
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening Metathesis Polymerisation derived poly(dicyclopentadiene) based materials
Sebastijan
Kovačič
 a and
Christian
Slugovc
*bc
a and
Christian
Slugovc
*bc
aNational Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, A 8010 Graz, Austria. E-mail: slugovc@tugraz
cChristian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, A 8010 Graz, Austria
First published on 4th June 2020
Abstract
During the last two decades, poly(dicyclopentadiene) prepared by Ring-opening Metathesis Polymerisation has emerged as an important competitor for epoxy- and polyester resins. This review article summarises the contributions from academia in this timeframe. The fields of initiator system development, polymer chemistry and physics, composites of poly(dicyclopentadiene), use in self-healing composites, novel processing opportunities and macro- as well as microporous materials are covered.
 Sebastijan Kovačič | Sebastijan Kovačič received his PhD in Chemistry from the University of Maribor, Slovenia, in 2011 working on hybrid polyHIPE materials. After a two-years postdoctoral stay at the Graz University of Technology with Prof. C. Slugovc, he received an Aly Kaufman Fellowship of Trust and joined the group of Prof. M. S. Silverstein at Technion, Israel Institute of Technology. Since 2014, he has been a research associate at the National Institute of Chemistry (NIC), Slovenia. His main research interests focus on emulsions, their generation and stability, and the various porous materials that can be synthesized therefrom including nanocomposites, hydrogels or carbons. |
 Christian Slugovc | Christian Slugovc received his PhD from Vienna University of Technology in 1998 under the supervision of Prof. K. Kirchner. From 1999 to 2000 he did a postdoctoral stay at the CSIC-Sevilla with Prof. E. Carmona. Afterwards he joined Graz University of Technology and habilitated in macromolecular chemistry in 2007. Currently he is an associate professor, deputy-head of Graz University of Technology's lead-project Porous Materials@Work and the director of the Christian Doppler Laboratory for Organocatalysis in Polymerisation. His current research strives towards advancing the efficacy in synthetic macromolecular chemistry and bringing (organic) materials to work. |
1. Introduction
Dicyclopentadiene (DCPD)1 is obtained as a by-product from steam cracking performed during the production of ethylene.2,3 DCPD finds applications in unsaturated polyester and hydrocarbon resins, in ethylene propylene diene monomer (EPDM) elastomers, as chemical feedstock, in cyclic olefin polymers and as a monomer for poly(dicyclopentadiene) (pDCPD) prepared via Ring-opening Metathesis Polymerisation (ROMP). The worldwide DCPD market is estimated to exceed 500 million Euro in 2019 and is expected to grow by annually 5% over the next 5 years.4 Especially the market segment of highly purified DCPD is gaining growth rate as pDCPD increasingly finds applications. ROMP of DCPD results in a cross-linked polymer that possesses high notch-impact strength and offers high temperature and corrosion resistant properties as well as high stability versus chemicals. Furthermore, the polymer's surface is quickly oxidized in air and thus a relatively polar surface is generated which can be easily painted. An additional corona treatment or flaming as required for many other polymers is thus not necessary. ROMP-derived pDCPD is used for example in the manufacturing of agricultural equipment, of body panels, and of cells for the chlor-alkali industry or in the petrochemical industry for piping. Furthermore, reinforced pDCPD parts became accessible and enabled new concepts for construction designs. pDCPD is mainly processed via reaction injection moulding (RIM), resin transfer moulding (RTM) and vacuum-assisted RTM (VTRTM).5–10 By these techniques large and complex parts can be manufactured in a quick and efficient way. Its comparatively low density combined with good mechanical properties makes it environmentally beneficial for applications in the automotive domain, where fuel efficacy is an important topic. Lately pDCPD based technology has additionally targeted, amongst others, the markets of downhole tools, subsea insulation, circuit boards, pressure vessels and parts for the energy generating industry.Herein the research on pDCPD-based materials conducted in roughly the last two decades in academia is summarized. Emphasis is laid on ruthenium based initiating systems.
2. Contemporary research directions
2.1. Processing of DCPD – the need for latent initiators
The initially implemented RIM processing of DCPD is based on mixing two monomer streams, one containing a pre-initiator and the second an activator, just before the combined resin is injected into the mould. Additionally, a retarder is added to adjust the (usually short) induction period, facilitating the filling of the mould.11 This implementation is by far not ideal when more time to fill the mould (big parts, parts exhibiting shapes with different aspect ratios, and fabrication of reinforced parts) is needed. Furthermore, classical initiation systems are sensitive to moisture and air, limiting also the use of many co-monomers and/or fillers.Therefore, a lot of attention has been devoted to the development of latent and insensitive initiation systems.12 Of particular interest is to enable so-called one-component systems. In such resins, the monomers and the initiator are already combined and polymerisation is then started after a predetermined time or by an external stimulus, in most cases elevated temperature. Such one-component resins facilitate RTM and VTRTM but also RIM processes in terms of providing long working time and better chemical compatibility with other ingredients as well as oxygen and moisture. Additionally, less sophisticated process machineries can be employed. Amongst thermally switchable initiators, also initiators which can be triggered by light, acids, redox-reactions and mechanical force have been investigated.12–15 Most initiator systems are based on ruthenium because of its high functional group tolerance and insensitivity towards oxygen and protic groups.
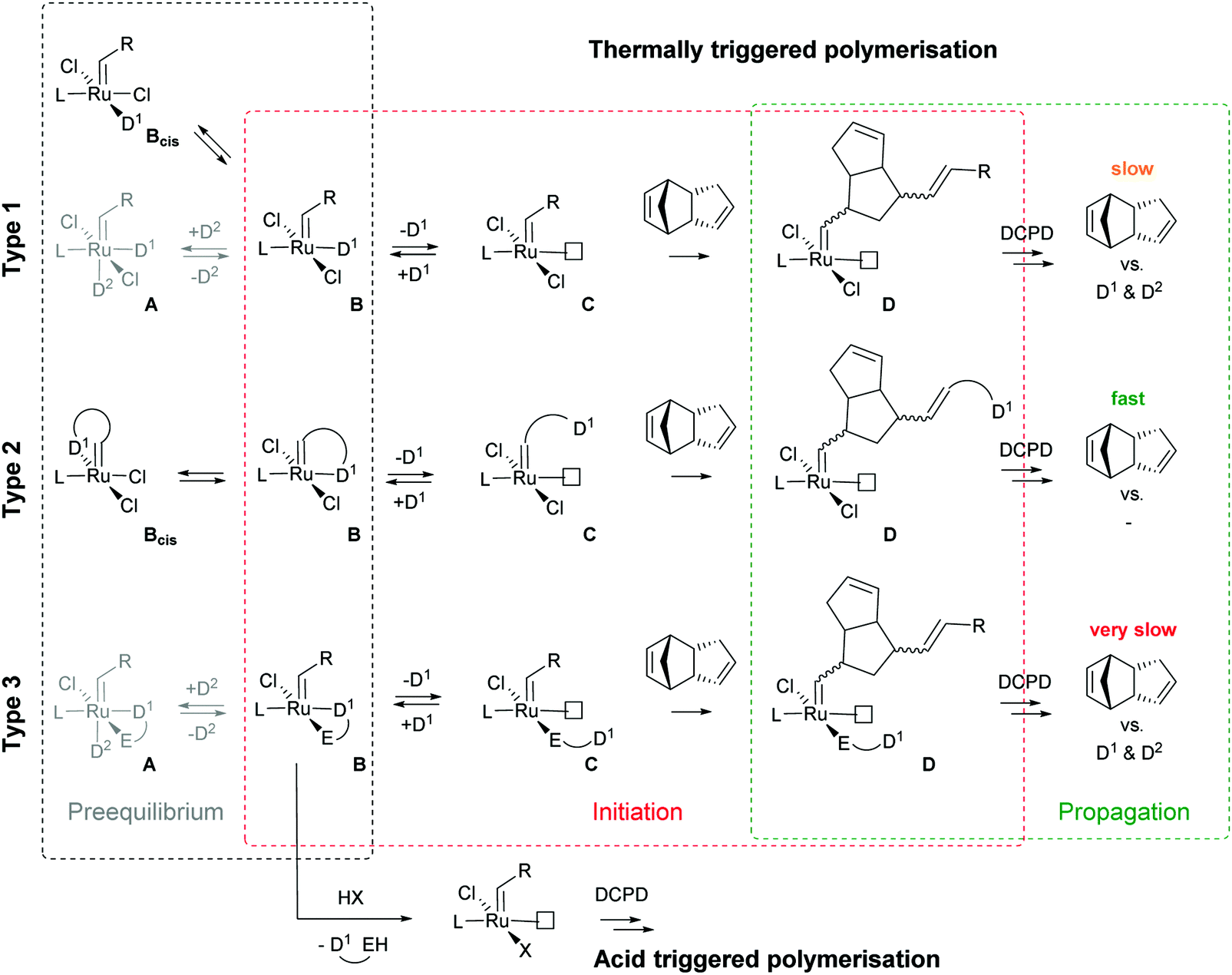 | ||
| Scheme 1 Most common avenues towards latent Ru-based initiators. L is typically a phosphine or an N-heterocyclic olefin; D1 and D2 are typically σ- or π-donor ligands such as amines, phosphines or N-heterocyclic carbenes; E is typically O or S; and R is typically an organic rest like a phenyl group. | ||
Subsequently the metathesis event takes place providing the initiation (Type 1, D). During propagation, the decoordinated ligand D1 is competing with DCPD for the vacant coordination site. Therefore, propagation is relatively slow. Upon adding more donor ligand (D2; D2 could be the same as D1), both initiation and propagation are further decelerated.17,18 In most publications the commercially available initiators G119,20 or G221,22 are used (Fig. 1) and, if necessary, the working time is adjusted upon addition of additional donor ligands like with amines,18 phosphines,23–25 N-heterocyclic carbenes26,27 or phosphite esters.28 Additionally, M2, an analogue of G2, is commercially available and exhibits a better latent behaviour than e.g.G2.29 Another way to create initiators that are distinctly more latent is to employ Type 1 pre-initiators exhibiting a cis-dichloro stereochemistry (Bcis). However, only a few cis-dichloro configured initiators of Type 1 are known.30 In this case, the metathetically inactive Bcis has to isomerize to the corresponding trans-dichloro form before initiation can start. As the only representative of this family, initiator M22 was benchmarked against G2 and M2 and showed by far the highest latency.29 Note that 10–30 ppm initiator (G2, M2 or M22) is sufficient to obtain pDCPD parts with similar mechanical characteristics to industrially manufactured samples.
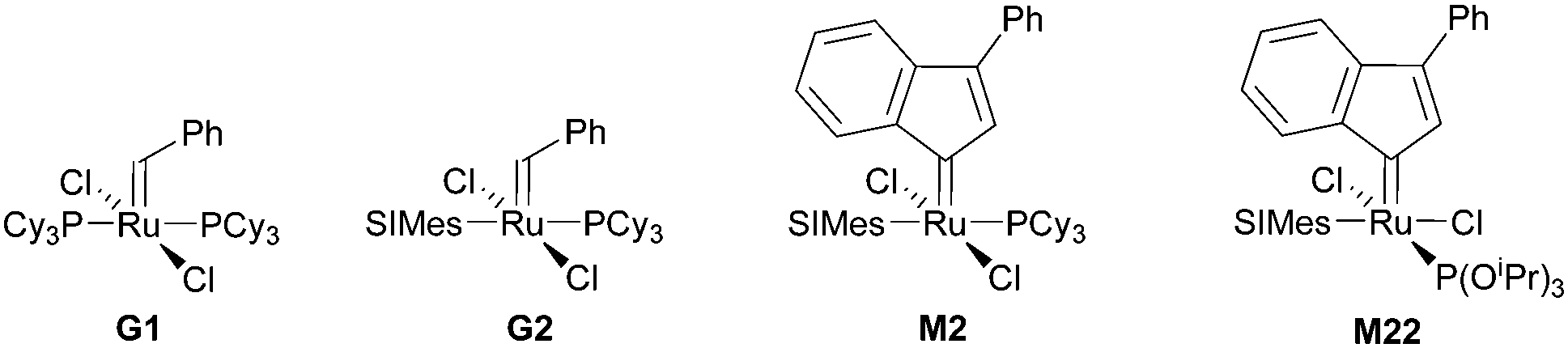 | ||
| Fig. 1 Commercial Type 1 initiators used for the polymerisation of DCPD. PCy3 = tricyclohexylphosphine; SIMes = 1,3-dimesitylimidazolidin-2-ylidene. | ||
In a second layout, the Type 2 system, a chelating carbene ligand is used (B). In that case, the decoordination of D1 is entropically disfavoured and, accordingly, initiation is slowed down. The effect becomes even bigger if the corresponding cis-dichloro isomer (Bcis) is used. Once initiated (Type 2, D), the chelate effect is cancelled because D1 is moved away from the metal centre and propagation is relatively fast. Note that addition of donor ligands (used to increase the processing window in Type 1 initiators) could have counterproductive effects because dechelation is accelerated in this case.31 As in the case of Type 1 initiators many potential latent compounds have been disclosed,32 but only a few have been described according to their performance in polymerising DCPD.33–38Fig. 2 shows exemplary Type 2 initiators which were tested. Most of them feature a nitrogen as the second ligand atom of the chelate ring (1–3), but also the sulphur-based variant 4 has been described.39 Although the different testing conditions used in the literature do not allow for identifying the best candidates, it is revealed that often subtle changes of the initiator structures cause big differences in their latency. Perhaps this is the reason why such Type 2 initiators are rarely applied in preparing pDCPD in the open research literature, although some of them are even commercialized.40–42
 | ||
| Fig. 2 Selected examples of Type 2 initiators tested in the polymerisation of DCPD. | ||
In a third layout, the donor ligand is tethered to an anionic ligand (Type 3, B).43,44 Variants featuring two such chelating ligands are also known.45,46 Similarly to Type 2 initiators, the chelate effect slows down the initiation, but also propagation is distinctly decelerated. Therefore, this layout is rather used in acid switchable systems (vide infra). Finally, a combination of Type 2 and 3, i.e. chelation takes place via the alkylidene and an anionic ligand, is known.47–49
In the area of potentially less toxic molybdenum and tungsten imido alkylidene complexes, the first latent and considerably air and moisture tolerant initiators for DCPD have been described recently.50,51 For more sensitive initiators a protection strategy by encapsulation in paraffin is available.52
As already emphasised, a meaningful comparison of the performance of the different initiators is hard to make. A standardized test protocol, as available for other olefin metathesis reactions, is missing.53 As a first step to establish such a protocol, it is suggested to study the curing of DCPD at low initiator loadings (e.g. 30 ppm with respect to DCPD) using thermogravimetric analysis coupled with differential scanning calorimetry (e.g. with a heating ramp of 3 °C).29 Additionally the quest towards latent initiators which are soluble in the monomer is underdeveloped. Solvents are undesirable in many processing technologies for DCPD and the solubility of many initiators in DCPD is limited.
