
Subtle substitution controls the rainbow chromatic behaviour of multi-stimuli responsive core-expanded pyrenes†
David T.
Hogan
 a,
Benjamin S.
Gelfand
a,
Denis M.
Spasyuk
b and
Todd C.
Sutherland
*a
a,
Benjamin S.
Gelfand
a,
Denis M.
Spasyuk
b and
Todd C.
Sutherland
*a
aDepartment of Chemistry, University of Calgary, 2500 University Dr NW, T2N 1N4, Calgary, Alberta, Canada. E-mail: todd.sutherland@ucalgary.ca
bCanadian Light Source Inc., 44 Innovation Blvd., S7N 2V3, Saskatoon, Saskatchewan, Canada
First published on 6th December 2019
Abstract
Materials that respond to multiple environmental stimuli by altering a physical property such as colour are fundamental to development of smart devices. Five core-expanded pyrene derivatives are presented to demonstrate that substitution by ring-fusion and addition of distal groups controls responsiveness to mechanical force, solvent, and acid in both solution and solid-state. Contained herein is the first direct observation of the subtle intermolecular forces that govern crystal-to-crystal transition in a new class of mechanochromic pyrene. Shearing the solid controls the aggregation state, altering the yellow-fluorescent J-type aggregated pristine material to orange-fluorescent individual monomers. X-ray diffraction analysis of each crystalline polymorph against a close structural analogue revealed that this force-induced behaviour was permitted by slight differences in distal substitution. In solution, the five core-expanded pyrenes display a rainbow of fluorescent colours in response to solvent polarity and acid. The breadth and location on the rainbow spectrum depends upon the style of ring-fusion as well as distal substitution on pyrene. Responsiveness to acid is mirrored in the solid-state – acid sensing is ambiently-reversible simply over time, without the need for additional base. Self-regenerating behaviour is permitted by only one of the two styles of ring-fusion borne by the core-expanded pyrenes.
Introduction
Stimulus-responsive materials are driving the growth of smart technology in the fields of molecular switches and machines,1 dynamic polymers,2 optical information storage,3 as well as biological probes4 and drug delivery.5 For a compound to be deemed ‘smart’, it must respond to the presence of an environmental stressor or external stimulus in an observable way. Chromatic materials are an important subclass which epitomise an observable response by relaying colours to the naked eye, forming the basis for smart electrochromic6 and thermochromic7 optics. The broad array of stimuli can include light,8 heat,9 solvent and vapours,10 acidity or basicity,11 and mechanical force.12,13 Due to the ability to sense polarity changes in their microenvironment and provide an optical response, solvatochromic molecules are being explored as dyes for cellular fluorescence microscopy14 and sensors for toxic organic vapours.10 When acidochromic compounds contact acid, Brønsted basic sites are protonated, which switches their character from electron-donating to electron-withdrawing and potentially redirects molecular conjugation pathways.15 The resulting colour change is used for contrast in write-erase encrypted inks16,17 and to mark the location of targeted acidic cellular organelles.18Research that deconvolutes the nature of molecular packing in the solid-state and its effects on macroscopic optical and electronic properties promises to aid the development of organic light emitting diodes, field-effect transistors, and photovoltaics.19 Such devices rely on hydrophobic dye molecules with large planar surfaces that interact with neighbours through a combination of dispersion and pi–pi interactions. The directionality of these weak forces rarely permits the existence of multiple polymorphs of a single compound.20 When mechanical force is used to change from one polymorph to another and the forms exhibit distinct visible and emitted colour, the material is termed mechanochromic.21,22 Shearing, crushing, straining, and hydrostatic pressure apply different stress to solids and each can result in the formation of new polymorphs, widening the scope of application for solid-state force sensors.23 The changes in structural properties associated with crystal-to-crystal transitions provide valuable information on the flexibility of forces that hold molecules together, which can be used to design better performing solid-state devices.24 If the transition is from crystalline to amorphous, then it is considerably less valuable because packing information must be indirectly inferred from electronic spectroscopy.
Considering the staggering number of environmental stimuli, modern technology is trending towards integrated sensing of multiple inputs.25 Such a strict design criterion requires that components respond to many individual stimuli, so the marketed devices are single-component and therefore economical to produce. The multi-stimuli responsive materials that emerge from this next-generation design might eventually be marketable as thermo-force26 sensors for wearable smart-skins.27
Pyrene is a polycyclic aromatic hydrocarbon (PAH) that has attracted attention from physical, biomedical, and synthetic chemists for many decades. Owing largely to its well-studied photophysical properties, pyrene has been incorporated into biological fluorescent probes,28 aggregation-induced emission fluorophores,29–32 molecular liquids,33 and two-photon absorbing dyes.34,35 This variety of application has been possible because of extensive research to control and document its reactivity.36–40 One desirable outcome of this reactivity is extension of pi-electron delocalisation through expansion of the core, also called ring-fusion.41 Core-expanded PAHs, including pyrene, are actively studied for organic field-effect transistors, light-emitting diodes, and photovoltaics because they absorb greater portions of the electromagnetic spectrum than their non-expanded analogues.42,43
By virtue of their promising absorption and emission profiles, core-expanded pyrenes can be strongly coloured.44–46 Incorporating such compounds into chromatic multi-stimuli responsive materials is therefore valuable to the smart materials community. A literature survey of all mechanochromic pyrenes in Section S1 of the ESI† uncovered 27 reports, 15 of which used simple monosubstituted pyrenes and only one explored core-expanded derivatives.47 All but one report involved transition from ordered or crystalline states into amorphous or poorly-ordered states.48 Packing information after mechanical force, grinding in this case, was inferred by spectroscopic measurements and thus failed to reveal any long-range structural details critical to solid-state materials design. There is only one report of crystal-to-crystal transition; however, X-ray structures for both polymorphs were not obtained, obscuring molecular-scale insight into the origins of the transition.
