
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn analogue of a kinase inhibitor exhibits subjective characteristics that contribute to its inhibitory activities as a potential anti-cancer candidate: insights through computational biomolecular modelling of UM-164 binding with lyn protein
Umar Ndagi *a,
Maryam Abdullahib,
Asmau N. Hamzac and
Mahmoud E. Solimanb
*a,
Maryam Abdullahib,
Asmau N. Hamzac and
Mahmoud E. Solimanb
aFaculty of Natural Sciences, Ibrahim Badamasi Babangida University, Lapai, Niger, Nigeria. E-mail: ndagiumar2@gmail.com; Tel: +2348036002456 Tel: +2347058739134
bMolecular Bio-Computation and Drug Design Research Group, School of Health Sciences, University of KwaZulu-Natal, Westville, Durban 4000, South Africa
cFaculty of Pharmaceutical Sciences, Ahmadu Bello University, Zaria, Kaduna, Nigeria
First published on 24th December 2019
Abstract
The recent emergence of lyn kinase as a driver of aggressive behaviour in triple-negative breast cancer (TNBC) remains a major concern posing a burden for people living with breast cancer and drug development. The binding of UM-164 to lyn protein has been noted to impact the conformational dynamics required for drug fitness. Herein, we provide the first account of the molecular impact of an experimental drug, UM-164 binding on lyn protein using various computational approaches including molecular docking and molecular dynamics simulation. These computational modelling methods enabled us to analyse parameters, for example principal component analysis (PCA), dynamics cross-correlation matrices (DCCM) analysis, hydrogen bond occupancy, thermodynamics calculation and ligand–residue interaction. Findings from these analyses revealed that UM-164 exhibited a higher binding affinity of −9.9 kcal mol−1 with lyn protein than Dasatinib, with a binding affinity of −8.3 kcal mol−1 on docking. It was observed that the binding of UM-164 to lyn protein decreases the capacity of its loop to fluctuate, influences the ligand optimum orientation on the conformational space of lyn protein, and increases the hydrogen bond formation in the lyn-UM-164 system. Also, an increase in drug binding energy of UM-164 was recorded with increasing residue correlation in the lyn-UM-164 system. It is quite informative to note that Met85 was a key stabilising factor in the binding of UM-164 to lyn protein. These findings can provide important insights that will potentially serve as a baseline in the design of novel lyn inhibitors. It could also stimulate further research into multidimensional approaches required to curb the influence of lyn protein in TNBC.
1. Introduction
In the recent past, breast cancer has been known to be the most frequently diagnosed and life-threatening cancer in women1 and a leading cause of cancer death among women.2 Breast cancers (BCs) ranked as the third leading cause of cancer death in the United States of America.2 BCs are classified into 6 genomic subtypes due to their genetic predispositions.3 The triple-negative subtypes are more heterogeneous with marked aggressiveness, more advanced frequency of relapse and lower survival in metastatic milieu relative to other forms of breast cancers.4 They are associated with the lack of oestrogen receptor (ER), progesterone receptor (PR) and lack of amplification of human epidermal growth factor receptor-2 (ErbB2/HER-2).4 They are characterised by well-defined ductal histology, high grade, high mitotic and cell proliferation rates.5,6The triple-negative breast cancer (TNBC) is commonly associated with poor prognosis, high risk of local reoccurrence rate (LRR), poor disease-free survival (PDFS) and cancer-specific survival (CSS).6 Reactive lymph nodes are often seen in local reoccurrence,6 and this is higher in patients with TNBC in the first 3 to 5 years after diagnosis compared to oestrogen receptor (ER) positive breast cancer.6 TNBCs are sometimes associated with genetic mutations,7,8 the distinct molecular features of TNBCs varies in both carriers and non-carriers of related genetic mutations.9 The clinicopathological characteristics of patients with TNBCs associated breast cancer gene 1 (BRCA1) mutation is quiet similar to patients diagnosed with other BRCA1 mutated tumours.10
There is an evidence that sporadic TNBCs and basal phenotypic tumours are linked to BRCA1 dysfunction, which is highly implicated in the development and progression of this disease.6 The aggressive behaviour of TNBCs may be attributed to cellular senescence and cytoprotective autophagy that can enhance the malignant phenotype responsible for this disease.11,12 The major histopathological changes resulting from a genetic mutation that occurs in TNBC include fibrosis and inflammation,13 hence the establishment of a panel of markers for the diagnosis of TNBCs metastasis is said to be a welcome idea. However, TNBCs usually exhibit a negative profile for most markers of breast origin13 consequently, routine diagnosis becomes a difficult tax.14 Efforts have been made in establishing a panel of makers with clinical usage for proper diagnosis of TNBCs metastases,14 this was mate with stiff challenges. However, there is research evidence for focal positivity to cytokeratin 7 (CK7) with heterogeneous properties.14,15
TNBCs is highly associated with basal-like breast carcinoma (BLBCs) and are considered as overlapping molecular entities, contributing to aggressive behaviour and poor breast cancer prognosis.16–18 Several potential therapeutic options have been recommended in TNBCs, but relative heterogeneity in TNBCs made it possible for us to consider that BRCA1/2 mutation with androgen receptor (AR) could be a potential drug target in the treatment of TNBCs.19 Other potential therapeutic targets may include, PIM-2 inhibitor and serine/threonine kinase.20
In the past, there is no approved targeted therapy for TNBCs in clinical practice.21,22 However, chemotherapeutic agents such as taxanes, anthracyclines and cyclophosphamide were used in the management of the patients with this form of cancer21 with little clearance. The need to have a good insight into the molecular features of primary TNBCs and their metastases is quite important in order to establish the stratification of TNBCs carcinogenesis towards the development of proper treatment strategy5 for the patients.