2.2. Influencing the properties of pDCPD
DCPD can exist in two stereoisomers, the endo- and the exo-isomer. Typically commercially available DCPD consists of >95% endo-DCPD and most studies and applications use endo-DCPD. However, exo-DCPD polymerises distinctly faster than endo-DCPD. The different polymerisation rates were ascribed to steric effects. In endo-DCPD the penultimate repeating unit is believed to interact with incoming DCPD, thus slowing down the polymerisation.72 However, we hypothesise that the higher steric crowding of the more reactive double bond in endo-DCPD (Fig. 3) impedes productive collisions with the metal centre necessary for coordinating the monomer. Proper coordination of the substrate to the metal alkylidene is known to be the first step in olefin metathesis and should therefore be more facile in the case of exo-DCPD.
 | ||
| Fig. 3 Space filling cartoons of endo- and exo-DCPD. | ||
Polymerising pDCPD produces in many cases and under most conditions a cross-linked, insoluble material. There has been much debate about the chemical nature of the crosslinks in many studies discussed in this article. Three cross-linking reactions are feasible (especially for initiators like G1 and G2). The most obvious is ring-opening metathesis of the second, less reactive double bond leading to cross-linking of the polymer.73 This mechanism operates already at low temperature. The second possibility is olefin addition, which rather takes place at elevated temperatures.74–76,78 A third option is oxidative cross-linking, which happens when pDCPD is aging under air (see below).77,78 Increased cross-linking is generally reflected in higher glass transition temperatures,79 which usually range from 140 to 165 °C (for cross-linked versions). However, even glass transition temperatures higher than 200 °C were recorded for pDCPD upon repeated heating–cooling cycles22 or prolonged heating.78
Cross-linking is a main factor for determining the mechanical properties of the material. Upon changing pDCPD's crosslinking density by copolymerisation of either chain extenders or cross-linkers, the impact of crosslinks on the mechanical properties was investigated.21,80,81 Typical mechanical properties of pDCPD comprise a Young's modulus in the range of 1.6–2.0 GPa, tensile stress at yield of about 35–70 MPa and elongation at break from 5–100%.21,29,71 Unlike other thermosetting materials such as epoxy resins, pDCPD lacks strong non-covalent interactions and thus cross-linking is only due to covalent bonds. This is the reason for the pronounced ductility of pDCPD compared to epoxy resins (with a similar glass transition temperature). The ductility results in the high toughness and remarkable impact performance of pDCPD.21 The ductility is conserved even at a temperature of 77 K.82 Digging deeper, these favourable properties are attributed to a facile plastic deformation and nano-scale void formation which accommodates the strain.83,84 The merit of the mechanical properties of pDCPD was impressively described in a publication on the ballistic performance of pDCPD in comparison with two epoxy–amine systems with comparable glass transition temperatures. pDCPD showed a 300–400% better penetration resistance in ballistic energy dissipation in a temperature range of −55 to 75 °C.85
The choice of the initiator (together with the processing conditions) not only determines the degree of cross-linking but also the microstructure of the polymer-backbone. While G1 produces rather trans-double bonds, a higher cis-double bond content is obtained with G2. The stereochemistry of the pDCPD backbone is another factor influencing the thermal and mechanical properties of the polymer. A nice showcase paper discussing all these aspects influencing the final properties of, in this case, pDCPD aerogels might be instructive for researchers who wish to become active in this field.86 Furthermore, in the context of cis and trans content, it is worth mentioning that molybdenum and tungsten (but no ruthenium) based initiators are known to provide stereospecific ROMP of DCPD, yielding in some cases crystalline polymers.87–89 Achieving stereospecific ROMP of DCPD with ruthenium based initiators might open the door for materials with better or even new properties.
Chain transfer agents are barely used in (materials directed) pDCPD chemistry because they impede the formation of the cross-linked structure or at least decrease the glass transition temperature of the thermoset. Two studies with monoterpenes demonstrate the principle. One publication reported the synthesis of hyperbranched (soluble) pDCPD upon using β-pinene, limonene oxide or d-limonene.98 Using less monoterpene the degree of crosslinking of pDCPD was altered and the thermoset was plasticised to a desired degree.99 Very low amounts of fluorescent chain transfer agents (not altering the thermal and mechanical properties) were used to study the polymerisation of pDCPD by operando fluorescence microscopy imaging. It was found that the morphology of the precipitated polymers at an early stage persists in the bulk polymer. Strand aggregation was found to be slow and irreversible relative to precipitation.100
Another type of copolymerisation makes use of the olefin addition occurring at elevated temperature. Substituted cyclopentadiene101 derivatives and substituted maleimide derivatives were employed.102,103 Rigid cross-linking monomers enhanced the thermal and dynamic mechanical properties.
Thermoset blends of pDCPD and epoxy resins resulted in macroscopically phase separated systems featuring two glass transition temperatures. In the blend the higher strength of the epoxy resin can be greatly retained while the fracture toughness was increased.104,105 The blends were used for gluing carbon fibre reinforced composites with steel. The epoxy–pDCPD blend adhesives realized much higher bond strengths compared to either neat epoxy or neat pDCPD, which was attributed to an increased shear toughness of the blend.106 Similarly, the blend of polyethylene wax and pDCPD was studied; here thermally induced cross-linking between pDCPD and the wax was postulated.107
Copolymerisation of polyhedral oligomeric silsesquioxanes with DCPD did not result in significant property changes of the composite material.108 However, the use of vinyl-functionalised silica particles (diameter 0.3–0.7 μm) led to a distinct enhancement of the toughness of the materials. Upon just adding 0.2 wt% of the particles the tensile toughness increased by a factor of 14, while the yield strengths decreased by about 10% compared to pDCPD without particles.109 In follow-up work, the silica particles have been differently functionalised and the effect of functionalisation on the mechanical properties of pDCPD composite materials was studied. Ethyl-functionalised particles were found to increase the strength and toughness.110–112 Poly(styrene) coated silica nanoparticles were shown to decrease the friction coefficient and the wear rate of pDCPD.113,114 An older study reports norbornene-capped silica particles and an improved wear resistance of the PDCPD/silica nanocomposite processed by RIM.115 Also MoS2 pDCPD nanocomposites were investigated for their wear resistance.116 The outstanding dielectric characteristics of pDCPD (which are similar to those of poly(propene)) motivated development of a RIM process allowing further improvement of them. By incorporation of 10% fumed silica, a permittivity of 2.8 was obtained and the mechanical properties were not compromised significantly.117 Another example aiming in the same direction comprised the use of BaTiO3 nanoparticles (5 wt%) for increasing the permittivity of pDCPD (1.7) to 2.4 whereby the breakdown strength was only reduced by 13%.118 Work on montmorillonite nanocomposites was performed.119–122 Most interesting is a contribution in which so-called intergallery-surface-initiated ROMP was used to obtain an exfoliated montmorillonite nanocomposite with 50% increased stiffness in compression.123
The most influential work on pDCPD nanocomposites was published in 2008. Norbornene functionalised multiwalled carbon nanotubes were used in low loadings (<0.5 wt%) and the resulting nanocomposites exhibited a slightly enhanced Young's modulus of 2.09–2.14 GPa (neat pDCPD 2.07 GPa) but a distinctly enhanced strain at break up to 51.8% (neat pDCPD 5.75%) resulting in a toughness of about 25 MPa (2.44 for the reference).124 A different carbon nanotube functionalisation route was disclosed and the derived material tested with DCPD.125,126 Non co-polymerisable carbon materials used as nanofillers comprise expanded graphite nanosheets,127 vapour grown carbon fibers and graphene oxide.128,129
Another type of filling is aimed at reducing the cost of pDCPD and its use in structural applications. Wollastonite, a cheap filler with a needle-like crystal shape and high thermal stability, was used to prepare composite materials with pDCPD. Up to 67 wt% of the CaSiO3 mineral was incorporated by planetary mixing and the composites’ high compressive strength of more than 100 MPa revealed them to be superior to high strength concrete (63 MPa).141 Recently, DCPD-based resins were tested for their ability to penetrate into rocks and to reinforce the rock's mechanical strength. Due to the low viscosity of the resin, the desired diffusion was obtained in dry and 50% water saturated rocks. Upon curing the storage modulus and the flexural strengths were improved, qualifying the resin for downhole applications such as sealants or plugs.142
2.3. Emerging processing of pDCPD
In 2001, the first report of frontal Ring-opening Metathesis Polymerisation was published. In this early report, high purity DCPD, G1 and triphenylphosphine as a retarder were introduced. The formulation was prepared above the melting point of DCPD and was then quickly cooled to 27 °C. The actual frontal polymerisation was then performed on solid mixtures. A maximum front temperature of about 205 °C propagating with a front velocity of 2.6 cm min−1 was obtained when the formulation contained 2000 equiv. of DCPD, 1 equiv. of G1 and 2.6 equiv. of triphenylphosphine.23 In follow-up work, G2 in combination with 4-dimethylaminopyridine as the retarder was introduced as the initiating system. Maximum front temperatures of around 200 °C were found when the formulation contained 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 equiv. of DCPD, 1 equiv. of G2 and between 1 and 8 equiv. of 4-dimethylaminopyridine. The variation of the retarder's concentration led to different front velocities, which were between 7.5 and 2.2 cm min−1. The pot life of the liquid formulation (toluene was used to keep DCPD liquid) was improved to about 20–30 min.160 As an alternative to 4-dimethylaminopyridine, limonene was tested as a retarding agent. Limonene is a chain transfer agent and thus, albeit frontal polymerisation was feasible, the desired mechanical properties of pDCPD were lost.161 Highly reactive exo-DCPD has been tested as a monomer in frontal polymerisation with G2 decelerated by 4-dimethylaminopyridine. Low G2 loadings of 10 ppm were sufficient to provide frontal polymerisation in this case. However, the pot life of these formulations was still about 20 min.162 The pot life issue was tackled by using aliphatic phosphite ester derivatives as retarders for G2. By varying the phosphite ester structure and concentration, a pot life of (endo-)DCPD-based formulations between 0.25 and 30 h was obtained while still allowing the frontal polymerisation to proceed. The velocity can be adjusted between 1 and 8 cm min−1 (Fig. 4).28 Such a high pot life is the key for industrial applicability and the same group demonstrated the rapid fabrication of parts with microscale features and carbon-fibre-reinforced polymer composites. The mechanical properties of such processed parts are similar to those of pDCPD cured under conventional conditions. Accordingly, frontal polymerisation processing allows for reducing energy requirements and cure times. Especially for the processing of fibre-reinforced polymer composites a huge saving of time and energy is achieved when compared to conventional epoxy-based parts. Furthermore, three-dimensional printed structures can be fabricated with these formulations. Upon tuning the retarder concentration, free-standing elastomeric gels containing prepolymerised DCPD are obtained. These gels can be extruded from a print head and frontally polymerised upon applying heat to the substrate. In such a process, simultaneous free-form printing of thermosets is achieved.163 Numerical studies of the initiation and propagation of the polymerisation front in fibre-reinforced composites were conducted to better understand frontal polymerisation of DCPD.164 Lately, the frontal copolymerisation of DCPD with other norbornene derivatives was disclosed with the aim to tune the material properties and simultaneously improve the efficiency in FROMP-based material manufacturing. In doing so, an unexpected increase in the front velocity when copolymerising DCPD with dinorbornenyl cross-linking monomers was found and rationalized.165
000 equiv. of DCPD, 1 equiv. of G2 and between 1 and 8 equiv. of 4-dimethylaminopyridine. The variation of the retarder's concentration led to different front velocities, which were between 7.5 and 2.2 cm min−1. The pot life of the liquid formulation (toluene was used to keep DCPD liquid) was improved to about 20–30 min.160 As an alternative to 4-dimethylaminopyridine, limonene was tested as a retarding agent. Limonene is a chain transfer agent and thus, albeit frontal polymerisation was feasible, the desired mechanical properties of pDCPD were lost.161 Highly reactive exo-DCPD has been tested as a monomer in frontal polymerisation with G2 decelerated by 4-dimethylaminopyridine. Low G2 loadings of 10 ppm were sufficient to provide frontal polymerisation in this case. However, the pot life of these formulations was still about 20 min.162 The pot life issue was tackled by using aliphatic phosphite ester derivatives as retarders for G2. By varying the phosphite ester structure and concentration, a pot life of (endo-)DCPD-based formulations between 0.25 and 30 h was obtained while still allowing the frontal polymerisation to proceed. The velocity can be adjusted between 1 and 8 cm min−1 (Fig. 4).28 Such a high pot life is the key for industrial applicability and the same group demonstrated the rapid fabrication of parts with microscale features and carbon-fibre-reinforced polymer composites. The mechanical properties of such processed parts are similar to those of pDCPD cured under conventional conditions. Accordingly, frontal polymerisation processing allows for reducing energy requirements and cure times. Especially for the processing of fibre-reinforced polymer composites a huge saving of time and energy is achieved when compared to conventional epoxy-based parts. Furthermore, three-dimensional printed structures can be fabricated with these formulations. Upon tuning the retarder concentration, free-standing elastomeric gels containing prepolymerised DCPD are obtained. These gels can be extruded from a print head and frontally polymerised upon applying heat to the substrate. In such a process, simultaneous free-form printing of thermosets is achieved.163 Numerical studies of the initiation and propagation of the polymerisation front in fibre-reinforced composites were conducted to better understand frontal polymerisation of DCPD.164 Lately, the frontal copolymerisation of DCPD with other norbornene derivatives was disclosed with the aim to tune the material properties and simultaneously improve the efficiency in FROMP-based material manufacturing. In doing so, an unexpected increase in the front velocity when copolymerising DCPD with dinorbornenyl cross-linking monomers was found and rationalized.165
