs exhibited HERs of 5100, 2400 and 650 μmol g−1 h−1, respectively. CTF-HUST-C1 possessed the highest photocatalytic activity towards hydrogen evolution among the three, which was higher than that of CTF-HUST-1 and other amorphous CTFs.
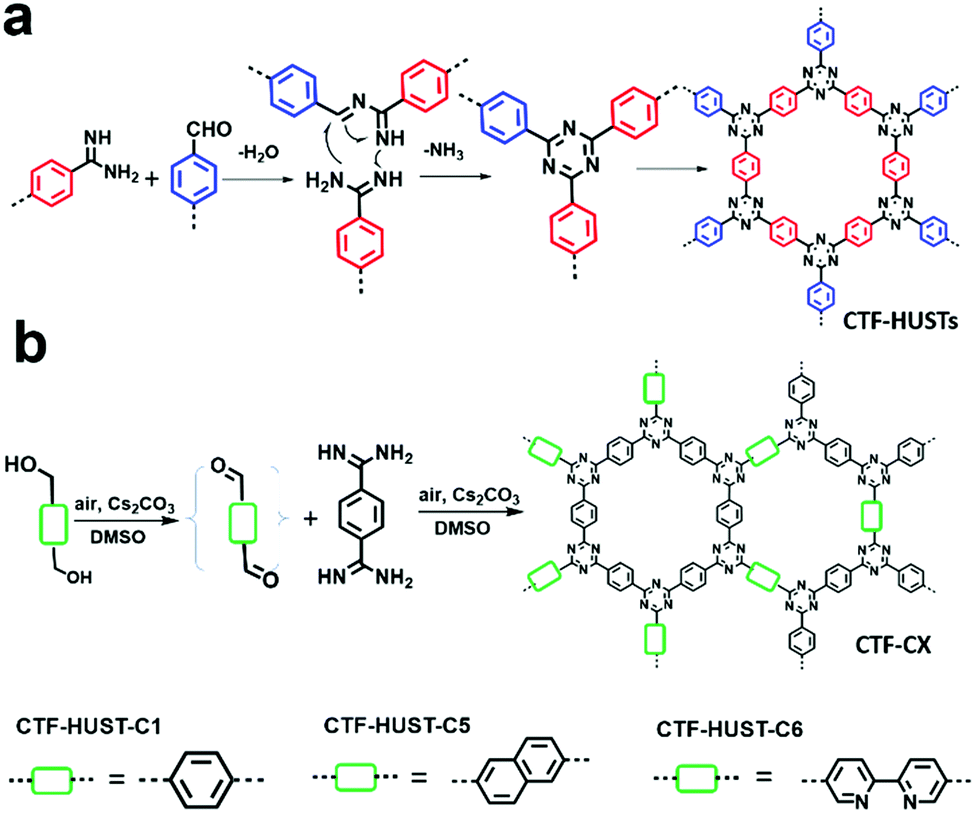 | ||
| Fig. 24 Reaction mechanisms for (a) CTF-HUSTs and (b) CTF-CX synthesis. Modified from ref. 72 and74 with permission from Wiley-VCH Verlag GmbH & Co. KGaA, Copyright 2017 and 2018, respectively. | ||
For better comparison, the photocatalytic activities of the above CMPs and COFs are summarized in Table 1.