Triple-negative breast cancer is highly associated with different proteins and has been well defined in a study that relates TNBCs with c-Src protein in an attempt to identify predictive markers response to chemotherapy.3 This study led to the discovery of dual kinase inhibitor UM-164 with profound activity against Src and p38 kinases.3 This inhibitor has been proved to be a promising lead compound for developing the first targeted therapeutic strategy against TNBC.23,24 In a related development, the therapeutic options in TNBC are said to be limited and chemotherapy is one of the best treatment strategies after surgery.
Recent research placed lyn protein as a driver of aggressive breast cancer that belongs to the family of protein kinase which is highly expressed in TNBC.25 This lyn kinase is a downstream effector of the c-KIT receptor in normal breast cells.25 Loss of BRCA1 function hyperactivates lyn via prolyl isomerase 1 upregulation.25 The lyn protein exists in isomeric form, its full-length promotes tumour cell invasion.25
Again, TNBC has a high risk of relapse26 this may be due to unidentified biomarkers that are responsible for aggression and progression of TNBC. This has made the finding of the recent discovery of the relationship between lyn and TNBC very lucrative in drug design. Fortunately, there exists a promising lead compound in the treatment of TNBC,23 the compound UM-164, is a potent dual kinase inhibitor of Src/p38 with established better potency profile than Dasatinib. Deriving the family advantage of protein kinase, UM-164 portend a greater inhibitory activity on lyn protein, therefore, UM-164 remain a potential drug to be tried on lyn protein. In addition, the protein tyrosine kinase (PTK) also known as lyn has been identified as the most important kinase in Src-family.27 It has significantly contributed to the development of certain solid tumour27 including colon cancer, prostate cancer, leukaemia and recently shown to be involved in TNBC.28
Approximately 15% of people living with breast cancer are triple-negative.29 This huge population is a global burden and source of serious concern because TNBC has a high risk of relapse.25 Therefore, the need for an in-depth molecular understanding of lyn in TNBC and perhaps, a better understanding of the conformational features and ligand binding landscape of UM-164 complex with lyn is highly imperative. This would forester good molecular understanding and detailed insight on the therapeutic effect of the lead compound UM-164 on lyn protein for successful management of TNBC.
Currently, no conformational studies have been carried out on lyn complex with UM-164. Herein, we aim to provide a detailed insight into the conformational features and ligand binding landscape of lyn complex with UM-164 thus providing a new dimension to future research on lyn protein. This will require long time-scale molecular dynamics simulations that will provide adequate information on the dynamics of the complex. To achieve this, we perform molecular docking and molecular dynamics (MD) simulations of; lyn-UM-164 complex, lyn-Dasatinib (wild type) system and unbound lyn (apo). Principal component analysis (PCA), dynamic cross-correlation (DCCA) and binding free energy calculations as well as ligand–residue interaction network profile, were done to understand the conformational dynamics resulting from the binding of UM-164 to lyn protein.
Different post dynamic techniques have been used to provide a molecular understanding of molecular dynamics. The principal component analysis (PCA), also known as essential dynamics analysis,30 is one of the most popular post-dynamics techniques30 that is widely applied to understand the changes in biological systems.30 PCA eliminates translational and rotational motions in molecular dynamic (MD) trajectory and correlated motions in atomic simulations of proteins.30,31
Based on our previous studies, we are sure that computational and molecular modelling tools as adopted in this study would provide the necessary information on the subject matter. These tools are known to facilitate the process of drug discovery,32 therefore, provide a platform for the discovery of novel therapeutics.32 Findings from this study would demonstrate the conformational and structural properties of lyn in complex with UM-164, may form the baseline for which other potential therapeutics targeting lyn can be developed.
2. Computational methods
2.1. System preparation and molecular docking
In one of the studies conducted in the past, an X-ray crystal structure of lyn was successfully elucidated in the apo and inhibitor-bound state,27 the kinase domain is represented in an active conformational state.27 Binding mode of inhibitors from this study reveals features accounting for their varying specificity to lyn,27 for this reason the inhibitors are increasingly becoming important class of therapeutic agent.27 With recent determination to profound solution to TNBC and wide acceptance of lyn as major player in solid and other forms of tumour,27 the X-ray crystal forms of lyn is said to be positioned for the design of the next generation of clinical therapeutics against lyn.27 It is known that all clinically used Src inhibitors act by binding the active conformation of the kinase.28 Therefore, the use of experimental compound UM-164 relative to Dasatinib that was originally designed as a selective inhibitor to the Src kinases is key to the termination of aggressive activities of lyn in TNBC.The X-ray crystal structure of lyn in complex with Dasatinib was obtained from the protein data bank (PDB) with code (2ZVA).27 This is an active conformational state of the lyn structure with a single chain (Chain A). However, missing residues were observed along the loop region and this protein was therefore properly generated/recovered with the aid of modeller in graphic user interphase (GUI) UCSF chimera.33 Before the replacement of the missing residues from the loop, the Dasatinib was separated from the protein. The existing chain A was used for docking; it reduces the computational cost.
Using the UCSF chimera,33 the water molecules were removed from the protein structure. This precedes the separation of Dasatinib from the protein and saved in mol2 and pdb format, respectively. In the same software, the hydrogen atom was added to the protein structure. The steepest descent method and MMFF94 force field in Avogadro34 software were then used to optimised Dasatinib and UM-164 for energy minimization. The resulting molecules were visualised using GUI of UCSF chimera33 and hydrogen atoms were then removed from these ligands. Provided in Fig. 1 is the 3D structure of lyn protein complex with UM-164.