 | ||
| Fig. 4 Frontal polymerisation of pDCPD according to ref. 28. | ||
Another processing method is surface-initiated ROMP from the vapour phase. The first report used, amongst other monomers, DCPD from the vapour phase for patterned growth of pDCPD on surfaces with microcontact printed and physisorbed G2.168 Later, surface initiated ROMP of exo- and endo-DCPD was studied using initiators covalently attached to gold and silicon substrates. Approx. 400 nm thick pDCPD films were obtained in less than 1 min of polymerisation and no significant differences in thickness were observed for the different stereoisomers. Film thicknesses ranging from tens of nanometres to hundreds of nanometres could be achieved upon adding decane as an unreactive diluent to the vapour phase. The films provide effective barriers to the diffusion of aqueous ions in excess of 1 × 106 Ω cm2 and exhibit a reduced Young's modulus of 15 GPa.70
Finally, the polymerisation of DCPD in a microemulsion leading to stable pDCPD latexes which can be further processed should be noted. Water soluble, acid switchable initiators were used to polymerise DCPD (and DCPD/cyclooctene mixtures) in an Emulgin® B3 stabilised microemulsion. With low initiator loadings of about 200 ppm, almost complete polymerisation was found and stable latexes with particle diameters ranging from 255 nm to 315 nm and low coagulate content were obtained.54 Prior work disclosed carbon nanotube loaded latexes.169 Another emulsion based approach is the preparation of particles featuring interfacially assembled nanoparticle-conjugated lipase. Such 10 μm big functional particles in which pDCPD serves as the glue displayed an increase in catalytic efficiency of the lipase when compared to the native enzyme. The particles could be magnetically recovered, thanks to the magnetic nanoparticles used. Moreover, the immobilized lipase exhibited enhanced thermal and pH stability170,171 and could be operated in deep eutectic solvents.172
2.4. Porous pDCPD
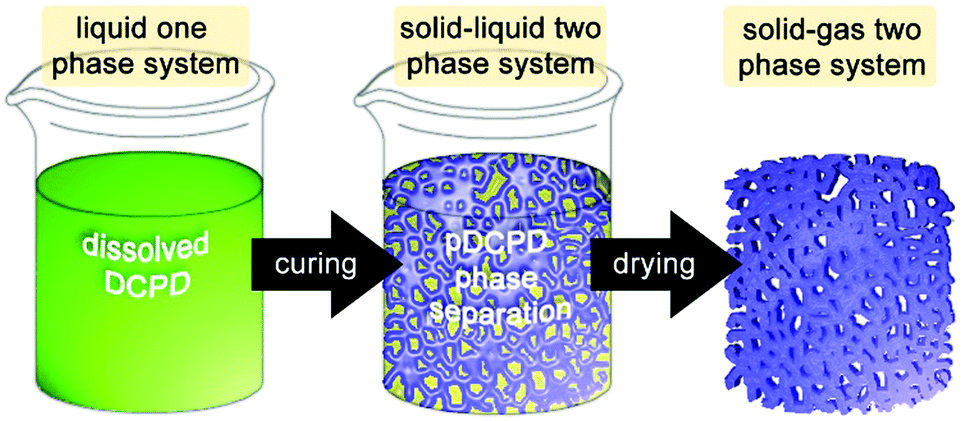 | ||
| Fig. 5 Cartoon of the process chemically induced phase separation for the preparation of macroporous pDCPD. | ||
For DCPD, 2-propanol is a suitable solvent for this purpose and allows for preparing closed or open cell porous monoliths depending of the 2-propanol share. Above 30 wt% of 2-propanol content, the porous structure consists of agglomerated spherical pDCPD particles (of about 3–4 μm). Porosities of up to 80% were obtained173 and monoliths exhibiting a gradient porosity were accessible.174 Additionally adding norbornene-functionalized poly(ethylene glycol) oligomers allowed for a decrease in the pore size and a related increase in the internal surface area.175
However, for many applications, the modification of the surface is desirable, if not necessary. The surface grafting of initiators for atom transfer radical polymerisation has been demonstrated and used to graft dense poly(methylmethacrylate) onto pDCPD. The process comprises three steps, with the thiol–ene reaction of mercaptoethanol with the double bonds of pDCPD being the key step.176 Furthermore, the double bonds can be brominated (forming a 100–200 nm thick brominated layer) and it has been demonstrated that subsequent nucleophilic substitution with amines yields monolayers on pDCPD.177 Epoxidation with m-chloroperoxybenzoic acid generates an approx. 10 nm thick layer of oxiranes. Subsequent ring opening with amines provides the installation of the desired functionality in a second step.178
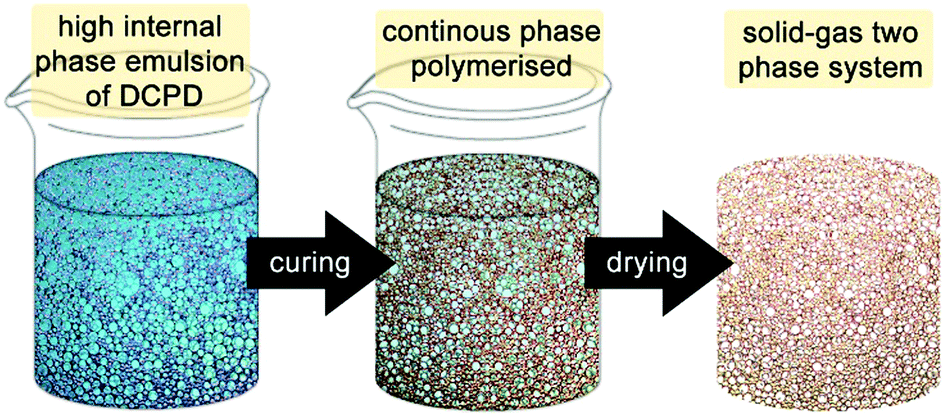 | ||
| Fig. 6 Cartoon of the high internal phase emulsion templating route to prepare open porous macroporous foams. | ||
The first attempt to template DCPD failed, presumably due to an insufficient emulsion stability at the elevated temperature needed for curing.180 However, using water as the majority phase, Pluronics L-121 as the surfactant and M2 as the initiator enables the preparation of high internal phase templated DCPD of about 80% porosity and exceptional mechanical properties compared to other high internal phase templated polymers.181 However, it was realized that the pDCPD monoliths quickly and fully oxidise at elevated temperature under ambient conditions yielding a polymer with an empiric sum formula of C5.2H6.4O1.9. The oxidation goes in hand with a stiffening of the foam. Nevertheless, the liability of oxidation constitutes the material's Achilles heel in many potential applications. Upon varying the DCPD to water ratio from 50:50 to 20:80 open porous monolithic polymers of different porosities could be prepared, whereby the openness of the foams of low porosity is a peculiarity within high internal phase templated polymers.182 The feature sizes of the foam, i.e. void and window sizes, can be determined upon variation of the surfactant amount (10–0.25 vol% with respect to DCPD). Void sizes between 3 and 70 μm can be obtained. Thereby it was revealed that the mechanical properties of the foams are distinctly dependent on the surfactant amount used. Especially when reducing the surfactant amount below generally accepted values in the field, ductile open porous polymer foams with 80% porosity and Young's moduli of about 110 MPa were prepared.183 A comprehensive investigation of the mechanical properties of unoxidised pDCPD foams of 90–50% porosity using tensile testing revealed Young's moduli between 31 and 416 MPa and moduli of toughness between 600 kJ m−3 and 2 MJ m−3.184 Similarly, membranes are easily accessible by doctor blading. Open-cellular macroporous membranes with about 80% porosity and 180 μm thickness are characterized by a Young's modulus of about 125MPa and an ultimate strength of 3.1MPa at an elongation of about 20%.185
Similar membranes obtained from copolymerizing norbornene and DCPD were used as separators in lithium-ion batteries. A comparison with cells made from conventional poly(ethylene)-based separators revealed their applicability despite the presence of unsaturation in the polymer backbone. The combination of the high porosity and good wettability of the separators suggests that such considerably thick separators might be useful in bigger lithium-ion batteries with increased safety issues.186 Like in the case of the separators, copolymerisation of other ROMP-able monomers has been studied and its effects on the mechanical properties187,188 and the foam's swelling behaviour have been disclosed.188
Amongst copolymerisation, the properties of the foams can be influenced upon the addition of inorganic nanoparticles. Magnetic foams were obtained by adding up to 30 wt% γFe2O3/Fe3O4 nanoparticles to the emulsion and subsequent curing. The oxides were mainly found at the surface of the polymeric phase and the good casting quality of the samples was maintained. Burning off the polymeric phase yielded macroporous Fe2O3, demonstrating a way towards high internal phase templated ceramics.189 Similarly, zinc oxide nanoparticles were used to prepare pDCPD nanocomposite foams. Subsequent calcination yielded macroporous ZnO foams, which showed an appealing catalytic performance in the catalytic wet air oxidation of bisphenol A.190 Furthermore, TiO2@pDCPD has been described and tested in the photocatalytic degradation of 4-nitrophenol.191 Later, the composite foam's mechanical properties with and without antioxidants have been disclosed.192 In another study, nanoparticular metal–organic frameworks (MOFs) were used to obtain 100 μm thick, fully open, hierarchically porous hybrid membranes. Up to 14 wt% of MIL-100(Fe) nanoparticles were incorporated, resulting in an increase of the microporosity from 5 to about 130 m2 g−1. The MIL-100(Fe)@pDCPD membranes showed an appealing catalytic activity in Friedel–Crafts alkylation in batch and flow-through mode, thereby demonstrating the preserved accessibility of Lewis acidic sites in the MOF.193 Another study utilized CuO and ZnO@pDCPD foams to grow MOFs (HKUST-1(Cu) and MOF-5(Zn)) upon addition of ligand solutions. In this way, up to about 75 wt% MOF loading of the foams was obtained and micropore accessibility was greatly retained. The hydrolytical instability of the MOFs was distinctly reduced in the MOF@pDCPD composites. This feature enabled the preservation of the composite's CO2 sorption capacity under humid conditions for at least 20 cycles.194 Another example of stabilization within macroporous pDCPD foams comes from encapsulating enzymes. Lipase was incorporated into pDCPD foams upon adding the enzyme to the aqueous phase of the high internal phase emulsion. Curing was done at room temperature and the encapsulated lipase was found to be active under harsh reaction conditions not complying with free lipase. Further, the lipase@pDCPD retains over 50% activity after 10 cycles of reuse.195 The above mentioned high internal phase emulsion approach was initiated to resolve recovery issues of 10 μm big pDCPD particles with interfacially assembled nanoparticle-conjugated lipase mentioned above.170
Post-polymerisation functionalization of emulsion templated pDCPD in the unoxidised state was performed with epoxidation,196 bromination,185 thiol–ene181 and inverse electron demand Diels–Alder reactions. In the latter approach, tetrazines (e.g. 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine) were reacted with the double bonds of pDCPD foams resulting in functionalisation of up to every second repeating unit with a potential metal chelating ligand.197 The same reaction was also used with linear pDCPD and the gelation of the resulting polymer with metal ions was studied.198 Epoxidation of pDCPD monoliths was performed in supercritical CO2. In that respect, it was revealed that the foams exhibit an unusually high uptake of scCO2 at zero swelling. The scCO2 treatment creates additional mesoporosity within the foam walls and a sequence of epoxidation followed by aminolysis with ethylenediamine yielded 11.4 mmol g−1 of nitrogen incorporated into the foam walls.196
As already mentioned, pristine pDCPD foams react with elemental oxygen from air yielding (amongst other functional groups) stable hydroperoxide moieties within the polymer scaffold. The hydroperoxide-containing pDCPD foams were then used to oxidise and sequester Demeton-S and 2-chloroethyl ethyl sulphide, employed as models for chemical warfare agents. The simplicity of the detoxification is believed to hold high promise for the development of self-decontaminating filter media.199,200 Another exploitation of the porous material's high tendency to oxidize was its use as an oxygen scavenger material. However, in this application emulsion templated poly(norbornadiene) proved superior to emulsion templated pDCPD.201 Oxidation in air leads to a stiffening of the macroporous monolithic specimens to such a degree that their cellular structure is not lost upon heating (up to 1400 °C) under an inert atmosphere. Thus, the ‘fixation’ upon oxidation allowed for the preparation of monolithic open macroporous carbons of 80–85% porosity featuring void diameters in the range of 87 to 2.5 μm. The porous carbon materials were characterized by carbon contents higher than 97%, electronic conductivities of up to 2800 S m−1, Young's moduli of up to 2.1 GPa and specific surface areas of up to 1200 m2 g−1. All these values were mainly depending on the carbonisation conditions.202
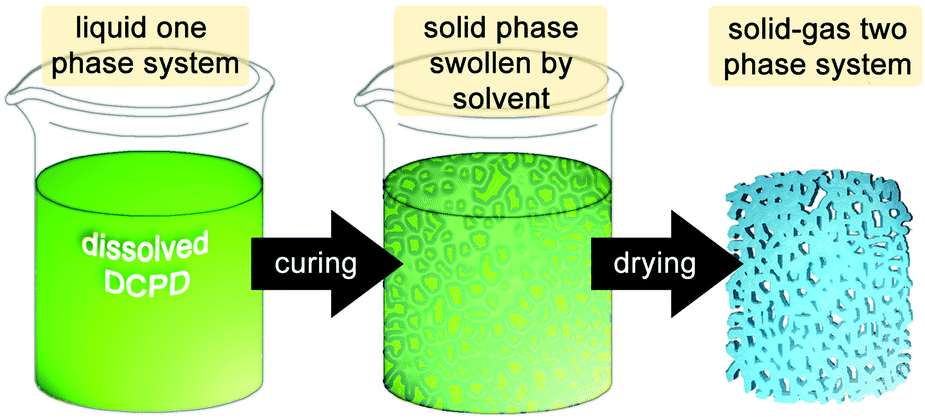 | ||
| Fig. 7 Schematic preparation of aerogels. | ||
Alternatively, ambient pressure drying processes are employed. In this case the volume shrinkage is much bigger (typically 50%) and pore structure damage occurs. High capillary forces are responsible for this phenomenon. The first pDCPD based aerogels were published in 2007 and were obtained by polymerizing a solution of DCPD with G1 targeting an aerogel density of 0.06 g cm−3. The system was further investigated by varying the target density, the initiator loading, the solvent and the drying conditions (supercritical CO2 drying vs. conventional drying). Typical pDCPD aerogels obtained within this study were characterized by a density of 0.06 g cm−3, a pore volume of 0.89 cm3 g−1, an average pore diameter of 15 nm, a surface area of 235 m2 g−1 and a thermal conductivity of 22.9 mW mK−1. However, the preparation of homogenous and uniform specimens was difficult to achieve.203 Key to improving this issue is precise control over the viscosity and gel time and gelation can be manipulated by reducing the amount of cross-linking through co-polymerisation with a non-crosslinking monomer. It was shown that only small additions of norbornene (1–10 wt%) increase the viscosity at the sol–gel transition by several orders of magnitude, resulting in better uniformity of the samples. The finding was applied to the coating of cylindrical and spherical surfaces with pDCDP aerogel films, which is particularly challenging because shear forces can additionally damage the growing gel network.204 This work was further extended towards other non-crosslinking but functionalized monomers (e.g.Fig. 8, R = CH2I) and the effect of functionalisation on the morphology of the aerogels was studied.205 Post-polymerisation iodation (and bromination) was tried in a forthcoming study. Because only limited control over the amount and the uniformity of halide incorporation was obtained, the incorporation of iodide (and also tributyltin) groups was preferably accomplished via copolymerisation of accordingly substituted norbornene derivatives. Such obtained aerogels were aimed at providing high-Z tracer element substituted organic aerogels for use in inertial confinement fusion.206
