
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitro-graphene oxide in iridium oxide hybrids: electrochemical modulation of N-graphene redox states and charge capacities†
E.
Pérez
,
N. M.
Carretero
,
S.
Sandoval
 ,
A.
Fuertes
,
G.
Tobias
and
N.
Casañ-Pastor
*
,
A.
Fuertes
,
G.
Tobias
and
N.
Casañ-Pastor
*
Institut de Ciencia de Materiales de Barcelona, CSIC, Campus UAB, 08193 Bellaterra, Barcelona, Spain. E-mail: nieves@icmab.es
First published on 17th February 2020
Abstract
Carbon plays a significant role in the development of electrode materials used in, for example, catalysis, energy storage and sensing. Graphene-based coatings and carbon nanotubes have expanded that role through nanostructuring of hybrids or the formation of composites. In particular, the formation of hybrids of nanocarbons with iridium oxide yield nanostructured materials through direct anodic deposition, which have substantially improved charge capacities vs. pure IrOx. Modification of the possible redox sites, new structuring of the hybrids and increased charge capacities are expected as a result. This work shows that nitrogen (N)-doped graphenes, as part of an IrOx hybrid, offer a new redox chemistry on graphene oxide through electrochemical modulation of the redox states of nitrogen in graphene, and yield stable nitro groups bound to carbon, which have, so far, the largest oxidation state reported in N-doped graphene. The hybrid materials are obtained in the form of coatings thanks to spontaneous adhesion of iridium oxo species on N-doped graphenes and further anodic electrodeposition of the mixture. While the oxidizing synthesis process already involves modification of the oxidation state of nitrogen, further electrochemical cycling evidences the electrochemical processes for both the IrOx and N groups attached to the graphene oxide. All the hybrids obtained present a wide range of nitrogen-based groups that include the nitro group, and a significant charge capacity that remains large upon electrochemical cycling and that involves all the faradaic processes from the iridium and graphene components. One hybrid, in particular, which includes the highest starting oxidation state, reaches a significantly higher charge capacity, higher even than the graphene oxide hybrid, and with 70% retention upon cycling. Although nitrogen doping of graphene is considered to be a reducing process, this study shows that an oxidizing range of nitrogen doping is also possible. IrOx, and the reversible redox processes that iridium offers, are thought to be essential in stabilizing an unusual nitro-carbon-oxide system and allowing a sustained high charge storage capacity that is twice that of pristine graphene or graphene oxide hybrids.
Introduction
Carbon has played a unique role in the design of electrochemical catalysis and energy storage systems or sensors. Its intrinsic capacitance has been explored in capacitors and batteries, both alone and in combination with other components,1 while additional faradaic contributions from other components in nanostructured hybrid materials have been crucial to the development of new nanostructures, and also for improving supercapacitor electrodes, as reviewed.2–4 Furthermore, in the form of graphene or carbon nanotubes (CNTs), nanocarbons show additional electrochemical features.5 When hybrids are formed, with IrOx, for example, a key component in O2 reduction and water oxidation,6 or in biocompatible electrodes,7,8 the charge capacities are found to increase by several orders of magnitude in the nanostructured material beyond the values expected from the charge attributed to the redox processes. In addition, the charge capacities remain stable during electrochemical cycling, while maintaining the redox properties and biocompatibility of the original material.9–12 The underlying nanostructure is significant from the basic point of view. In the case of carbon nanotubes, the carbon tubes sustain the oxide, as in a reinforced concrete structure,9 allowing thicker coatings and very stable electrodes compared with pure IrOx. Nanocarbon hybrids are also remarkably different from graphite hybrids in terms of reversibility, since they retain most of their charge capacity beyond 1000 cycles, while for graphite hybrids the charge capacity drops during the first 100 cycles to the original value of IrOx. The large size of the graphite particles may favor carbon loss during cycling, probably through a smaller interaction with IrOx.10 Within that framework, a further increase in charge capacities is envisaged if nanocarbons include redox species that can contribute to the pseudocapacity within the potential window available in aqueous solutions, without becoming hydrophobic. In that sense, of the many types of possible graphene-based materials, N-containing graphenes are good candidates for hybrid formation in terms of chemical interactions. Several strategies allow the introduction of heteroatoms into graphene, both during its synthesis and via post-treatment of the already synthesized material. Thin films and individual layers of nitrogen-doped graphene can, for instance, be prepared by low-energy N2+ ion sputtering,13 laser irradiation14 and chemical vapor deposition.15 The introduction of nitrogen into previously synthesized samples can be achieved by a variety of protocols, with graphene oxide widely employed as the starting material. These include microwave treatment,16 hydrothermal synthesis,17 laser-assisted reaction,18 and ammonolysis of graphene oxide.19 In general, for the same deposition method, the microstructure of hybrids with graphene-derived phases has been seen to be highly correlated with the type of graphene used.10,11N-doped graphene hybrids have been explored by using direct chemical processes with a variety of metals and metal oxides,20–23 including cobalt and manganese oxides. Their intrinsic electrochemical properties, however, have been mainly explored in terms of oxygen reduction catalysis in pure-form N-graphenes.24 Within the context of hybrid formation, and in particular with IrOx for ease of comparison with the hybrids mentioned above, studying the electrochemical modulation of N-doping species in graphene oxide could offer a new opportunity in terms of fundamental modification of graphene in oxidizing conditions and also in a range of charge capacities.
In particular, significant difficulties arise when developing materials as coatings on substrates with different shapes for electrode applications, if nanocarbons are desired as part of the coating. Although other metal oxides have been hybridized with graphenes, IrOx–CNT and IrOx–graphene (exfoliated graphene and graphene oxide, GO) hybrids represent the only case where electrodeposition driven by the oxide phase allows a final conducting coating. Anodically deposited IrOx is an amorphous conducting material, with iridium in mixed-valence form and the empirical formula K1.7IrO0.8(OH)2.7·0.4H2O. This structure deintercalates K+ ions when immersed in aqueous solutions that are poor in K+, undergoes redox processes that activate water oxidation, and increases charge capacity (CSC) in bioelectrodes when used as a coating, whilst also decreasing the impedance of the electrode.8–12 The increased capacity yields a better biological response under electric field stimulation, but further increase is desired in electrostimulation if using low-frequency electric fields. In IrOx–C hybrids, electrodeposition of the material coating is not self-limited as it occurs when IrOx is deposited alone from solution,9–11 possibly due to better conductivity of the deposited hybrid material. Significantly, neither graphene, nor graphene oxide, nor carbon nanotubes are electrodeposited independently within the available potential range of the aqueous solution. However, in the presence of iridium oxo species in solution both materials deposit together, as if the iridium precursor species carry the carbon nano- and microparticles.9–11 This suggests a direct chemical interaction in solution between the carbon species and the iridium ions, through chemical interaction in functionalized CNTs and graphene oxide, and has been observed in other cases (Ni–Co double hydroxides).25 Such interaction, however, also exists when using graphene obtained from electrochemical exfoliation of graphite without any added oxygen, possibly by means of labile adsorbed oxygen from the exfoliation process.11
On the basis of these previous observations, it would be of interest to explore further the possible chemistry related to nitrogen doping groups in graphene, and the subsequent increase in charge capacity of other graphene species through interaction with IrOx, as well as the stability and reversibility of the hybrids as electrode coatings. Based on the large variety of possible graphenes, the variety of resulting hybrid materials is even larger in terms of nano- and microstructures, and, in turn, their electrical and electrochemical properties. In particular, electrodeposition of nanostructured graphene hybrids is largely an unexplored area, with the exception of our previous work on mille-feuille IrOx–graphenes and CNT hybrids.10,11 Although different oxidation conditions or use of high-energy physical methods results in different electrochemical and capacitive behavior26–28 in graphenes, oxidizing hybrid formation is rather unusual. This work shows the first hybrid coatings of iridium oxide and N-containing graphene in oxidative conditions, in which various types of nitrogen are observed, including the highest oxidation state of nitro-graphene. Oxidative conditions create a type of N-graphene hybrid with oxidized nitrogen or nitro groups, which are new in the field of graphene. N-Containing graphene precursors obtained at low temperatures, up to 300 °C have been chosen, to achieve hydrophilic phases. This is a key feature for the interaction with IrOx precursors in aqueous media, but is also important in their use in aqueous electrochemical cells and in electrostimulation electrodes. The redox contribution from iridium and nitrogen groups has been studied, as well as the resulting changes in faradaic and charge capacity properties, despite the difficulty of having complex redox behavior that includes nitrogen groups and mixed-valence iridium. This work shows a remarkable increase in charge capacity related to formation of the hybrids, which is much higher than the values achieved for the corresponding IrOx–graphene oxide, CNT–COOH or pristine graphene hybrids. Chemical transformation of the N groups is observed during anodic deposition of the hybrids and also during further electrochemical cycling. The most stable N-graphene oxide–IrOx hybrid corresponds to the N-graphene prepared at higher temperatures, presumably because of a stronger N–C bond or through a stronger N–Ir interaction. Nitrogen redox processes are considered to add to the global charge capacity of the hybrid, reflecting the chemistry of the amine, amide and also the most significant nitro groups in graphene, while the intrinsic mixed-valence behavior of Ir interacts directly in the formation of the hybrid and its final electrochemical processes.