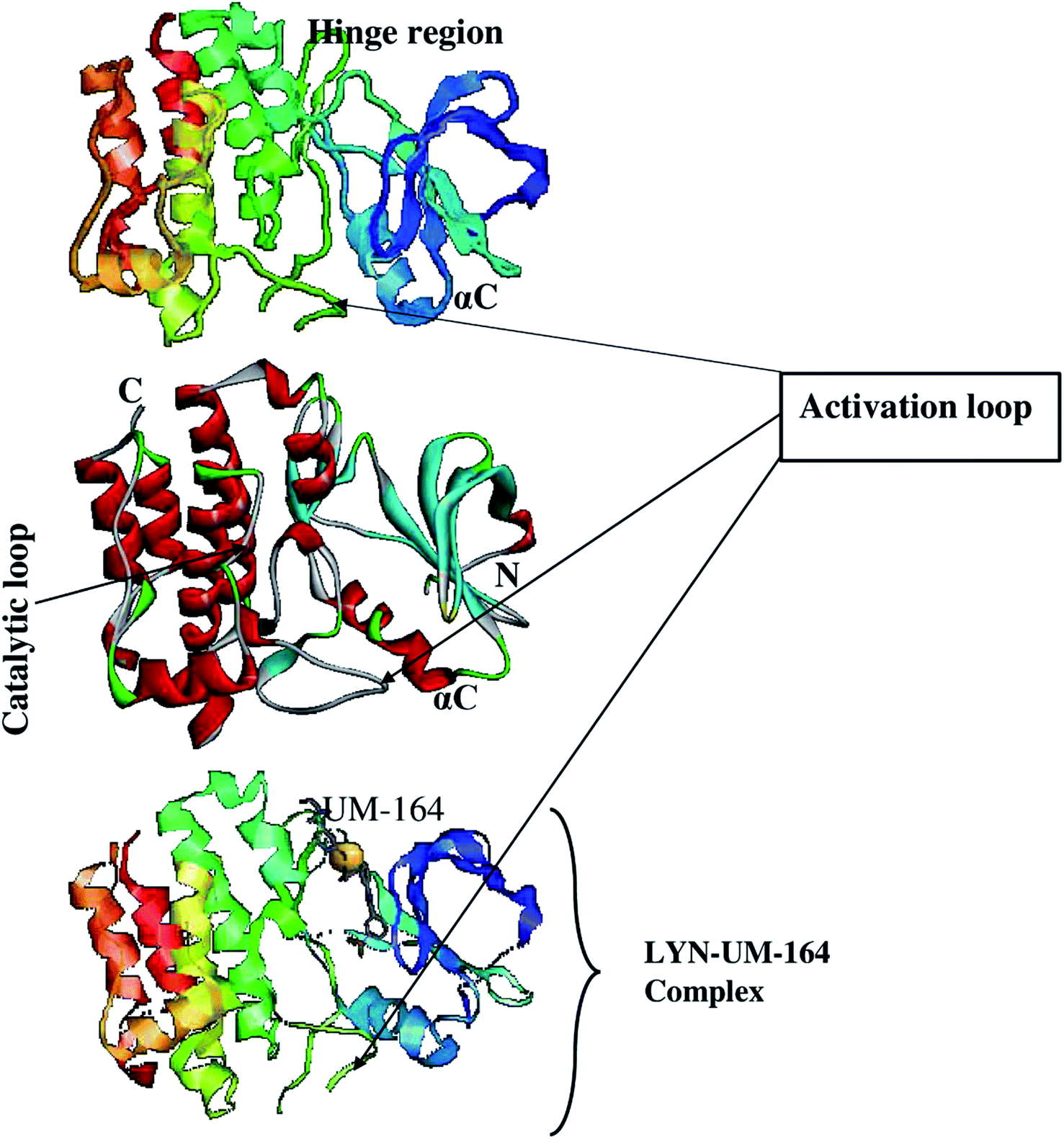 | ||
| Fig. 1 The 3D structure of LYN protein27 investigated in this study. | ||
2.2. Molecular docking
To access the docking pose and corresponding binding energy, molecular docking was conducted using Autodock vina.35 The Gasteiger partial charges36 were used to allocate the atomic charges to both the protein and the ligands. AutoDock graphical user interphase (AGUI) provided by MGL tools was used to outline AutoDock atom type. The grid box was set out with corresponding grid parameters being x = 54 Å, y = 80 Å, z = 76 Å, for the dimension while x = 17.43 Å, y = −7.57 Å, z = 26.25 Å, for the centre and 8 exhaustiveness. The selected dimension houses the whole Dasatinib plus UM-164 binding sites in the protein complex. Five (5) docking runs was performed, the Lamarckian genetic algorithm36 in AutoDock vina was used to generate different docked conformation and dynamic ligand positions within the protein conformational space. The binding energy for the docked conformations was between −8.2 kcal mol−1 to −9.8 kcal mol−1 for each docking run conducted. The two ligands used in this study is presented in Fig. 2.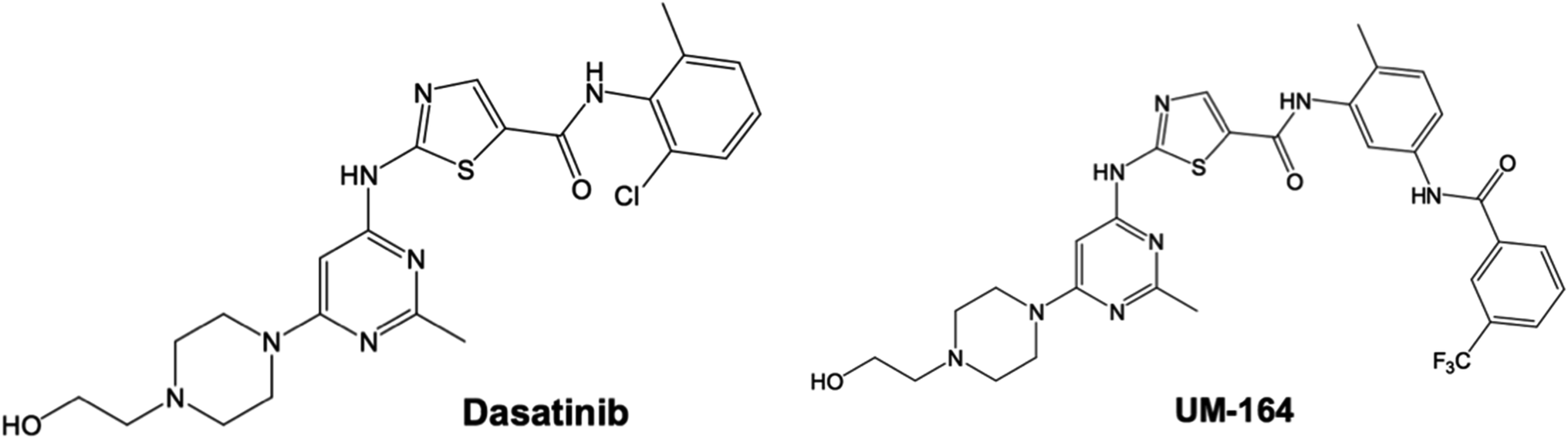 | ||
| Fig. 2 2D structure of Dasatinib and experimental Src inhibitor (UM-164). | ||
2.3. System preparation for MD
Three systems were used for the MD simulation, the docked complex of lyn-UM-164, lyn-Dasatinib complex and apo. The apo system was prepared by uploading the X-ray crystal structure of unbound lyn with pdb code 2ZV7,27 which is an active conformation of lyn structure available in the PDB with a single chain. In this study, we use the same chain for MD simulation after recovering the missing residues with the UCSF Chimera software.33 The modeller tool in Chimera was used to generate the missing loop residues and the best model was isolated from the pool as the most negative score. Hydrogen was deleted from the apo structure and H++ server37 was used to assign the correct protonation state to the protein structure.The ligand from the docked complex of lyn-UM-164 and lyn-Dasatinib (wild type) were both separated from the protein and hydrogen atoms were added to it using Chimera software.33 Gaussian 09 (ref. 38) calculations at HF/6-31G* level of theory was executed to optimise the geometry of Dasatinib and UM-164. The ligand charges were then fitted with R.E.D server,39 this produces ligand parameters in mol2 format for subsequent MD simulations.