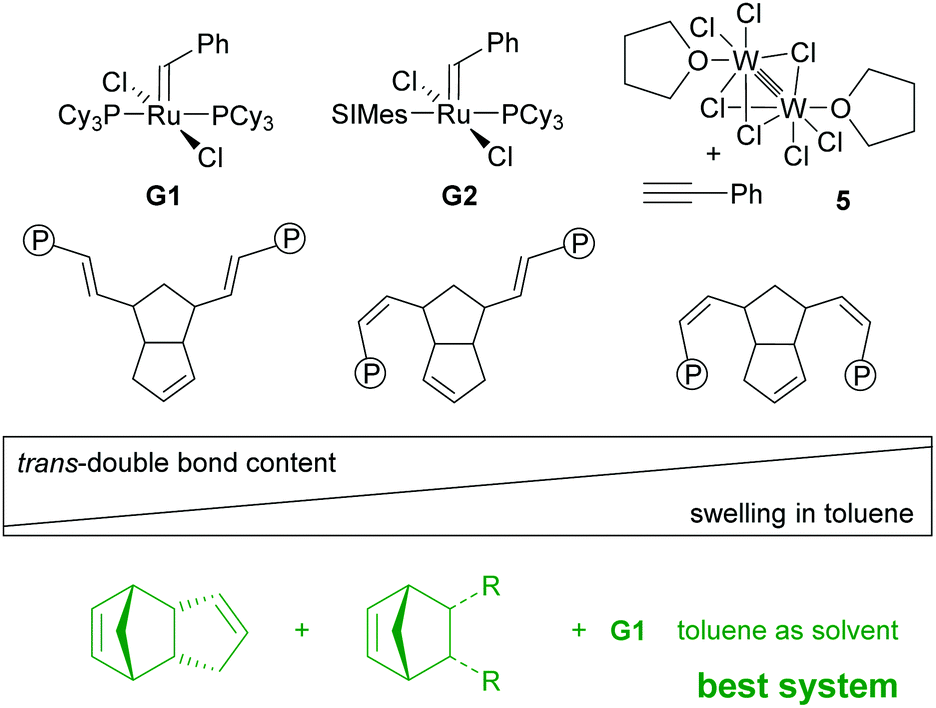 | ||
| Fig. 8 Initiators used for the preparation of aerogels and the preferred stereochemistry of the double-bond in the repeating unit, being decisive for the aptitude in aerogel synthesis; the best systems are late gelating mixtures of DCPD with less than 10 mol% non-crosslinking monomer (R = H, CH2I, Sn(nBu)3) cured with G1 in toluene. | ||
Switching from G1 to G2 as the initiator caused severe deformation of the wet gels during processing (swelling in toluene) yielding aerogels unsuitable for any application. This issue was resolved by performing free radical polymerisation of methyl methacrylate in the pores of the wet gel and well-defined aerogels could be obtained.207 The phenomenon is explained by a synergism related to the nano-topology of the two components. It was shown that upon drying the macroscopic deformation of wet gels is accompanied by coalescence of initially formed polymer-nanoparticles that form non-mass-fractal secondary particles. While in the case of neat pDCPD it is believed that these secondary particles further rearrange in such a way that the secondary particles move in the empty space of the others, this movement is impeded in the presence of poly(methyl methacrylate) and thus the structure is better conserved.208 Operando fluorescence microscopy imaging supported the findings.100 On a molecular level, this behaviour was attributed to the higher trans-content of pDCPD polymerised with G1 when compared to pDCPD made with G2. It was concluded that higher trans-content makes pDCPD more rigid, while higher cis-content results in increased malleability.86 Accordingly, the swelling of cis-enriched pDCPD (in a nonpolar solvent) should be higher than of trans-enriched pDCPD. Indeed, preparing xerogels with the initiator-systems Na[W2(μ-Cl)3Cl4(THF)2] (THF)3/phenylacetylene (5) yielded heavily deformed specimens with high cis-content. Their swelling in toluene was studied and compared to samples prepared with G1. The xerogels prepared with 5 increased their volume by more than a factor of 100, while xerogels obtained from G1 did not swell at all.209 The swelling behaviour of the 5-retrieved pDCPD xerogel was further studied using 44 different solvents. From these data, the Hansen solubility parameters and the Flory–Huggins parameter χ12 of mostly cis pDCPD were derived. The xerogels were particularly useful for separating chloroform from water.210,211
As is the case for all other porous pDCDP variants, also the aerogels are prone to oxidative degradation under ambient conditions. Although the oxidized variants have never been studied, an approach to mitigate this issue was published. Upon hydrogenation with p-toluenesufonyl hydrazide, hydrogenated pDCPD aerogels became accessible. The improved oxidative stability was proven by thermolysis studies at 135 °C for 2 h under ambient conditions. While the pDCPD samples turned brownish and shrank and oxidation products were identified, the hydrogenated versions stayed colourless and no typical pDCPD oxidation products were observed in the infrared spectra. However, also the hydrogenated aerogels shrank considerably during the thermal treatment.212
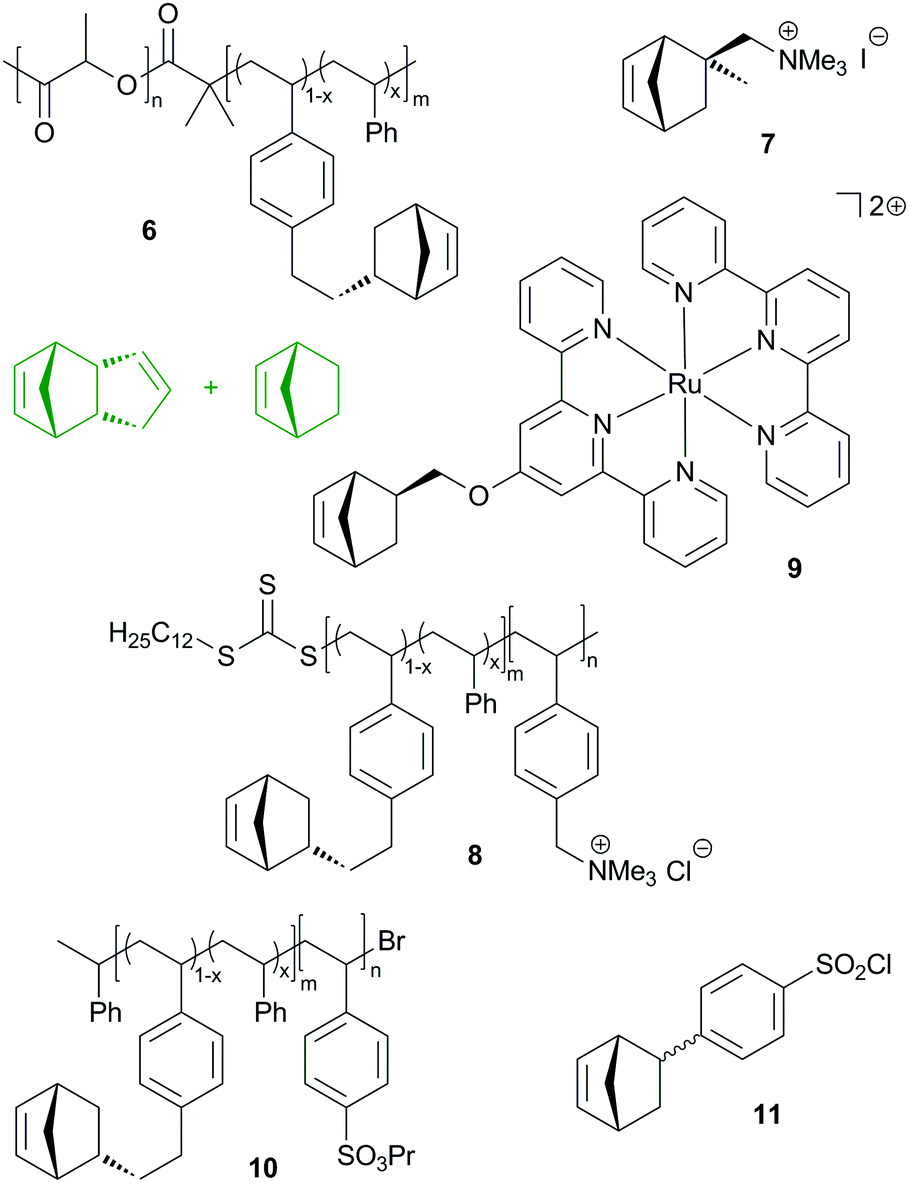 | ||
| Fig. 9 Structural (in green) and functional (macro)monomers used for the preparation of nanoporous membranes. | ||
Similarly to the reactive diblock-copolymer discussed above, macromonomer 8 was used in the copolymerisation with a mixture of DCPD and cyclooctene, yielding membranes that have a bicontinous morphology. In other words, a hydrophobic network featuring hydrophilic conduction paths was established. The resulting membranes exhibited hydroxide conduction as high as 120 mS cm−1 in water at 60 °C. However, their mechanical properties were not sufficient for practical applications.222 This issue was resolved by roughly doubling the molecular mass of the diblock-copolymer. In this case, the conductivity dropped to a still high 98 mS cm−1. Controlled membrane swelling led to about 120% wt% water uptake, whereby the membrane's mechanical integrity prevented excessive water uptake. Qualifications in fuel cells were performed and resulted in increased power and current density compared to a commercial Tokuyama A201 membrane tested under the same conditions.223
Divalent bis(terpyridine)ruthenium(II) based monomer 9 and DCPD were used to prepare membranes as outlined in the previous studies. The DCPD to 9 molar ratios employed were 2:1, 5:1 and 10:1. Hydrogen carbonate and hydroxide conductivities (19.6, 28.6 and 27.0 mS cm−1 at 30 °C) were measured as well as the mechanical properties (ultimate strength between 0.6 and 27 MPa at an elongation of roughly 53–87%). Swelling tests in aqueous methanol suggested good methanol tolerance.224 In a next step, the same group made clear that the conductivity and hydration are dominated by the extent of cross-linking of the membranes.225 In a further study, the polymer architecture was slightly changed by using norbornene as an additional co-monomer and, most importantly, ruthenium, nickel and cobalt bis(terpyridine) complexes were employed. While the water uptake, mechanical properties and chemical stability were greatly uninfluenced by the use of the different metal complexes, a high impact on conductivity was found. More earth abundant nickel was identified as a competitive alternative to ruthenium.226 This discovery was further investigated with the aim to explain differences in anion exchange membranes in general. Isothermal titration calorimetry was employed and indicated a weaker cation–counterion association in the case of the nickel complex.227
More recently, benzenesulfonyl chloride bearing norbornene (11), DCPD and norbornene were copolymerized yielding upon hydrolysis 100–120 nm thick membranes. DCPD served as a crosslinker and increasing crosslinking decreased the proton conductivity, water uptake and swelling ratio, while the selectivity (proton conductivity vs. methanol permeability) increased with increasing crosslinking.230 Another group additionally incorporated carboxylic acid groups into the membrane.231
In summary of this section, it can be stated that several highly promising nanoporous membranes based on DCPD have been disclosed. However, in the face of the high tendency of DCPD based porous materials to oxidize, their long-term performance is in question. Studies on the oxidation of nanoporous DCPD are definitely missing.
2.5. DCPD derivatives
In general, it is not easy to functionalise DCPD without compromising its polymerisability via ROMP and only two functionalized polymerisable DCPD derivative families have been disclosed so far (Fig. 10). The first example (12) is derived from heterodimerisation of carboxylated cyclopentadiene with cyclopentadiene. ROMP with G2 yields a linear homopolymer, which can be thermally cross-linked in a subsequent step. The cross-linked polymer exhibits glass transition temperatures higher than 170 °C.232 Based on extensive studies, a radical process was identified to be responsible for the crosslinking reaction.233 The processability by RIM of the liquid monomer was demonstrated and similar storage and loss moduli to those for pDCPD were found.234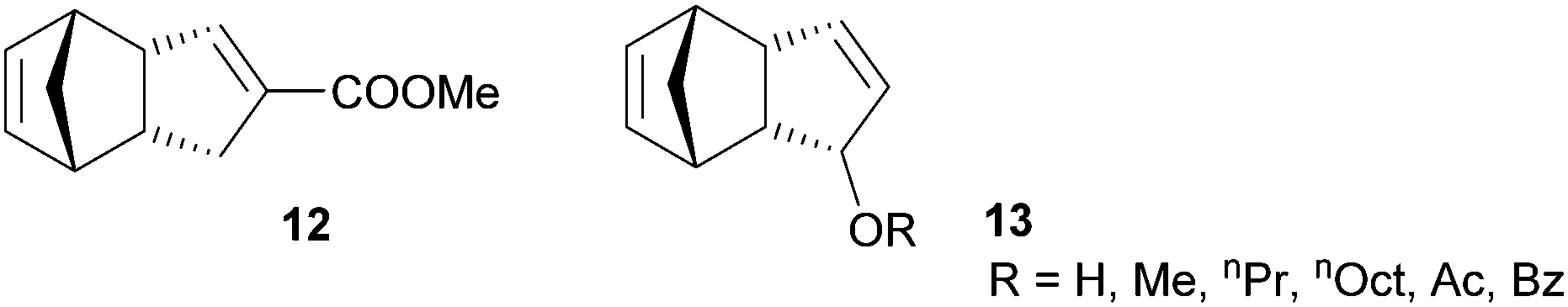 | ||
| Fig. 10 Functionalised DCPD derivatives. | ||
The other monomer family (13) is based on oxidizing the methylene group of the cyclopentene ring in DCPD. Initially, the acetylated monomer was reported in the polymerisation with G1 yielding a linear polymer characterized by higher glass transition temperatures but lower thermal stability when compared to pDCPD.235 Later, the parent alcohol, alkyl ethers and esters were polymerised (now with G2 in a solvent free variant) and the obtained cross-linked polymers were characterised via differential scanning calorimetry, thermogravimetric analysis and dynamic mechanical analysis.236,237 Generally, the polymerisation of the functionalised DCPD monomers is decelerated in comparison with DCPD. Taking the tedious preparation of both monomer types into consideration, weighty reasons for their application (instead of DCPD) are needed. Their main advantage might be their reduced volatility and smell with respect to DCPD. In some applications, the use of DCPD is limited because of its unpleasant odour.238,239 This issue might be resolved with functionalized DCPD derivatives.240
3. Challenges and perspectives
One of the main challenges in pDCPD chemistry and technology is, according to the judgement of the authors, to get control over the olefin addition based cross-linking at high temperatures. This process leads to steadily growing embrittlement and is particularly problematic in fibre based composite materials. The means to ‘freeze’ this process at a desired point (i.e. the desired cross-linking density) would be highly desirable. While oxidation seems not to be a big issue for many applications – in fact, the self-passivation of pDCPD is often rather an advantage – it has been ascertained as a core problem for many porous variants of pDCPD. Membranes and monoliths will drastically change their properties upon oxidation and will most probably lose their applicability. Although several examples of making use of oxidised porous pDCPD have been shown, the means to protect pDCPD from oxidising is desperately needed.As regards initiators, gaining control over the stereochemistry of the double bonds and the tacticity with robust initiators is desirable. Additionally, initiators should be well soluble in DCPD, facilitating preparation. Photoinitiators operating in the visible at low loadings should be another target.
The fields of nanocomposites and fibre reinforced composites are going their way. Emphasis, however, should also be laid on finding cheap and practical solutions as soon as the principles are nailed down.
Frontal polymerisation of pDCPD is an emerging processing technology with great potential to revolutionise manufacturing. The same applies to the far less mature additive manufacturing processes not relying on frontal polymerisation. Fast stereolithographic fabrication of pDCPD parts would certainly spark great interest. Another emerging issue comprises research towards reprocessing of densely cross-linked thermoset pDCPD. Recently, a first study on the triggered degradation of pDCPD copolymerised with cyclic silyl ether-based monomers was uploaded to a preprint server. It is expected that the recyclability of pDCPD will attract increasing attention in the near future.241
4. Conclusions
In recent years, it became clear that pDCPD can compete effectively with epoxy- and polyester resins in many applications. Thanks to the intense research work conducted in the last two decades, environmentally robust initiators and the means to adjust the pot life of resins became available. With this achievement, the design freedom of creating resins was greatly enhanced. The use of many co-monomers or fillers unimaginable to employ with conventional initiators became feasible and new processing possibilities emerged. pDCPD based micro- and macroporous materials with intriguing mechanical properties became accessible. Silica-reinforced nanocomposites emerged and fibre reinforced composites became accessible. The latter field of activity is of particular technological and economic interest and DCPD resins can now demonstrate their strengths. pDCPD is characterised by thermal and many mechanical properties which are comparable to competing resins but it shows a distinctly higher toughness. Its density is lower than that of many competing thermosets. DCPD-resins feature a low viscosity, allowing for faster and more flexible processing in consistently high quality. With the advent of new processing possibilities, such as frontal polymerisation or stereolithography, in the last few years, DCPD based resins offer another unique feature. Moreover, a company is advertising its DCPD technology with a considerably lower carbon footprint than epoxy resins. Although the underlying study for the claim has not been disclosed, the statement appears plausible.Despite the tremendous discoveries and developments of the last decades, the authors are convinced that the real potential of pDCPD in materials chemistry is by far not uncovered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CS is indebted to the financial support through the Lead project LP-03 “Porous Materials@Work” of Graz University of Technology and SK acknowledges the financial support from the Slovenian Research Agency (grant P2-0145).Notes and references
- Register entry for DCPD at the European Chemical Agency see: https://echa.europa.eu/registration-dossier/-/registered-dossier/15412.
- T. T. P. Cheung, Cyclopentadiene and Dicyclopentadiene in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, 2001, DOI:10.1002/0471238961.0325031211050514.a01.pub2.
- D. Hönicke, R. Födisch, P. Claus and M. Olson, Cyclopendadiene and Cyclopentene in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012 Search PubMed.
- Market size estimations issued before the outbreak of the COVID-19 pandemic, https://marketersmedia.com/over-45-cagr-dicyclopentadiene-market-size-will-bypass-800-million-by-2024/88898782 retrieved on April 13th, 2020.
- A. Nickel and B. D. Edgecombe, Industrial Applications of ROMP in Polymer Science: A Comprehensive Reference, Elsevier, 2012, vol. 4, p. 749 Search PubMed.
- L. Matejka, C. Houtman and C. W. Macosko, Polymerization of Dicyclopentadiene: A New Reaction Injection Molding System, J. Appl. Polym. Sci., 1985, 30, 2787–2803 CrossRef CAS.
- J. C. Mol, Industrial applications of olefin metathesis, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- C. Slugovc, in Industrial Applications of Olefin Metathesis Polymerization in Olefin Metathesis: Theory and Practice, ed. K. Grela, John Wiley & Sons, 2014, pp. 329–334 Search PubMed.
- Z. Yao, L. W. Zhou, B.-B. Dai and K. Cao, Ring-opening metathesis copolymerization of dicyclopentadiene and cyclopentene through reaction injection molding process, J. Appl. Polym. Sci., 2012, 125, 2489–2493 CrossRef CAS.