Here, the direct crystal-to-crystal transformation of core-expanded pyrenes is reported along with an investigation of the subtle intermolecular forces that govern solid-state stimulus-responsiveness in a new class of mechanochromic substance. The solid-state aggregation of a quinoxaline-fused pyrene could be controlled by shearing the solid, inducing a crystalline change from yellow-fluorescent, J-type aggregates to orange-fluorescent weakly-interacting monomers. Other core-expanded pyrenes were crystallographically characterised to explore the crystal-to-crystal transition, which relies upon the nature of distal substituents on pyrene. By happenstance, the core-expanded pyrenes also exhibited intense blue-to-red solvatochromic fluorescence, blue-to-red fluorescent acidochromism in solution, and self-regenerable acidochromism in the solid-state. This rainbow chromatic, multi-responsiveness molecule in solution and solid-state was discovered to depend upon the nature of core-expansion, as quinoxaline-fusion differed from dicyanopyrazine-fusion.
Results and discussion
Synthetic procedures
Commercially available pyrene served as the scaffold for construction of all core-expanded pyrenes. Diversification of substituted pyrene diketones 1, 2, and 3 following Path A in Scheme 1 yielded quinoxaline-fused pyrenes H2quin, Ph2quin, and NMe2quin. Diaminomaleonitrile was condensed with 1 and 2 to yield dicyanopyrazine-fused pyrenes H2pyr and Ph2pyr.45 Pyrene diketone 3 failed to yield NMe2pyr under the same conditions, likely due to poor electrophilicity of diketone 3, compounded by poor nucleophilicity of diaminomaleonitrile. Ph2quin, NMe2quin and Ph2pyr have not been reported because the synthetic methods to achieve this substitution pattern on pyrene have only recently been developed.49 The complete synthetic route, synthetic details and characterizations of all compounds are in Section S3 of the ESI.†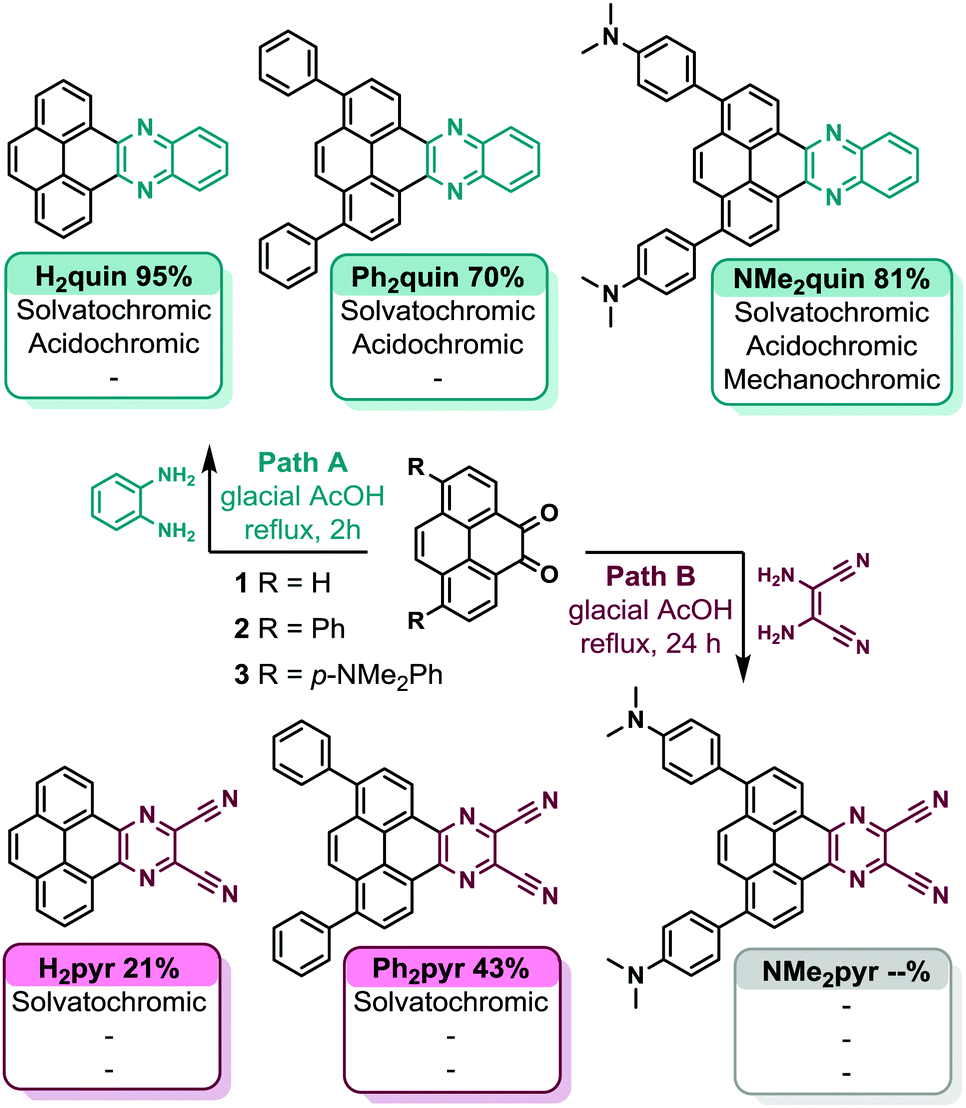 | ||
| Scheme 1 Divergent synthesis of core-expanded pyrenes. Path A yields quinoxaline-fused pyrenes highlighted in teal, and Path B yields dicyanopyrazine-fused pyrenes in cerise. NMe2pyr could not be synthesized. | ||
Mechanochromism of NMe2quin
Following isolation from a flask, shearing the saffron-yellow NMe2quin with a metal rod produced red-orange streaks that fluoresced orange, distinct from the yellow-fluorescent pristine solid. More solid was ground using an agate mortar and pestle, the proton nuclear magnetic resonance (1H NMR) spectrum of which in Section S4b of the ESI† is identical to pristine NMe2quin, ruling out mechanochemical transformation. When preparing single-crystals, yellow-fluorescent yellow prisms and orange-fluorescent orange rods, shown in Fig. 1b and c, grew from the same solution. Comparing the two crystalline polymorphs to bulk NMe2quin powder X-ray diffraction (PXRD) in Fig. 1a, the orange rods (NMe2quin-grou) match the ground sample. The yellow prisms (NMe2quin-pris) match pristine powder, with the diffraction peak at 6.3° originating from minor inclusion of the ground polymorph. Given this information, the origin of the mechanochromism lies in transition from one crystal form to another. This appears to be the only report for pyrene where analysis of crystal structures before and after grinding leads to identification of packing features responsible for mechanochromism. | ||
| Fig. 1 (a) PXRD patterns of pristine (yellow) and ground (red) NMe2quin, compared to the simulated patterns of NMe2quin-pris and NMe2quin-grou crystals. Polarized optical microscopy images of (b) NMe2quin-pris and (c) NMe2quin-grou crystals viewed with 200× magnification. | ||
While some materials change from crystalline to amorphous in response to external force,50–52 the exact molecular orientation in a crystal-to-crystal transition is inspectable by X-ray diffraction. Observing a mechanochromic event at the molecular level is critical for identifying changes in packing motifs and rationally designing the next generation of force-responsive materials. As shown in Fig. 2a, pristine solid NMe2quin-pris is composed of vertically stacked pairs with near-planar pyrene cores offset by ca. 28° in J-type aggregates,53 and inter-pyrene pi–pi mean-plane distances of 3.601 Å. Quantitative analysis of the short-contacts reveals the directionality of the intermolecular forces and the overall packing motifs. Of the 88 short-contacts in the unit cell shown in Fig. 2b, 68% are oriented vertically between molecules in a stack while 32% are horizontal between two parallel stacks. Noteworthy among these are the 16 C⋯C and C⋯N interactions at 3.148–3.286 Å long, resulting from close inter-pyrene pi–pi stacking. The remaining 72 short-contacts are made up of 8 N⋯H hydrogen bonds, 48 C–H⋯pi and 16 H⋯H interactions.