Experimental
Synthesis of graphene oxide
GO was prepared using a modified Hummers’ method.29 Briefly, 5 g of graphite powder (<20 μm, Sigma-Aldrich), 115 mL of H2SO4 (Panreac, 98%) and 2.5 g of NaNO3 (Sigma-Aldrich, <99%) were mixed together, keeping the temperature at 0 °C for 30 minutes. Afterwards, 15 g of KMnO4 (Panreac, 99%) was added slowly and the mixture was continuously stirred for 30 min. Then, 230 mL of distilled water was slowly added, maintaining the reaction temperature at 98 °C for 2 h. Finally, 1 L of distilled water and 5 mL of H2O2 (Acros Organic, 30%) were added and the reaction mixture was cooled down. The resulting graphene oxide was purified by subsequent centrifugation and washing with distilled water until the pH of the solution was neutral.Ammonolysis treatment of GO
In order to obtain N-containing, reduced graphene oxide samples, 100 mg of GO was spread in a sintered alumina boat and subsequently annealed over 1 h at temperatures ranging between 100 °C and 300 °C with an ammonia flow rate of 300 mL min−1 (Carburos Metálicos, 99.99%).30 The nitrogen-doped graphene oxides were named, respectively, GN100, GN220 and GN300 according to the temperature used in their preparation.Anodic electrodeposition of IrOx hybrid films of N-containing graphene oxides
Deposition of IrOx was carried out by dynamic potential electrodeposition methods, using a three-electrode electrochemical cell. The pre-deposition iridium solution for hybrid formation was a modification of the one used for pure IrOx,8 and was prepared from 5.5 mg mL−1 of N-containing GO precursors dispersed by sonication over 30 minutes in 10 mL of MilliQ water, and further addition of 2 × 10−4 mol IrCl3·H2O (Aldrich 99.9%) and 5 × 10−3 mol of K2CO3 (Aldrich 99%), with a final pH of 10. The solution was aged at 37 °C for 7 days and stored at 4 °C until use to allow for iridium hydrolysis. The working electrode, where the sample was deposited, was a transparent soda-lime glass slide (38 × 12 mm2, AFORA), which had been previously coated with 5 nm of titanium (as the adhesion layer) and 12 nm of platinum, by thermal evaporation.8 A platinum sheet (Goodfellow, 99.9%) of the same geometric shape and frontal area was used as a counter electrode, and was placed in a parallel arrangement, with a separation of 1 cm from the working electrode. A platinum wire, 0.5 mm in diameter (Goodfellow, 99.9%) was used as the pseudo-reference electrode, with its potential vs. Ag/AgCl being 0.02 V.8 Thin films were obtained at room temperature by a potentiodynamic sweep involving 50 potential cycles, and observations were also made at 25 cycles. The cyclic potential sweep rate was 5 mV s−1, and the potential limits were from equilibrium (near 0.0 V) to 0.85 V vs. Pt for all graphene hybrids. The maximum potential for cycling was optimized empirically since the potential used for bare IrOx did not yield any deposit. A VMP potentiostat (Biologic) was used for electrodeposition control. Bare IrOx coatings used for comparison were synthesized using the same substrates and with a similar potentiodynamic protocol: scan rate of 10 mV s−1, potential limit to 0.55 V and 50 cycles, using a pre-deposition solution described by Cruz et al.8 Electrodepositions using only N-graphenes in the suspensions did not yield coatings in the potential window in water.Coating characterization
IrOx–N-graphene hybrids (named GNIr100, GNIr220 and GNIr300 from GN100, GN220 and GN300 N-graphene and iridium suspensions, respectively) were characterized using several techniques. Scanning electron microscopy (SEM), using a FEI Magellan 400L high-resolution scanning electron microscope at 10–30 kV in high vacuum conditions, was performed for all samples. The resulting images were used to characterize roughness using Mountains™ software since atomic force microscopy (AFM) experiments failed for the large range of roughness (RMS) observed. RMS values obtained are the square root of the average squares of altitudes vs. the average profile. The thickness of the coatings was also measured by SEM, by placing the transverse-fractured substrate in parallel with the electron beam. X-ray photoelectron spectroscopy (XPS) measurements were performed at room temperature using a Kratos AXIS ultra DLD spectrometer with a monochromatic AlKα X-ray source, and a PHI 5500 Multitechnique System (from Physical Electronics) with a monochromatic X-ray source (AlKα: 1486.6 eV energy and 350 W) placed perpendicular to the analyzer axis. The analyzed area was a circle of 0.8 mm diameter, and the selected resolution was 187.5 eV of the pass energy (0.8 eV per step) for the general spectra, and 11.75 eV of the pass energy (0.05 eV per step) for the high-resolution spectra. The C 1s aliphatic peak at 284.8 eV was used as reference for all spectra. Fourier transform-infrared (FT-IR) spectra of the coatings were recorded with a FT/IR-4700 (JASCO) instrument in transmission mode. Deconvolution was performed using a Gaussian–Lorentzian mode. The electrochemical behavior of the resulting coatings was studied by cyclic voltammetry (CV) using the same potentiostat and cell system as described above, with 10 mV s−1 sweeps. For easy comparison with previous studies,8–11 a neutral sodium phosphate buffer containing sodium and potassium chlorides as electrolyte (PBS, 0.137 M NaCl, 0.0027 M KCl, 0.010 M Na2HPO4, 0.0018 M KH2PO4 at pH 7.4) was used. The area of the samples was 1 cm2 and the separation between electrodes was 1 cm. The cathodic charge storage capacity (CSCc) was determined from integration of the cathodic current during a potential sweep between −0.6 V and 0.8 V vs. Pt, at a sweep rate of 10 mV s−1, and was defined per unit volt. Stability tests were carried out to evaluate the change in CSCc over 1000 cycles, as described previously for IrOx and GOIr.8–12Results and discussion
Using aged oxo-iridium solutions with suspended N-containing reduced graphene oxide (N-graphene) it was possible to deposit coatings of N-graphene–IrOx hybrid materials. N-Graphenes were obtained by treatment of graphene oxide with ammonia gas at 100 °C (GN100), 220 °C (GN220) and 300 °C (GN300). Taking into account that pure suspensions of N-graphenes do not deposit, it seems that the driving force for such deposition is the IrOx hybrid formation. For ease of nomenclature, the hybrid coatings are named GNIr100, GNIr220 and GNIr300 (depending on the precursor used). Visual inspection reveals the presence of uniform coatings (see Fig. 1, optical and SEM low-resolution images), and shows that the first stages of deposition involve blue IrOx. Only after the electrode is coated with IrOx do the GN–IrOx hybrids also deposit. Transverse cutting of the coatings shows a layered mille-feuille microstructure, but large grains related to the graphene precursors are apparent. SEM images show cracks clearly related to dehydration under vacuum and are more visible for the thickest coatings (those of GNIr220 and GNIr300), which also contain more highly hydrated IrOx. Coating adhesion is greatly improved by using potential values from a rest potential (near 0 V vs. Pt (equivalent to vs. Ag/AgCl)) to 0.8 V vs. Ag/AgCl for the dynamic potential sweep for electrodeposition. Profiles of cut samples show that, for the same electrodeposition time, GNIr100 systematically yields a thinner coating, while GNIr220 and GNIr300 result in thicker coatings than that reported for GOIr (see Table 1), although GNIr220 tends to detach after electrochemical cycling. | ||
| Fig. 1 (a) Representative I (mA) vs. E (V) vs. Ag/AgCl (E(Pt) = E(Ag/AgCl)) for the dynamic anodic deposition process. (b) Naked view image of initial deposition stages (25 cycles) and final (50 cycles) coatings from GNIr220 electrodeposition. (c) Detail of layered structure in the coatings. (d) SEM surface and profile images for GN powder precursor SEM images at various magnifications. (e) SEM images for GNIr hybrid coatings. (f) Transverse cut images of GNIr coatings. | ||
It is worth noting that the large particles observed in the SEM images from powdered GN precursors (Fig. 1d) do not represent the final dispersed particles in the iridium-containing solutions from which electrodeposition was performed, as is clearly seen when comparing Fig. 1d and e. The morphology of the resulting deposits is globular and resembles that obtained when GO is used, although individual particles follow the mille-feuille-layered nanostructure observed for IrOx-exfoliated graphene hybrids (see high-resolution SEM images in Fig. 1, Fig. S1 (ESI†) and ref. 11 for details). On the other hand, the size and morphology of the dispersed graphene particles seems to define the morphology of the hybrid material, which results in a very uniform coating at all scales tested (as shown in the backscattering SEM images in Fig. S1 in the ESI†). Previously characterized suspensions8,10 evidence Ir–O species of 10 nm size, according to dynamic light scattering, adhered to graphene platelets, which develop into 2 nm Ir metal particles under the conditions for transmission electron microscopy measurements. The size of the graphene platelets, reported previously as ∼10 μm in the xy-plane, is much larger than the size of the IrOx agglomerates, which explains the final microstructure and roughness of the coating (see Table 1). In addition, dehydration under SEM conditions creates the cracks observed in Fig. 1e, and thus the roughness derived from the SEM images is overestimated, while no AFM was possible for these rough coatings (Table 1).