- H.-G. Kim, H. J. Son, D.-K. Lee, D.-W. Kim, H. J. Park and D.-H. Cho, Optimization and analysis of reaction injection molding of polydicyclopentadiene using response surface methodology, Korean, J. Chem. Eng., 2017, 34, 2099–2109 CAS.
- D. S. Breslow, Metathesis polymerization, Prog. Polym. Sci., 1993, 18, 1141–1195 CrossRef CAS.
- S. Monsaert, A. L. Vila, R. Drozdak, P. V. D. Voort and F. Verpoort, Latent olefin metathesis catalysts, Chem. Soc. Rev., 2009, 38, 3360–3372 RSC.
- S. Naumann and M. R. Buchmeiser, Latent and delayed action polymerization systems, Macromol. Rapid Commun., 2014, 35, 682–701 CrossRef CAS PubMed.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Switchable Polymerization Catalysts, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- A. J. Teator and C. W. Bielawski, Remote control Grubbs catalysts that modulate ring-opening metathesis polymerizations, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2949–2960 CrossRef CAS.
- D. J. Nelson, S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Key processes in ruthenium-catalysed olefin metathesis, Chem. Commun., 2014, 50, 10355–10375 RSC.
- C. Slugovc, S. Demel and F. Stelzer, Ring opening metathesis polymerisation in donor solvents, Chem. Commun., 2002, 2572–2573 RSC.
- S. J. P’Poo and H.-J. Schanz, Reversible Inhibition/Activation of Olefin Metathesis: A Kinetic Investigation of ROMP and RCM Reactions with Grubb's Catalyst, J. Am. Chem. Soc., 2007, 129, 14200–14212 CrossRef PubMed.
- M. R. Kessler and S. R. White, Cure kinetics of the ring-opening metathesis polymerization of dicyclopentadiene, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2373–2383 CrossRef CAS.
- X. Liu, X. Sheng, J. K. Lee and M. R. Kessler, Isothermal cure characterization of dicyclopentadiene, J. Therm. Anal. Calorim., 2007, 89, 453–457 CrossRef CAS.
- G. Yang and J. K. Lee, Curing Kinetics and Mechanical Properties of endo-Dicyclopentadiene Synthesized Using Different Grubb's Catalysts, Ind. Eng. Chem. Res., 2014, 53, 3001–3011 CrossRef CAS.
- Y. Vidavsky, Y. Navon, Y. Ginzburg, M. Gottlieb and N. G. Lemcoff, Thermal properties of ruthenium alkylidene-polymerized dicyclopentadiene, Beilstein, J. Org. Chem., 2015, 11, 1469–1474 CAS.
- A. Mariani, S. Fiori, Y. Chekanov and J. A. Pojman, Frontal Ring-Opening Metathesis Polymerization of Dicyclopentadiene, Macromolecules, 2001, 34, 6539–6541 CrossRef CAS.
- T. C. Mauldin and M. R. Kessler, Latent catalytic systems for ring-opening metathesis-based thermosets, J. Therm. Anal. Calorim., 2009, 96, 705–713 CrossRef CAS.
- H. M. Yoo, J. H. Jeon, M.-X. Li, W. I. Lee and S. W. Choi, Analysis of curing behavior of endo-dicyclopentadiene using different amounts of decelerator solution, Composites, Part B, 2019, 161, 439–454 CrossRef CAS.
- T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, A Novel Class of Ruthenium Catalysts for Olefin Metathesis, Angew. Chem., Int. Ed., 1998, 37, 2490–2493 CrossRef CAS PubMed.
- R. T. M. Jakobs and R. P. Sijbesma, Mechanical Activation of a Latent Olefin Metathesis Catalyst and Persistence of its Active Species in ROMP, Organometallics, 2012, 31, 2476–2481 CrossRef CAS.
- I. D. Robertson, L. M. Dean, G. E. Rudebusch, N. R. Sottos, S. R. White and J. S. Moore, Alkyl Phosphite Inhibitors for Frontal Ring-Opening Metathesis Polymerization Greatly Increase Pot Life, ACS Macro Lett., 2017, 6, 609–612 CrossRef CAS.
- A. Leitgeb, J. Wappel, C. A. Urbina-Blanco, S. Strasser, C. Wappl, C. S. J. Cazin and C. Slugovc, Two commercially available initiators for the retarded ring-opening metathesis polymerization of dicyclopentadiene, Monatsh. Chem., 2014, 145, 1513–1517 CrossRef CAS.
- X. Bantreil and C. S. J. Cazin, Phosphite ligands in Ru-based olefin metathesis catalysts, Monatsh. Chem., 2015, 146, 1043–1052 CrossRef CAS.
- B. Trzaskowski and K. Grela, Structural and Mechanistic Basis of the Fast Metathesis Initiation by a Six-Coordinated Ruthenium Catalyst, Organometallics, 2013, 32, 3625–3630 CrossRef CAS.
- Y. Vidavsky, A. Anabya and N. G. Lemcoff, Chelating alkylidene ligands as pacifiers for ruthenium catalysed olefin metathesis, Dalton Trans., 2012, 41, 32–43 RSC.
- P. A. van der Schaaf, R. Kolly, H.-J. Kirner, F. Rime, A. Mühlebach and A. Hafner, Synthesis and reactivity of novel ruthenium carbene catalysts. X-ray structures of [RuCl2(CHSC6H5)(PiPr3)2] and [RuCl2(CHCH2CH2-C,N-2-C5H4N)(PiPr3)], J. Organomet. Chem., 2000, 606, 65–74 CrossRef CAS.
- T. Ung, A. Hejl, R. H. Grubbs and Y. Schrodi, Latent Ruthenium Olefin Metathesis Catalysts That Contain an N-Heterocyclic Carbene Ligand, Organometallics, 2004, 23, 5399–5401 CrossRef CAS.
- N. M. Shcheglova, V. D. Kolesnik, R. V. Ashirov and E. A. Krasnokutskaya, Latent ruthenium carbene complexes with six-membered N- and S-chelate rings, Russ. Chem. Bull., 2016, 65, 490–497 CrossRef CAS.
- A. A. Lyapkov, L. S. Soroka, R. V. Ashirov, D. I. Zemlyakov, D. A. Rusakov and F. Verpoort, Kinetics of Dicyclopentadiene Polymerization in the Presence of the Second Generation Hoveyda-Grubbs Catalyst with N-Chelating Ligand, Polym. Sci. C, 2019, 61, 41–48 CAS.
- V. V. Afanas’ev, T. M. Yumasheva and N. B. Bespalova, Promising Catalytic Processes of Dicyclopentadiene Metathesis Polymerization to Obtain a New Class of Polymeric and Polymer–Composite Materials, Polym. Sci. C, 2019, 61, 31–40 Search PubMed.
- D. Sivanesan, B. Seo, C.-S. Lim, D. Choi, T. Kima and H.-G. Kim, An additional potential donor-bearing alkylidene-containing latent NHC–ruthenium-based catalyst for olefin metathesis polymerization, Polym. Chem., 2020, 11, 2511–2518 RSC.
- Y. Ginzburg, A. Anaby, Y. Vidavsky, C. E. Diesendruck, A. Ben-Asuly, I. Goldberg and N. G. Lemcoff, Widening the Latency Gap in Chelated Ruthenium Olefin Metathesis Catalysts, Organometallics, 2011, 30, 3430–3437 CrossRef CAS.
- A. Hejl, M. W. Day and R. H. Grubbs, Latent Olefin Metathesis Catalysts Featuring Chelating Alkylidenes, Organometallics, 2006, 25, 6149–6154 CrossRef CAS.
- E. Pump, A. Leitgeb, A. Kozłowska, A. Torvisco, L. Falivene, L. Cavallo, K. Grela and C. Slugovc, Variation of the Sterical Properties of the N-Heterocyclic Carbene Coligand in Thermally Triggerable Ruthenium-Based Olefin Metathesis Precatalysts/Initiators, Organometallics, 2015, 34, 5383–5392 CrossRef CAS.
- S. J. Czarnocki, I. Czeluśniak, T. K. Olszewski, M. Malinska, K. Woźniak and K. Grela, Rational and Then Serendipitous Formation of Aza Analogues of Hoveyda-Type Catalysts Containing a Chelating Ester Group Leading to a Polymerization Catalyst Family, ACS Catal., 2017, 7, 4115–4121 CrossRef CAS.
- B. Allaert, N. Dieltiens, N. Ledoux, C. Vercaemst, P. Van Der Voort, C. V. Stevens, A. Linden and F. Verpoort, Synthesis and activity for ROMP of bidentate Schiff base substituted second generation Grubbs catalysts, J. Mol. Catal. A: Chem., 2006, 260, 221–226 CrossRef CAS.
- S. Monsaert, N. Ledoux, R. Drozdzak and F. Verpoort, A highly controllable latent ruthenium Schiff base olefin metathesis catalyst: Catalyst activation and mechanistic studies, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 302–310 CrossRef CAS.
- J. S. M. Samec, B. K. Keitz and R. H. Grubbs, Latent ruthenium olefin metathesis catalysts featuring a phosphine or an N-heterocyclic carbene ligand, J. Organomet. Chem., 2010, 695, 1831–1837 CrossRef CAS.
- J. Wappel, R. C. Fischer, L. Cavallo, C. Slugovc and A. Poater, Simple activation by acid of latent Ru-NHC-based metathesis initiators bearing 8-quinolinolate co-ligands, Beilstein J. Org. Chem., 2016, 12, 154–165 CrossRef CAS PubMed.
- R. Gawin, A. Makal, K. Woźniak, M. Mauduit and K. Grela, A dormant ruthenium catalyst bearing a chelating carboxylate ligand: in situ activation and application in metathesis reactions, Angew. Chem., Int. Ed., 2007, 46, 7206–7209 CrossRef CAS PubMed.
- A. Kozłowska, M. Dranka, J. Zachara, E. Pump, C. Slugovc, K. Skowerski and K. Grela, Chelating Ruthenium Phenolate Complexes-Synthesis, General Catalytic Activity and Applications in Olefin Metathesis Polymerisation, Chem. – Eur. J., 2014, 20, 14120–14125 CrossRef PubMed.
- A. Gawin, E. Pump, C. Slugovc, A. Kajetanowicz and K. Grela, Ruthenium-amide Complexes – Synthesis and Catalytic Activity in Olefin Metathesis and in Ring Opening Polymerization, Eur. J. Inorg. Chem., 2018, 1766–1774 CrossRef CAS.
- J. Beerhues, S. Sen, R. Schowner, G. M. Nagy, D. Wang and M. R. Buchmeiser, Tailored molybdenum imido alkylidene N-heterocyclic carbene complexes as latent catalysts for the polymerization of dicyclopentadiene, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3028–3033 CrossRef CAS.
- I. Elser, B. R. Kordes, W. Frey, K. Herz, R. Schowner, L. Stöhr, H. J. Altmann and M. R. Buchmeiser, Latent and Air-Stable Pre-Catalysts for the Polymerization of Dicyclopentadiene: From Penta- to Hexacoordination in Molybdenum Imido Alkylidene N-Heterocyclic Carbene Complexes, Chem. – Eur. J., 2018, 24, 12652–12659 CrossRef CAS PubMed.
- L. Ondi, G. M. Nagy, J. B. Czirok, A. Bucsai and G. E. Frater, From Box to Bench: Air-Stable Molybdenum Catalyst Tablets for Everyday Use in Olefin Metathesis, Org. Process Res. Dev., 2016, 20, 1709–1716 CrossRef CAS.
- T. Ritter, A. Hejl, A. G. Wenzel, T. W. Funk and R. H. Grubbs, A Standard System of Characterization for Olefin Metathesis Catalysts, Organometallics, 2006, 25, 5740–5745 CrossRef CAS.
- S. L. Balof, K. O. Nix, M. S. Olliff, S. E. Roessler, A. Saha, K. B. Müller, U. Behrens, E. J. Valente and H.-J. Schanz, Hexacoordinate Ru-based olefin metathesis catalysts with pH-responsive N-heterocyclic carbene (NHC) and N-donor ligands for ROMP reactions in non-aqueous, aqueous and emulsion conditions, Beilstein J. Org. Chem., 2015, 11, 1960–1972 CrossRef CAS PubMed.
- O. Eivgi and N. G. Lemcoff, Turning the Light On: Recent Developments in Photoinduced Olefin Metathesis, Synthesis, 2018, 49–63 CAS.
- T. Karlen, A. Ludi, A. Mühlebach, P. Bernhard and C. Pharisa, Photoinduced ring opening metathesis polymerization (PROMP) of strained bicyclic olefins with ruthenium complexes of the type [(η6-arene1)Ru(η6-arene2)]2+ and [Ru(Nc-R)6]2+, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1665–1674 CrossRef CAS.
- A. Hafner, A. Mühlebach and P. A. van der Schaaf, One-Component Catalysts for Thermal and Photoinduced Ring Opening Metathesis Polymerization, Angew. Chem., Int. Ed. Engl., 1997, 36, 2121–2124 CrossRef CAS.
- D. Wang, K. Wurst, W. Knolle, U. Decker, L. Prager, S. Naumov and M. R. Buchmeiser, Cationic RuII Complexes with N-Heterocyclic Carbene Ligands for UV-Induced Ring-Opening Metathesis Polymerization, Angew. Chem., Int. Ed., 2008, 47, 3267–3270 CrossRef CAS PubMed.
- D. Wang, K. Wurst and M. R. Buchmeiser, Cationic versus Neutral RuII N-Heterocyclic Carbene Complexes as Latent Precatalysts for the UV-Induced Ring-Opening Metathesis Polymerization, Chem. – Eur. J., 2010, 16, 12928–12934 CrossRef CAS PubMed.
- J. Pinaud, T. K. H. Trinh, D. Sauvanier, E. Placet, S. Songsee, P. Lacroix-Desmazes, J.-M. Becht, B. Tarablsi, J. Lalevée, L. Pichavant, V. Héroguez and A. Chemtob, In Situ Generated Ruthenium–Arene Catalyst for Photoactivated Ring-Opening Metathesis Polymerization through Photolatent N-Heterocyclic Carbene Ligand, Chem. – Eur. J., 2018, 24, 337–341 CrossRef CAS PubMed.
- T. K. H. Trinh, G. Schrodj, S. Rigolet, J. Pinaud, P. Lacroix-Desmazes, L. Pichavant, V. Héroguez and A. Chemtob, Combining a ligand photogenerator and a Ru precatalyst: a photoinduced approach to cross-linked ROMP polymer films, RSC Adv., 2019, 9, 27789–27799 RSC.
- D. Wang, J. Unold, M. Bubrin, I. Elser, W. Frey, W. Kaim, G. Xu and M. R. Buchmeiser, Ruthenium–Triazene Complexes as Latent Catalysts for UV-Induced ROMP, Eur. J. Inorg. Chem., 2013, 5462–5468 CrossRef CAS.
- O. Eivgi, S. Guidone, A. Frenklah, S. Kozuch, I. Goldberg and N. G. Lemcoff, Photoactivation of Ruthenium Phosphite Complexes for Olefin Metathesis, ACS Catal., 2018, 8, 6413–6418 CrossRef CAS.
- O. Eivgi, A. Vaisman, N. B. Nechmad, M. Baranov and N. G. Lemcoff, Latent Ruthenium Benzylidene Phosphite Complexes for Visible-Light-Induced Olefin Metathesis, ACS Catal., 2020, 10, 2033–2038 CrossRef CAS.
- C. Theunissen, M. A. Ashley and T. Rovis, Visible-Light-Controlled Ruthenium-Catalyzed Olefin Metathesis, J. Am. Chem. Soc., 2019, 141, 6791–6796 CrossRef CAS PubMed.
- A. Goetz and A. J. Boydston, Metal-Free Preparation of Linear and Cross-Linked Polydicyclopentadiene, J. Am. Chem. Soc., 2015, 137, 7572–7575 CrossRef CAS PubMed.