 | ||
| Fig. 2 Packing in NMe2quin-pris crystals in the upper panels viewed along the a-axis, highlighting (a) the close intermolecular distances and angles of J-type aggregated molecules in three separate stacks and (b) the vertically dominant short-contacts in the unit cell. Packing in NMe2quin-grou crystals in the lower panels viewed along the b-axis, highlighting (c) the large intermolecular distances between four separate sheets and (b) the horizontally dominant short-contacts in the unit cell. All ellipsoids are shown with 50% probability. In (b) and (d), C and N atoms are shown in grey and blue, respectively, while H atoms have been omitted for clarity. | ||
The packing in ground solid NMe2quin-grou in Fig. 2c is instead composed of alternating sheets with the pyrene core mean-planes separated by at least 4.283 Å. Intermolecular pi–pi interactions are not significant at this distance54,55 so the composition can be likened to individual weakly-interacting monomers. There are 78 short-contacts in the unit cell in Fig. 2d, and they are switched relative to NMe2quin-pris – 67% are horizontal between molecules in a sheet and 33% are vertical between two parallel sheets. There are also no heavy atom-heavy atom interactions; short-contacts are composed of 6 N⋯H hydrogen bonds, 64 C–H⋯pi and 8 H⋯H interactions. This is likely a consequence of the greater inter-pyrene distances in NMe2quin-grou. The difference in directionality of the packing supports the dominance of J-type aggregation in NMe2quin-pris which is forcefully suppressed to form monomeric NMe2quin-grou.
Considering the disparity between the number of short-contacts and pi–pi interactions, the relative stability of the two polymorphs was explored by dispersion energy-corrected density functional theory (DFT) calculations. As an approximation, the DFT energy of crystal structures containing twelve molecules of NMe2quin-pris and NMe2quin-grou was calculated and then compared to twelve isolated NMe2quin molecules from those crystals. This method models the dispersive stabilization energy from putting molecules close together. The data in Table S8 (ESI†) indicate that NMe2quin-pris is the more stable form of NMe2quin and thus mechanical force is required to overcome the strong intermolecular forces in the pristine form. Full results are available in Section S5c of the ESI,† while computational specifications are in the Computational ESI.†
Curiously, NMe2quin is the only pyrene derivative of the five to exhibit mechanochromism, which presents an opportunity to explain why this phenomenon appears selective. Crystal-to-crystal mechanochromism would only occur if the molecules can pack into stable polymorphs, otherwise the order would be destroyed as the solid enters an amorphous phase. Analysing the packing in NMe2quin-grou in Fig. 2b, most stabilizing short-contacts are between molecules in the same sheet (vide supra). What's more, 24 of the 78 short-contacts involve the dimethylamino groups, meaning this functional group contributes greatly to packing.
It is hypothesized that these groups balance the energetic cost of separating the pyrene cores with extra dispersion interactions, a strategy employed to promote dimerization of super-bulky trityl radicals.56,57 This extra dispersion energy would be necessary to allow NMe2quin to access a polymorph where the pyrene cores only weakly interact. The closest structural analogue, Ph2quin, lacks dimethylamino groups on the phenyl rings and is not mechanochromic. Shown in Fig. S27 (ESI†), these molecules pack into close antiparallel sheets separated by 3.375 Å. Intermolecular pi–pi interactions dominate the crystal structure – visual inspection reveals strongly directional inter-sheet short-contacts. Of the 134 short-contacts in the unit cell, 66% are vertical between sheets while 34% are between molecules in a sheet. The prevalence of pi–pi interactions between sheets in Ph2quin, coupled with the absence of a dispersion energy donating group,56,57 as in NMe2quin, suggests that it would not be able to occupy a polymorph akin to NMe2quin-grou. This subtle substitution effect is a potential origin for the shear-controllable aggregation in NMe2quin.
Solid NMe2quin responds to mechanical force by changing visible and emitted colour. The absorption spectrum of yellow NMe2quin-pris in Fig. 3a is composed of structured peaks around 490 nm, corresponding to restricted molecular motion in the excited state from tight J-aggregation. When ground, the bands coalesce into a broad peak at 483 nm without vibrational fine structure because the molecules have freedom to vibrate and rotate. Even though NMe2quin-grou appears red-orange, implying a redshift of the absorption bands when in fact there is a 7 nm blueshift, the increased band breadth accounts for the new colour. This is accompanied by dramatic changes in fluorescence, from yellow at 556 nm to orange at 614 nm in Fig. 3a, c and d. Spectroscopic means are commonly used to identify aggregated states of molecular dyes.53 The pristine form bears the hallmarks of J-aggregation – a redshifted absorption peak and lesser Stokes shift when compared to those of the monomeric ground form. The correlation between pristine/ground material and the crystal polymorphs NMe2quin-pris/NMe2quin-grou was further strengthened by acquiring single-crystal fluorescence microscopy images. In Fig. 3e, the NMe2quin-pris prisms exhibited the highest intensity on the green channel (520–570 nm) while the NMe2quin-grou rods were most intense in the orange (545–675 nm) and red channels (555–705 nm). This distinction in fluorescence wavelengths mimics that of the pristine and ground powders.