2.4. Molecular dynamics simulations
The three stipulated systems, lyn-Dasatinib (wild type), lyn-UM-164 and apo systems were simulated using graphics processing unit (GPU) version of the Particle Mesh Ewald Molecular Dynamics (PMEMD)40 package with Sander module of Amber14. The atomic partial charges of the ligand were generated with the aid of antechamber module using restrained electrostatic potential (RESP) and force field parameters of general amber force field (gaff).41 Amber force field ff14SB42 was used to set the protein parameters. The LEaP module implemented in Amber14 was used to add hydrogen atoms to the protein and to add counter ions for system neutralization. Each system is enclosed in an orthorhombic TIP3P water box with protein atoms located 10 Å between the protein surface and box boundary to allow for adequate solvation of the systems throughout the simulation time.All the systems had cubic periodic boundary conditions implemented, long-range electrostatic interaction was treated using the petric-mesh Ewald40 method implemented in Amber14 with a nonbonding cut-off distance of 12 Å. The systems were subjected to two minimization process, partial and full minimizations. The restrained potential of 500 kcal mol−1 Å−2 was applied to the solute for 1000 steps, this was used for initial energy minimization process while unrestrained conjugated gradient minimization for 1000 steps was conducted for the entire system using the SANDER module of the Amber14 program.
Canonical ensemble (NVT) MD simulations were performed for 50 ps and system was gradually heated from 0 to 300 K, with the aid of the Langevin thermostat43 to achieve harmonic restraint of 5 kcal mol−1 Å−2 for solute atoms with a random collision frequency of 1 ps. The systems were equilibrated at 300 K with 2 fs time step in the NPT ensemble for 500 ps without any restraint and a Berendsen barostat44 was used to maintain the pressure at 1 bar. The hydrogen bonds in the systems were constrained with the aid of SHAKE45 algorithm.
The 2 fs timescale and SPFP precision model were used for the MD runs. In the absence of restraints, a production run of 300 ns MD simulations was performed in an isothermal–isobaric (NPT) ensemble using a Berendsen barostat at a pressure of 1 bar and a pressure-coupling constant of 2 ps. For every 1 ps of simulation, coordinates were saved and trajectories were analysed. Post-MD analyses were performed, this includes principal component analysis (PCA), root mean square deviation (RMSD), root mean square fluctuations (RMSF), the radius of gyration (Rg), hydrogen bond occupancy and dynamic cross-correlation (DCCR) using PTRAJ and CPPTRAJ46 module in Amber14, as well as ligand–residue interactions. Trajectories were visualised using Chimera software. The results were analysed and plots were generated with the aid of Origin software.47
2.5. Thermodynamic calculations
The binding free energy calculation is an important thermodynamic parameter that offers detailed information on the interaction between ligand and protein.48 It also provides a good understanding of the mechanism of binding, including their various energy contributions, such as enthalpy and entropy, to molecular recognition.48 Molecular Mechanics/Generalized-Born Surface Area method (MM/GBSA)48 is a popular method that can be used to estimate the binding free energy of small ligands to the biological macromolecule.48 The binding free energy of lyn-UM-164, lyn-Dasatinib (wild type) and apo systems were calculated using MM/GBSA.48 For a 300 ns trajectory, 1000 snapshots were considered during the calculation of the binding free energy. The following equations described binding free energy calculation:| ΔGbind = Gcomplex − Greceptor − Gligand | (1) |
| ΔGbind = Egas + Gsol − TS | (2) |
| Egas = Eint + EvdW + Eele | (3) |
| Gsol = GGB + GSA | (4) |
From the equation above, Egas is the energy of the gas phase, Eint represents internal energy, Eele represents Coulomb while EvdW is the van der Waals energies. Egas is estimated directly from the ff14SB42 force field. Gsol which is the solvation free energy can be broken down to polar and non-polar forms of contribution. The contribution of polar solvation (GGB) is assessed by resolving GGB equation and non-polar solvation (GSA) is determined from the solvent-accessible surface area, which can be estimated from water probe radius of 1.4 Å with temperature (T) and total solute entropy (S). The MM/GBSA binding free energy method in Amber14 was used to calculate the contribution of each residue to the total binding free energy.