Surface XPS characterization
In all cases, the XPS spectra of the hybrid coatings obtained by anodic electrodeposition show the elements expected from the graphene precursors and IrOx (Fig. 2–4; see Fig. S1–S3 for general spectra, ESI†). K, Ir, and O are always present for the mixed-valence electrodeposited phase K1.7IrO0.8(OH)2.7·0.4H2O (named “IrOx” usually)8 and C, N and O for the N-graphene oxides studied previously29 (see ESI,† Fig. S2–S4 for general spectra). For ease of comparison, the XPS C 1s, N 1s and O 1s data of the N-graphene precursors are also included in the figures. Table 2 shows the atomic quantifications observed for each coating and the relevant atomic ratios, in comparison with the precursors used, which facilitates identification of the significant data. The resulting K/Ir ratio equals that of electrodeposited IrOx in all cases, evidencing the existence of the unchanged iridium oxohydroxide phase described as “IrOx”, as previously reported.8 Also, a freshly prepared GO–IrOx (GOIr) hybrid shows reproducible values with respect to those previously reported.10 The C/Ir ratio, compared with the endemic carbon seen in IrOx, evidences a large proportion of graphene per Ir atom. Thus, iridium oxide anodic deposition is able to carry a substantial amount of carbon graphene in the process. As reported previously for GO–IrOx hybrids (GOIr), adhesion of iridium phases to graphene oxide already exists in solution, and explains the resulting global deposition.10,11 As expected, the oxygen contribution from IrOx raises the O/C ratio for all hybrids with respect to graphene precursors, while the oxygen from graphene is also included in the hybrid materials. The notable increase in O/C ratios in the hybrids, however, goes beyond the contribution from IrOx alone (O/Ir ca. 4), reaching values of O/Ir = 10, which is much more than the O content from graphene precursors and suggests extended hydration. All N-graphenes used here as precursors contain remaining oxygen, with lower temperature processing yielding higher contents. Thus, the GN100 precursor shows the smallest N/C ratio and the largest O/C.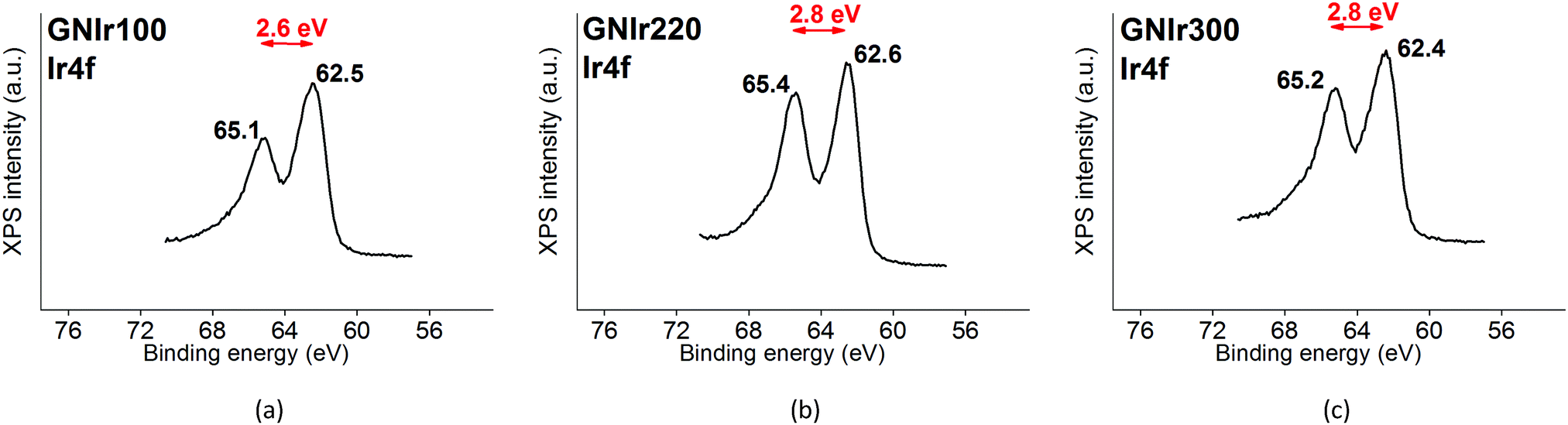 | ||
| Fig. 2 Ir 4f signal in each of the GNIr hybrids: (a) GNIr100, (b) GNIr220, (c) GNIr300. | ||
 | ||
| Fig. 3 C 1s, N 1s and O 1s XPS signals for the (a) GN100 precursor and (b) the hybrid GNIr100. For ease of comparison, C 1s, N 1s and O 1s for the electrochemically cycled coating (c) is also included. | ||
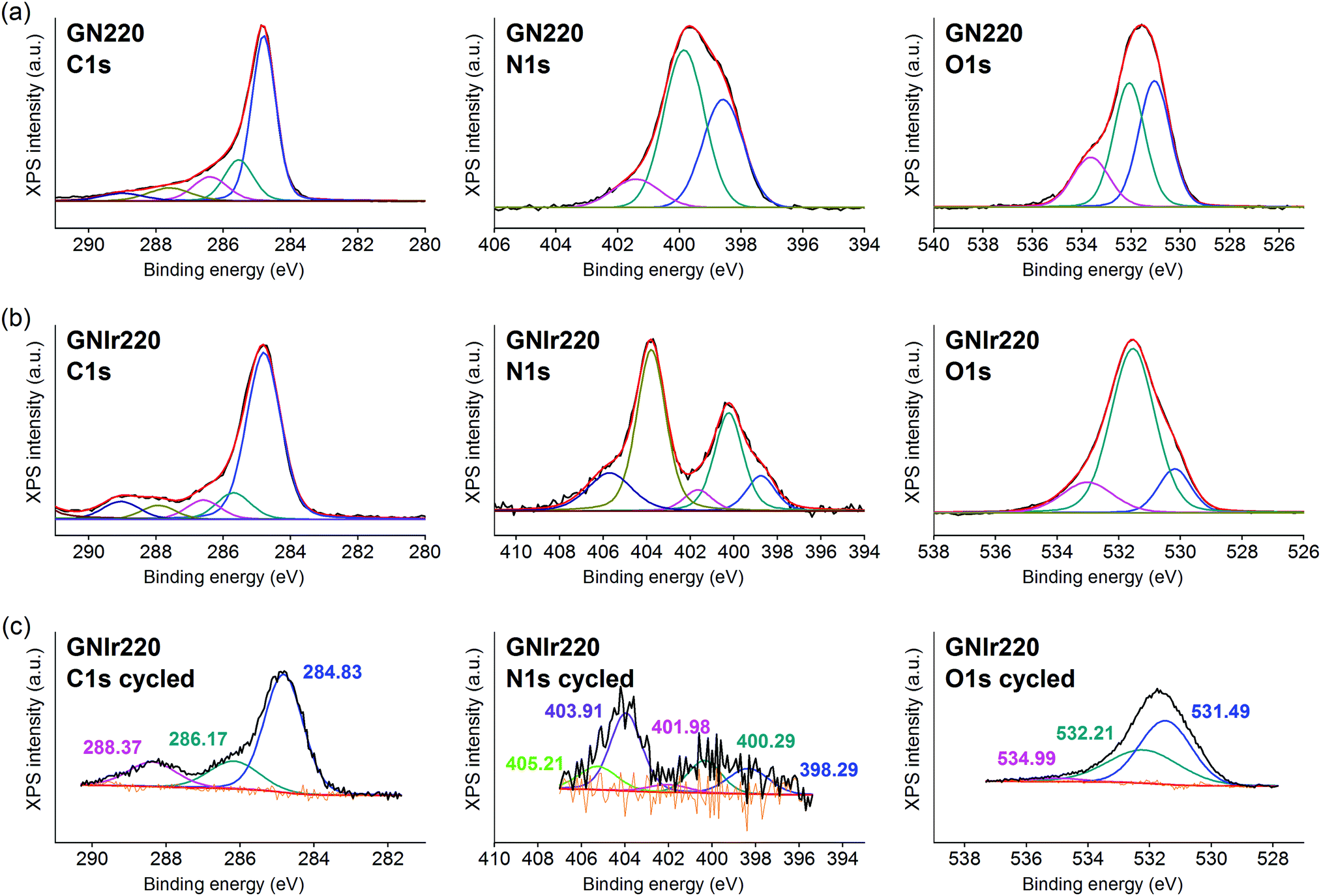 | ||
| Fig. 4 C 1s, N 1s and O 1s XPS signals for (a) GN220, (b) GNIr220. For ease of comparison, C 1s, N 1s and O 1s for the electrochemically cycled coating (c) is also included. | ||
| C 1s | N 1s | O 1s | K 2p | Ir 4f | C/Ir | O/Ir | K/Ir | N/Ir | N/C | O/C | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Coating detaches systematically after cycling. | |||||||||||
| GNIr100 | 25.4 | 11.4 | 50.4 | 7.9 | 4.9 | 5.1 | 10.2 | 1.6 | 2.3 | 0.45 | 1.98 |
| GNIr220 | 21.6 | 7.7 | 55.1 | 9.8 | 5.9 | 3.7 | 9.4 | 1.7 | 1.3 | 0.36 | 2.55 |
| GNIr300 | 24.1 | 4.4 | 55.4 | 10.0 | 6.0 | 4.0 | 9.2 | 1.7 | 0.7 | 0.18 | 2.29 |
| IrOx8 | 15.7 | — | 47.6 | 23.5 | 13.2 | 1.2 | 3.6 | 1.8 | — | — | 3.03 |
| GOIr | 27.3 | — | 47.4 | 16.0 | 9.3 | 2.9 | 5.1 | 1.7 | — | — | 1.74 |
| GN100 precursor | 73.0 | 7.6 | 19.4 | — | — | — | — | — | 0.10 | 0.27 | |
| GN220 precursor | 78.5 | 11.1 | 10.3 | — | — | — | — | — | 0.14 | 0.13 | |
| GN300 precursor | 82.0 | 11.0 | 7.0 | — | — | — | — | — | 0.13 | 0.09 | |
| After electrochemical cycling | |||||||||||
| GNIr100 c | 21.3 | 2.0 | 67.2 | 5.3 | 4.1 | 5.2 | 16.3 | 1.3 | 0.5 | 0.1 | 3.2 |
| GNIr220 ca | 29.5 | 3.9 | 47.0 | 12.1 | 7.5 | 4.0 | 6.3 | 1.6 | 0.5 | 0.1 | 1.6 |
| GNIr300 c | 20.2 | 2.8 | 64.1 | 7.4 | 5.4 | 3.7 | 11.8 | 1.4 | 0.5 | 0.1 | 3.2 |
The existence of endemic carbon even in IrOx precludes some conclusions, but the comparisons between phases taken in a semiquantitative way and, in particular the N/Ir ratios, show clear features.