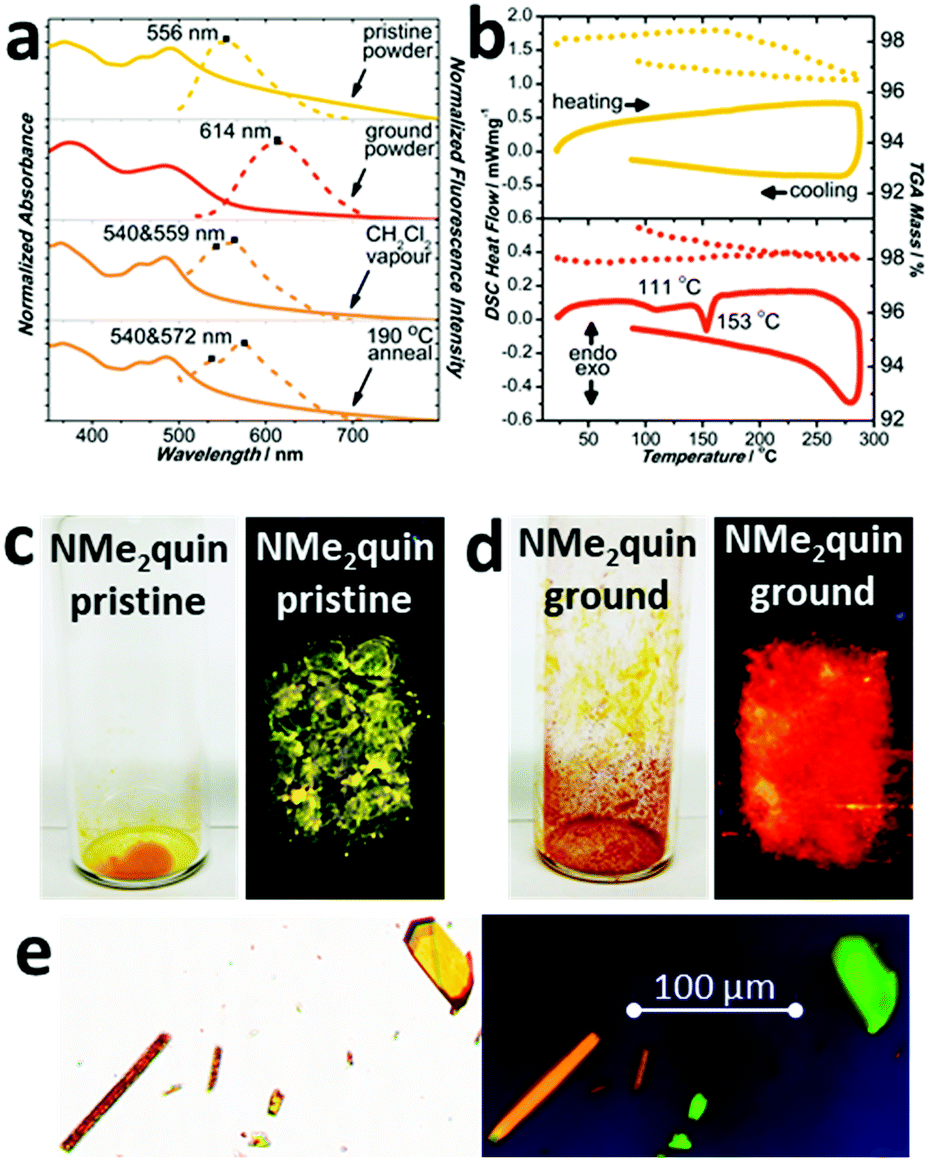 | ||
| Fig. 3 (a) UV-visible absorption and fluorescence profiles of pristine (yellow, λexc = 480 nm) and ground (red, λexc = 490 nm) NMe2quin, and those after annealing with CH2Cl2 vapour or at 190 °C (λexc = 480 nm). (b) Differential scanning calorimetry profiles of pristine (yellow) and ground (red) NMe2quin accompanied by the dotted curves of simultaneous thermogravimetric analysis. Photographs under ambient and 365 nm UV light of (c) pristine and (d) ground solid NMe2quin. Images of prismatic NMe2quin-pris and rod-like NMe2quin-grou from (e) visible light optical microscopy and fluorescence microscopy both at 200× magnification with the green (λexc = 430–510 nm, λem = 475–575 nm) and orange (λexc = 520–570 nm, λem = 545–675 nm) channels overlaid onto the image. | ||
Reversing the transition from NMe2quin-pris to NMe2quin-grou can be accomplished in a few different ways. Differential scanning calorimetry in Fig. 3b on NMe2quin-grou showed two exothermic transitions – a broad, cold crystallisation at 111 °C and a sharp phase transition at 153 °C, with no mass loss observed by thermal gravimetric analysis. The same analysis in Fig. 3b on pristine solid showed no thermal events. Variable temperature PXRD in Fig. S37 (ESI†) indicated that these thermal events are both associated with gradual reversion back to NMe2quin-pris, serving to thermally re-aggregate the sample. That this transition is exothermic adds to the validity of conclusions from DFT calculations on the greater stability of J-aggregated NMe2quin-pris crystals. The same re-aggregation can be induced by exposing the ground solid to dichloromethane vapours. After heating to 190 °C or vapour-exposure, the absorption spectra in Fig. 3a regain their fine structure and the emission blueshifts back to the yellow region. The change is also apparent in Fig. S113 (ESI†) by visual inspection of the solid before and after regeneration. However, the peaks in the regenerated spectra differ by a few nanometres. The variable temperature and post vapour-exposure PXRD in Fig. S38 (ESI†) indicate that only NMe2quin-pris forms at the expense of NMe2quin-grou; there are no extraneous peaks. Any spectral differences can therefore be attributed to generation of minor quantities of amorphous material during reversible control of the aggregation.