| Co-catalyst | SED | Solvent | Light (nm) | HER (μmol g−1 h−1) | AQE | Ref. | |
|---|---|---|---|---|---|---|---|
| CP-CMP10 | — | 20 vol% DEA | Water | >420 | 174 (6 h) | — | 146 |
| PCP4e | — | 20 vol% TEA | Water | >400 | 1914 (3 h, r.t.) | 0.34% (350 nm) | 153 |
| PCP10 | — | 20 vol% TEA | Water | >400 | 2642 (2 h) | 1.05% (400 nm) | 149 |
| PyBT-2 | 3 wt% Pt | 20 vol% TEOA | Water | >420 | 296 (8 h) | — | 150a |
| PhBT-4 | 3 wt% Pt | 20 vol% TEOA | Water | >420 | 228 (8 h) | — | 150a |
| PTEPB | — | — | Water pH = 7 | >420 | 218 (8 h) | 10.3% (420 nm) | 150b |
| PTEB | — | — | Water PH = 7 | >420 | 102 (8 h) | 7.6% (420 nm) | 150b |
| DBTD-CMP1 | 3 wt% Pt | 20 vol% TEOA | Water | >420 | 4600 (6 h, 10 °C) | 3.3% (400 nm) | 151 |
| DBTD-CMP2 | 3 wt% Pt | 20 vol% TEOA | Water | >420 | 440 (6 h, 10 °C) | — | 151 |
| DBTD-CMP3 | 3 wt% Pt | 20 vol% TEOA | Water | >420 | 140 (6 h, 10 °C) | — | 151 |
| DBTD-CMP1 | — | 20 vol% TEOA | Water | >420 | 2460 (6 h, 10 °C) | — | 151 |
| DBTD-CMP2 | — | 20 vol% TEOA | Water | >420 | 186 (6 h, 10 °C) | — | 151 |
| DBTD-CMP3 | — | 20 vol% TEOA | Water | >420 | 116 (6 h, 10 °C) | — | 151 |
| TFPT | 2.2 wt% Pt | 10 vol% TEOA | Water | >420 | 1970 (3 h) | 2.2–3.9% (500 nm) | 154 |
| N0-COF | 8 wt% H2PtCl6 | 1 vol% TEOA | PBS buffer pH = 7 | >420 | 23 (8 h, 25 °C) | 0.001% (450 nm) | 68 |
| N1-COF | 8 wt% H2PtCl6 | 1 vol% TEOA | PBS buffer pH = 7 | >420 | 90 (8 h, 25 °C) | 0.077% (450 nm) | 68 |
| N2-COF | 8 wt% H2PtCl6 | 1 vol% TEOA | PBS buffer pH = 7 | >420 | 438 (8 h, 25 °C) | 0.19% (450 nm) | 68 |
| N3-COF | 8 wt% H2PtCl6 | 1 vol% TEOA | PBS buffer pH = 7 | >420 | 1703 (8 h, 25 °C) | 0.44% (450 nm) | 68 |
| TP-BDDA | 3 wt% Pt | 10 vol% TEOA | Water | >395 | 324 (10 h, 30 °C) | 1.8% (520 nm) | 157 |
| TP-EDDA | 3 wt% Pt | 10 vol% TEOA | Water | >395 | 30 (10 h, 30 °C) | — | 157 |
| FS-COF | 8 wt% H2PtCl6 | 0.1 M ascorbic acid | Water pH = 2.6 | >420 | 10100 (5 h) |
3.2% (420 nm) | 158 |
| S-COF | 8 wt% H2PtCl6 | 0.1 M ascorbic acid | Water pH = 2.6 | >420 | 4440 (5 h) | — | 158 |
| TP-COF | 8 wt% H2PtCl6 | 0.1 M ascorbic acid | Water pH = 2.6 | >420 | 1600 (5 h) | — | 158 |
| FS-COF | — | 0.1 M ascorbic acid | Water pH = 2.6 | >420 | 1320 (5 h) | — | 158 |
| S-COF | — | 0.1 M ascorbic acid | Water pH = 2.6 | >420 | 600 (5 h) | — | 158 |
| FS-COF-WS5F | 8 wt% H2PtCl6 | 0.1 M ascorbic acid | Water pH = 2.6 | >420 | 16300 (5 h) | — | 158 |
| sp2c-COF | 3 wt% Pt | 10 vol% TEOA | Water | ≥420 | 1360 (5 h, r.t.) | — | 159 |
| sp2c-COFERDN | 3 wt% Pt | 10 vol% TEOA | Water | ≥420 | 2120 (5 h, r.t.) | — | 159 |
| g-C40N3-COF | 3 wt% Pt | 10 vol% TEOA | Water pH = 10.3 | >420 | 2596 (4 h) | 4.84% (420 nm) | 160 |
| g-C31N3-COF | 3 wt% Pt | 10 vol% TEOA | Water pH = 10.3 | >420 | 542 (4 h) | — | 160 |
| g-C37N3-COF | 3 wt% Pt | 10 vol% TEOA | Water pH = 10.3 | >420 | 396 (4 h) | — | 160 |
| CTF-HUST-1 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 1072 (3 h, r.t.) | — | 74 |
| CTF-HUST-2 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 2647 (3 h, r.t.) | — | 74 |
| CTF-HUST-3 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 1238 (3 h, r.t.) | — | 74 |
| CTF-HUST-4 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 1582 (3 h, r.t.) | — | 75 |
| CTF-HUST-C1 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 5100 (5 h, 25 °C) | — | 72 |
| CTF-HUST-C5 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 2400 (5 h, 25 °C) | — | 72 |
| CTF-HUST-C6 | 3 wt% Pt | 10 vol% TEOA | Water | >420 | 650 (5 h, 25 °C) | — | 72 |
6. Outlook and future perspectives
POPs, as a new type of heterogeneous photocatalysts, possess the advantage of easy separation for recycling in comparison to their homogeneous counterparts. Among them, CMPs and COFs known as representatives of amorphous and crystalline POPs, possess unique features both in structure and functions modification to favor photocatalysis.161 Indeed, great advances have been achieved in recent years; however, the catalytic performances still cannot meet the demand for practical applications due to the unsatisfactory photocatalytic activity. Donor–acceptor structures are favorable for the separation of photoexcited electron-hole pairs, and thus enhance photocatalytic activity. In addition, many aforementioned aspects such as specific nanostructures, composition, dipole orientation and connectivity of the building blocks also exert significant impacts on the eventual performance. However, there are several challenges that need to be addressed before POP-based photocatalysts can be developed further for real applications. Among them, we mainly consider the following two aspects.6.1 Design and synthesis