2.6. Principal component analysis
Principal component analysis (PCA) is also referred to as essential dynamics of the protein.49 It is a multivariate statistical technique applied to motions in protein49 to systematically reduce the number of dimensions needed to describe the protein dynamics.49 This is usually done through the decomposition process that screen observed motions from largest to smallest spatial scale.49 Therefore, PCA can be used to describe the atomic displacement and conformational changes of proteins complex by extracting different modes of conformation of protein complex during dynamic simulations. PCA can also be used to describe the direction of motion (eigenvectors) and the extent of motion (eigenvalues) of the biological system.In this study, 300 ns of MD trajectories were stripped off their solvent molecules and the ions using the CPPTRAJ46 module in Amber14. This is a predetermined step before processing the MD trajectory for PCA. The principal component analysis was conducted on Cα atoms on 1000 snapshots at 100 ps time interval each. Using the in-house script, the first two principal components (PC1 and PC2) were computed and 2 × 2 covariance matrices were generated using Cartesian coordinates of Cα atoms. PC1 and PC2 correspond to the first two eigenvectors of a covariant matrix. The PC plots were constructed with Origin software.47
2.7. Dynamic cross-correlation matrices (DCCM)
The cross-correlation is a 3D matrix representation that graphically provides time-correlated information among the residues of the proteins.50 Visual pattern recognition is one of the commonest method used in the analyses of residue-based time-correlated data. To probe into dynamics of lyn-UM-164, lyn-Dasatinib and apo structures, a DCCM was generated to determine cross-correlated displacements of backbone Cα atoms in the trajectories, using the following equation| Cij = 〈ΔriΔrj〉/(〈Δri2〉〈Δrj2〉)1/2 | (5) |
where i and j represent ith and jth residues and Δri and Δrj correspond to the displacement of ith and jth atom from the mean, respectively. The coefficient of cross-correlation Cij, varies between the range −1 to +1, where the upper and lower limits correspond to strong correlated (+) and anti-correlated (−) motions within the period of simulations. The DCCM analysis was conducted using CPPTRAJ module in Amber14.46 The matrix generated were analysed using Origin software.47
3. Results and discussion
3.1. Docking studies and structural validation
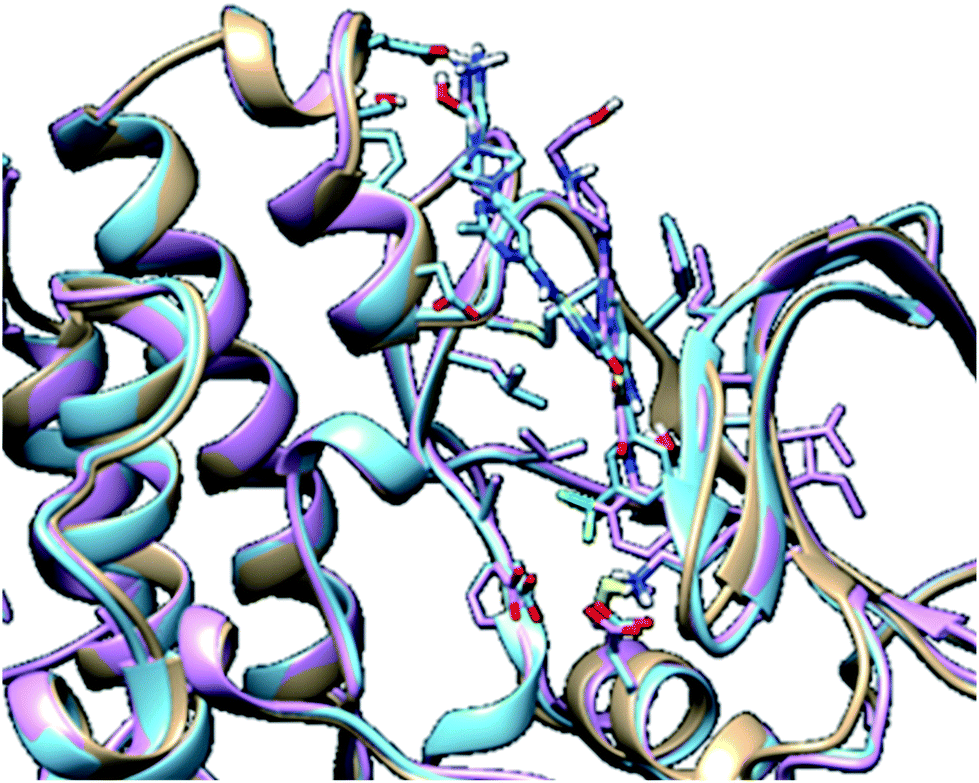
In molecular modelling, docking of a ligand to the protein active site is one of the commonly used methods to determine the preferred orientation of each of the entity that forms the complex (ligand and receptor) when bound together. However, results from docking may sometimes remain unreliable. This is true because even the best-docked conformation can shift from the active site of the protein within a short time interval of MD simulation. Therefore, we embarked on MD simulations to be sure that the docked complex remained in the active site of protein within a specific time scale. Fig. 3 shows the apo, docked complex of lyn-Dasatinib and docked complex of lyn-UM-164 superimposed on each other. This was done to validate the orientation of the docked ligand on the active site of the receptor.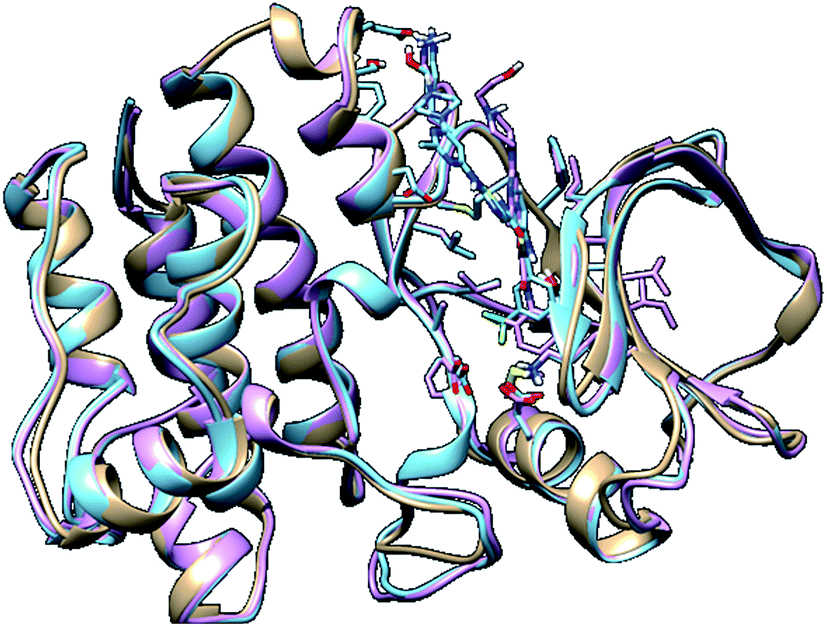 | ||
| Fig. 3 A 3D depiction of apo (light-brown), docked lyn-Dasatinib (light-blue) and lyn-UM-164 (light-pink) superimposed to validate the docking. | ||
It is important to note that, the docked complex was used to run 300 ns of molecular dynamics simulation. The result of docking shows that the UM-164 demonstrated higher binding affinity of −9.9 kcal mol−1 than Dasatinib with a binding affinity of −8.3 kcal mol−1. This relative high binding affinity exhibited by UM-164 can greatly contribute to the displacement of Dasatinib by UM-164 in the hierarchy of antineoplastic potency, particularly against the new target (lyn protein).