Solvatochromism of core-expanded pyrenes
During purification, solutions of the core-expanded pyrenes displayed a variety of emissive colours upon exposure to 365 nm ultra-violet (UV) light simply by changing the organic solvent. Such colourful behaviour prompted a spectroscopic investigation of the compounds’ absorptive and emissive properties in solution, and it became clear that the range of colours was affected by substitution pattern and style of ring-fusion. The absorption spectra of all five pyrenes in Fig. S44–S51 (ESI†) show no peak shifting trends in solvents of varying polarity, from cyclohexane to dimethylsulfoxide, and each remains visibly yellow. DFT calculations, shown in Fig. S52–S56 (ESI†), reproduce the absorption spectra with moderate to excellent accuracy, yielding from 1 nm to 46 nm wavelength deviation. The spatial separation of the highest-occupied molecular orbital (HOMO) from the lowest unoccupied molecular orbital (LUMO) in the Kohn–Sham orbital surfaces in Fig. S1–S9 (ESI†) suggest that each core-expanded pyrene has donor–acceptor character. Detailed results of computational methods are available in the ESI,† while information on the computational procedures is in the Computational ESI.†Although the visible colours do not change, the colours emitted under UV light in Fig. 4c–g display dramatic solvatochromism depending on the nature of the solvent.14 Each colour of the rainbow, except purple, is represented by the five pyrene derivatives. The quinoxaline-fused pyrenes H2quin and Ph2quin span the colours from deep-blue to sea-green, shifting fluorescence peak wavelength by 49 nm and 52 nm in Fig. S57 and S61 (ESI†). In similar fashion the dicyanopyrazine-fused derivatives H2pyr and Ph2pyr span deep-blue to orange and the peaks shift by 97 nm and 99 nm in Fig. 4a and Fig. S65 (ESI†). These two sets demonstrate that while appending simple phenyl groups hardly affects the solvatochromism, the nature of ring-fusion has a more dramatic effect. Dicyanopyrazine is more electron-withdrawing than quinoxaline and creates more pronounced donor–acceptor character,45 which increases the influence of solvents on the fluorescence energy.58,59 The fluorescence of NMe2quin spans 137 nm as shown in Fig. S69 (ESI†), the most of the five pyrenes due to its dimethylamino groups, resulting in sea-green to orange-red emission. For all compounds, fluorescent efficiencies (Φf range from 0.03 to 0.4) are in Table S15 (ESI†) and their absorption and excitation spectra are compared in Fig. S75–S79 (ESI†).
 | ||
| Fig. 4 (a) Fluorescence spectra of 10 μM Ph2pyr (λexc = 450 nm) in solvents of increasing polarity. (b) Plots of Ph2pyr fluorescence peak wavelength against Catalán's SdP (closed squares) and SP parameters (open squares); solid and dashed lines show least-squares linear regression with goodness-of-fit. (c–g) Photographs of 10 μM solutions of H2pyr, Ph2pyr, H2quin, NMe2quin and Ph2quin under 365 nm UV light in solvents of increasing polarity. | ||
Analysis of solvent–solute interactions using the method proposed by Catalán60 suggests that non-specific interactions dominantly affect the core-expanded pyrenes. The superior linear fitting to dipolarity in Fig. 4b and Fig. S58, S62, S66, S70 and the very poor correlation to specific solvent–solute effects in Fig. S59, S63, S67, S71 and S73 (ESI†) show that this is true for all five pyrene derivatives. The Stokes shifts also correlated to the empirical ET(30) scale58 and to the Lippert–Mataga equation61,62 in Fig. S60, S64, S68, S72 and S74 (ESI†) supporting the presence of intra-molecular charge transfer. Increasingly polar solvents stabilize the charge-transfer state, leading to red-shifted fluorescence. This experimental observation is in accord with the locations of DFT-calculated Kohn–Sham orbitals in Fig. S1–S9 (ESI†). It is this pronounced donor–acceptor character – strongly affected by the choice of ring-fusion – which bestows rainbow deep-blue to orange-red responsiveness to solvent dipolarity upon the core-expanded pyrenes.
Acidochromism of core-expanded pyrenes
To continue investigating the effects of p-dimethylaniline, quinoxaline and dicyanopyrazine moieties on the stimulus-responsiveness of core-expanded pyrenes, the environmental acidity was modified. Monitoring the absorption profiles as trifluoroacetic acid (TFA) is added to H2quin and Ph2quin in CH2Cl2, in Fig. 5a, c and Fig. S83 (ESI†), the original peaks decrease as new green and yellow-absorbing peaks appear over 100 nm away. The cyan and sea-green fluorescence of H2quin and Ph2quin in Fig. 5b, d and Fig. S84, S85 (ESI†) also fade as weak redshifted fluorescence appears 200 nm and 160 nm. Matching with DFT-calculated absorption spectra in Fig. S94 and S95 (ESI†), supports protonation of the quinoxaline nitrogen, which decreases the energy of the LUMO. The excitation spectra are recorded in Fig. S98 and S99 (ESI†), matching the absorption spectra. Simple protonation reactions are often reversed in the presence of base and addition of triethylamine (NEt3) regenerates the original absorption and fluorescence peaks of H2quin and Ph2quin to their pre-TFA positions, in Fig. S80–S82 and S86–S88 (ESI†). | ||
| Fig. 5 Solution acidochromism in the upper panels. Titrations of TFA into 4 μM CH2Cl2 solutions of H2quin monitored by (a) UV-visible absorption and (b) fluorescence spectroscopy (λexc1 = 415 nm, λexc2 = 535 nm). Arrows indicate the direction of peak growth/decay, initial traces are light orange and final traces are dark orange, and the asterisk marks amplified baseline from ×15 magnification. Photographs of H2quin and Ph2quin TFA titrated-CH2Cl2 solutions under (c) ambient and (d) 365 nm UV light. Solid acidochromism in the lower panels. (e) UV-visible spectroscopy profile of self-regenerating solid H2quin, post exposure to HCl vapours. (f) Recycling fluorescence spectra of solid H2quin after exposure and evaporation of HCl vapour. Photographs of solid H2quin under (g) ambient and (h) 365 nm UV light before and after exposure to HCl vapour. | ||