A general strategy for the design of POP-based photocatalysts is required to integrate proper photosensitizers into the frameworks. Some typical moieties used in optoelectronics with strong electron-withdrawing properties are often used. For example, benzothiadiazoles and their derivatives are usually utilized as electron acceptors for the construction of CMPs. On the other hand, nitrogen-rich triazines are frequently applied as building blocks for COFs. Most CMPs are generally prepared via metal-assisted C–C cross-coupling reactions (e.g., Suzuki,80,82,146,147,149–151 Sonogashira,79,81,100,111,117 Heck,162 Glaser,150b and Yamamoto163 cross-coupling). The residue of metal species may cause potential interference and detrimental contamination for photocatalytic applications.164 In comparison, COFs are usually synthesized under conditions without noble metal catalysts and are mostly constructed based on dynamic covalent chemistry (DCC). The reversible formation of covalent bonds guarantees high crystallinity, while this will consequently lead to low stability of the COF skeleton. Hence, as outlined above, although the photocatalytic activity of COFs remains nearly unchanged after several catalytic cycles, their crystallinity often decreases significantly after photocatalysis. Thus, a remaining challenge is to enhance the robustness of COFs. The utilization of irreversible chemical transformations (e.g., tautomerism and oxidation)63,165,166 or intramolecular hydrogen bonding is an effective way to prepare robust COFs. Another alternative strategy is connecting building blocks using more stable CC linkages62,95–98 instead of the relatively labile –CN–167,168 and –B–O– bonds.169 However, in both cases, only limited examples have been realized, and they still suffer from either complicated preparation procedures or harsh reaction conditions. Therefore, there is much room for the further development of new synthetic strategies for POPs, including exploring novel building blocks, new linkages, and facile synthetic techniques.
6.2 Applications for photocatalysis
Benefiting from flexible structures, the band gaps of POPs may fall in the visible-light range through structural adjustment, holding great promise for solar-energy utilization. POP-based photocatalysts are able to mediate a series of reactions under visible light irradiation, such as the selective oxidation of organic sulfides,82,170 oxidation coupling of primary amines,93,94,170 aza-Henry reaction,102,104 hydroxylation of arylboronic acids,63,118,164 C-3 formylation and thiocyanation of indoles,108 selective bromination of aromatic compounds,171 reductive dehalogenation of organic bromide,113,114,118,172 and free radical polymerization of methyl methacrylate (MMA).116 Also, usually high transformation efficiency and excellent tolerance to a wide scope of substrates have been achieved in these cases. In addition, a variety of reactive species are produced, including singlet oxygen, which can be used in photodynamic therapy.173 However, the photocatalytic application of most POPs for chemical transformation is limited to a narrow scope of redox reactions. Thus, to explore the scope of their applications, POPs with extremely high redox potentials are desirable, which may come at a cost in terms of visible-light utilization efficiency.161 Thanks to their large porosity and excellent photocatalytic activity, POPs can be used not only as adsorbents but also as photocatalysts for the treatment of water pollutants. Moreover, POP-based photocatalysts have attracted increasing research interests in photocatalytic hydrogen evolution. Recently, it has been reported that DBTD-containing CMPs (DBTD-CMP1) and COFs (FS-COF) exhibited the highest HER among two types of POPs.151,158 These photocatalysts are highly competitive with the existing catalysts and have excellent stability and recyclability.151,158In the case of photocatalytic applications, the most important issue is to improve the efficiency of photocatalysts. The whole photocatalytic process involves a number of elementary photochemical processes including light harvesting, photoinduced electron–hole separation and charge migration to the surface of the photocatalyst. Thus, it is vital to increase the efficiency of each step and reduce the energy loss among these steps for through solar energy conversion.174
(1) The chemical structures of POPs determine their efficiency of sunlight utilization. Highly conjugated structures with narrow band gaps enabling absorption covering the visible-light range or even near-infrared region are desired. Besides the structures of building blocks, it has been demonstrated that the geometry of knots affects the conjugation over the 2D sheet of 2D COFs also. For example, an azine-linked COF with tetragonal topology possessed extended conjugation over the 2D sheet, while that with a hexagonal structure yielded a finite degree of conjugation.68,174
