
A difluoroboron β-diketonate-based luminescent material with tunable solid-state emission and thermally activated delayed fluorescence†
Han
Zhang
,
Peng-Zhong
Chen
,
Li-Ya
Niu
 * and
Qing-Zheng
Yang
*
* and
Qing-Zheng
Yang
*
Key Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: niuly@bnu.edu.cn; qzyang@bnu.edu.cn
First published on 28th November 2019
Abstract
We demonstrate a solid luminescent material based on difluoroboron β-diketonate containing a triphenylamine moiety manifesting tunable solid-state luminescence and thermally activated delayed fluorescence (TADF). The compound shows strong fluorescence in both organic solutions and solid states. It exhibits significantly different emission colours in different aggregation states: bright orange emission in its crystalline solid state and red emission in the amorphous state. The orange and red emissive states are able to be reversibly switched by repetitive grinding–fuming processes with little fatigue. More importantly, the aggregation state plays a key role in the TADF properties. Its solid exhibits TADF properties in the amorphous state but not in the crystalline solid state due to the different ΔEST values in the relative morphologies.
Introduction
Organic fluorescent materials have received increasing interest in recent years due to their potential applications in organic light-emitting diodes (OLEDs), bioimaging, sensing and photoelectronic devices.1–8 Controlling and tuning the emission properties of organic molecules are essential for the exploration of fluorophores with excellent optical properties. The conventional fluorophores with tunable emissions were typically achieved through covalent modification of molecular structures.9–12 Alternatively, supramolecular strategies through self-assembly of fluorophores are attractive to tune the emission properties of the fluorescent materials, and avoid sophisticated synthesis.13,14 The photophysical properties of supramolecular organic materials are determined by not only their molecular structures, but also their conformations and aggregation states. Most traditional fluorophores show weak emission in aggregated states due to the strong π–π stacking interaction, known as aggregation-caused quenching (ACQ). By contrast, aggregation-induced-emission (AIE)-type fluorophores are weakly or practically nonfluorescent in solution, but highly fluorescent in aggregated states, thus attracting great attention to construct supramolecular fluorescent materials.15–18Different from either ACQ or AIE materials, difluoroboron β-diketonate dye is an important luminescent material which shows strong fluorescence in both solutions and solid states.19–24 Furthermore, the fluorescence of difluoroboron β-diketonate dyes in the solid-state strongly depends on the molecular structure, conformation and intermolecular interactions.25–27 Thus, they usually show reversible fluorescence switching in response to external stimuli (e.g. mechanical force, heat and solvent vapor). These stimuli could alter the molecular arrangement, conformation and non-covalent intermolecular interactions, thus resulting in a change in the photophysical properties, such as emission colors and intensities.28–32 Difluoroboron β-diketonate dyes show great potential in thermally activated delayed fluorescence (TADF) materials due to their D–A structures and excellent emission properties in the aggregation state.8,33 TADF materials are promising third-generation luminescent materials for use in the construction of highly efficient OLEDs since they can harvest both singlet and triplet excitons through an efficient reversed intersystem crossing (RISC) process.1,34–36 Although considerable progress has been made in the preparation of TADF materials,37–42 the influence of their molecular aggregation states on the luminescent properties has been rarely explored, especially the aggregation state-controllable properties.43–45
In this work, we report a difluoroboron β-diketonate compound 1 with D–π–A structure which exhibits tunable solid-state luminescence, mechanochromism, and TADF properties. Compound 1 is composed of triphenylamine as the electron donor, a dioxaborine ring as the electron acceptor and a phenyl group as a bridge unit. It manifests bright fluorescence in both solution and the solid state, and exhibits reversible mechanochromic properties between orange emission in the crystalline state and red emission in the amorphous state. The different nanostructures including nanospheres, nanosheets, and nanorods are obtained by modulating the conditions for assembly. The TADF properties of compound 1 are tuned by changing their aggregation state. It manifests delayed fluorescence in the amorphous state, but does not exhibit delayed fluorescence in the crystalline state since the singlet-to-triplet energy gap, ΔES1–T1 (ΔEst) is changed by modulation of the aggregation state.
Results and discussion
Molecular design and synthesis
The combination of strong electron donor–acceptor interaction and extended π-conjugation with good electron delocalization in a D–π–A structure is used to design fluorophores with bright, red-shifted emission with a large Stokes shift.46–49 We designed difluoroboron β-diketonate 1 by connection of a strong electron-withdrawing dioxaborine moiety with a good electron-donating triphenylamine group. The dioxaborine moiety as an electron-deficient group has been used to prepare difluoroboron β-diketonate fluorophores with intramolecular charge transfer (ICT) character.19 The triphenylamine group is a widely used moiety in organic luminescent materials due to its strong electron-donating ability.50–54 The two phenyl rings in the triphenylamine moiety with twist conformation would generate diverse intermolecular π–π interactions, leading to tunability of the emission properties in the solid state. We anticipate that the combination of dioxaborine and triphenylamine moieties could afford compound 1 with rich photophysical properties.We synthesize compound 1 through the procedure shown in Scheme 1. Compound S2, obtained by a Suzuki reaction of S1 with 4′-bromoacetophenone, reacts with methyl acetate by Claisen condensation to afford β-diketone S3. Compound 1 is synthesized by the complexation of S3 with boron trifluoride. It has been fully characterized by 1H NMR, 13C NMR, and HRMS and further confirmed by single-crystal X-ray analysis.