If the solids were also to behave in this reversible manner, they would be more easily applied into vapour-sensing devices. Solid H2quin in Fig. 5g initially appears lemon yellow under ambient light and fluoresces bluish green in Fig. 5h; optical characterization is presented in Fig. S103 and S104 (ESI†). Molecules of H2quin in both the crystalline (Fig. S29 and S30, ESI†) and bulk powder (Fig. S39, ESI†) forms are aligned in helical stacks of overlapping pyrene cores, separated by intermolecular distances of 3.422 Å. Upon exposure to HCl vapours for 2 hours the sample darkens to purple-red, which is marked by appearance of a broad shoulder at 535 nm in the absorption spectrum in Fig. S105 (ESI†) and loss of fluorescence in Fig. S106 (ESI†). Intriguingly, in Fig. 5e and f the original spectra are recovered after only 5 minutes by allowing the HCl to evaporate. No external base is required to revert the acidochromism, which is promising for self-regenerating recyclability. The vapour-sensing process leaves the molecular and solid-state packing structures of H2quin unperturbed, by 1H NMR in Section S4b (ESI†) and PXRD analyses in Fig. S40 (ESI†). The reversible acidochromic response is demonstrated by unchanged absorption and fluorescence profiles in Fig. 5f and Fig. S107 (ESI†) after five HCl treatment cycles. Contrary to H2quin, solid Ph2quin does not respond to the presence of acidic vapours even though it bears a quinoxaline structural unit, which endowed it with reversible acidochromism in solution. Fig. S108 (ESI†) shows the unchanged absorption spectrum after 16 hour exposure to HCl vapours. Inspection of the solid-state structure in the crystalline and powder forms in Fig. S27 and S41 (ESI†) reveals no obvious motif that might shield the nitrogen atoms from reactivity with acid, although an elusive explanation based on molecular packing is likely at the heart of this phenomenon.
When in CH2Cl2 solution, NMe2quin exhibits 2-stage acidochromism unlike the other quinoxaline-fused pyrenes, which is shown in Fig. 6a–d. The first stage results in a 30 nm blueshift in the absorption peaks, which also blueshifts the fluorescence 130 nm from orange-red to green. The spectra closely resemble those of neutral Ph2quin, suggesting that the p-dimethylaniline groups become protonated and lose their electron-donating capability. In the second stage, the longest wavelength absorption peak redshifts 100 nm and the green fluorescence redshifts by 150 nm to red. This trend is mimicked by the excitation spectra, in Fig. S100 (ESI†). The spectra take on the appearance of Ph2quin with TFA, suggesting that the quinoxaline nitrogen is now protonated. Both stages can be generated in reverse by gradual addition of NEt3, shown in Fig. S89–S93 (ESI†). Experimental absorption and 1H NMR spectroscopy of NMe2quin during both protonation stages, in Section S4b (ESI†), are matched by rigorous DFT-calculated spectra of the possible protonated species, found in Fig. S10–S24 (ESI†). These observations correspond to pKa values; dimethylaniline nitrogen atoms (conjugate acid pKa = 5.0)63 are more basic than those of quinoxaline (conjugate acid pKa = 0.8)64 and would be protonated first.
 | ||
| Fig. 6 Solution acidochromism in the upper panels. Two-stage titrations of TFA into 4 μM CH2Cl2 solutions of NMe2quin monitored by (a) UV-visible absorption and (b) fluorescence spectroscopy (λexc1 = 425 nm, λexc2 = 545 nm). Arrows indicate peak growth/decay, initial traces are light purple and final traces are dark purple, and the asterisk marks amplified baseline from ×15 magnification. Photographs of NMe2quin TFA titrated-CH2Cl2 solutions under (c) ambient and (d) 365 nm UV light. Solid acidochromism in the lower panels. (e) UV-visible spectroscopy profile of self-regenerating solid NMe2quin, post exposure to HCl vapours. (f) Recycling fluorescence spectra of solid NMe2quin after exposure and evaporation of HCl vapour. Photographs of solid NMe2quin under (g) ambient and (h) 365 nm UV light before and after exposure to HCl vapour. | ||
Solid NMe2quin also responds to acidic vapours; however, the reversibility is not as complete as for H2quin. Initially in Fig. 6g and h, the yellow-orange solid fluoresces yellow and optical spectroscopy is presented in Fig. S103 and S104 (ESI†). Upon 30 minute exposure to HCl vapours the absorption profile in Fig. S109 (ESI†) gains a broad shoulder from 500 nm to 700 nm, granting black-purple colouration to the solid, which also loses fluorescence in Fig. S110 (ESI†). As shown in Fig. 6e, after 30 minutes the HCl fully evaporates without added base as the peaks past 500 nm collapse. The greater affinity for HCl relative to H2quin (3 min, versus 30 min evaporation time) is hypothesized to originate from the additional basic sites in NMe2quin, suggesting that the distal p-dimethylamino groups allow fine-tuning of the reversible acidochromism. After evaporation, the original absorption and fluorescence spectral features are not regained, in Fig. 6e and Fig. S110 (ESI†). The chemical structure is unchanged as judged by 1H NMR in Section S4b (ESI†) but the packing structure from PXRD in Fig. S38 (ESI†) has been perturbed from its original state. This new packing structure still exhibits self-regenerating acidochromism, and both absorption in Fig. S111 (ESI†) and fluorescence profiles in Fig. 6f are unchanged after five HCl treatment cycles.