A direct interaction between IrOx and N-graphenes, even if the identity of each component remains distinct, may explain the type of changes observed in some fractions, such as N/C. Thus, if we were to assume invariance of IrOx, changes in the O content in the hybrid would be attributed to modification of the N-graphenes, but the results show more complex data than this. This is evidenced by the fact that N/C increases up to five times with hybrid formation according to the XPS quantification. Furthermore, the N/C relative quantifications are inverted with respect to the corresponding N-graphene precursors, and the GNIr100 hybrid is the one with significantly large N/C and N/Ir values, and the smallest O/C. In IrOx, O is easily exchanged, in the form of OH−, since it is a true oxohydroxide, with intercalated K+ and mixed-valence properties that allow further ionic intercalations/deintercalations.8 Thus, part of the oxygen in the IrOx fraction of the hybrid may come from the N-graphene derivative. But it is also highly probable that during anodic deposition, functional oxygenated C groups (such as COO−, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or C–OH) are oxidized to CO2, involving loss of the functional C in graphenes and of functional oxygen. Indeed, it has been confirmed previously8 that the oxalate ion present in the Ir precursor solution is oxidized to CO2 and has redox processes in this potential region (see below). If that is the case, the remaining oxidized graphene will be enriched in N with respect to the corresponding precursor. Other effects, such as segregation of some N-based groups at the surface, are also possible. All these considerations evidence that each component is chemically modified to some extent in the hybrid.
O or C–OH) are oxidized to CO2, involving loss of the functional C in graphenes and of functional oxygen. Indeed, it has been confirmed previously8 that the oxalate ion present in the Ir precursor solution is oxidized to CO2 and has redox processes in this potential region (see below). If that is the case, the remaining oxidized graphene will be enriched in N with respect to the corresponding precursor. Other effects, such as segregation of some N-based groups at the surface, are also possible. All these considerations evidence that each component is chemically modified to some extent in the hybrid.
Furthermore, oxygen exchange between graphenes and iridium ions has been observed before. While graphene from electrochemical exfoliation of graphite has not shown extra oxygen in the hybrid,11 with O/Ir remaining close to 4 as in IrOx, the GO–IrOx hybrid shows an O/Ir value close to 5.0 (for a GO with O/C ratio of 0.4).10 This involves 2.5 GO per IrOx. In this work, the final O/Ir value of ca. 9–10 and C/Ir of around 4 units in excess of the endemic carbon cannot derive from the O/C ratios in the graphene precursors (values of 0.1–0.3). Only the large degree of hydration shown by the cracks observed in the SEM image might explain this value. Such increased hydration may come from aqueous pockets favored by the presence of oxoiridium and oxalic acid.
Using only quantitative information from XPS, the formation of IrOx hybrids also shows different chemical reactivity and possible changes in the functional and doping N groups. On the other hand, high-resolution XPS for each element yields significant information with respect to the chemical moieties present upon hybrid formation. See Fig. 2–5 for Ir 4f, C 1s, N 1s and O 1s spectra for each hybrid compared with the graphene precursors, and after electrochemical cycling.
 | ||
| Fig. 5 C 1s, N 1s and O 1s XPS signals for (a) GN300 precursor, (b) GN300Ir hybrid. For ease of comparison, C 1s, N 1s and O 1s have also been included for the electrochemically cycled coating (c). | ||
The Ir 4f signal remains as previously reported for IrOx8 and is constant in all cases, with an asymmetric doublet for the 4f7/2 and 4f5/2 signals at 62.5 and 65.1 eV, which contain Ir satellite peaks (shown in Fig. 2). Therefore, in all cases the signal corresponds to a similar oxidation state.