- B. K. Keitz and R. H. Grubbs, A Tandem Approach to Photoactivated Olefin Metathesis: Combining a Photoacid Generator with an Acid Activated Catalyst, J. Am. Chem. Soc., 2009, 131, 2038–2039 CrossRef CAS PubMed.
- W. Joo, C. H. Chen, J. P. Moerdyk, R. P. Deschner, C. W. Bielawski and C. G. Willson, Photoinitiated ring-opening metathesis polymerization, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1791–1795 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Living ring-opening metathesis polymerization, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- I. Njoroge, P. A. Kempler, X. Deng, S. T. Arnold and G. K. Jennings, Surface-Initiated Ring-Opening Metathesis Polymerization of Dicyclopentadiene from the Vapor Phase, Langmuir, 2017, 33, 13903–13912 CrossRef CAS PubMed.
- G. Yang and J. K. Lee, Curing Kinetics and Mechanical Properties of endo-Dicyclopentadiene Synthesized Using Different Grubb's Catalysts, Ind. Eng. Chem. Res., 2014, 53, 3001–3011 CrossRef CAS.
- J. D. Rule and J. S. Moore, ROMP reactivity of endo- and exo-dicyclopentadiene, Macromolecules, 2002, 35, 7878–7882 CrossRef CAS.
- R. A. Fisher and R. H. Grubbs, Ring-opening metathesis polymerization of exo-dicyclopentadiene: reversible crosslinking by a metathesis catalyst, Macromol. Chem. Macromol. Symp., 1992, 63, 271–277 CrossRef CAS.
- T. A. Davidson, K. B. Wagener and D. B. Priddy, Polymerization of Dicyclopentadiene: A Tale of Two Mechanisms, Macromolecules, 1996, 29, 786–788 CrossRef CAS.
- T. A. Davidson and K. B. Wagener, The polymerization of dicyclopentadiene: an investigation of mechanism, J. Mol. Catal. A: Chem., 1998, 133, 67–74 CrossRef CAS.
- D. Dimonie, M. Dimonie, V. Munteanu, H. Iovu, J. Couve and M. J. Abadie, Nature of the first exothermic peak on the DSC curve of linear polydicyclopentadiene obtained with a special catalytic system, Polym. Degrad. Stab., 2000, 70, 319–324 CrossRef CAS.
- D. Dimonie, M. Dimonie, S. Stoica, V. Munteanu and M. J. Abadie, Some aspects of the thermal stability of linear polydicyclopentadiene (L-PDCPD), Polym. Degrad. Stab., 2000, 67, 167–170 CrossRef CAS.
- P. Y. Le Gac, D. Choqueuse, M. Paris, G. Recher, C. Zimmer and D. Melot, Durability of polydicyclopentadiene under high temperature, high pressure and seawater (Offshore Oil Production Conditions), Polym. Degrad. Stab., 2013, 98, 809–817 CrossRef CAS.
- S. Bhuyan, S. Sundararajan, X. Sheng and M. Kessler, Influence of crosslinking density on the tribological behaviour of norbornene-based polymeric materials, Wear, 2011, 270, 550–554 CrossRef CAS.
- X. Sheng, J. K. Lee and M. R. Kessler, Influence of cross-link density on the properties of ROMP thermosets, Polymer, 2009, 50, 1264–1269 CrossRef CAS.
- T. R. Long, R. M. Elder, E. D. Bain, K. A. Masser, T. W. Sirk, J. H. Yu, D. B. Knorr, Jr. and J. L. Lenhart, Influence of molecular weight between crosslinks on the mechanical properties of polymers formed via ring-opening metathesis, Soft Matter, 2018, 14, 3344–3360 RSC.
- V. J. Toplosky and R. P. Walsh, Thermal and Mechanical Properties of Poly-Dicyclopentadiene (DCPD) at Cryogenic Temperatures, AIP Conf. Proc., 2006, 824, 219–224 CrossRef CAS.
- R. M. Elder, D. B. Knorr Jr, J. W. Andzelm, J. L. Lenhart and T. W. Sirk, Nanovoid formation and mechanics: a comparison of poly(dicyclopentadiene) and epoxy networks from molecular dynamics simulations, Soft Matter, 2016, 12, 4418–4434 RSC.
- R. M. Elder, A. Zaccone and T. W. Sirk, Identifying Nonaffine Softening Modes in Glassy Polymer Networks: A Pathway to Chemical Design, ACS Macro Lett., 2019, 8, 1160–1165 CrossRef CAS.
- D. B. Knorr Jr, K. A. Masser, R. M. Elder, T. W. Sirk, M. D. Hindenlang, J. H. Yu, A. D. Richardson, S. E. Boyd, W. A. Spurgeon and J. L. Lenhart, Overcoming the structural versus energy dissipation trade-off in highly crosslinked polymer networks: Ultrahigh strain rate response in polydicyclopentadiene, Compos. Sci. Technol., 2015, 114, 17–25 CrossRef.
- A. Bang, D. Mohite, A. M. Saeed, N. Leventis and C. Sotiriou-Leventis, Polydicyclopentadiene Aerogels from First- versus Second-Generation Grubb's Catalysts: A Molecular versus a Nanoscopic Perspective, J. Sol-Gel Sci. Technol., 2015, 75, 460–474 CrossRef CAS.
- S. Hayano, T. Sugawara and Y. Tsunogae, Synthesis, characterization, and properties of crystalline poly(endo-dicyclopentadiene) resin made via cis- and isoselective bulk ring-opening metathesis polymerization, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3153–3158 CrossRef.
- S. Hayano, Y. Takeyama, Y. Tsunogae and I. Igarashi, Hydrogenated Ring-Opened Poly(endo-dicyclopentadiene)s Made via Stereoselective ROMP Catalyzed by Tungsten Complexes: Crystalline Tactic Polymers and Amorphous Atactic Polymer, Macromolecules, 2006, 39, 4663–4670 CrossRef CAS.
- B. Autenrieth, H. Jeong, W. P. Forrest, J. C. Axtell, A. Ota, T. Lehr, M. R. Buchmeiser and R. R. Schrock, Stereospecific Ring-Opening Metathesis Polymerization (ROMP) of endo-Dicyclopentadiene by Molybdenum and Tungsten Catalysts, Macromolecules, 2015, 48, 2480–2492 CrossRef CAS.
- A. Mühlebach, P. A. van der Schaaf, A. Hafner and F. Setiabudi, Thermal stability and degradation of hydrocarbon metathesis polymers, J. Mol. Catal. A: Chem., 1998, 132, 181–188 CrossRef.
- V. Defauchy, P.-Y. Le Gac, A. Guinault, J. Verdu, G. Recher, R. Drozdzak and E. Richaud, Kinetic analysis polydicyclopentadiene oxidation, Polym. Degrad. Stab., 2017, 142, 169–177 CrossRef CAS.
- J. Huang, W. Minne, R. Drozdzak, G. Recher, P.-Y. Le Gac and E. Richaud, Thermal oxidation of poly(Dicyclopentadiene)-Decomposition of Hydroperoxides, Polym. Degrad. Stab., 2020, 174, 109102 CrossRef.
- E. Richaud, P. Y. Le Gac and J. Verdu, Thermooxidative aging of polydicyclopentadiene in glassy state, Polym. Degrad. Stab., 2014, 102, 95–104 CrossRef CAS.
- J. Huang, A. David, P.-Y. Le Gac, C. Lorthioir, C. Coelho and E. Richaud, Thermal oxidation of poly(dicyclopentadiene)-kinetic modeling of double bond consumption, Polym. Degrad. Stab., 2019, 166, 258–271 CrossRef CAS.
- B. Wang, K. Mireles, M. Rock, Y. Li, V. K. Thakur, D. Gao and M. R. Kessler, Synthesis and Preparation of Bio-Based ROMP Thermosets from Functionalized Renewable Isosorbide Derivative, Macromol. Chem. Phys., 2016, 217, 871–879 CrossRef CAS.
- R. Ding, Y. Xia, T. C. Mauldin and M. R. Kessler, Biorenewable ROMP-based thermosetting copolymers from functionalized castor oil derivative with various cross-linking agents, Polymer, 2014, 55, 5718–5726 CrossRef CAS.
- H. Cui and M. R. Kessler, Composition-dependent fracture toughness of ROMP-based Dilulin/dicyclopentadiene copolymers, J. Mater. Sci., 2014, 49, 4880–4890 CrossRef CAS.
- R. T. Mathers, K. Damodaran, M. G. Rendos and M. S. Lavrich, Functional Hyperbranched Polymers Using Ring-Opening Metathesis Polymerization of Dicyclopentadiene with Monoterpenes, Macromolecules, 2009, 42, 1512–1518 CrossRef CAS.
- J. M. Delancey, M. D. Cavazza, M. G. Rendos, C. J. Ulisse, S. G. Palumbo and R. T. Mathers, Controlling crosslinking in thermosets via chain transfer with monoterpenes, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3719–3727 CrossRef CAS.
- E. M. Hensle and S. A. Blum, Phase Separation Polymerization of Dicyclopentadiene Characterized by In Operando Fluorescence Microscopy, J. Am. Chem. Soc., 2013, 135, 12324–12328 CrossRef CAS PubMed.
- F. Hu, Y. Zheng, Y. Fang, X. Ren and X. Liu, Preparation and properties of cyclopentadiene-containing monomer modified polydicyclopentadiene, Polymer, 2014, 55, 2809 CrossRef CAS.
- F. Hu, Y. Zheng, Y. Fang, X. Ren and X. Liu, Preparation and properties of high performance phthalide – containing bismaleimide reinforced polydicyclopentadiene, J. Appl. Polym. Sci., 2014, 131, 40474 CrossRef.
- J. Kuang, N. Zheng, C. Liu and Y. Zheng, Manipulating the thermal and dynamic mechanical properties of polydicyclopentadiene via tuning the stiffness of the incorporated monomers, e-Polymers, 2019, 19, 355–364 CAS.
- B. J. Rohde, M. L. Robertson and R. Krishnamoorti, Concurrent curing kinetics of an anhydride-cured epoxy resin and polydicyclopentadiene, Polymer, 2015, 69, 204–214 CrossRef CAS.
- B. J. Rohde, K. M. Le, R. Krishnamoorti and M. L. Robertson, Thermoset Blends of an Epoxy Resin and Polydicyclopentadiene, Macromolecules, 2016, 49, 8960–8970 CrossRef CAS.
- M. Liu, B. J. Rohde, R. Krishnamoorti, M. L. Robertson and M. Dawood, Bond behavior of epoxy resin–polydicyclopentadiene phase separated interpenetrating networks for adhering carbon fiber reinforced polymer to steel, Polym. Eng. Sci., 2020, 60, 104–112 CrossRef CAS.
- B. Pan, J. Du, C. Zhang, S. Du, J. Zhang, J. Liu, Y. Zhang and Y. Zhang, Reinforcing effect of low polyethylene wax content on polydicyclopendadiene, J. Macromol. Sci., Part B: Phys., 2014, 53, 440–451 CrossRef CAS.
- G. S. Constable, A. J. Lesser and E. B. Coughlin, Morphological and Mechanical Evaluation of Hybrid Organic−Inorganic Thermoset Copolymers of Dicyclopentadiene and Mono- or Tris(norbornenyl)-Substituted Polyhedral Oligomeric Silsesquioxanes, Macromolecules, 2004, 37, 1276–1282 CrossRef CAS.
- Y. Wang, L. Zhang, J. Sun, J.-B. Bao, Z. Wang and L. Ni, Dramatic Toughness Enhancement of Polydicyclopentadiene Composites by Incorporating Low Amounts of Vinyl-Functionalized SiO2, Ind. Eng. Chem. Res., 2017, 56, 4750–4757 CrossRef CAS.
- Z.-L. He, J.-K. Xu, L. Zhang, H.-Y. Ren and S.-Y. Fu, Dramatically enhanced tensile strength and impact toughness of polydicyclopentadiene composites by covalent bond formation between phenyl-functionalized silica and dicyclopentadiene, Composites, Part B, 2019, 170, 31–40 CrossRef CAS.
- H.-Y. Ren, Z.-L. He, D.-L. Li, L. Zhang, L.-N. Chen, Y.-W. Lou and M.-D. Xu, Synergistic enhanced yield strength, tensile ductility and impact toughness of polydicyclopentadiene nanocomposites by introducing low loadings of di-functionalized silica, Polym. Test., 2019, 79, 106052 CrossRef CAS.
- Z.-L. He, L.-N. Chen, L. Zhang, H.-Y. Ren, M.-D. Xu and Y.-W. Lou, Effect of filler functional groups on the mechanical properties and relevant mechanisms of polydicyclopentadiene nanocomposites, J. Appl. Polym. Sci., 2020, 49010 CrossRef CAS.
- S. Peng, K. Chang, J. Ma, B. Pan, J. Zhang and Q. Niu, In-situ polymerization of silica/polydicyclopentadiene nanocomposites: Improved tribological properties and evaluation of interfacial compatibility, Ferroelectrics, 2018, 523, 22–31 CrossRef CAS.
- S. Peng, K. Chang, J. Ma, B. Pan, J. Zhang and Q. Niu, Silica/polydicyclopentadiene nanocomposites: Preparation, characterization, and mechanical properties, Ferroelectrics, 2018, 523, 32–40 CrossRef CAS.
- B. Pan, Y. Cheng, Y. Wang, Y. Feng, W. Ye, Y. Tian and X. Wang, Polydicyclopentadiene Reinforced with Grafted Silica Nanoparticles, Polym.-Plast. Technol. Eng., 2013, 52, 586–591 CrossRef CAS.
- S. G. Peng, Y. P. Niu and X. J. Fan, Enhancement of Mechanical and Tribological Properties in Ring-Opening Metathesis Polymerization Functionalized Molybdenum Disulfide/Polydicyclopentadiene Nanocomposites, J. Appl. Polym. Sci., 2013, 129, 1045–1052 CrossRef CAS.
- W. Yin, S. Kniajanski and B. Amm, Dielectric properties of polydicyclopentadiene and polydicyclopentadiene-silica nanocomposite, IEEE Int. Symp. Electr. Insul., San Diego, CA, 2010, pp. 1–5.
- P. Culver, C. W. Beier, J. P. Rafson and R. L. Brutchey, Surface modification of BaTiO3 inclusions in polydicyclopentadiene nanocomposites for energy storage, J. Appl. Polym. Sci., 2014, 131, 40290 CrossRef.
- M. Yoonessi, H. Toghiani, W. L. Kingery and C. U. Pittman, Preparation characterization and properties of exfoliated/delaminated organically modified clay/dicyclopentadiene resin nanocomposites, Macromolecules, 2004, 37, 10365–10371 CrossRef.
- M. Yoonessi, H. Toghiani, T. L. Daulton, J.-S. Lin and C. U. Pittman, Clay Delamination in Clay/Poly(Dicyclopentadiene) Nanocomposites Quantified by Small Angle Neutron Scattering and High-Resolution Transmission Electron Microscopy, Macromolecules, 2005, 38, 818–831 CrossRef CAS.
- B. L. Pan, Y. L. Xing, C. F. Zhang and Y. Q. Zhang, Study on Erosion Wear Behavior of PDCPD/MMT Nanocomposite, Adv. Mater. Res., 2010, 123, 231–234 Search PubMed.
- Y. X. He, Y. N. Yan, L. Zhang, S. B. Zhu and Y. Q. Zhang, Preparation and Properties of Polydicyclopentadiene/MMt Nanocomposites Using Supported Tungsten Catalyst, Polym.-Plast. Technol. Eng., 2011, 50, 1103–1108 CrossRef CAS.
- R. Simons, S. N. Guntari, T. K. Goh, G. G. Qiao and S. A. Bateman, Poly(dicyclopentadiene)-montmorillonite nanocomposite formation via simultaneous intergallery-surface initiation and chain crosslinking using ROMP, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 89–97 CrossRef CAS.