Probing acidochromism in this way also revealed a surprising dichotomy between the five pyrenes. Quinoxaline-fused structures respond to the presence of acid, while H2pyr and Ph2pyr bearing dicyanopyrazine-fusion could not. Absorption and fluorescence spectra of H2pyr and Ph2pyr in solution with even 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 equivalents of TFA do not show any new peaks, in Fig. S101 and S102 (ESI†). The CH2Cl2 solutions remain visibly yellow with orange fluorescence. The solids in Fig. S112 (ESI†) are both yellow-orange fluorescent, and the absorption and fluorescence spectra are in Fig. S103 and S104 (ESI†). Powdered H2pyr matches the single crystal structure in Fig. S42 (ESI†), and the molecular packing is described in Fig. S32 (ESI†). The crystal structure of Ph2pyr in Fig. S34 (ESI†) has included solvent, so molecular arrangement in the bulk powder based on PXRD in Fig. S43 (ESI†) cannot be described. The solids are not responsive to HCl vapours, akin to the behaviour in solution. Lack of new peaks and therefore new chemical species suggests that the electron-withdrawing cyano groups render the neighbouring nitrogen atoms less basic, deactivating the molecules for acid responsiveness. Cyano groups are absent from the quinoxaline-fused pyrenes, and therefore they retain full activity.
000 equivalents of TFA do not show any new peaks, in Fig. S101 and S102 (ESI†). The CH2Cl2 solutions remain visibly yellow with orange fluorescence. The solids in Fig. S112 (ESI†) are both yellow-orange fluorescent, and the absorption and fluorescence spectra are in Fig. S103 and S104 (ESI†). Powdered H2pyr matches the single crystal structure in Fig. S42 (ESI†), and the molecular packing is described in Fig. S32 (ESI†). The crystal structure of Ph2pyr in Fig. S34 (ESI†) has included solvent, so molecular arrangement in the bulk powder based on PXRD in Fig. S43 (ESI†) cannot be described. The solids are not responsive to HCl vapours, akin to the behaviour in solution. Lack of new peaks and therefore new chemical species suggests that the electron-withdrawing cyano groups render the neighbouring nitrogen atoms less basic, deactivating the molecules for acid responsiveness. Cyano groups are absent from the quinoxaline-fused pyrenes, and therefore they retain full activity.
Conclusions
Subtle substitution has been shown to influence the multi-stimuli responsive behaviour of core-expanded pyrenes when exposed to mechanical force, solvents and acid. Unprecedented analysis of a crystal-to-crystal transformation for a pyrene derivative in response to mechanical force resulted in a shift from yellow to orange fluorescence. The dimethylamino groups on the distal positions of NMe2quin uniquely allowed the solid-state aggregation to be controlled from J-type to individual monomers. This change was reversible upon exposure to solvent vapours or by thermal treatment. Substitution pattern also dramatically affected the solvatochromic fluorescence. The dicyanopyrazine-fusion endowed H2pyr and Ph2pyr with greater donor–acceptor character than quinoxaline-fused H2quin and Ph2quin, which in turn made them more sensitive to solvent dipolarity. The various types of nitrogen atoms in the five pyrenes also made the molecules behave differently when exposed to acid. Quinoxaline-fused H2quin, Ph2quin and NMe2quin respond to the presence of acid, whereas the responsiveness is deactivated when cyano groups are adjacent to the basic sites. Solid-state acidochromism was even reversible under ambient conditions, without the need for external base. Given the subtlety with which substitution pattern influences responsiveness in solution and the solid-state, core-expanded pyrenes are currently being explored for aggregation-induced emission29 properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council (NSERC) of Canada for a Discovery Grant, Dr George Shimizu and Racheal Huynh for TGA and DSC analysis, and Dr Michelle Thibault for mass spectrometric analysis. Acquisition and structure solution for Ph2pyr and Ph2quin crystals were performed on the Macromolecular Beamline CMCF-BM (O8BM) at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.Notes and references
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194 RSC.
- Y. Kobayashi and J. Abe, Adv. Opt. Mater., 2016, 4, 1354–1357 CrossRef CAS.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo and A. Concheiro, Chem. Commun., 2014, 50, 7743–7765 RSC.
- C. Reus, M. Stolar, J. Vanderkley, J. Nebauer and T. Baumgartner, J. Am. Chem. Soc., 2015, 137, 11710–11717 CrossRef CAS PubMed.
- W. Yang, Y. Feng, Q. Si, Q. Yan, P. Long, L. Dong, L. Fu and W. Feng, J. Mater. Chem. A, 2019, 7, 97–106 RSC.
- Y. Zhao, S. Bertolazzi and P. Samorì, ACS Nano, 2019, 13, 4814–4825 CrossRef CAS PubMed.
- A. Chu, F. K.-W. Hau, L.-Y. Yao and V. W.-W. Yam, ACS Mater. Lett., 2019, 1, 277–284 CrossRef CAS.
- M. Mohar, ChemistrySelect, 2019, 4, 8061–8067 CrossRef CAS.
- R. J. Das and K. Mahata, Org. Lett., 2018, 20, 5027–5031 CrossRef CAS PubMed.
- E. Nagata, S. Takeuchi, T. Nakanishi, Y. Hasegawa, Y. Mawatari and H. Nakano, ChemPhysChem, 2015, 16, 3038–3043 CrossRef CAS PubMed.
- S. Zeng, D. Zhang, W. Huang, Z. Wang, S. G. Freire, X. Yu, A. T. Smith, E. Y. Huang, H. Nguon and L. Sun, Nat. Commun., 2016, 7, 11802 CrossRef CAS PubMed.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- Y. U. Kim, G. E. Park, S. Choi, C. G. Park, M. J. Cho and D. H. Choi, Dyes Pigm., 2019, 160, 372–377 CrossRef CAS.
- T. Qin, L. Sheng and S. X.-A. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 40838–40843 CrossRef CAS PubMed.
- P. S. Hariharan, J. Pitchaimani, V. Madhu and S. P. Anthony, Opt. Mater., 2017, 64, 53–57 CrossRef CAS.
- Y. Zhang, S. Tang, E. R. Thapaliya, L. Sansalone and F. M. Raymo, Chem. Commun., 2018, 54, 8799–8809 RSC.
- G. Han, Y. Yi and Z. Shuai, Adv. Energy Mater., 2018, 8, 1702743 CrossRef.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
- X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed.
- R. Tan, S. Wang, H. Lan and S. Xiao, Curr. Org. Chem., 2016, 21, 236–248 CrossRef.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Chem. – Eur. J., 2015, 21, 13164–13174 CrossRef CAS PubMed.