On the other hand, formation of the hybrids modifies the deconvolutions of the C 1s, O 1s and N 1s peaks (see Fig. 3–5), and the relative amounts of the components. In the case of C 1s, the main peak for all samples corresponds to the so-called aliphatic carbon, and it is used to normalize all spectra, and also coincides with the graphitic signals. The GN100 phase C 1s deconvolution renders three other peaks related to carbons C–O (and C–N), CO and COO− at 286, 288 and 290 eV, respectively. Upon anodic hybrid formation with IrOx, in GNIr100 the signal intensity increases at larger binding energies (288 eV) corresponding to higher oxidation species, such as carboxylates, involving oxidation of functionalized carbons, and a substantial decrease of the 286 eV peak (C–N) occurs. This is similar for GNIr200 and GNIr300 hybrids vs. their precursors GN220 and GN300, although in these cases the peak at 286 eV was originally smaller.
Especially significant are the N 1s signals. The low relative sensitivity of this element in XPS renders a noisy signal for the hybrids with lower N content (see Table 2), although significant features may still be observed. All precursors have two main signals between 399 and 400 eV, assigned to amide, pyridinic-N (∼398.6 eV), and amine, pyrrolic-N (400 eV), respectively.30 Pyridinic and pyrrolic peaks are usually correlated to N atoms at the edge of the graphene planes, with each of them bonded to two C atoms and donating electrons to the aromatic π system. However, their binding energy overlaps with that of N-based aliphatic groups, which have been shown to exist in these samples. A peak at 401.3 eV, corresponding to quaternary-N or graphitic-N in which N atoms are incorporated into the graphene layer and replace C atoms directly,29,31,32 is also observed. However, upon anodic deposition of the hybrid, two new peaks appear at very high binding energy, 403 and 406 eV, which can only be ascribed to oxidized nitrogen, or nitrogen oxide groups, such as pyridine-N-oxide and nitro groups,33 respectively, and/or to nitrogen bound to Ir, although the Ir 4f signal is insensitive to such an interaction. The only reference we have found for chemical adhesion of Ir species to graphene does not show such high-energy peaks,34 evidencing that no oxidation occurs by direct chemical reaction with iridium in the precursor solutions. Therefore, oxidation of the nitrogen in graphene occurs by means of oxidative synthesis of the hybrid. This result is relevant since it may allow modulation of the N electronic states in graphene, and its use as a redox reservoir.
The oxidation process and the interaction or coordination to iridium is not equal for all N-graphenes as the hybrids are formed. GNIr100 and GNIr220 show a larger ratio of nitro N 1s signals at 403–406 eV, while GNIr300 still contains larger relative amounts of the lower energy N 1s signals, as if hybrid formation has not involved as much oxidation. GN300 has been described as containing more N-dopant species within the carbon graphitic structure than GN100 and GN220 (and indeed it should be more stable against oxidation), while GN100 and GN220 contain more reactive functionalization, with N species attached to graphene but not being part of the basal structure. As seen in the next section, such added reactivity does not translate into additional electrochemical charge storage capacity compared with GN300.
Although the conducting character of the coating introduces significant modification to the baseline infrared spectroscopy and signal relaxation, an attempt was made to identify functional groups (see Fig. S4, ESI†). However, no peaks corresponding to nitro groups (either nitrite or nitrate (near 1500 and 1300 cm−1)) were found, while N–Ir bonds would be hidden in the IrOx oxide low-energy frequencies (around 500 cm−1). The only relevant peak, at 2360 cm−1, corresponds to CO2.
The O 1s XPS signals in the hybrids have the usual three components related to O2−, OH− and H2O, at 530, 531 and 532 eV, respectively, as found for IrOx,8 which overlap with the signals for amide and carbonyl or carboxyl groups from N-graphenes, which are usually found at binding energies of 534, 532 and 531 eV, respectively. Once the hybrid is formed, the relative amount of the signal near 532–531 eV increases with respect to IrOx, evidencing this overlap of water and organic-derived signals. The hybrids also reflect the high intensity of the 531 eV binding energy peak (ascribed to OH−) that is usually found in IrOx, and the large degree of hydration, 532 eV.8 It is notable, however, that the GN precursors had more intense high-energy peaks, which are not present in the hybrid GN–IrOx and this is probably due to oxidation during anodic deposition.
Electrochemical behavior of hybrids
Cyclic voltammetry of GNIr hybrid coatings in saline phosphate buffer show the wide waves previously reported for IrOx (Emax anodic near 0 and 0.55 V vs. Pt, and corresponding Emax cathodic near −0.5 and 0.0 V vs. Pt related to intercalation/deintercalation processes in IrOx)8 (Fig. 6). On the other hand, each hybrid shows an intrinsically different change in the intensities of the waves in CV depending on the graphene precursor used, which suggests additional contributions from graphene groups that are not visible at first glance. In general, it is known that different electrochemical behaviors are observed for GO depending on the preparation method used.27 Given the redox complexity of the O and N-graphene functionalities possible in GNIr hybrids, where pyrrolic, pyridinic, phenolic, amines and amides could show electrochemical oxidation waves35,36 in addition to the intrinsic behavior of IrOx mixed-valence redox processes,8,35 an empirical approach to the observed CVs is the most suitable. Thus, it is remarkable that an increase in global current density is observed for all hybrid coatings compared with pure IrOx, although only in the second wave for GNIr100, and also that a significant increase in currents and lower potentials for oxygen evolution with respect to IrOx are observed. Within these general features that are common to all the hybrids, notable differences are also observed. GNIr100 shows an increase in intensity of the second oxidation wave for Ir (maximum at 0.5 V vs. Ag/AgCl), but not in the first (near 0 V), while the other hybrids, including non-nitrogenated GOIr, show an increase in intensity of both waves upon formation of the hybrid material. Pyrrolic, pyridinic and phenolic groups would contribute, with redox waves near 0 V, 0.2 V and 0.6 vs. Pt, respectively,35,36 while O functionalities, such as COOH, CO, and C–O, may contribute at 0.2, 0.6 and −0.6 V vs. Ag/AgCl.17 Therefore, changes in these potential regions could be related to the O and N ring groups and to modifications of the Ir redox activity in the hybrid. Significantly, only GNIr220 seems to have an extra anodic redox wave and larger intensity in the cathodic region, for which only electrolysis itself may help to identify the process responsible.
 | ||
| Fig. 6 Cyclic voltammetry (10th cycle) in sodium phosphate buffer, pH 7.4, of GNIr hybrid coatings in comparison with GOIr and IrOx coatings (10 mV s−1). | ||
The global current increase is largest for GNIr300 and correspondingly the charge capacity is also largest, while waves are less resolved involving a larger capacitive effect from carbon. The catalytic effect of IrOx on oxygen evolution6 also seems to be enhanced in this particular hybrid. As expected, the increased intensity effects in N-graphene hybrids correlate to a large extent with the thickness of the coating (see Table 3), with different thicknesses for each hybrid electrodeposition process, even under the same conditions. It also correlates with the larger proportions of graphene in the hybrids GNIr220 and GNIr300 shown in Table 2, derived from the spontaneously larger adhesion of GN220 and GN300 to IrOx. However, it is clear that more variables are involved. In terms of charge storage capacities (see Table 3), previously GOIr had been shown10,11 to have the same charge capacity as the graphene exfoliated–IrOx hybrid, while the latter was much thinner (see Table 3) and with a smaller C content. Here, exfoliated graphene–IrOx (eGIr) and GNIr110 show practically the same thickness, but the latter has a much lower surface charge capacity, even with larger roughness (and therefore surface area).