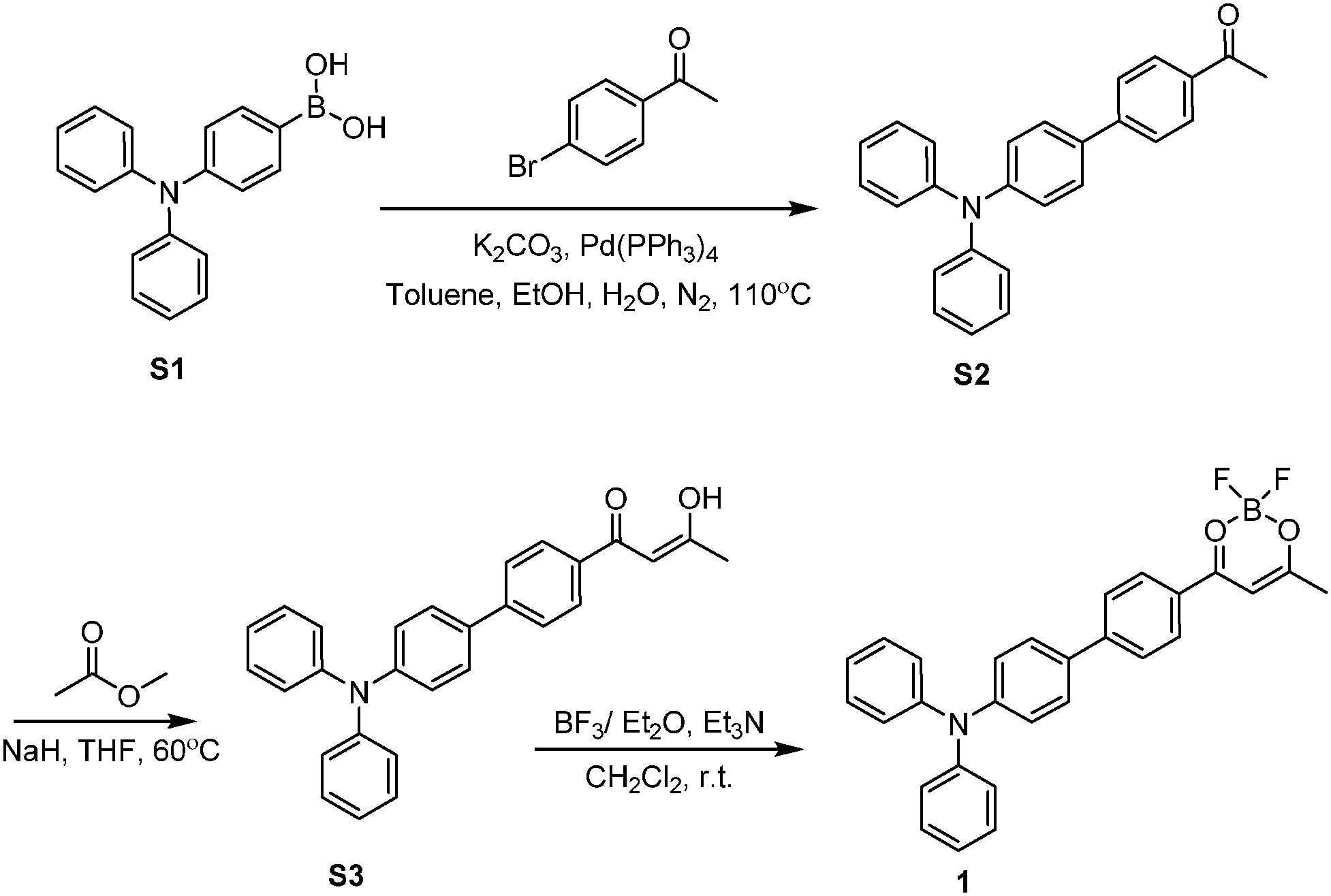 | ||
| Scheme 1 Synthetic route to compound 1. | ||
The photophysical property of 1 in solution
We investigate the influence of solvents with different polarities on the photophysical properties of compound 1 and list the detailed data in Table 1 and Table S2 (ESI†). As shown in Fig. 1, it shows an intense absorption band centered at around 440 nm which is ascribed to intramolecular charge transfer (ICT) transition from the electron-donating triphenylamine units to the electron-accepting dioxaborine ring. The absorption spectra are slightly influenced by the polarity of the solvents, whereas its emission manifests remarkable positive solvatochromism.55,56 With the increase of the polarity of the solvent, the emission peaks red-shift from 481 nm in hexane to 761 nm in acetone, indicating its excited state has an intramolecular charge transfer (ICT) character. Compound 1 displays a strong fluorescence emission in nonpolar solvents with a quantum yield of 0.98. The quantum yield decreases with increasing polarity of the solvent. The emission peak of acetone at short wavelength is assigned to the locally excited (LE) state, while the peak at long wavelength is ascribed to the intramolecular charge transfer (ICT) state. In nonpolar solvents, the emission from the ICT state is much more intense than that from the LE state; so we could only find one emission band. As the polarity of the solvent increases, the emission from ICT is more sensitive to the solvent and decreases drastically. The solvent shows less influence on the emission from the LE state. The ICT emission with the relatively stronger emission from the LE state makes up the two emission bands in acetone. Exceptionally, the photophysical properties of 1 in dichloromethane do not follow the same trend as the other solvents. Compound 1 exhibits deviant red-shifted emission and low quantum yield in dichloromethane, which is probably due to the specific interactions between the π-electrons of the fluorophores with surrounding dichloromethane molecules.| 1 | λ abs/nm | ε/cm−1 M−1 | λ em /nm | Φ f | τ f /ns | k f /109 s−1 | k nr /109 s−1 |
|---|---|---|---|---|---|---|---|
| a The compounds are excited at 438 nm. b Measured using an integrating sphere method in an air atmosphere. c Measured using a single-photo-counting method in an air atmosphere. d Radiative rate constant (kf = Φf/τf). e Nonradiative rate constant (knr = (1 − Φf)/τf). | |||||||
| Hexane | 425 | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
481 | 0.98 | 2.49 | 0.39 | 0.01 |
| Toluene | 438 | 21000 |
556 | 0.98 | 3.4 | 0.29 | 0.01 |
| THF | 430 | 22100 |
671 | 0.53 | 1.4 | 0.38 | 0.34 |
| CH2Cl2 | 442 | 23000 |
727 | 0.01 | 0.8 | 0.01 | 1.24 |
| CHCl3 | 447 | 20800 |
660 | 0.69 | 3.8 | 0.18 | 0.08 |
| Acetone | 429 | 21200 |
761 | 0.03 | 0.5 | 0.06 | 1.94 |
| EA | 425 | 22000 |
661 | 0.21 | 1.4 | 0.15 | 0.56 |
| CH3OH | 378 | 13700 |
— | — | — | — | — |
| CH3CN | 429 | 19900 |
— | — | — | — | — |
| DMSO | 444 | 18400 |
— | — | — | — | — |
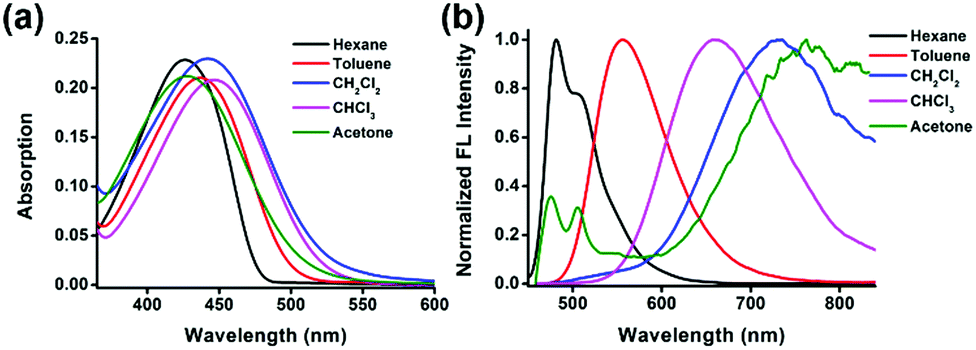 | ||
| Fig. 1 (a) Normalized absorption and (b) emission spectra (λex = 438 nm) of compound 1 (10 μM) in different solvents. | ||
Emission in solid states and reversible mechanochromic luminescence