- J. Zhang, A. Li, H. Zou, J. Peng, J. Guo, W. Wu, H. Zhang, J. Zhang, X. Gu, W. Xu, S. Xu, S. H. Liu, A. Qin, J. W. Y. Lam and B. Z. Tang, Mater. Horiz., 2019 10.1039/C9MH01041F.
- S. Zeng, H. Sun, C. Park, M. Zhang, M. Zhu, M. Yan, N. Chov, E. Li, A. T. Smith, G. Xu, S. Li, Z. Hou, Y. Li, B. Wang, D. Zhang and L. Sun, Mater. Horiz., 2019 10.1039/C9MH00851A.
- G. Bains, A. B. Patel and V. Narayanaswami, Molecules, 2011, 16, 7909–7935 CrossRef CAS PubMed.
- Md. M. Islam, Z. Hu, Q. Wang, C. Redshaw and X. Feng, Mater. Chem. Front., 2019, 3, 762–781 RSC.
- C.-Z. Wang, Y. Noda, C. Wu, X. Feng, P. Venkatesan, H. Cong, M. R. J. Elsegood, T. G. Warwick, S. J. Teat, C. Redshaw and T. Yamato, Asian J. Org. Chem., 2018, 7, 444–450 CrossRef CAS.
- S. Sasaki, S. Suzuki, K. Igawa, K. Morokuma and G. Konishi, J. Org. Chem., 2017, 82, 6865–6873 CrossRef CAS PubMed.
- X. Feng, Z. Xu, Z. Hu, C. Qi, D. Luo, X. Zhao, Z. Mu, C. Redshaw, J. W. Y. Lam, D. Ma and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 2283–2290 RSC.
- J. C. Walsh, D. T. Hogan, K. M. Williams, S. D. Brake, G. Venkataramana, T. A. Misener, B. J. Wallace, R. P. Johnson, D. W. Thompson, Y. Zhao, B. D. Wagner and G. J. Bodwell, ChemPlusChem, 2019, 84, 754–765 CrossRef CAS.
- Y. Niko, H. Moritomo, H. Sugihara, Y. Suzuki, J. Kawamata and G. Konishi, J. Mater. Chem. B, 2015, 3, 184–190 RSC.
- C.-Z. Wang, R. Zhang, K. Sakaguchi, X. Feng, X. Yu, M. R. J. Elsegood, S. J. Teat, C. Redshaw and T. Yamato, ChemPhotoChem, 2018, 2, 749–756 CrossRef CAS.
- A. Mateo-Alonso, Chem. Soc. Rev., 2014, 43, 6311 RSC.
- A. M. Dmytrejchuk, S. N. Jackson, R. Meudom, J. D. Gorden and B. L. Merner, J. Org. Chem., 2018, 83, 10660–10667 CrossRef CAS PubMed.
- P. Jin, T. Song, J. Xiao and Q. Zhang, Asian J. Org. Chem., 2018, 7, 2130–2146 CrossRef CAS.
- X. Feng, J.-Y. Hu, C. Redshaw and T. Yamato, Chem. – Eur. J., 2016, 22, 11898–11916 CrossRef CAS PubMed.
- J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Org. Biomol. Chem., 2014, 12, 212–232 RSC.
- U. H. F. Bunz and J. Freudenberg, Acc. Chem. Res., 2019, 52, 1575–1587 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- S. N. Keller, A. D. Bromby and T. C. Sutherland, Eur. J. Org. Chem., 2017, 3980–3985 CrossRef CAS.
- R. García, M. Melle-Franco and A. Mateo-Alonso, Chem. Commun., 2015, 51, 8037–8040 RSC.
- C.-Z. Wang, X. Feng, M. R. J. Elsegood, T. G. Warwick, S. J. Teat, C. Redshaw, Y.-S. Bi and T. Yamato, Asian J. Org. Chem., 2019, 8, 155–160 CrossRef CAS.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981–9988 RSC.
- Q. Kong, W. Zhuang, G. Li, Y. Xu, Q. Jiang and Y. Wang, New J. Chem., 2017, 41, 13784–13791 RSC.
- S. N. Keller, N. L. Veltri and T. C. Sutherland, Org. Lett., 2013, 15, 4798–4801 CrossRef CAS PubMed.
- Y. Yin, Z. Chen, C. Fan, G. Liu and S. Pu, ACS Omega, 2019, 4, 14324–14332 CrossRef CAS PubMed.
- K. Zheng, H. Yang, F. Ni, Z. Chen, S. Gong, Z. Lu and C. Yang, Adv. Opt. Mater., 2019, 1900727 CrossRef CAS.
- M. Liu, Y. Han, W. Yuan, C. Guo, S. Shi, X. Liu and Y. Chen, Dalton Trans., 2019, 48, 14626–14631 RSC.
- B. Heyne, Photochem. Photobiol. Sci., 2016, 15, 1103–1114 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- S. Rösel, C. Balestrieri and P. R. Schreiner, Chem. Sci., 2017, 8, 405–410 RSC.
- S. Rösel, J. Becker, W. D. Allen and P. R. Schreiner, J. Am. Chem. Soc., 2018, 140, 14421–14432 CrossRef PubMed.
- V. G. Machado, R. I. Stock and C. Reichardt, Chem. Rev., 2014, 114, 10429–10475 CrossRef CAS PubMed.
- Y. Niko, S. Sasaki, K. Narushima, D. K. Sharma, M. Vacha and G. Konishi, J. Org. Chem., 2015, 80, 10794–10805 CrossRef CAS.
- J. Catalán, J. Phys. Chem. B, 2009, 113, 5951–5960 CrossRef PubMed.
- E. Lippert, Z. Naturforsch., A: Phys. Sci., 1955, 10, 541–545 Search PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- M. M. Fickling, A. Fischer, B. R. Mann, J. Packer and J. Vaughan, J. Am. Chem. Soc., 1959, 81, 4226–4230 CrossRef CAS.
- A. Albert, R. Goldacre and J. Phillips, J. Chem. Soc., 1948, 2240 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, full compound characterization, density functional calculations, UV-visible absorption and fluorescence spectra, single crystal and powder X-ray diffraction. CCDC 1959736–1959741. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00710e |
| This journal is © the Partner Organisations 2020 |