| Sample | Thickness (μm) | RMS (μm) | N/Ir and Cgrap/Ir | CSCc (mC cm−2) | CSCc (mC cm−2) per Cgraph | CSCc (F g−1) | CSCc (F g−1)/Cgraph |
|---|---|---|---|---|---|---|---|
| GNIr100 | 0.7 | 2.2 | 2.3/3.9 | 21.6 | 5.5 | 110.4 | 28 |
| GNIr220 | 2.9 | 5.3 | 1.3/2.5 | 132.7 | 53 | 163.4 | 65 |
| GNIr300 | 4.0 | 4.1 | 0.7/2.8 | 176.5 | 63 | 157.6 | 56 |
| IrOx8 | 0.14 | 0.002 | —/0 | 22 | 22 | 392.9 | — |
| GOIr10 | 1.5 | 2.4 | —/1.7 | 108 | 64 | 257.1 | 151 |
| eGIr11 | 0.7 | 0.3 | —/0.4 | 94 | 235 | 479.6 | 1200 |
The largest charge capacity for the N-graphene oxide hybrids, of 177 mC cm−2, was found for the GNIr300 hybrid (1 V range in CV), and it was also larger than that found for GOIr, pristine graphene–IrOx (eGIr) and IrOx. On the other hand, the specific capacity (F g−1 based on an approximated density of 2 g cm−3 found for carbon and IrOx8) suggests that GNIr220 has a similar capacity to GNIr300, and similar values to those reported for N-doped graphenes,37 where the largest contribution corresponds to N in the graphene basal planes. These values, however, are lower than for other IrOx hybrids with GO or for pristine graphene reported previously,10,11 or even IrOx.8 It is worth discussing here that neither of the parameters for surface or specific capacity is fully representative of the actual use of the hybrid, since the area exposed at the interface depends on roughness and wettability, and the active chemical groups being exposed may vary. Furthermore, previous experiments with coatings have shown that surface CSCc is a good indicator of the limits of charge delivery before radical formation starts, and cultures of neural cells have shown that the cell response changes for charges above or below the CSCc values.12 Taking this biological probe as a good indicator of surface electrochemistry helps to attribute significance to each parameter for the electrodes.
In general, as observed in Tables 2 and 3 for the N-graphene–IrOx hybrids discussed here, the absolute value of CSCc is larger where there is larger graphene content, but it does not correlate with the increase in total N. This indicates that basal graphene has a crucial role in nitrogen redox behavior, and that only some types of nitrogen add to the CSCc; for example, the type of nitrogen present in GNIr300 doping the graphene basal planes. On the other hand, if we consider the effect per carbon atom (with respect to iridium), the largest CSCc value is still the one found for exfoliated graphene (reaching 235 mC cm−2), which does not contribute any added oxygen (nor N, of course) to the hybrid.11 GNIr220 and GNIr300 have absolute CSCc values that are almost double those observed for GO and pristine graphene hybrids (up to 177 vs. 106 mC cm−2), although the values are similar to GOIr if considered per C atom (50–60 mC cm−2). This is due to the larger amount of N-graphene spontaneously attached to IrOx in the hybrid. Thus, from a practical point of view, these hybrids would be the choice for electrodes of larger capacity, while the correlations among the types of graphene contribute to our fundamental understanding of the role of nanocarbons in hybrids. The actual increase in carbon content from GNIr100, to GNIr220 and GNIr300, on the other hand, must be related to a larger interaction with IrOx in the latter cases through N groups in the structure, and this could be developed further.
The evolution of electrode hybrid coatings upon electrochemical cycling is also of great significance. Two types of behavior are found for the N-graphene hybrids (see Fig. 7). Upon cycling, the CVs still show IrOx anodic waves, but the first wave, near 0 V vs. Ag/AgCl, decreases in intensity except for GNIr100.
 | ||
| Fig. 7 Cyclic value. GNIr220 detaches from the electrode for voltammetry for each preparation of GNIr hybrids after 1000 cycles, in comparison with cycled IrOx and GOIr hybrid (10 mV s−1). | ||
On the other hand, the observed current density for GNIr100 increases notably in the region of both iridium peaks, and also at potentials for O2 evolution, while the first wave seems to disappear for GNIr220 and GNIr300, as it was also found for pure IrOx. The first oxidation wave in pure IrOx corresponds to mass loss due to K+ deintercalations, as found from electrochemical quartz microbalance studies,8 and its disappearance may indicate an alternative intercalation process with larger potential after a certain number of cycles. Also of significance is the lower O2 evolution potential for GNIr220 and GNIr300, which overlaps with the maximum of the second oxidation wave of Ir.
Beyond possible implications in the catalysis of O2 evolution, the evolution of materials during electrochemical cycling is significant in itself, and is clearly observed to correspond with CSCc values, in comparison with IrOx or graphene oxide hybrids (see Fig. 8). Usually, for most materials, a decrease in CSCc is observed with consecutive cycles.9–11 Previously reported IrOx hybrids with CNT, GO or exfoliated graphene show a retention of 70 to 80% of the initial capacity at 1000 cycles, while the hybrid prepared with graphite shows a rapid decrease, reaching about 20% of the original CSCc in the first 100 cycles.9–11 Nanostructuring therefore has a very significant effect. In this work, GNIr220 and GNIr300 follow the same type of behavior as other graphene–IrOx hybrids, having a large initial capacity that decreases upon cycling to about 50–75% of the original, possibly through oxidation and CO2 formation. This was observed previously for oxalate at room temperature during IrOx deposition,8 and suggests that the 50% loss may be due to the smaller amount of material as the cycling progresses in the case of GNIr220. Neither GNIr100 nor GNIr300 detach, however, evidencing better adhesion to the Pt substrate. Significantly, GNIr300, with a larger capacity than GOIr, also retains a CSC value above the maximum value for other hybrids after 1000 cycles, with large currents in the oxidation scans. Curiously, GNIr100 increases its charge capacity during the first few cycles, as it is oxidized (as described below), and slowly reaches the same CSCc as the GNIr220 coating. Such an increase must be related to the observed N oxidation described in the XPS data, as the final hybrid in both cases could be similar.
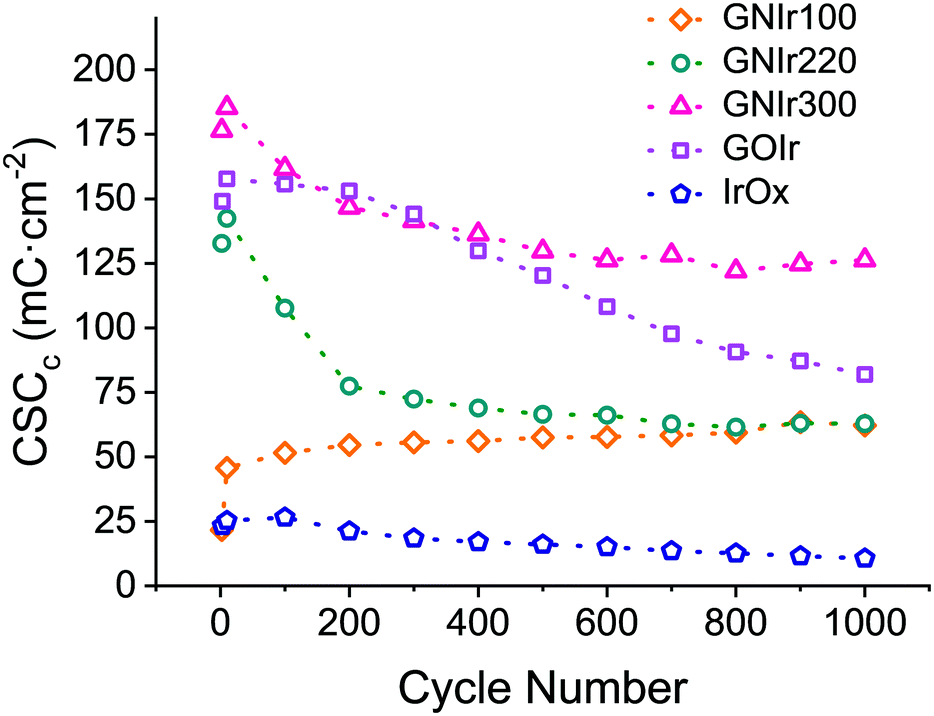 | ||
| Fig. 8 Cathodic CSC evolution during cycling for IrOx (blue), GOIr (grey), GNIr100 (yellow), GNIr220 (green) and GNIr300 (red). | ||
The large electrochemical stability found for GNIr300 suggests a large resistance to oxidation that could be crucial in its use as an electrode. Examples where carbon electrodes are used despite being oxidized, as in M–O2 batteries or in vanadium redox flow cells, would benefit from such an electrode.38–40
The actual redox processes involved during electrochemical treatment may be envisaged using XPS. Thus, after the 1000 voltammetric cycles (see Fig. 3–5, part c), XPS analysis shows the same type of deconvolution in N 1s binding energies for GNIr100 and GNIr220, but with larger intensity in the peaks from N–O and nitro groups (403 and 405 eV). Both hybrids differ in peak quantification from GNIr300, which still contains the maximum peak for the lower energy pyrrolic-, pyridinic- and quaternary-N (398–401 eV).