- W. Jeong and M. R. Kessler, Toughness Enhancement in ROMP Functionalized Carbon Nanotube/Polydicyclopentadiene Composites, Chem. Mater., 2008, 22, 7060–7068 CrossRef.
- G. Yang, S. C. Lee and J. K. Lee, Reinforcement of norbornene-based nanocomposites with norbornene functionalized multi-walled carbon nanotubes, Chem. Eng. J., 2016, 288, 9–18 CrossRef CAS.
- Z. He, J. Sun, L. Zhang, Y. Wang, H. Ren and J.-B. Bao, Synergistic reinforcing and toughening of polydicyclopentadiene nanocomposites with low loadings vinyl-functionalized multi-walled carbon nanotubes, Polymer, 2018, 153, 287–294 CrossRef CAS.
- B. L. Pan, C. F. Zhang, J. Zhao, Y. Q. Zhang and Y. Z. Zhang, Mechanical properties of polydicyclopentadiene/graphite nanosheet composites prepared by reaction injection moulding, Plast., Rubber Compos., 2012, 41, 319–325 CrossRef CAS.
- F. Y. Hu, J. Du and Y. B. Zheng, Morphological effect of fillers on graphite reinforced polydicyclopentadiene based composites, Polym. Compos., 2014, 35, 1918–1925 CrossRef CAS.
- F. Y. Hu, J. Du, Y. Fang, X. Ren, X. Liu and Y. B. Zheng, In situ formation of mildly oxidized graphene oxid/polydicyclopentadiene composite and reinforced mechanical and thermal performances, J. Macromol. Sci., 2014, 51, 3246 Search PubMed.
- J. H. Jeon, W. I. Lee, J. M. Choi and S. W. Choi, Analysis of Cryogenic Impact Properties for a Glass-Fiber-Reinforced Dicyclopentadiene with a Different Amount of Decelerator Solution, Materials, 2019, 12, 439–454 CrossRef PubMed.
- K. A. M. Vallons, R. Drozdzak, M. Charret, S. V. Lomov and I. Verpoest, Assessment of the mechanical behaviour of glass fibre composites with a tough polydicyclopentadiene (PDCPD) matrix, Composites, Part A, 2015, 78, 191–200 CrossRef CAS.
- J.-H. Kim, D.-J. Kwon, P.-S. Shin, H.-S. Park, Y.-M. Baek, K. L. DeVries and J.-M. Park, Evaluation of interfacial and mechanical properties of glass fiber and p-DCPD composites with surface treatment of glass fiber, Composites, Part B, 2018, 153, 420–428 CrossRef CAS.
- H. Cui and M. R. Kessler, Glass fiber reinforced ROMP-based bio-renewable polymers: Enhancement of the interface with silane coupling agents, Compos. Sci. Technol., 2012, 72, 1264–1272 CrossRef CAS.
- H. M. Yoo, D. J. Kwon, J. M. Park, S. H. Yum and W. I. Lee, Mechanical properties of norbornene-based silane treated glass fiber reinforced polydicyclopentadiene composites manufactured by the S-RIM process, e-Polymers, 2017, 17, 159–166 CAS.
- Y. Hu, A. W. Lang, X. Li and S. R. Nutt, Hygrothermal aging effects on fatigue of glass fiber/polydicyclopentadiene composites, Polym. Degrad. Stab., 2014, 110, 464–472 CrossRef CAS.
- Y. Hu, X. Li, A. W. Lang, Y. Zhang and S. R. Nutt, Water immersion aging of polydicyclopentadiene resin and glass fiber composites, Polym. Degrad. Stab., 2016, 124, 35–42 CrossRef CAS.
- Y. Hu, Y. Zhang and S. Nutt, Thermal oxidation aging of polydicyclopentadiene and composites, Polym. Compos., 2018, 39, 1742–1751 CrossRef CAS.
- E. Devaux, C. Caze, G. Recher and D. Bielarski, Characterization of interfacial adhesion in a ultra-high-molecular-weight polyethylene reinforced polydicyclopentadiene composite, Polym. Test., 2002, 21, 457–462 CrossRef CAS.
- H. M. Yoo, M. S. Kim, B. S. Kim, D. J. Kwon and S. W. Choi, Impact and shear properties of carbon fabric/poly-dicyclopentadiene composites manufactured by vacuum-assisted resin transfer molding, e-Polymers, 2019, 19, 437–443 CAS.
- W. Xin, J. Severino, A. Venkert, H. Yu, D. Knorr, J.-M. Yang, L. Carlson, R. Hicks and I. De Rosa, Fabrication and Characterization of Solid Composite Yarns from Carbon Nanotubes and Poly(dicyclopentadiene), Nanomaterials, 2020, 10, 717 CrossRef CAS PubMed.
- H. A. Colorado, W. Yuan, Z. Guo, J. Juanri and J.-M. Yang, Poly-dicyclopentadiene-wollastonite composites toward structural applications, J. Compos. Mater., 2014, 48, 2023–2031 CrossRef.
- Z. Chen and S. S. Zhu, In-situ polymerization of dicyclopentadiene for rock reinforcement, J. Pet. Sci. Eng., 2019, 173, 912–921 CrossRef CAS.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Self-healing Materials: A Review of Advances in Materials, Evaluation, Characterization, and Monitoring Techniques, Composites, Part B, 2016, 87, 92–119 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Autonomic healing of polymer composites, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- G. O. Wilson, J. S. Moore, S. R. White, N. R. Sottos and H. M. Andersson, Autonomic Healing of Epoxy Vinyl Esters via Ring Opening Metathesis Polymerization, Adv. Funct. Mater., 2008, 18, 44–52 CrossRef CAS.
- E. N. Brown, N. R. Sottos and S. R. White, Fracture testing of a self-healing polymer composite, Exp. Mech., 2002, 42, 372–379 CrossRef CAS.
- M. R. Kessler, N. R. Sottos and S. R. White, Self-healing structural composite materials, Composites, Part A, 2003, 34, 743–753 CrossRef.
- E. N. Brown, S. R. White and N. R. Sottos, Retardation and repair of fatigue cracks in a microcapsule toughened epoxy composite-Part I: Manual infiltration, Compos. Sci. Technol., 2005, 65, 2466–2473 CrossRef CAS.
- E. N. Brown, S. R. White and N. R. Sottos, Retardation and repair of fatigue cracks in a microcapsule toughened epoxy composite-Part II: In situ self-healing, Compos. Sci. Technol., 2005, 65, 2474–2480 CrossRef CAS.
- D. Rule, E. N. Brown, N. R. Sottos, S. R. White and J. S. Moore, Wax-Protected Catalyst Microspheres for Efficient Self-Healing Materials, Adv. Mater., 2005, 17, 205–208 CrossRef.
- J. D. Rule, N. R. Sottos and S. R. White, Effect of microcapsule size on the performance of self-healing polymers, Polymer, 2007, 48, 3520–3529 CrossRef CAS.
- G. O. Wilson, M. M. Caruso, N. T. Reimer, S. R. White, N. R. Sottos and J. S. Moore, Evaluation of Ruthenium Catalysts for Ring-Opening Metathesis Polymerization-Based Self-Healing Applications, Chem. Mater., 2008, 20, 3288–3297 CrossRef CAS.
- L. Guadagno, P. Longo, M. Raimondo, C. Naddeo, A. Mariconda, V. Vittoria, G. Iannuzzo and S. Russo, Use of Hoveyda-Grubb's second generation catalyst in self-healing epoxy mixtures, Composites, Part B, 2011, 42, 296–301 CrossRef.
- B. Özgün, Ö. Solmaz and K. Şehitoğlu, Applications of ruthenium indenylidene catalysts on ROMP-based self-healing epoxy systems, Polymer, 2015, 69, 343–348 CrossRef.
- L. Guadagno, A. Mariconda, A. Agovino, M. Raimondo and P. Longo, Protection of graphene supported ROMP catalyst through polymeric globular shell in self-healing materials, Composites, Part B, 2017, 116, 352–360 CrossRef CAS.
- P. Longo, A. Mariconda, E. Calabrese, M. Raimondo, C. Naddeo, L. Vertuccio, S. Russo, G. Iannuzzo and L. Guadagno, Development of a new stable ruthenium initiator suitably designed for self-repairing applications in high reactive environments, Ind. Eng. Chem. Res., 2017, 54, 234–251 CrossRef CAS.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Self-Healing Materials With Microvascular Networks, Nat. Mater., 2007, 6, 581–585 CrossRef CAS PubMed.
- R. Shen, R. S. Amano, G. Lewinski, A. Kumar and K. Matt, A New Vascular System Highly Efficient in the Storage and Transport of Healing Agent for Self-Healing Wind Turbine Blades, J. Energy Resour. Technol., 2019, 141, 051212 CrossRef CAS.
- R. Shen, R. S. Amano and G. Lewinski, Self-Healing Performance Comparison Between Two Promising Vascular Vessel Systems of the Wind Turbine Blade, J. Energy Resour. Technol., 2019, 141, 111203 CrossRef CAS.
- A. Ruiu, D. Sanna, V. Alzari, D. Nuvoli and A. Mariani, Advances in the frontal ring opening metathesis polymerization of dicyclopentadiene, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2776–2780 CrossRef CAS.
- V. Alzari, D. Nuvoli, D. Sanna, A. Ruiu and A. Mariani, Effects of limonene on the frontal ring opening metathesis polymerization of dicyclopentadiene, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 63–68 CrossRef CAS.
- I. D. Robertson, E. L. Pruitt and J. S. Moore, Frontal Ring-Opening Metathesis Polymerization of Exo-Dicyclopentadiene for Low Catalyst Loadings, ACS Macro Lett., 2016, 5, 593–596 CrossRef CAS.
- I. D. Robertson, M. Yourdkhani, P. J. Centellas, J. E. Aw, D. G. Ivanoff, E. Goli, E. M. Lloyd, L. M. Dean, N. R. Sottos, P. H. Geubelle, J. S. Moore and S. R. White, Rapid Energy-Efficient Manufacturing of Polymers and Composites via Frontal Polymerization, Nature, 2018, 557, 223–227 CrossRef CAS PubMed.
- E. Goli, I. D. Robertson, P. H. Geubelle and J. S. Moore, Frontal Polymerization of Dicyclopentadiene: A Numerical Study, J. Phys. Chem. B, 2018, 122, 4583–4591 CrossRef CAS PubMed.
- H. Liu, H. Wei and J. S. Moore, Frontal Ring-Opening Metathesis Copolymerization: Deviation of Front Velocity from Mixing Rules, ACS Macro Lett., 2019, 8, 846–851 CrossRef CAS.
- B. Wang, K. F. Arias, Z. Zhang, Y. Liu, Z. Jiang, H.-J. Sue, N. Currie-Gregg, S. Bouslog, Z. J. Pei and S. Wang, 3D printing of in-situ curing thermally insulated thermosets, Manuf. Lett., 2019, 21, 1–6 CrossRef.
- L. M. Bellan, G. W. Coates and H. G. Craighead, Poly(dicyclopentadiene) submicron fibers produced by electrospinning, Macromol. Rapid Commun., 2006, 27, 511–515 CrossRef CAS.
- D. Fu, L.-T. Weng, B. Du, O. K. C. Tsui and B. Xu, Solventless Polymerization at the Gas-Solid Interface to Form Polymeric Thin Films, Adv. Mater., 2002, 14, 339–343 CrossRef CAS.
- H. T. Ban, M. Shigeta, T. Nagamune and M. Uejima, Synthesis of cyclo-olefin copolymer latexes and their carbon nanotube composite nanoparticles, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4584–4591 CAS.
- J. N. Talbert, L.-S. Wang, B. Duncan, Y. Jeong, S. M. Andler, V. M. Rotello and J. M. Goddard, Immobilization and Stabilization of Lipase (CaLB) through Hierarchical Interfacial Assembly, Biomacromolecules, 2014, 15, 3915–3922 CrossRef CAS PubMed.
- S. M. Andler, L.-S. Wang, J. M. Goddard and V. M. Rotello, Preparation of Biocatalytic Microparticles by Interfacial Self-Assembly of Enzyme–Nanoparticle Conjugates Around a Cross-Linkable Core, Methods Enzymol., 2016, 571, 1–17 CAS.
- S. M. Andler, L.-S. Wang, V. M. Rotello and J. M. Goddard, Influence of Hierarchical Interfacial Assembly on Lipase Stability and Performance in Deep Eutectic Solvent, J. Agric. Food Chem., 2017, 65, 1907–1914 CrossRef CAS PubMed.
- A. Della Martina, J. G. Hilborn and A. Mühlebach, Macroporous Cross-Linked Poly(dicyclopentadiene), Macromolecules, 2000, 33, 2916–2921 CrossRef CAS.
- A. Della Martina and J. G. Hilborn, Gradient porosity poly(dicyclopentadiene), J. Mater. Res., 2001, 16, 2045–2052 CrossRef CAS.
- A. Della Martina, L. Garamszegi and J. G. Hilborn, Pore size modification of macroporous crosslinked poly(dicyclopentadiene), J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2036–2046 CrossRef CAS.
- A. Della Martina, L. Garamszegi and J. G. Hilborn, Surface functionalization of cross-linked poly(dicyclopentadiene), React. Funct. Polym., 2003, 57, 49–55 CrossRef CAS.
- M. Perring and N. B. Bowden, Assembly of Organic Monolayers on Polydicyclopentadiene, Langmuir, 2008, 24, 10480–10487 CrossRef CAS PubMed.
- M. Perring, T. R. Longa and N. B. Bowden, Epoxidation of the surface of polydicyclopentadiene for the self-assembly of organic monolayers, J. Mater. Chem., 2010, 20, 8679–8685 RSC.
- T. Zhang, R. A. Sanguramath, S. Israel and M. S. Silverstein, Emulsion Templating: Porous Polymers and Beyond, Macromolecules, 2019, 52, 5445–5479 CrossRef CAS.
- K. Benmachou, H. Deleuze and V. Heroguez, Ring opening polymerisation of highly concentrated inverse emulsions to obtain microcellular foams, React. Funct. Polym., 2003, 55, 211–217 CrossRef CAS.
- S. Kovačič, P. Krajnc and C. Slugovc, Inherently Reactive polyHIPE Material from Dicyclopentadien, Chem. Commun., 2010, 46, 7504–7506 RSC.
- S. Kovačič, K. Jeřabek, P. Krajnc and C. Slugovc, Ring Opening Metathesis Polymerisation of emulsion templated dicyclopentadiene giving open porous materials with excellent mechanical properties, Polym. Chem., 2012, 3, 325–328 RSC.
- S. Kovačič, N. B. Matsko, K. Jeřabek, P. Krajnc and C. Slugovc, On the Mechanical Properties of HIPE Templated Macroporous poly(Dicyclopentadiene) Prepared with Low Surfactant Amounts, J. Mater. Chem. A, 2013, 1, 487–490 RSC.
- S. Kovačič, E. Žagar and C. Slugovc, Strength versus toughness of emulsion templated poly(dicyclopentadiene) foams, Polymer, 2019, 169, 58–65 CrossRef.
- S. Kovačič, F. Preishuber-Pflügl and C. Slugovc, Macroporous Polyolefin Membranes from Dicyclopentadiene High Internal Phase Emulsions: Preparation and Morphology Tuning, Macromol. Mater. Eng., 2014, 299, 843–850 CrossRef.
- S. Kovačič, H. Kren, P. Krajnc, S. Koller and C. Slugovc, The use of an emulsion templated microcellular poly(dicyclopentadiene-co-norbornene) membrane as separator in lithium-ion batteries, Macromol. Rapid Commun., 2013, 34, 581–587 CrossRef PubMed.
- E. H. Mert, C. Slugovc and P. Krajnc, Tailoring the Mechanical and Thermal Properties of Dicyclopentadiene PolyHIPEs with the Use of a Comonomer, Express Polym. Lett., 2015, 9, 344–353 CrossRef CAS.