3.2. System stability MD simulations
To explore the dynamics of a well relaxed and equilibrated system, 300 ns of MD simulation of the three systems was carried out before the MD trajectory analysis. This is to allow an adequate investigation into the stability of 3D backbone atoms of the protein structure by assessing the various RMSD fluctuations relative to their initial structure during the period of simulation. The RMSD was calculated to assess the overall dynamics, stability and convergence of the various systems and the results are presented in Fig. 4.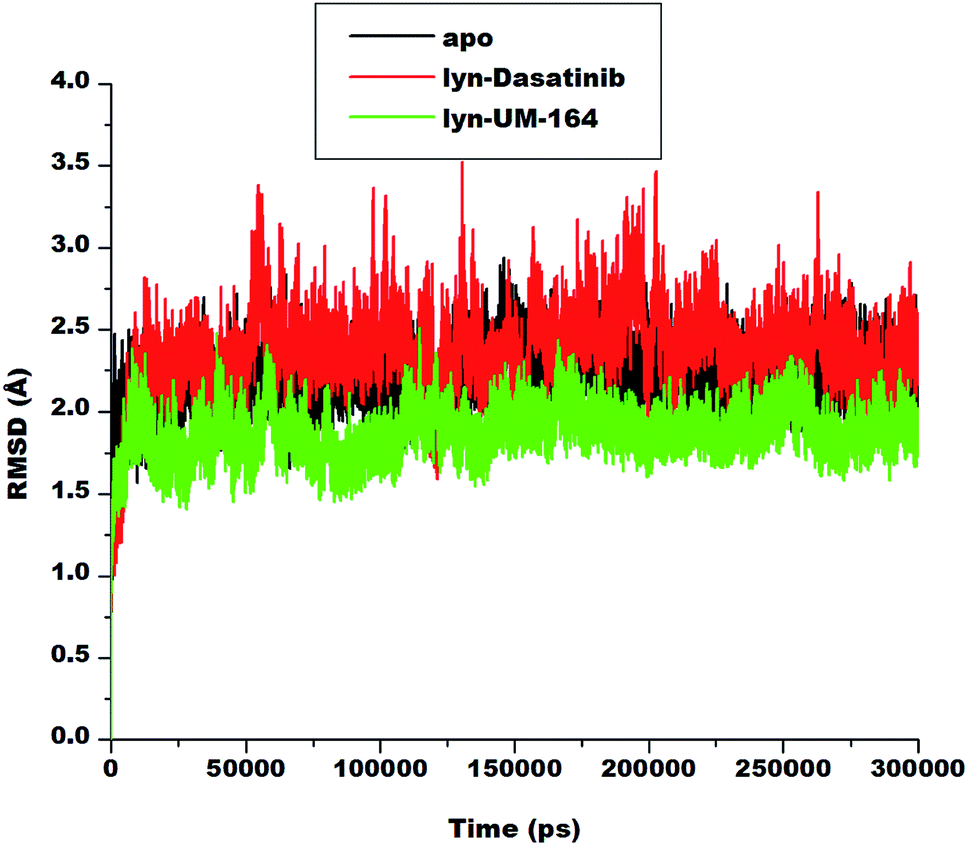 | ||
| Fig. 4 RMSD plot of Cα atoms of apo (black), lyn-Dasatinib (red) and lyn-UM-164 (green) systems, respectively. | ||
The RMSD of apo, lyn-Dasatinib and lyn-UM-164 protein structure graphically elucidate the convergence of the respective systems. Apo system is characterised with RMSD fluctuations with an initially low RMSD fluctuation of 2.5 Å within 0 and 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ps, this is followed by a relative increase in RMSD at intervals with highest peak RMSD of 2.7 Å at 75000 ps. These fluctuations reflect the changes associated with protein expansion within this period, thereby allowing the solvent molecules to infiltrate the hydrophobic sites. However, after approximately 150000 ps, the RMSD trajectories converged and the fluctuation rested below 2.50 Å.
000 ps, this is followed by a relative increase in RMSD at intervals with highest peak RMSD of 2.7 Å at 75000 ps. These fluctuations reflect the changes associated with protein expansion within this period, thereby allowing the solvent molecules to infiltrate the hydrophobic sites. However, after approximately 150000 ps, the RMSD trajectories converged and the fluctuation rested below 2.50 Å.