As mentioned above, Ir 4f5/2 and 4f7/2 doublets remain at the same binding energies, with only a slight modification of the asymmetry in the GNIr100 hybrid related to satellite peaks, which does not imply any energy shift and/or changes in oxidation states. The Ir 4f signals are rather insensitive to possible coordination changes and do not yield information on possible modifications of N–Ir interactions. It is known that K+ is exchanged in and out of pure IrOx without redox processes by immersion in aqueous solutions with no K+ ions present, presumably by exchange with H+ from the media. On the other hand, several intercalation phenomena are possible in IrOx (Na+ intercalation and OH− deintercalation at cathodic potentials, K+ redox deintercalation at anodic potentials from IrOx). In fact, the K content in the hybrids after cycling is lowered to K/Ir 1.3 vs. 1.6 in GNIr100, for example (see Table 2), and Na 1s and Na KLL signals appear in the spectra only after cycling in sodium-containing electrolyte (not related to direct wetting), evidencing the existing redox reactions in the hybrids, which are similar to those expected in pure IrOx.
On the other hand, along with a decrease in K/Ir upon cycling, depletion is observed at N/Ir ratios for GN100Ir and GN220Ir, the hybrids originally having larger values, while the O/Ir has increased dramatically. The significant decrease in N/Ir ratio, with stable C/Ir ratios, involves oxidation and liberation of nitrogen oxide groups. After cycling, the N/Ir values for these two hybrids, GNIr100 and GNIr220, become similar to those observed for GNIr300, as if only N-doping at the basal plane remains. Therefore, N-containing graphene hybrids with IrOx evolve to the most stable N-doping found in GNIr300, possibly through irreversible oxidation of the N groups to an oxidized NO-C-type entity, while other nitrogen species are eliminated.
The O 1s XPS spectra after cycling also show a significant shift towards lower binding energies, with the maximum at the signal corresponding to OH− (531 eV), which is also consistent with the oxidation and elimination of some C–O and CO, COO− groups (as CO2 or other soluble species) for which larger shifts had been observed.
In summary, although anodic formation of the hybrid involves oxidation for all hybrids, the lower temperature cases, GNIr100 and GNIr220, have further oxidation changes in N signals during electrochemical cycling, while the GNIr300 phase remains closer to the original material and maintains the highest value for CSCc. All processes involved suggest a large range of states in the oxidizing range.
Conclusions
Chemical interaction between iridium oxo species and N-containing graphene oxide allows anodic deposition of IrOx–N-graphene hybrids to different extents and with different Cgraphene/Ir ratios, depending on the graphene precursor used. The hybrids show up to seven times larger charge storage capacity than IrOx, and the higher values correspond to larger N-graphene content in the hybrid, and also to the greatest oxidation to nitro groups. The anodic hybrid formation is responsible for the stability of the nitro-graphene oxide in the final material and modulation of the redox state of N after cycling. The most stable hybrid corresponds to the one with the largest amount of N-doping in the graphene structure, and large relative amounts of graphene, i.e. GNIr300, which actually contains pyridinic-, pyrrolic-, graphitic-N and remaining oxygen. The CSCc values for GNIr300 remain high after 1000 cycles of electrochemical sweeping, and are higher than other hybrids tested previously and in this work. In terms of the contribution of carbons in the hybrids, if calculated per carbon atom, the pristine graphene hybrids with IrOx still show the highest CSCc, but N-doped graphene oxide (GNIr300 here) forms a hybrid with larger carbon amounts, and results in the largest increase in absolute CSCc value. The observed roughness correlates directly with the particle size of the precursor N-graphenes, and does not define charge capacity. Lower temperature treatment in the N-graphenes yields hybrids with redox states available from N that are oxidized during formation and during electrochemical treatment, without achieving the final CSC of the GNIr300 hybrid. In all the hybrids, oxidized nitrogen and nitro groups are present, which correlate with a large resistance to oxidation in GNIr300 and stable CSCc values after 1000 cycles. The redox modulation observed during preparation of the hybrid material and during electrochemical cycling evidences the large stability of the nitro groups present in the hybrid, a remarkable finding that opens a new pathway in the chemistry of graphene.Authors contributions
EP performed most of the hybrid material electrodeposition and XPS studies. GT, AF and SS designed and prepared the graphene precursors. NC started the first hybrid within the frame of her PhD thesis. NCP directed the main work of hybrid formation and electrochemical work. GT and NCP handled the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financing from the Ministry of Science, Innovation and Universities, through MAT2015-65192-R, MAT2017-86616-R, RTI2018-097753-B-I00, Fundació Marató TV3 (110130/31) and Severo Ochoa Program (SEV-2015-0496).References
- E. Frackowiak and F. Beguin, Carbon materials for the electrochemical storage of energy in capacitors, Carbon, 2001, 39, 937 CrossRef CAS.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Nanoporous metal/oxide hybrid electrodes for electrochemical supercapacitors, Nat. Nanotechnol., 2011, 6, 232 CrossRef CAS PubMed.
- X. Deng, J. Li, L. Ma, J. Sha and N. Zhao, Three-dimensional porous carbon materials and their composites as electrodes for electrochemical energy storage systems, Mater. Chem. Front., 2019, 3, 2221 RSC.
- L. Lyu, K.-d. Seong, D. Ko, J. Choi, C. Lee, T. Hwang, Y. Cho, X. Jin, W. Zhang, H. Pang and Y. Piao, Recent development of biomass-derived carbons and composites as electrode materials for supercapacitors, Mater. Chem. Front., 2019, 3, 2543 RSC.
- G. Zhang, Y. Han, C. Shao, N. Chen, G. Sun, X. Jin, J. Gao, B. Ji, H. Yang and L. Qu, Processing and manufacturing of graphene-based microsupercapacitors, Mater. Chem. Front., 2018, 2, 1750 RSC.
- M. Yagi, E. M. I. Tomita and T. Kuwabara, Remarkably high activity of electrodeposited IrO2 film for electrocatalytic water oxidation, J. Electroanal. Chem., 2005, 579, 83 CrossRef CAS.
- S. Gawad, M. Giugliano, M. Heuschkel, B. Wessling, H. Markram, U. Schnakenberg, P. Renaud and H. Morgan, Substrate arrays of iridium oxide microelectrodes for in vitro neuronal interfacing, Front. Neuroeng., 2009, 2, 1 CAS.
- A. Cruz, Ll. Abad, N. M. Carretero, J. Moral-Vico, J. Fraxedas, P. Lozano, G. Subías, V. Padial, M. Carballo, J. Collazos-Castro and N. Casan-Pastor, Iridium Oxohydroxide, a Significant Member in the Family of Iridium Oxides. Stoichiometry, Characterization, and Implications in Bioelectrodes, J. Phys. Chem. C, 2012, 116, 5155 CrossRef CAS.
- N. M. Carretero, M. P. Lichtenstein, E. Pérez, L. Cabana, C. Suñol and N. Casañ-Pastor, IrOx-Carbon Nanotubes Hybrid: A Nanostructured Material for Electrodes with Increased Charge Capacity in Neural systems, Acta Biomater., 2014, 10, 4548 CrossRef CAS PubMed.