Compound 1 exhibits different emission colors in crystalline and amorphous solid states with emission bands centered at 596 nm and 670 nm, respectively. The crystals of compound 1 are grown from a solution of mixed solvent of hexane and dichloromethane by a slow evaporation method. As shown in Fig. 2, the sheet-like crystals of compound 1 show bright orange emission centered at 596 nm with a fluorescence quantum yield of 40%. By contrast, through a fast precipitation process by injecting a THF solution of compound 1 into water, an amorphous solid of compound 1 is obtained. The amorphous powder of 1 exhibits broadened and red-shifted emission of 670 nm with fluorescence quantum yield of 25%, which is a 74 nm bathochromic shift compared with that of crystals of 1. The absorption of the amorphous solid also shows a bathochromic shift compared to the crystalline solid (Fig. S7, ESI†).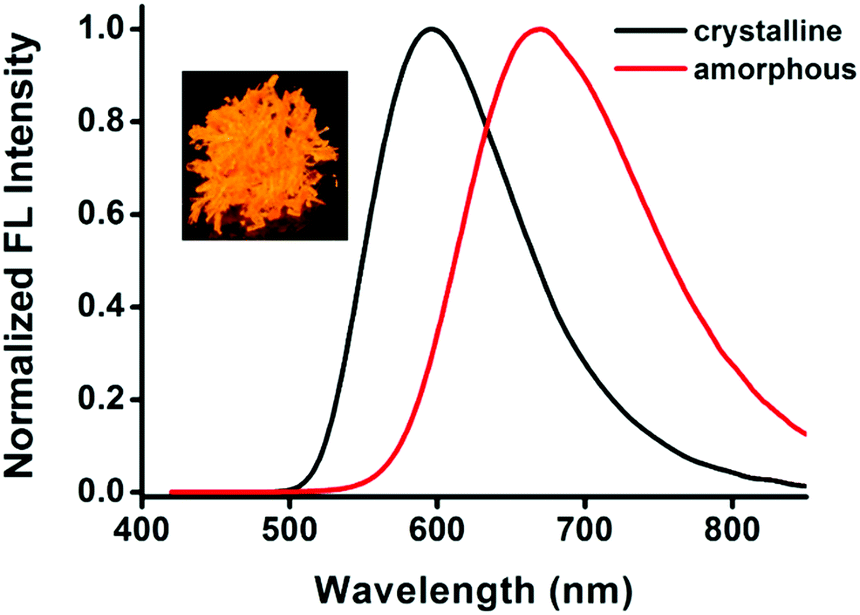 | ||
| Fig. 2 The fluorescence emission spectra of compound 1 in the solid state. Inset: Picture of crystals of compound 1 (under 365 nm UV light). | ||
The single-crystal X-ray diffraction analysis is carried out to gain insight into the relationship among the luminescent properties, molecular conformation and intermolecular interactions. From the crystallographic conformation shown in Fig. 3, the molecular geometry of 1 is relatively coplanar except for the lateral phenyl rings of the triphenylamine unit. The crystals belong to a crystal system of triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with unit cell parameters of a = 9.6654(19), b = 10.237(2), and c = 23.230(5) Å. Each unit cell contains four molecules, adopting a face-to-tail stacking mode at a separation of 3.6 Å (Fig. S8, ESI†). The crystal structure analysis reveals that the planarity of compound 1 results in the good delocalization of the electron in the whole molecule to benefit bright emission in the solid state. Also, two phenyl rings in the triphenylamine segment fan out from the central dioxaborine plane extending the intermolecular distance between the two parallel planes, remarkably reducing the intermolecular π–π interaction, and essentially preventing emission quenching in its aggregated state. The molecular overlap between adjacent molecules in Fig. 3b also indicates that there are weak π–π interactions in the crystal. In addition, each of the lateral phenyl rings of the triphenylamine form close interactions with the rings of the neighbouring molecules. Abundant intermolecular interactions in the crystal lattice strongly restrict intramolecular rotations, rigidify the molecular conformation, and block the non-radiative release of energy in the aggregated state. As for the amorphous solids, the intermolecular interactions are destroyed to some extent, the aryl rings of the molecules can undergo rotation or vibration, and the emission in the amorphous state becomes weaker than that in the crystalline state probably because of the relative enhancement of the non-radiative transition. Besides, the amorphous solids might adopt a more planar conformation compared with crystalline solid and exhibit a red-shifted emission to the crystalline solids.57 In addition, there are other factors that may influence the fluorescence of the solid, like defects and self-trapped excitons.