In lyn-Dasatinib system, the highest fluctuation observed was 3.5 Å at 125000 ps, after which the fluctuation rested below 2.5 Å at one point. Similarly, in lyn-UM-164 the peak RMSD of 2.5 Å was reached at about 50000 ps. The average RMSD of 1.89 Å and 2.19 Å was observed in apo and lyn-Dasatinib systems respectively, while in lyn-UM-164, an average RMSD of 1.79 Å was observed. These account for the system stability since the standard parameter for a stable system is RMSD of 2 Å and below.
3.3. Root mean square fluctuation (RMSF)
Amino acids aggregates to form a protein51 and play an important role in the dynamics and conformational features of the protein. Changes associated with protein conformation can occur during a chemical reaction or mechanical events.52 Consequently, ligand interaction with active site residue may induce conformational changes in the structure of the protein and alter its functions. These conformational changes are more pronounced during ligand binding as a result of ligand-induced motion mirroring the chemical activity within the residues and the ligand.53Knowledge of ligand-induced conformational changes in the protein structure is critical to the structure-based rational drug design. The RMSF determines the average atomic mobility of backbone atoms (N, Cα and C) during MD simulation.54 To understand the conformational changes and to explore the effect of ligand binding on protein dynamics, the RMSF of the studied systems was calculated from MD trajectories and presented in Fig. 5. The core of the protein appears to be more rigid relative to the loops (solvent-exposed) with very high fluctuations.
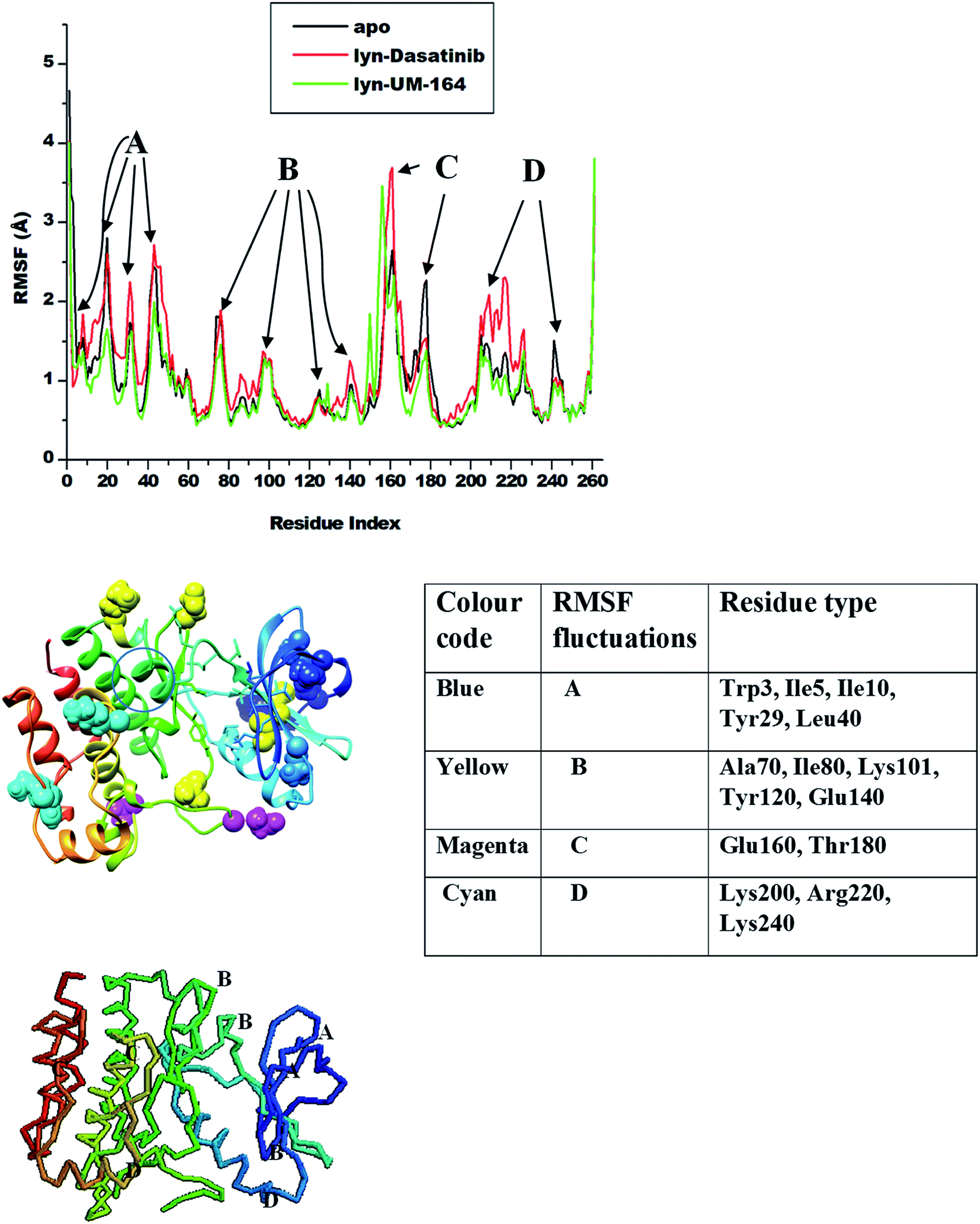 | ||
| Fig. 5 RMSF plot of Cα atoms of apo (black), lyn-Dasatinib (red) and lyn-UM-164 (green) systems, respectively. | ||
The highest fluctuations were observed in the loop region C involving residue Glu160 and Thr180 with RMSF values of 4.0 Å and 2.4 Å respectively. In loop region B, fluctuations are not extensive relative to other loops, this involves residues Ala70, Ile80, Lys101, Tyr120 and Glu140 with RMSF values of 1.8 Å, 1.2 Å, 0.8 Å, 1.1 Å and 1.0 Å in apo, lyn-Dasatinib and lyn-UM-164 systems, respectively. Loop region A involving residues Trp3, Ile5, Ile10, Tyr29 and Leu40 exhibits fluctuations with RMSF values of 1.8 Å, 2.7 Å, 2.4 Å, 2.7 Å and 1.1 Å. The RMSF values of 2.0 Å, 2.3 Å and 1.4 Å were observed in loop region D with residues Lys200, Arg220 and Lys240 in apo, lyn-Dasatinib and lyn-UM-164 systems.