- E. Vakalopoulou and C. Slugovc, The effects of enhancing the crosslinking degree in high internal phase emulsion templated poly(dicyclopentadiene), Macromol. Chem. Phys., 2019, 220, 1900423 CrossRef CAS.
- S. Kovačič, N. B. Matsko, G. Ferk and C. Slugovc, Macroporous poly(dicyclopentadiene) γFe2O3/Fe3O4 nanocomposite foams by high internal phase emulsion templating, J. Mater. Chem. A, 2013, 1, 7971–7978 RSC.
- S. Kovačič, A. Anžlovar, B. Erjavec, G. Kapun, N. B. Matsko, M. Zigon, E. Zagar, A. Pintar and C. Slugovc, Macroporous ZnO Foams by High Internal Phase Emulsion Technique: Synthesis and Catalytic Activity, ACS Appl. Mater. Interfaces, 2014, 6, 19075–19081 CrossRef PubMed.
- E. Yüce, E. H. Mert, P. Krajnc, F. N. Parın, N. San, D. Kaya and H. Yıldırım, Photocatalytic Activity of Titania/Polydicyclopentadiene PolyHIPE Composites, Macromol. Mater. Eng., 2017, 302, 1700091 CrossRef.
- E. Yüce, P. Krajnc, H. H. Mert and E. H. Mert, Influence of nanoparticles and antioxidants on mechanical properties of titania/polydicyclopentadiene polyHIPEs: A statistical approach, J. Appl. Polym. Sci., 2019, 136, 46913 CrossRef.
- S. Kovačič, M. Mazaj, M. Jeselnik, D. Pahovnik, E. Žagar, C. Slugovc and N. Zabukovec Logar, Synthesis and Catalytic Performance of Hierarchically Porous MIL-100(Fe)@polyHIPE Hybrid Membranes, Macromol. Rapid Commun., 2015, 36, 1605–1611 CrossRef PubMed.
- M. Mazaj, N. Zabukovec Logar, E. Žagar and S. Kovačič, A facile strategy towards a highly accessible and hydrostable MOF-phase within hybrid polyHIPEs through in situ metal-oxide recrystallization, J. Mater. Chem. A, 2017, 5, 1967–1971 RSC.
- S. M. Andler and J. M. Goddard, Stabilization of Lipase in Polymerized High Internal Phase Emulsions, J. Agric. Food Chem., 2018, 66, 3619–3623 CrossRef CAS PubMed.
- N. Trupej, Z. Novak, Ž. Knez, C. Slugovc and S. Kovačič, Supercritical CO2 Mediated Functionalization of Highly Porous Emulsion-Derived Foams: scCO2 Absorption and Epoxidation, J. CO2 Util., 2017, 21, 336–341 CrossRef CAS.
- A.-C. Knall, S. Kovačič, M. Hollauf, D. P. Reishofer, R. Saf and C. Slugovc, Inverse electron demand Diels-Alder (iEDDA) functionalisation of macroporous poly(dicyclopentadiene) foams, Chem. Commun., 2013, 49, 7325–7327 RSC.
- Z. Yao, Z. Wang, Y. Yu, C. Zeng and K. Cao, Facile synthesis and properties of the chemo-reversible and highly tunable metallogels based on polydicyclopentadiene, Polymer, 2017, 119, 98–106 CrossRef CAS.
- C. L. McGann, G. C. Daniels, S. L. Giles, R. B. Balow, J. L. Miranda-Zayas, J. G. Lundin and J. H. Wynne, Air Activated Self-Decontaminating Polydicyclopentadiene PolyHIPE Foams for Rapid Decontamination of Chemical Warfare Agents, Macromol. Rapid Commun., 2018, 39, 1800194 CrossRef PubMed.
- R. B. Balow, S. L. Giles, C. L. McGann, G. C. Daniels, J. G. Lundin, P. E. Pehrsson and J. H. Wynne, Rapid Decontamination of Chemical Warfare Agent Simulant with Thermally Activated Porous Polymer Foams, Ind. Eng. Chem. Res., 2018, 57, 8630–8634 CrossRef CAS.
- E. Vakalopoulou, S. M. Borisov and C. Slugovc, Fast oxygen scavenging of macroporous poly(norbornadiene) prepared by Ring-opening Metathesis Polymerization, Macromol. Rapid Commun., 2020, 41, 1900581 CrossRef CAS PubMed.
- S. Kovačič, N. B. Matsko, K. Gruber, S. Koller and C. Slugovc, Ring-Opening Metathesis Polymerization Derived Hierarchically Porous Carbon-Foams, ChemRxiv, 2020 DOI:10.26434/chemrxiv.11674041.v1.
- J. K. Lee and G. L. Gould, Polydicyclopentadiene based aerogel: a new insulation material, J. Sol-Gel Sci. Technol., 2007, 44, 29–40 CrossRef CAS.
- C. Dawedeit, S. H. Kim, T. Braun, M. A. Worsley, A. L. Stephan, K. J. Wu, C. C. Walton, A. A. Chernov, J. H. Satcher Jr., A. V. Hamza and J. Biener, Tuning the rheological properties of sols for low-density aerogel coating applications, Soft Matter, 2012, 8, 3518–3521 RSC.
- S. H. Kim, M. A. Worsley, C. A. Valdez, S. J. Shin, C. Dawedeit, T. Braun, T. F. Baumann, S. A. Letts, S. O. Kucheyev, K. J. J. Wu, J. Biener, J. H. Satcher, Jr. and A. V. Hamza, Exploration of the versatility of ring opening metathesis polymerization: an approach for gaining access to low density polymeric aerogels, RSC Adv., 2012, 2, 8672–8680 RSC.
- S. H. Kim, S. J. Shin, J. M. Lenhardt, T. Braun, J. D. Sain, C. A. Valdez, R. N. Leif, S. O. Kucheyev, K. J. J. Wu, J. Biener, J. H. Satcher, Jr. and A. V. Hamza, Deterministic control over high-Z doping of polydicyclopentadiene-based aerogel coatings, ACS Appl. Mater. Interfaces, 2013, 5, 8111–8119 CrossRef CAS PubMed.
- D. P. Mohite, S. Mahadik-Khanolkar, H. Luo, H. Lu, C. Sotiriou-Leventis and N. Leventis, Polydicyclopentadiene aerogels grafted with PMMA: I. Molecular and interparticle crosslinking, Soft Matter, 2013, 9, 1516–1530 RSC.
- D. P. Mohite, S. Mahadik-Khanolkar, H. Luo, H. Lu, C. Sotiriou-Leventis and N. Leventis, Polydicyclopentadiene aerogels grafted with PMMA: II. Nanoscopic characterization and origin of macroscopic deformation, Soft Matter, 2013, 9, 1531–1539 RSC.
- G. Raptopoulos, G. C. Anyfantis, D. Chriti and P. Paraskevopoulou, Synthesis and structural characterization of poly(dicyclopentadiene) gels obtained with a novel ditungsten versus conventional W and Ru mononuclear catalysts, Inorg. Chim. Acta, 2017, 460, 69–76 CrossRef CAS.
- D. Chriti, G. Raptopoulos, G. C. Anyfantis and P. Paraskevopoulou, An Extreme Case of Swelling of Mostly cis-Polydicyclopentadiene by Selective Solvent Absorption-Application in Decontamination and Environmental Remediation, ACS Appl. Polym. Mater., 2019, 1, 1648–1659 CrossRef CAS.
- D. Chriti, G. Raptopoulos, B. Brandenburg and P. Paraskevopoulou, Large, Rapid Swelling of High-cis Polydicyclopentadiene Aerogels Suitable for Solvent-Responsive Actuators, Polymers, 2020, 12, 1033 CrossRef CAS PubMed.
- J. M. Lenhardt, S. H. Kim, A. J. Nelson, P. Singhal, T. F. Baumann and J. H. Satcher Jr, Increasing the oxidative stability of poly(dicyclopentadiene) aerogels by hydrogenation, Polymer, 2013, 54, 542–547 CrossRef CAS.
- T. R. Long, A. Gupta, A. L. Miller, D. G. Rethwisch and N. B. Bowden, Selective flux of organic liquids and solids using nanoporous membranes of polydicyclopentadiene, J. Mater. Chem., 2011, 21, 14265–14276 RSC.
- A. Gupta, T. R. Long, D. G. Rethwisch and N. B. Bowden, Retention of palladium and phosphine ligands using nanoporous polydicyclopentadiene thimbles, Chem. Commun., 2011, 47, 10236–10238 RSC.
- A. Gupta and N. B. Bowden, Separation of cis-fatty acids from saturated and trans-fatty acids by nanoporous polydicyclopentadiene membranes, ACS Appl. Mater. Interfaces, 2013, 5, 924–933 CrossRef CAS PubMed.
- L. Chen, W. A. Phillip, E. L. Cussler and M. A. Hillmyer, Robust Nanoporous Membranes Templated by a Doubly Reactive Block Copolymer, J. Am. Chem. Soc., 2007, 129, 13786–13787 CrossRef CAS PubMed.
- M. A. Amendt, L. Chen and M. A. Hillmyer, Formation of Nanostructured Poly(dicyclopentadiene) Thermosets Using Reactive Block Polymers, Macromolecules, 2010, 43, 3924–3934 CrossRef CAS.
- W. A. Phillip, M. Amendt, B. O’Neill, L. Chen, M. A. Hillmyer and E. L. Cussler, Diffusion and Flow Across Nanoporous Polydicyclopentadiene-Based Membranes, ACS Appl. Mater. Interfaces, 2009, 1, 472–480 CrossRef CAS PubMed.
- L. Chen and M. A. Hillmyer, Mechanically and Thermally Robust Ordered Nanoporous Monoliths Using Norbornene-Functional Block Polymers, Macromolecules, 2009, 42, 4237–4243 CrossRef CAS.
- M. A. Amendt, M. Roerdink, S. Moench, W. A. Phillip, E. L. Cussler and M. A. Hillmyer, Functionalized Nanoporous Membranes from Reactive Triblock Polymers, Aust. J. Chem., 2011, 64, 1074–1082 CrossRef CAS.
- T. J. Clark, N. J. Robertson, H. A. Kostalik IV, E. B. Lobkovsky, P. F. Mutolo, H. D. Abruna and G. W. Coates, A ring-opening metathesis polymerization route to alkaline anion exchange membranes: development of hydroxide-conducting thin films from an ammonium-functionalized monomer, J. Am. Chem. Soc., 2009, 131, 12888–12889 CrossRef CAS PubMed.
- S. C. Price, X. M. Ren, A. C. Jackson, Y. S. Ye, Y. A. Elabdand and F. L. Beyer, Bicontinuous Alkaline Fuel Cell Membranes from Strongly Self-Segregating Block Copolymers, Macromolecules, 2013, 46, 7332–7340 CrossRef CAS.
- X. Ren, S. C. Price, A. C. Jackson, N. Pomerantz and F. L. Beyer, Highly Conductive Anion Exchange Membrane for High Power Density Fuel-Cell Performance, ACS Appl. Mater. Interfaces, 2014, 6, 13330–13333 CrossRef CAS PubMed.
- Y. Zha, M. L. Disabb-Miller, Z. D. Johnson, M. A. Hickner and G. N. Tew, Metal-Cation-Based Anion Exchange Membranes, J. Am. Chem. Soc., 2012, 134, 4493–4496 CrossRef CAS PubMed.
- M. L. Disabb-Miller, Y. Zha, A. J. DeCarlo, M. Pawar, G. N. Tew and M. A. Hickner, Water Uptake and Ion Mobility in Cross-Linked Bis(terpyridine)ruthenium-Based Anion Exchange Membranes, Macromolecules, 2013, 46, 9279–9287 CrossRef CAS.
- M. T. Kwasny and G. N. Tew, Expanding metal cation options in polymeric anion exchange membranes, J. Mater. Chem. A, 2017, 5, 1400–1405 RSC.
- M. T. Kwasny, L. Zhu, M. A. Hickner and G. N. Tew, Thermodynamics of Counterion Release Is Critical for Anion Exchange Membrane Conductivity, J. Am. Chem. Soc., 2018, 140, 7961–7969 CrossRef CAS PubMed.
- L. Chen, D. T. Hallinan, Jr., Y. A. Elabd and M. A. Hillmyer, Highly Selective Polymer Electrolyte Membranes from Reactive Block Polymers, Macromolecules, 2009, 42, 6075–6085 CrossRef CAS.
- W. A. Phillip, E. Martono, L. Chen, M. A. Hillmyer and E. L. Cussler, Seeking an ammonia selective membrane based on nanostructured sulfonated block copolymers, J. Membr. Sci., 2009, 337, 39–46 CrossRef CAS.
- X. Li, Y. Zhao, Z. Feng, X. Xiang, S. Wang, X. Xie and V. K. Ramani, Ring-opening metathesis polymerization for the preparation of polynorbornene-based proton exchange membranes with high proton conductivity, J. Membr. Sci., 2017, 528, 55–63 CrossRef CAS.
- C. Wang, Z. Feng, Y. Zhao, X. Li, W. Li, X. Xie, S. Wang and H. Hou, Preparation and properties of ion exchange membranes for PEMFC with sulfonic and carboxylic acid groups based on polynorbornenes, Int. J. Hydrogen Energy, 2017, 42, 29988–29994 CrossRef CAS.
- T. J. Cuthbert, J. Chen, F. P. Burns, M. G. Moffitt and J. E. Wulff, Thermally Crosslinked Functionalized Polydicyclopentadiene with a High Tg and Tunable Surface Energy, ACS Omega, 2016, 1, 532–540 CrossRef PubMed.
- T. J. Cuthbert, T. Li, A. W. H. Speed and J. E. Wulff, Structure of the Thermally Induced Cross-Link in C-Linked Methyl Ester-Functionalized Polydicyclopentadiene (fPDCPD), Macromolecules, 2018, 51, 2038–2047 CrossRef CAS.
- T. J. Cuthbert, T. Li and J. E. Wulff, Production and Dynamic Mechanical Analysis of Macro-Scale Functionalized Polydicyclopentadiene Objects Facilitated by Rational Synthesis and Reaction Injection Molding, ACS Appl. Polym. Mater., 2019, 1, 2460–2471 CrossRef CAS.
- L. Gong, K. Liu, E. Ou, F. Xu, Y. Lu, Z. Wang, T. Gao, Z. Yang and W. Xu, ROMP of acetoxy-substituted dicyclopentadiene to a linear polymer with a high Tg, RSC Adv., 2015, 5, 26185–26188 RSC.
- S. Saha, Y. Ginzburg, I. Rozenberg, O. Iliashevsky, A. Ben-Asuly and N. G. Lemcoff, Cross-linked ROMP polymers based on odourless dicyclopentadiene derivatives, Polym. Chem., 2016, 7, 3071–3075 RSC.
- R. S. Phatake, A. Masarwa, N. G. Lemcoff and O. Reany, Tuning thermal properties of cross-linked DCPD polymers by functionalization, initiator type and curing methods, Polym. Chem., 2020, 11, 1742–1751 RSC.
- J. E. Amoore and E. Hautala, Odor as an aid to chemical safety: Odor thresholds compared with threshold limit values and volatilities for 214 industrial chemicals in air and water dilution, J. Appl. Toxicol., 1983, 3, 272–290 CrossRef CAS PubMed.
- L. G. Stehmeier, M. M. Francis, T. R. Jack and G. Voordouw, Biodegradation of Dicyclopentadiene in the Field, Biodegradation, 1999, 10, 135–148 CrossRef CAS.
- K. M. Kransler, Results of a 90-day inhalation study of dicyclopentadiene in B6C3F1 mice, Toxicol. Ind. Health, 2014, 30, 459–466 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, A Comonomer Strategy for Triggered Degradation and Re/Upcycling of High-Performance Thermoset Plastics, ChemRxiv, 2019 DOI:10.26434/chemrxiv.11328242.v1.
| This journal is © the Partner Organisations 2020 |