with unit cell parameters of a = 9.6654(19), b = 10.237(2), and c = 23.230(5) Å. Each unit cell contains four molecules, adopting a face-to-tail stacking mode at a separation of 3.6 Å (Fig. S8, ESI†). The crystal structure analysis reveals that the planarity of compound 1 results in the good delocalization of the electron in the whole molecule to benefit bright emission in the solid state. Also, two phenyl rings in the triphenylamine segment fan out from the central dioxaborine plane extending the intermolecular distance between the two parallel planes, remarkably reducing the intermolecular π–π interaction, and essentially preventing emission quenching in its aggregated state. The molecular overlap between adjacent molecules in Fig. 3b also indicates that there are weak π–π interactions in the crystal. In addition, each of the lateral phenyl rings of the triphenylamine form close interactions with the rings of the neighbouring molecules. Abundant intermolecular interactions in the crystal lattice strongly restrict intramolecular rotations, rigidify the molecular conformation, and block the non-radiative release of energy in the aggregated state. As for the amorphous solids, the intermolecular interactions are destroyed to some extent, the aryl rings of the molecules can undergo rotation or vibration, and the emission in the amorphous state becomes weaker than that in the crystalline state probably because of the relative enhancement of the non-radiative transition. Besides, the amorphous solids might adopt a more planar conformation compared with crystalline solid and exhibit a red-shifted emission to the crystalline solids.57 In addition, there are other factors that may influence the fluorescence of the solid, like defects and self-trapped excitons.
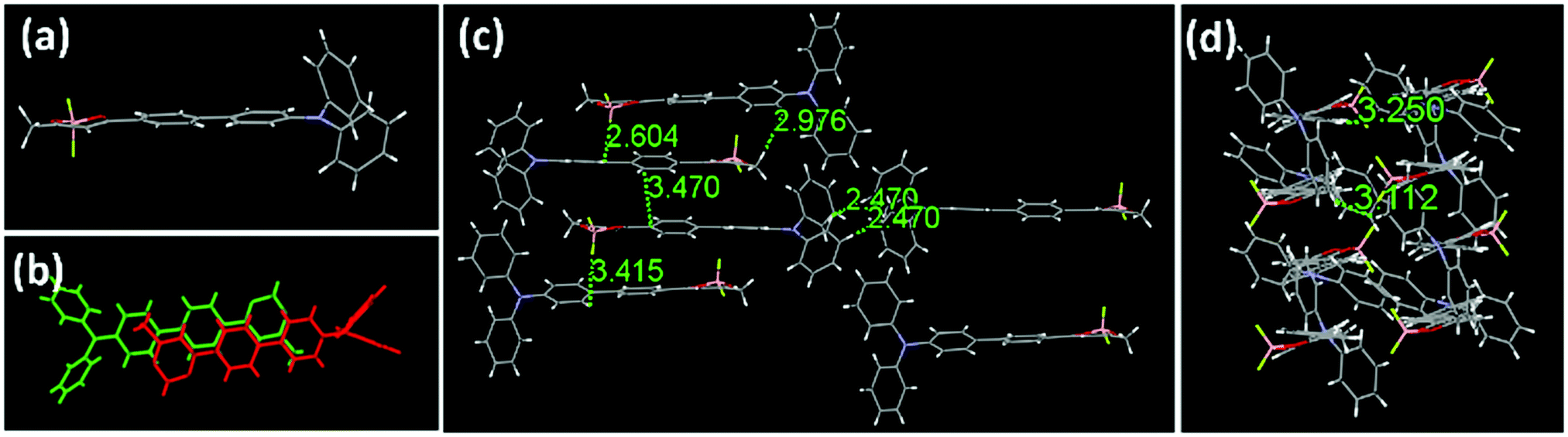 | ||
| Fig. 3 The molecular structures and stacking in the single crystal of compound 1. (a) The molecular conformation of compound 1 in side view. (b) The molecular overlap between adjacent molecules. (c and d) Molecular stacking structures. The solvent molecules are omitted for clarity. | ||
The different emission properties of 1 in crystalline and amorphous solid states encourage us to study the response of solid materials of compound 1 to external stimuli.58–62 After the crystalline powder of compound 1 is thoroughly ground on a glass substrate, the emission maximum red-shifts from 596 nm to 670 nm. The broadened peak and decreased quantum yield suggest its disordered amorphous nature. To verify the hypothesis, powder X-ray diffraction (PXRD) analysis is performed on the crystalline solid before and after grinding. The initial crystalline powder of compound 1 exhibits a distinctive crystalline diffraction pattern due to the well-ordered crystalline structures. By contrast, the ground powder shows no pronounced diffraction, demonstrating the amorphous packing (Fig. S9, ESI†). This result indicates that a crystalline–amorphous phase transition is responsible for the mechanochromic behavior of compound 1.