(2) During the migration process, electrons and holes may recombine, and thus lower the solar conversion efficiency, and therefore, the lifetime of excitons is another important element that needs to be considered. Great efforts have been devoted to enhancing the electric conductivity and shortening the charge transfer distance to reduce the recombination of excitons.14b Over the past years, many strategies have been developed, such as placing some type of electron/hole sink (e.g., sacrificial electron donor) at the surface to remove the electron/hole from the recombination arena, constructing nanostructures to shorten the charge transport distance,150b reducing defects inside the POPs to ensure that excitons migrate to the surface of photocatalysts, and forming donor–acceptor structures.79,112
(3) The last step for photocatalysis is the redox reaction at the interface of catalysts and the reaction mixture. Sufficient surface area is needed for the exposure of more active sites for redox reaction. Whereas, the accumulation of carriers on the surface would definitely increase the charge recombination, and meanwhile damage the long-term stability of photocatalysts. Thus, the surface kinetics should be accelerated, while the introduction of co-catalysts is also an effective way to address this problem.14b
Finally, photocatalysts need to be stable and robust for long-term photocatalytic cycling processes.
As an important aspect of the photocatalytic application, photosensitized singlet oxygen (1O2) has attracted tremendous research interest. The application of POPs as a photosensitizer for 1O2 generation involves an intersystem crossing (ISC) from the lowest singlet excited state (S1) to the lowest triplet excited state (T1), and enhancing the ISC process is vital for enhancing the performance of photosensitizers. The introduction of heavy atoms that enhancing spin–orbit coupling will promote this process, but at the expense of safety. Another effective method is to lower the energy gap between S1 and T1. The “polymerization-enhanced photosensitization” concept was recently proposed to design highly effective photosensitizers since conjugated polymers possess many energy levels in each energy band.175 Therefore, the conjugation of POPs should be well adjusted for better performances as photosensitizers.
An urgent problem that needs to be solved is the dispersion of photocatalysts. Especially for water pollution treatment and photocatalytic hydrogen production, catalysts need to have good water wettability.161,176 B. V. Lotsch et al. highlighted this issue in the report entitled “The wetter the better”.177 F. Vilela et al. provided a valid solution of post-modification strategies to address this problem.83 Introducing water-compatible groups within the polymer is another approach to achieve the target. In a recent perspective,178 D. Jiang et al. scrutinized the design concept and synthetic strategy for COFs used for light-driven hydrogen production, and a glorious vision was proposed for overall water splitting to produce both hydrogen and oxygen by a single and scalable COF photocatalyst. Currently, the reported CMPs are all amorphous, while most of the COFs are obtained as microcrystalline powders. The structures of COFs are mainly deduced by combining theoretical PXRD simulation results with the experimental data. However, exact structure determinations are crucial for better understanding the structure–property relationship for further design and synthesis. Although a few COFs with resolved single crystal structures were recently reported,167,179,180 it is highly desirable to develop reliable strategies to construct COF crystals,75 especially 2D COFs. POP-based devices are another important direction that requires further development for realizing real applications in sensing, emitting and catalysis, etc. Nevertheless, due to their insolubility in most common solvents, powdered POPs are difficult for processing. A. I. Cooper et al.181 and A. Hu et al.182 proposed novel strategies of hyperbranched structuring and size control to improve the “solubility” of POPs, respectively. While D. Jiang et al.183 and S. Dai et al.184 separately prepared POP-based thin films or membranes by electropolymerization or in situ polymerization. Thus, more efforts should be devoted to developing new methods for processing POPs or fabricating POP-based films for device-directed applications. Finally, the real charge transfer pathway during the photocatalysis process is unclear, and therefore, more photophysical analysis and molecular simulation are required for better understanding the reaction mechanism.