- N. M. Carretero, M. P. Lichtenstein, E. Pérez, S. Sandoval, G. Tobias, C. Suñol and N. Casan-Pastor, Enhanced Charge Capacity in Iridium Oxide-Graphene Oxide Hybrids, Electrochim. Acta, 2015, 157, 369 CrossRef CAS.
- E. Pérez, M. P. Lichtenstein, C. Suñol and N. Casan-Pastor, Coatings of Nanostructured Pristine Graphene-IrOx Hybrids for Neural Electrodes: Layered Stacking and the role of non-oxygenated Graphene, Mater. Sci. Eng., C, 2015, 55, 218 CrossRef PubMed.
- M. P. Lichtenstein, E. Pérez, L. Ballesteros, C. Suñol and N. Casañ-Pastor, Short term electrostimulation enhancing neural repair in vitro using large charge capacity intercalation electrodes, Appl. Mater. Today, 2017, 6, 29 CrossRef.
- L. Zhang, Y. Ye, D. Cheng, W. Zhang, H. Pan and J. Zhu, Simultaneous reduction and N-doping of graphene oxides by low-energy N2+ ion sputtering, Carbon, 2013, 62, 365 CrossRef CAS.
- I. Choi, H. Y. Jeong, D. Y. Jung, M. Byun, C.-G. Choi, B. H. Hong, S.-Y. Choi and K. J. Lee, Laser-Induced Solid-Phase Doped Graphene, ACS Nano, 2014, 8, 7671 CrossRef CAS PubMed.
- N. Wei, L. Yu, Z. Sun, Y. Song, M. Wang, Z. Tian, Y. Xia, J. Cai, Y.-y. Li, L. Zhao, Q. Li, M. H. Rümmeli, J. Sun and Z. Liu, Scalable Salt-Templated Synthesis of Nitrogen-Doped Graphene Nanosheets toward Printable Energy Storage, ACS Nano, 2019, 13, 7517 CrossRef CAS PubMed.
- X. Chen, X. Bo, W. Ren, S. Chen and C. Zhao, Microwave-assisted shock synthesis of diverse ultrathin graphene-derived materials, Mater. Chem. Front., 2019, 3, 1433 RSC.
- B. Jiang, C. Tian, L. Wang, L. Sun, C. Chen, X. Nong, Y. Qiao and H. Fu, Highly concentrated, stable nitrogen-doped graphene for supercapacitors: Simultaneous doping and reduction, Appl. Surf. Sci., 2012, 258, 3438 CrossRef CAS.
- D. Kepić, S. Sandoval, Á. Pérez del Pino, E. György, L. Cabana, B. Ballesteros and G. Tobias, Nanosecond Laser-Assisted Nitrogen Doping of Graphene Oxide Dispersions, ChemPhysChem, 2017, 18, 935 CrossRef PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, Simultaneous nitrogen doping and reduction of graphene oxide, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- X. Lang, A. Hirata, T. Fujita and M. Chen, Nanoporous metal/oxide hybrid electrodes for electrochemical supercapacitors, Nat. Nanotechnol., 2011, 6, 232 CrossRef CAS PubMed.
- C. Xu, M. Lu, B. Yan, Y. Zhan, P. Balaya, L. Lu and J. Y. Lee, Electronic Coupling of Cobalt nanoparticles to Nitrogen-doped Graphene for Oxygen reduction and evolution reactions, ChemSusChem, 2016, 9, 3067 CrossRef CAS PubMed.
- A. Sarapuu, E. Kibena-Poldsepp, M. Borghei and T. Tammeveski, Electrocatalysis of oxygen reduction on heteroatom-doped nanocarbons and transition metal-nitrogen-carbon catalysts for alkaline membrane fuel cells, J. Mater. Chem. A, 2018, 6, 776 RSC.
- K. Choi, S. Lee, Y. Shim, J. Oh, S. Kim and S. Park, Electrocatalytic performances of N-doped graphene with anchored iridium species in oxygen reduction reaction, 2D Mater., 2015, 2, 034019 CrossRef.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, Nitrogen-doped graphene and its electrochemical applications, J. Mater. Chem., 2010, 20, 7491 RSC.
- Y. J. Yang and W. Li, Hierarchical Ni-Co double hydroxide nanosheets on reduced graphene oxide self-assembled on Ni foam for high-energy hybrid supercapacitors, J. Alloys Compd., 2019, 776, 543–553 CrossRef CAS.
- N. M. Latiff, C. Mayorga-Martinez, L. Wang, Z. Sofer, A. C. Fischer and M. Pumera, Microwave irradiated N- and B,Cl-doped graphene: Oxidation method has strong influence on capacitive behavior, Appl. Mater. Today, 2017, 9, 204 CrossRef.
- A. Y. S. Eng, A. Ambrosi, C. K. Chua, F. Sanek, Z. Sofer and M. Pumera, Unusual Inherent Electrochemistry of Graphene Oxides Prepared using Permanganate oxidants, Chem. – Eur. J., 2013, 19, 12673 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Electrochemistry of Graphene and Related Materials, Chem. Rev., 2014, 114, 7150 CrossRef CAS PubMed.
- S. Sandoval, N. Kumar, A. Sundaresan, C. N. R. Rao, A. Fuertes and G. Tobias, Enhanced Thermal Oxidation Stability of Reduced Graphene Oxide by Nitrogen Doping, Chem. – Eur. J., 2014, 20, 11999 CrossRef CAS PubMed.
- S. Sandoval, N. Kumar, J. Oró-Solé, A. Sundaresan, C. N. R. Rao, A. Fuertes and G. Tobias, Tuning the nature of nitrogen atoms in N-containing reduced graphene oxide, Carbon, 2016, 96, 594 CrossRef CAS.
- X. Wang, Z. Hou, T. Ikeda, M. Oshima, M.-a. Kakimoto and K. Terakura, Theoretical characterization of X-ray absorption, emission, and photoelectron spectra of nitrogen doped along graphene edges, J. Phys. Chem. A, 2012, 117, 579–589 CrossRef PubMed.
- G. Gabriel, G. Sauthier, J. Fraxedas, M. Moreno-Manas, M. T. Martínez and C. Miravitlles, et al., Preparation and characterization of single-walled carbon nanotubes functionalized with amines, Carbon, 2006, 44, 1891 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, Review on recent progress in nitrogen doped graphene: synthesis, characterization, and its potential applications, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- J. Baltrusaitis, P. M. Jayaweera and V. H. Grassian, XPS study of nitrogen dioxide adsorption on metal oxide particle surfaces under different environmental conditions, Phys. Chem. Chem. Phys., 2009, 11, 8295 RSC.
- Q. Fulian and R. G. Compton, Laser-Activated Voltammetry: Measurement of the Diffusion Coefficients of Electropassivating Species. Application to Pyrrole and Phenol in Aqueous Solution, Anal. Chem., 2000, 72, 1830 CrossRef CAS PubMed.
- A. M. Khattak, Z. A. Ghazi, B. Liang, N. A. Khan, A. Iqbal, L. Li and Z. Tang, A redox-active 2D covalent organic framework with pyridine moieties capable of faradaic energy storage, J. Mater. Chem. A, 2016, 4, 16312 RSC.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nitrogen-Doped Graphene for High-Performance Ultracapacitors and the Importance of Nitrogen-Doped Sites at Basal Planes, Nano Lett., 2011, 11, 2472 CrossRef CAS PubMed.
- J. Jin, F. Fu Xiaogang, Y. Liu, Z. Wei, K. Niu and J. Zhang, Identifying the active site in Nitrogen-doped graphene for the VO+2/VO2+ Redox reaction, ACS Nano, 2013, 7, 4764 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Nitrogen and Oxygen Dual-Doped Carbon Hydrogel Film as a Substrate-Free Electrode for Highly Efficient Oxygen Evolution Reaction, Adv. Mater., 2014, 26, 2925 CrossRef CAS PubMed.
- Y. Yang, F. Zheng, G. Xia, Z. Luna and Q. Chen, Experimental and theoretical investigations of nitro-group doped porous carbon as a high performance lithium-ion battery anode, J. Mater. Chem. A, 2015, 3, 18657 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, general XPS survey scan and IR data. See DOI: 10.1039/c9qm00752k |
| This journal is © the Partner Organisations 2020 |