The ground sample manifests a fast response to the vapor of dichloromethane at room temperature. In a few seconds, its emission color changes from red (670 nm) to orange (596 nm) which is identical to the emission color of the initial crystalline solid (Fig. 4). The PXRD pattern of the fumed sample shows partially reversed-back diffraction peaks to those of the crystalline pattern, which verifies the phase transition. The emission is switchable reversibly between orange and red with a high contrast of 74 nm through the repetitive grinding–fuming processes with little fatigue, as shown in Fig. 4c. Similarly, after being fumed by different organic solvents (chloroform, ethyl acetate, and methyl alcohol), the emission of the ground film switches back to the initial color, suggesting a recrystallization process instead of interaction with the solvent molecules (Fig. 4b). Moreover, the fully ground amorphous solid shows self-recovery properties, and the spontaneous reversal from the amorphous state to crystalline state is visualized through the change of its fluorescence. After 8 hours at room temperature, the ground red emission solid turns back to bright orange emission at 610 nm (Fig. S10a, ESI†).63
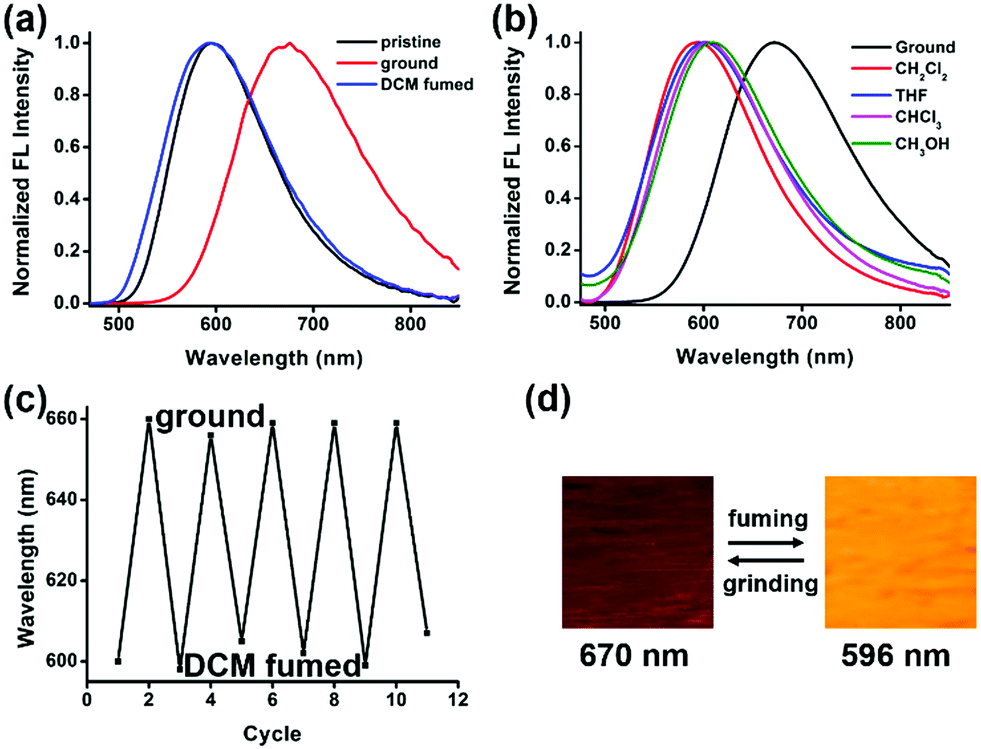 | ||
| Fig. 4 (a) Normalized emission spectra of compound 1 upon external stimuli. (b) Normalized emission spectra of compound 1 upon grinding and fuming by different organic solvents. (c) The reversible switching emission of compound 1 by repeated grinding–fuming. (d) Photographs of compound 1 upon external stimuli. | ||
The differential scanning calorimetry (DSC) study on compound 1 in the crystalline state is shown in Fig. S11 (ESI†). It manifests two endothermic peaks. One sharp peak at 218 °C corresponds to the melting point, and the other one at 86 °C suggests the occurrence of a phase transition. There is an exothermic peak for the amorphous solid at 70 °C, manifesting a phase transition from amorphous to crystalline states. The phase transition is probably because the crystalline solid is thermodynamically more stable than the amorphous solid. This result may also explain the self-recovery process of the amorphous state at room temperature (Fig. S10, ESI†).
Self-assembly behaviour
We obtain aggregates with different morphologies by injecting the solution of compound 1 in THF into water with or without surfactants at room temperature with stirring. By tuning the fraction of THF, the type of surfactants and the concentration of surfactants, we obtain three different nanostructures: nanospheres, nanosheets and nanorods64–68 (Fig. 5b–d). We obtain nanospheres in water and different fractions of THF with a diameter of around 150 nm, while other nanostructures are obtained with the assistance of surfactants. The increase of the fraction of THF (fTHF) is beneficial to form nanocrystals. For example, nanospheres are formed when fTHF = 1/11 in CTAB aqueous solutions, while nanosheets are formed when fTHF is increased to 1/6 (Fig. S6, ESI†). We choose three typical surfactants, including an anionic (lauryl sodium sulfate, SDS), a cationic (cetyltrimethyl ammonium bromide, CTAB) and a nonionic (pluronic F127) surfactant to study the influence of the surfactant on the assembly of 1. With both anionic and cationic surfactants, compound 1 yields thin nanosheets with irregular sizes, while with nonionic surfactant, nanorods with smooth surface and edges are obtained. Scanning electron microscopy (SEM) images reveal that the nanorods are about 100–150 nm in width. The concentration of the surfactants has little influence on the morphology of the nanostructures. The detailed morphology of the nanostructures in different growth conditions is shown in Fig. S6 (ESI†).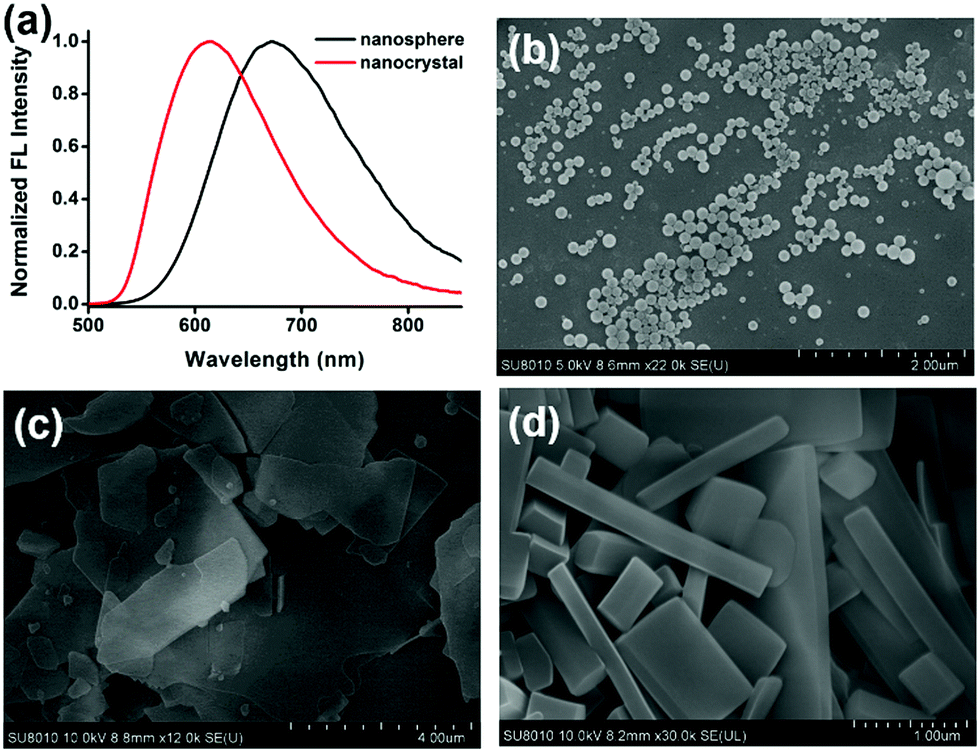 | ||
| Fig. 5 (a) Normalized emission spectra of the nanosphere and nanocrystal dispersion of compound 1. (b–d) The SEM images of compound 1 nanospheres (water), nanosheets (CTAB aqueous solution), and nanorods (F127 aqueous solution), respectively. | ||
The emission of different nanostructures is investigated. The different emission properties of amorphous nanospheres and crystalline nanostructures enable the in situ and real time exploration of the self-assembly process by using fluorescence spectroscopy rapidly and conveniently.64,69 The spectral change during the self-assembly demonstrates the morphological transition process. As shown in Fig. S12 (ESI†), initially, the emission peak at ∼670 nm corresponds to that of the amorphous state, suggesting the formation of nanospheres at the start. Then the emission peak gradually blue-shifts to ∼600 nm that represents the emission of crystals, suggesting the transformation into nanorods with the help of the surfactants. The results are in good agreement with the SEM images (Fig. 5).