In summary, with excellent photocatalytic performance and good reusability, POPs (especially CMPs and COFs) represent a promising platform for efficient photocatalysis. We anticipate that POPs have a promising future with more efficient photocatalytic POPs emerging for to practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0207500), National Natural Science Foundation of China (51973153), and the Natural Science Foundation of Tianjin (17JCJQJC44600).Notes and references
- Y. L. Wong, J. M. Tobin, Z. Xu and F. Vilela, J. Mater. Chem. A, 2016, 4, 18677 RSC.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- J. D. Xiao and H. L. Jiang, Acc. Chem. Res., 2019, 52, 356 CrossRef CAS.
- I. F. Teixeira, E. C. M. Barbosa, S. C. E. Tsang and P. H. C. Camargo, Chem. Soc. Rev., 2018, 47, 7783 RSC.
- H. Yamashita, K. Mori, Y. Kuwahara, T. Kamegawa, M. Wen, P. Verma and M. Che, Chem. Soc. Rev., 2018, 47, 8072 RSC.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997 CrossRef CAS.
- H. Yi, D. Huang, L. Qin, G. Zeng, C. Lai, M. Cheng, S. Ye, B. Song, X. Ren and X. Guo, Appl. Catal., B, 2018, 239, 408 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782 RSC.
- Y. Li, C. Gao, R. Long and Y. Xiong, Mater. Today Chem., 2019, 11, 197 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- K. Kailasam, M. B. Mesch, L. Möhlmann, M. Baar, S. Blechert, M. Schwarze, M. Schröder, R. Schomäcker, J. Senker and A. Thomas, Energy Technol., 2016, 4, 744 CrossRef CAS.
- (a) Y.-K. Peng and S. C. E. Tsang, Nano Today, 2018, 18, 15 CrossRef CAS; (b) M. Xiao, B. Luo, S. Wang and L. Wang, J. Energy Chem., 2018, 27, 1111 CrossRef.
- J. Di, C. Yan, A. D. Handoko, Z. W. Seh, H. Li and Z. Liu, Mater. Today, 2018, 21, 749 CrossRef CAS.
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401 CrossRef CAS.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1 CrossRef CAS.
- F. A. Frame, E. C. Carroll, D. S. Larsen, M. Sarahan, N. D. Browning and F. E. Osterloh, Chem. Commun., 2008, 2206 RSC.
- M. B. Tahir, G. Nabi, M. Rafique and N. R. Khalid, Int. J. Environ. Sci. Technol., 2017, 14, 2519 CrossRef CAS.
- P. Dong, G. Hou, X. Xi, R. Shao and F. Dong, Environ. Sci.: Nano, 2017, 4, 539 RSC.
- G.-J. Lee and J. J. Wu, Powder Technol., 2017, 318, 8 CrossRef CAS.
- S. G. Kumar and K. S. R. K. Rao, RSC Adv., 2015, 5, 3306 RSC.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536 CrossRef CAS.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428 CrossRef CAS.
- T. Stoll, C. E. Castillo, M. Kayanuma, M. Sandroni, C. Daniel, F. Odobel, J. Fortage and M.-N. Collomb, Coord. Chem. Rev., 2015, 304, 20 CrossRef.
- C. Herrero, B. Lassallekaiser, W. Leibl, A. Rutherford and A. Aukauloo, Coord. Chem. Rev., 2008, 252, 456 CrossRef CAS.
- H. Takeda and O. Ishitani, Coordin, Chem. Rev., 2010, 254, 346 CAS.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 689 RSC.
- G.-Q. Zhang, W. Ou and Y.-S. Xu, Sci. China Mater., 2018, 61, 1001 CrossRef CAS.
- H. Liu, W. Feng, C. W. Kee, Y. Zhao, D. Leow, Y. Pan and C.-H. Tan, Green Chem., 2010, 12, 953 RSC.
- W. Fan, Q. Yang, F. Xu and P. Li, J. Org. Chem., 2014, 79, 10588 CrossRef CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819 CrossRef CAS.
- C. Xu, W. Zhang, J. Tang, C. Pan and G. Yu, Front. Chem., 2018, 6, 592 CrossRef CAS.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083 RSC.
- X. Zou, H. Ren and G. Zhu, Chem. Commun., 2013, 49, 3925 RSC.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2006, 66, 768 CrossRef CAS.
- J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670 RSC.
- C. D. Wood, B. Tan, A. Trewin, H. J. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mater., 2007, 19, 2034 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230 RSC.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS.