TADF property
We observe emission quenching of compound 1 by oxygen in solution. As shown in Fig. S13a (ESI†), bubbling oxygen into the nitrogen saturated solution of 1 in chloroform decreases the fluorescence. After removing oxygen by bubbling nitrogen, the emission is recovered and is more intense than that in the air saturated solution. The lifetime of emission in the presence of oxygen is 3.8 ns. However, the time-resolved emission spectrum of the degassed solution exhibits biexponential decay curves with emission lifetimes of 4.3 ns and 18.0 μs. The above results suggest that the emission might be thermally activated delayed fluorescence (Fig. S12c and S13b, ESI†).To further prove the TADF character of its emission, we study its emission properties in solid states. Compound 1 in the amorphous state exhibits both prompt fluorescence with lifetime of 34.0 ns and a delayed fluorescence with a long lifetime of 13.5 μs (Fig. 6a). To further confirm the delayed fluorescence is TADF, we measure the temperature-dependent lifetime of the amorphous sample. The TADF involves an ISC from S1 to T1 and RISC from T1 to S1, which generates the final emissive singlet excitons. The energy level of the S1 state is located above that of the T1 state, thus the RISC in the TADF process is an endothermal process. This thermally activated process is more and more efficient with increasing temperature. With decreasing temperature from 200 K to 100 K, a decrease of the lifetime of long-lived emission is observed and disappears at last, because there is not enough thermal energy to supply for the RISC process. The temperature-dependent lifetime provides direct evidence for its character of TADF (Fig. 6c).70 By contrast, the crystal of compound 1 exhibits only monoexponential decay with prompt fluorescence decay of 32 ns (Fig. 6b).
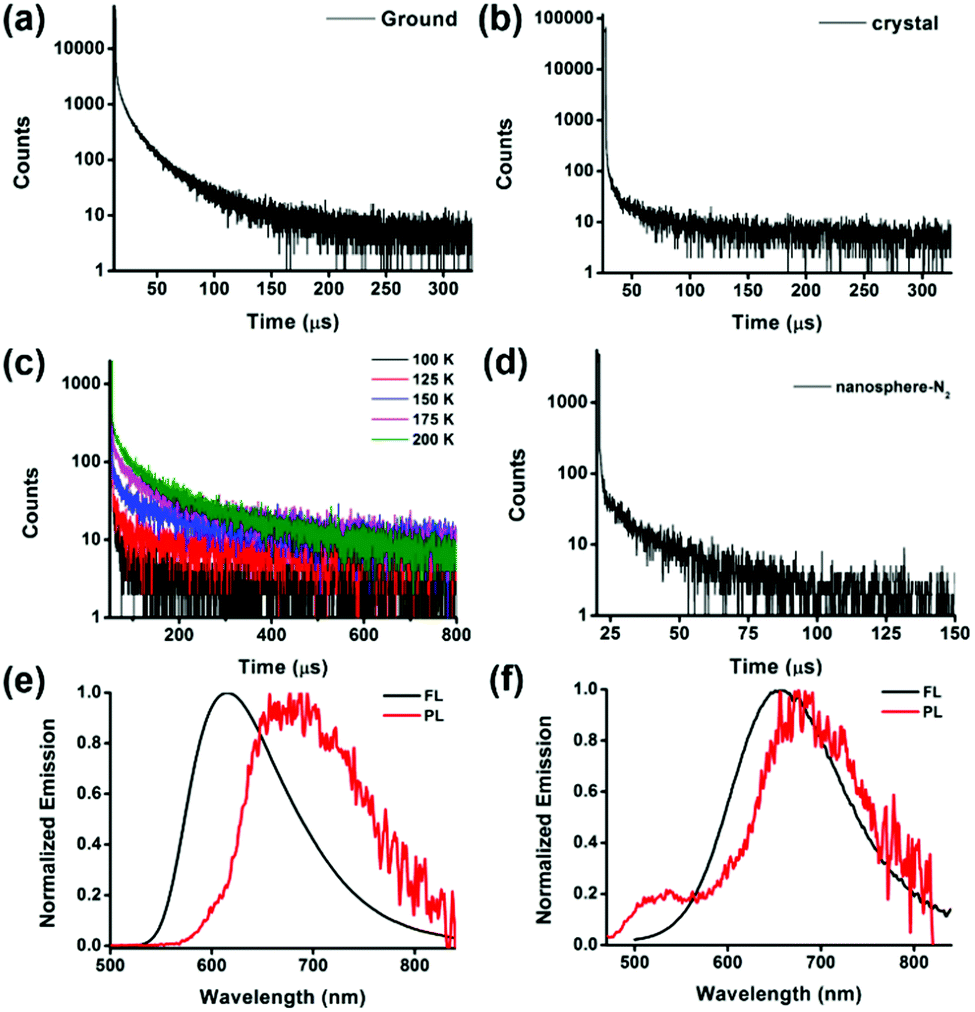 | ||
| Fig. 6 The transient decay of compound 1 in (a) amorphous and (b) crystalline states. (c) The transient decay of compound 1 in the amorphous state at different temperatures. (d) The transient decay of the nanosphere dispersion in aqueous solution under a nitrogen atmosphere. The fluorescence (room temperature) and phosphorescence (77 K) of compound 1 in the crystalline (e) and amorphous (f) state (1 ms delay). | ||
In addition, the nanosphere of compound 1 is in the amorphous aggregated state, and it exhibits a prompt fluorescence at 670 nm with a lifetime of 4.6 ns in an air atmosphere. After bubbling with nitrogen, a delayed lifetime of 12.5 μs appears, as shown in Fig. 6d. However, when it forms crystalline aggregations as nanoslices or nanorods, the delayed fluorescence is not detected even after bubbling with nitrogen. From the above observation, we speculate that the TADF property is not only related to the molecular structure or the environment, but also influenced by the aggregated morphology of the solids.
To investigate how the different morphology of the same molecule could influence the property of excited states to generate different emissions, we compare the fluorescence at room temperature and phosphorescence at 77 K of the crystalline and amorphous states of compound 1, respectively. As exhibited in Fig. 6e and f, the two different solid states show similar phosphorescence, while the amorphous state shows red-shifted fluorescence, suggesting that the decrease in singlet energy level results in a smaller ΔEST from 0.178 eV to 0.045 eV by breaking the organized molecular packing in the crystalline state. The small ΔEST could enhance the RISC and lead to TADF character.
The TADF materials are promising emitters for application in the OLED field. Our study demonstrates that not only the chemical structure, but also the morphology of the molecules would influence the TADF properties, thus affecting the final performance of the OLED devices.
Conclusions
In summary, we develop a triphenylamine-based difluoroboron β-diketonate compound 1 with strong fluorescence in both solutions and solid states, mechanochromic luminescence, and morphology-dependent TADF properties. Compound 1 with D–π–A structure is a typical ICT molecule and exhibits solvatochromism in solution. It shows different emissions in crystalline and amorphous solid states, and the emission is sensitive to external stimuli, such as grinding, heating, and solvent fuming. This compound forms various nanostructures by self-assembly in THF and water with or without the surfactants. Compound 1 in the amorphous state showed a TADF, which is not observed in the crystalline state due to the much larger ΔEST of the crystalline state than that of the amorphous state. This result indicates that the TADF could be tuned through controlling the aggregation states of the fluorophores. Our study may provide a new strategy for the investigation and application of TADF emitters to maximize the device efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21525206, 21971023).Notes and references
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC.
- T.-H. Han, Y. Lee, M.-R. Choi, S.-H. Woo, S.-H. Bae, B. H. Hong, J.-H. Ahn and T.-W. Lee, Nat. Photonics, 2012, 6, 105 CrossRef CAS.
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114 CrossRef CAS PubMed.
- B. Gu, W. Wu, G. Xu, G. Feng, F. Yin, P. H. J. Chong, J. Qu, K.-T. Yong and B. Liu, Adv. Mater., 2017, 29, 1701076 CrossRef PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC.
- F. A. Mann, Z. Lv, J. Großhans, F. Opazo and S. Kruss, Angew. Chem., Int. Ed., 2019, 58, 11469 CrossRef CAS PubMed.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed.
- D.-H. Kim, A. D’Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98 CrossRef CAS.
- T. Butler, M. Zhuang and C. L. Fraser, J. Phys. Chem. C, 2018, 122, 19090 CrossRef CAS.
- J. Huang, N. Sun, J. Yang, R. Tang, Q. Li, D. Ma and Z. Li, Adv. Funct. Mater., 2014, 24, 7645 CrossRef CAS.
- J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577 RSC.
- Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654 CrossRef CAS PubMed.
- X.-L. Ni, S. Chen, Y. Yang and Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177 CrossRef CAS PubMed.
- M. L. Saha, X. Yan and P. J. Stang, Acc. Chem. Res., 2016, 49, 2527 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC.
- J. Mei, Y. Huang and H. Tian, ACS Appl. Mater. Interfaces, 2018, 10, 12217 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC.
- P.-Z. Chen, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, Coord. Chem. Rev., 2017, 350, 196 CrossRef CAS.
- C. A. DeRosa, S. A. Seaman, A. S. Mathew, C. M. Gorick, Z. Fan, J. N. Demas, S. M. Peirce and C. L. Fraser, ACS Sens., 2016, 1, 1366 CrossRef CAS PubMed.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 15728 CrossRef CAS.
- N. Liu, P.-Z. Chen, J.-X. Wang, L.-Y. Niu and Q.-Z. Yang, Chin. Chem. Lett., 2019, 30, 1939 CrossRef CAS.
- J.-Y. Zhu, C.-X. Li, P.-Z. Chen, Z. Ma, B. Zou, L.-Y. Niu, G. Cui and Q.-Z. Yang, Mater. Chem. Front., 2020 10.1039/c9qm00518h.
- H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS PubMed.
- P.-Z. Chen, H. Zhang, L.-Y. Niu, Y. Zhang, Y.-Z. Chen, H.-B. Fu and Q.-Z. Yang, Adv. Funct. Mater., 2017, 27, 1700332 CrossRef.
- L. Wilbraham, M. Louis, D. Alberga, A. Brosseau, R. Guillot, F. Ito, F. Labat, R. Métivier, C. Allain and I. Ciofini, Adv. Mater., 2018, 30, 1800817 CrossRef PubMed.