- (a) P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450 CrossRef CAS; (b) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC; (c) C. S. Diercks and O. M. Yaghi, Science, 2017, 355, e1585 CrossRef.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580 CrossRef CAS.
- A. F. M. El-Mahdy, C. Young, J. Kim, J. You, Y. Yamauchi and S. W. Kuo, ACS Appl. Mater. Interfaces, 2019, 11, 9343 CrossRef CAS.
- X. Yuan, Y. Wang, G. Deng, X. Zong, C. Zhang and S. Xue, Polym. Adv. Technol., 2019, 30, 417 CrossRef CAS.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905 CrossRef CAS.
- X. Wang, X. Han, J. Zhang, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 12332 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821 CrossRef CAS.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 6814 CrossRef CAS.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430 CrossRef CAS.
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623 CrossRef CAS.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS.
- R. W. Tilford, W. R. Gemmill, H.-C. zur Loye and J. J. Lavigne, Chem. Mater., 2006, 18, 5296 CrossRef CAS.
- R. W. Tilford, S. J. Mugavero III, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741 CrossRef CAS.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS.
- Z. Li, X. Feng, Y. Zou, Y. Zhang, H. Xia, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 13825 RSC.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968 CrossRef CAS.
- M. Liu, K. Jiang, X. Ding, S. Wang, C. Zhang, J. Liu, Z. Zhan, G. Cheng, B. Li, H. Chen, S. Jin and B. Tan, Adv. Mater., 2019, 31, 1807865 CrossRef.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. I. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149 CrossRef CAS.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351 CrossRef.
- M. J. Davies, Biochem. Biophys. Res. Commun., 2003, 305, 761 CrossRef CAS.
- C. S. Foote and S. Wexler, J. Am. Chem. Soc., 1964, 86, 3879 CrossRef CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432 CrossRef CAS.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158 RSC.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177 RSC.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770 CrossRef CAS.
- S.-I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443 CrossRef CAS.
- R. D. Patil and S. Adimurthy, Asian J. Org. Chem., 2013, 2, 726 CrossRef CAS.
- R. W. Layer, Chem. Rev., 1963, 63, 489 CrossRef CAS.
- R. D. Patil and S. Adimurthy, Adv. Synth. Catal., 2011, 353, 1695 CrossRef CAS.
- H. Huang, J. Huang, Y.-M. Liu, H.-Y. He, Y. Cao and K.-N. Fan, Green Chem., 2012, 14, 930 RSC.
- L. Liu, S. Zhang, X. Fu and C. H. Yan, Chem. Commun., 2011, 47, 10148 RSC.
- R. Neumann and M. Levin, J. Org. Chem., 1991, 56, 5707 CrossRef CAS.
- A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162 CrossRef CAS.
- H. Li, P. Tang, Y. Zhao, S.-X. Liu, Y. Aeschi, L. Deng, J. Braun, B. Zhao, Y. Liu, S. Tan, W. Meier and S. Decurtins, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2935 CrossRef CAS.
- N. Kang, J. H. Park, K. C. Ko, J. Chun, E. Kim, H. W. Shin, S. M. Lee, H. J. Kim, T. K. Ahn, J. Y. Lee and S. U. Son, Angew. Chem., Int. Ed., 2013, 52, 6228 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. Zhang, Adv. Mater., 2015, 27, 6265 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673 CrossRef CAS.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849 CrossRef CAS.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 417 RSC.
- E. Jin, J. Li, K. Geng, Q. Jiang, H. Xu, Q. Xu and D. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef.
- Y. Wei, W. Chen, X. Zhao, S. Ding, S. Han and L. Chen, Polym. Chem., 2016, 7, 3983 RSC.
- J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779 CrossRef CAS.
- W. Liu, Q. Su, P. Ju, B. Guo, H. Zhou, G. Li and Q. Wu, ChemSusChem, 2017, 10, 664 CrossRef CAS.
- Y. Zhi, Z. Li, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2017, 5, 22933 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Synth. Catal., 2016, 358, 2576 CrossRef CAS.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef.
- W. Wu and W. Su, J. Am. Chem. Soc., 2011, 133, 11924 CrossRef CAS PubMed.
- X. Li, X. Gu, Y. Li and P. Li, ACS Catal., 2014, 4, 1897 CrossRef CAS.
- H. Jiang, W. Yu, X. Tang, J. Li and W. Wu, J. Org. Chem., 2017, 82, 9312 CrossRef CAS.
- W. Zhang, J. Tang, W. Yu, Q. Huang, Y. Fu, G. Kuang, C. Pan and G. Yu, ACS Catal., 2018, 8, 8084 CrossRef CAS.