- T. Xie, B. Zhang, X. Zhang and G. Zhang, Mater. Chem. Front., 2017, 1, 693 RSC.
- H. V. Humeniuk, A. Rosspeintner, G. Licari, V. Kilin, L. Bonacina, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 10559 CrossRef CAS PubMed.
- S. Varughese, J. Mater. Chem. C, 2014, 2, 3499 RSC.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168 CrossRef CAS PubMed.
- Z. Xie, T. Yu, J. Chen, E. Ubba, L. Wang, Z. Mao, T. Su, Y. Zhang, M. P. Aldred and Z. Chi, Chem. Sci., 2018, 9, 5787 RSC.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654 CrossRef CAS PubMed.
- M. L. Daly, C. A. DeRosa, C. Kerr, W. A. Morris and C. L. Fraser, RSC Adv., 2016, 6, 81631 RSC.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2014, 14, 330 CrossRef PubMed.
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777 CrossRef CAS PubMed.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908 CrossRef CAS PubMed.
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, P.-Y. Huang, M.-J. Huang, C.-Z. Ren-Wu, C.-Y. Yang, M.-J. Chiu, L.-K. Chu, H.-W. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2016, 138, 628 CrossRef CAS PubMed.
- S. Shao, J. Hu, X. Wang, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2017, 139, 17739 CrossRef CAS PubMed.
- K. Wang, C.-J. Zheng, W. Liu, K. Liang, Y.-Z. Shi, S.-L. Tao, C.-S. Lee, X.-M. Ou and X.-H. Zhang, Adv. Mater., 2017, 29, 1701476 CrossRef PubMed.
- K. Wu, T. Zhang, Z. Wang, L. Wang, L. Zhan, S. Gong, C. Zhong, Z.-H. Lu, S. Zhang and C. Yang, J. Am. Chem. Soc., 2018, 140, 8877 CrossRef CAS PubMed.
- K. Isayama, N. Aizawa, J. Y. Kim and T. Yasuda, Angew. Chem., Int. Ed., 2018, 57, 11982 CrossRef CAS PubMed.
- R. Pashazadeh, P. Pander, A. Lazauskas, F. B. Dias and J. V. Grazulevicius, J. Phys. Chem. Lett., 2018, 9, 1172 CrossRef CAS PubMed.
- L. Yang, X. Wang, G. Zhang, X. Chen, G. Zhang and J. Jiang, Nanoscale, 2016, 8, 17422 RSC.
- X. Peng, F. Song, E. Lu, Y. Wang, W. Zhou, J. Fan and Y. Gao, J. Am. Chem. Soc., 2005, 127, 4170 CrossRef CAS PubMed.
- F. Qian, C. Zhang, Y. Zhang, W. He, X. Gao, P. Hu and Z. Guo, J. Am. Chem. Soc., 2009, 131, 1460 CrossRef CAS PubMed.
- Y. Suzuki and K. Yokoyama, J. Am. Chem. Soc., 2005, 127, 17799 CrossRef CAS PubMed.
- D. Zhang, V. Martín, I. García-Moreno, A. Costela, M. E. Pérez-Ojeda and Y. Xiao, Phys. Chem. Chem. Phys., 2011, 13, 13026 RSC.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348 RSC.
- H. Li, Z. Chi, B. Xu, X. Zhang, X. Li, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2011, 21, 3760 RSC.
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frère and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459 CrossRef CAS PubMed.
- Y. Tang, Y. Wang, X. Li, H. Ågren, W.-H. Zhu and Y. Xie, ACS Appl. Mater. Interfaces, 2015, 7, 27976 CrossRef CAS PubMed.
- P. Xue, P. Chen, J. Jia, Q. Xu, J. Sun, B. Yao, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569 RSC.
- G. Signore, R. Nifosì, L. Albertazzi, B. Storti and R. Bizzarri, J. Am. Chem. Soc., 2010, 132, 1276 CrossRef CAS PubMed.
- S. Uchiyama, K. Kimura, C. Gota, K. Okabe, K. Kawamoto, N. Inada, T. Yoshihara and S. Tobita, Chem. – Eur. J., 2012, 18, 9552 CrossRef CAS PubMed.
- Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429 CrossRef CAS PubMed.
- J. Han, J. Sun, Y. Li, Y. Duan and T. Han, J. Mater. Chem. C, 2016, 4, 9287 RSC.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302 CrossRef CAS PubMed.
- Y. Li, L. Chen, Y. Ai, E. Y.-H. Hong, A. K.-W. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 13858 CrossRef CAS PubMed.
- Y. Sagara, K. Kubo, T. Nakamura, N. Tamaoki and C. Weder, Chem. Mater., 2017, 29, 1273 CrossRef CAS.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed.
- J. Liu, C. Xing, D. Wei, Q. Deng, C. Yang, Q. Peng, H. Hou, Y. Li and K. Li, Mater. Chem. Front., 2019, 3, 2746 RSC.
- Y. Li, T. Liu, H. Liu, M.-Z. Tian and Y. Li, Acc. Chem. Res., 2014, 47, 1186 CrossRef CAS PubMed.
- A. Shao, Y. Xie, S. Zhu, Z. Guo, S. Zhu, J. Guo, P. Shi, T. D. James, H. Tian and W.-H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 7275 CrossRef CAS PubMed.
- J. Wu, X. Zhu, Y. Guan, Y. Wang, F. Jin, R. Guan, F. Liu, M. Chen, Y. Tian and S. Yang, Angew. Chem., Int. Ed., 2019, 58, 11350 CrossRef CAS PubMed.
- C. Yang, Q. T. Trinh, X. Wang, Y. Tang, K. Wang, S. Huang, X. Chen, S. H. Mushrif and M. Wang, Chem. Commun., 2015, 51, 3375 RSC.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 2022 CrossRef CAS PubMed.
- P.-Z. Chen, L.-Y. Niu, H. Zhang, Y.-Z. Chen and Q.-Z. Yang, Mater. Chem. Front., 2018, 2, 1323 RSC.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1962413. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00672a |
| This journal is © the Partner Organisations 2020 |