- A. Hornung, S. Donner, A. Balabanovich and H. Seifert, J. Cleaner Prod., 2005, 13, 525 CrossRef.
- C. Schnabel, M. Worner, B. Gonzalez, I. del Olmo and A. M. Braun, Electrochim. Acta, 2001, 47, 719 CrossRef CAS.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2014, 2, 18720 RSC.
- Z. Li, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng, X. Chen, Z. Shi and X. Liu, Appl. Catal., B, 2019, 245, 334 CrossRef CAS.
- M. Hasegawa, Chem. Rev., 1983, 83, 507 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307 CrossRef CAS.
- Z. J. Wang, K. Landfester and K. A. I. Zhang, Polym. Chem., 2014, 5, 3559 RSC.
- (a) Y. Zhao, H. Liu, C. Wu, Z. Zhang, Q. Pan, F. Hu, R. Wang, P. Li, X. Huang and Z. Li, Angew. Chem., Int. Ed., 2019, 58, 5376 CrossRef CAS; (b) Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46 CrossRef CAS; (c) Y. Ma, Z. Wang, X. Xu and J. Wang, Chin. J. Catal., 2017, 38, 1956 CrossRef CAS; (d) L. Yuan and Y.-J. Xu, Appl. Surf. Sci., 2015, 342, 154 CrossRef CAS; (e) R. K. Yadav, A. Kumar, N.-J. Park, K.-J. Kong and J.-O. Baeg, J. Mater. Chem. A, 2016, 4, 9413 RSC.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250 CrossRef CAS.
- I. Ali, Chem. Rev., 2012, 112, 5073 CrossRef CAS PubMed.
- A. Dabrowski, Z. Hubicki, P. Podkoscielny and E. Robens, Chemosphere, 2004, 56, 91 CrossRef CAS PubMed.
- A. Mills, R. H. Davies and D. Worsley, Chem. Soc. Rev., 1993, 22, 417 RSC.
- S. Wang and Y. Peng, Chem. Eng. J., 2010, 156, 11 CrossRef CAS.
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172 CrossRef CAS.
- V. K. Gupta, B. Gupta, A. Rastogi, S. Agarwal and A. Nayak, J. Hazard. Mater., 2011, 186, 891 CrossRef CAS.
- M. Minceva, R. Fajgar, L. Markovska and V. Meshko, Sep. Sci. Technol., 2008, 43, 2117 CrossRef CAS.
- G. H. Ning, Z. Chen, Q. Gao, W. Tang, Z. Chen, C. Liu, B. Tian, X. Li and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 8897 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786 CrossRef CAS PubMed.
- N. Huang, L. Zhai, H. Xu and D. Jiang, J. Am. Chem. Soc., 2017, 139, 2428 CrossRef CAS.
- Y. Yuan, H. Huang, L. Chen and Y. Chen, Macromolecules, 2017, 50, 4993 CrossRef CAS.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031 CrossRef CAS PubMed.
- A. Li, H.-X. Sun, D.-Z. Tan, W.-J. Fan, S.-H. Wen, X.-J. Qing, G.-X. Li, S.-Y. Li and W.-Q. Deng, Energy Environ. Sci., 2011, 4, 2062 RSC.
- Y. Li, W. Chen, W. Hao, Y. Li and L. Chen, ACS Appl. Nano Mater., 2018, 1, 4756 CrossRef CAS.
- Y. Li, M. Liu and L. Chen, J. Mater. Chem. A, 2017, 5, 13757 RSC.
- B. Wang, Z. Xie, Y. Li, Z. Yang and L. Chen, Macromolecules, 2018, 51, 3443 CrossRef CAS.
- S. He, B. Yin, H. Niu and Y. Cai, Appl. Catal., B, 2018, 239, 147 CrossRef CAS.
- A. D. Bokare and W. Choi, Environ. Sci. Technol., 2010, 44, 7232 CrossRef CAS.
- C.-C. Wang, X.-D. Du, J. Li, X.-X. Guo, P. Wang and J. Zhang, Appl. Catal., B, 2016, 193, 198 CrossRef CAS.
- M. Costa, Toxicol. Appl. Pharmacol., 2003, 188, 1 CrossRef CAS.
- X. Yang, Y. Xiang, Y. Qu, X. Ding and H. Chen, J. Catal., 2017, 345, 319 CrossRef CAS.
- W. Chen, Z. Yang, Z. Xie, Y. Li, X. Yu, F. Lu and L. Chen, J. Mater. Chem. A, 2019, 7, 998 RSC.
- M. Bowker, Green Chem., 2011, 13, 2235 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef CAS.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753 CrossRef CAS.
- D. J. Martin, P. J. Reardon, S. J. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- 149 L. Li, W.-Y. Lo, Z. Cai, N. Zhang and L. Yu, Macromolecules, 2016, 49, 6903 CrossRef CAS.
- (a) Y. Xu, N. Mao, C. Zhang, X. Wang, J. Zeng, Y. Chen, F. Wang and J.-X. Jiang, Appl. Catal., B, 2018, 228, 1 CrossRef; (b) L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef.
- Z. Wang, X. Yang, T. Yang, Y. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J.-X. Jiang, ACS Catal., 2018, 8, 8590 CrossRef CAS.
- T. Ishi-i, K. Yaguma, T. Thiemann, M. Yashima, K. Ueno and S. Mataka, Chem. Lett., 2004, 33, 1244 CrossRef.
- Z.-S. Li, J.-X. Chen, Y.-B. Huang, G.-R. Chen and T.-Y. Lan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, 777 CrossRef.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789 RSC.
- (a) Z. Zhao, H. Su, P. Zhang, Y. Cai, R. T. K. Kwok, Y. Chen, Z. He, X. Gu, X. He, H. H. Y. Sung, I. D. Willimas, J. W. Y. Lam, Z. Zhang and B. Z. Tang, J. Mater. Chem. B, 2017, 5, 1650 RSC; (b) M. Planells, A. Abate, D. J. Hollman, S. D. Stranks, V. Bharti, J. Gaur, D. Mohanty, S. Chand, H. J. Snaith and N. Robertson, J. Mater. Chem. A, 2013, 1, 6949 RSC; (c) S. Ghosh, N. A. Kouame, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505 CrossRef CAS.
- (a) B. J. Smith, N. Hwang, A. D. Chavez, J. L. Novotney and W. R. Dichtel, Chem. Commun., 2015, 51, 7532 RSC; (b) E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. UribeRomo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416 CrossRef CAS.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomacker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180 CrossRef CAS PubMed.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632 CAS.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712 CrossRef.
- L. Sun, Z. Liang, J. Yu and R. Xu, Polym. Chem., 2013, 4, 1932 RSC.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426 CrossRef CAS.
- Z. J. Wang, R. Li, K. Landfester and K. A. I. Zhang, Polymer, 2017, 126, 291 CrossRef CAS.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524 CrossRef CAS PubMed.
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L. H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48 CrossRef CAS PubMed.
- Z. F. Pang, T. Y. Zhou, R. R. Liang, Q. Y. Qi and X. Zhao, Chem. Sci., 2017, 8, 3866 RSC.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594 CrossRef CAS.
- R. Li, Z. J. Wang, L. Wang, B. C. Ma, S. Ghasimi, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2016, 6, 1113 CrossRef CAS.
- W. Huang, B. C. Ma, D. Wang, Z. J. Wang, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2017, 5, 3792 RSC.
- C. J. G. Kenneth, R. Weishaupt and T. J. Dougherty, Cancer Res., 1976, 36, 2326 Search PubMed.
- T. He, K. Geng and D. Jiang, ACS Mater. Lett., 2019, 1, 203 CrossRef CAS.
- W. Wu, D. Mao, S. Xu, Kenry, F. Hu, X. Li, D. Kong and B. Liu, Chem, 2018, 4, 1937 CAS.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819 RSC.
- T. Banerjee and B. V. Lotsch, Nat. Chem., 2018, 10, 1175 CrossRef.
- T. He, K. Geng and D. Jiang, ACS Mater. Lett., 2019, 1, 203 CrossRef CAS.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830 CrossRef CAS.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52 CrossRef CAS.
- G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 12727 CrossRef CAS.
- S. Deng, J. Zhi, X. M. Zhang, Q. Q. Wu, Y. Ding and A. G. Hu, Angew. Chem., Int. Ed., 2014, 53, 14144 CrossRef CAS PubMed.
- C. Gu, N. Huang, J. Gao, F. Xu, Y. Xu and D. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4850 CrossRef CAS.
- Z. A. Qiao, S. H. Chai, K. Nelson, Z. Bi, J. Chen, S. M. Mahurin, X. Zhu and S. Dai, Nat. Commun., 2014, 5, 3705 CrossRef PubMed.
| This journal is © the Partner Organisations 2020 |