These activation loops are important in the conversion of the inactive conformational state of tyrosine kinase to active conformation state. However, the lyn-Dasatinib complex used in this study is an active form of tyrosine kinase. This is because dasatinib is one of the first kinase inhibitor identified to be potent in active form of the protein.27 Similarly, inactive form of tyrosine kinase has low basal activity55 because its activation loop can interconvert with the phosphorylated active state only at a lower frequency.56 Based on this, an analogue of Dasatinib called UM-164 is well position to bind favourably to lyn protein.
The binding of UM-164 rigidified the residues thereby decreasing the tendency of loop fluctuations as seen in Fig. 5. This conforms with experimental evidence narrating that the catalytic activity of protein tyrosine kinase can be regulated by the phosphorylation state of the activation loop Tyr residue (Tyr120) and is correlated with movements in the loop and Cα. The region of the activation loop spanning this residue is highly flexible.27 Note that the binding of UM-164 to lyn protein causes maximum rigidity around this region Try120. This perhaps is due to the presence of fluorinated benzene ring that prevents the flexible loops from fluctuating, thereby stabilising the conformational state of the protein as seen in Fig. 6.
 | ||
| Fig. 6 Structure of lyn-UM-164 complex with a fluorinated benzene ring. | ||
This relative reduction in fluctuation decrease the tendency of phosphorylation that would otherwise increase the catalytic activity of lyn. Higher fluctuations are observed in the lyn-Dasatinib system meaning that UM-164 is more stable in the active site of lyn protein and may have better pharmacological activity as a chemotherapeutic agent targeting lyn protein.
3.4. Radius of gyration (RoG)
The radius of gyration is a moment of inertia of Cα atoms from their centre of mass. It is frequently used in molecular dynamics simulations to gain insight into the molecular stability of biological systems. The RoG of apo, lyn-Dasatinib and lyn-UM-164 was assessed and the result is presented in Fig. 7.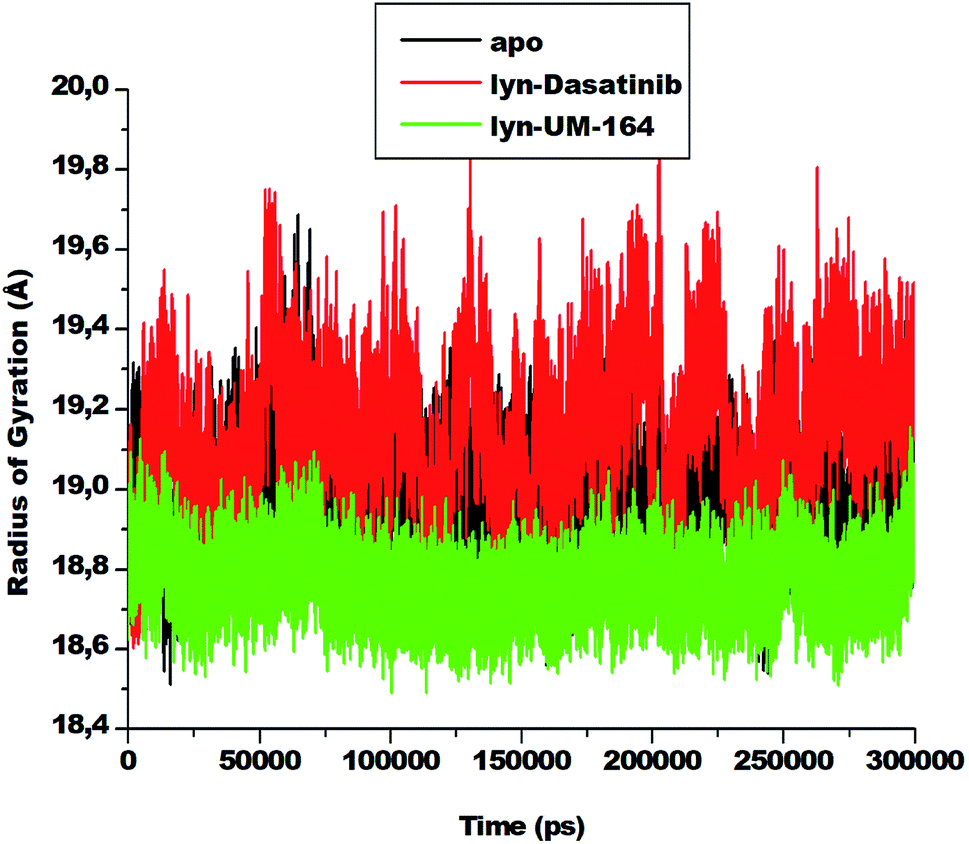 | ||
| Fig. 7 The radius of gyration plot of Cα atoms of apo (black), lyn-Dasatinib (red) and lyn-UM-164 (green) protein structures, respectively. | ||
There is a relatively similar level of compactness in apo and lyn-Dasatinib systems. However, lyn-UM-164 system exhibits a higher level of compactness and a lower average RoG of 18.839 Å compare to the first two systems. The apo has average RoG of 19.015 Å while the lyn-Dasatinib has a higher RoG of 19.178 Å. The relative high RoG in apo and lyn-Dasatinib systems agreed with the experimental evidence that state that the binding of Dasatinib and Imatinib to lyn causes only minor changes in overall structure from the unliganded state.27 The conformational stability achieved in lyn-UM-164 can be compared to the stability of lyn after binding of UM-164.
3.5. Principal component analysis (PCA)
Protein conformation plays a crucial role in the determination of its function,57 and PCA is one of the essential tools used in determining the flexibility of each atom during the simulation.30 Fig. 8 represent PCA plot of the three systems used in this study. For significant representation in this study, we adopt the clustering method of principal component (PC). This method can describe different conformational states sampled during a simulation by grouping molecular structures into a subset based on their conformational similarities.58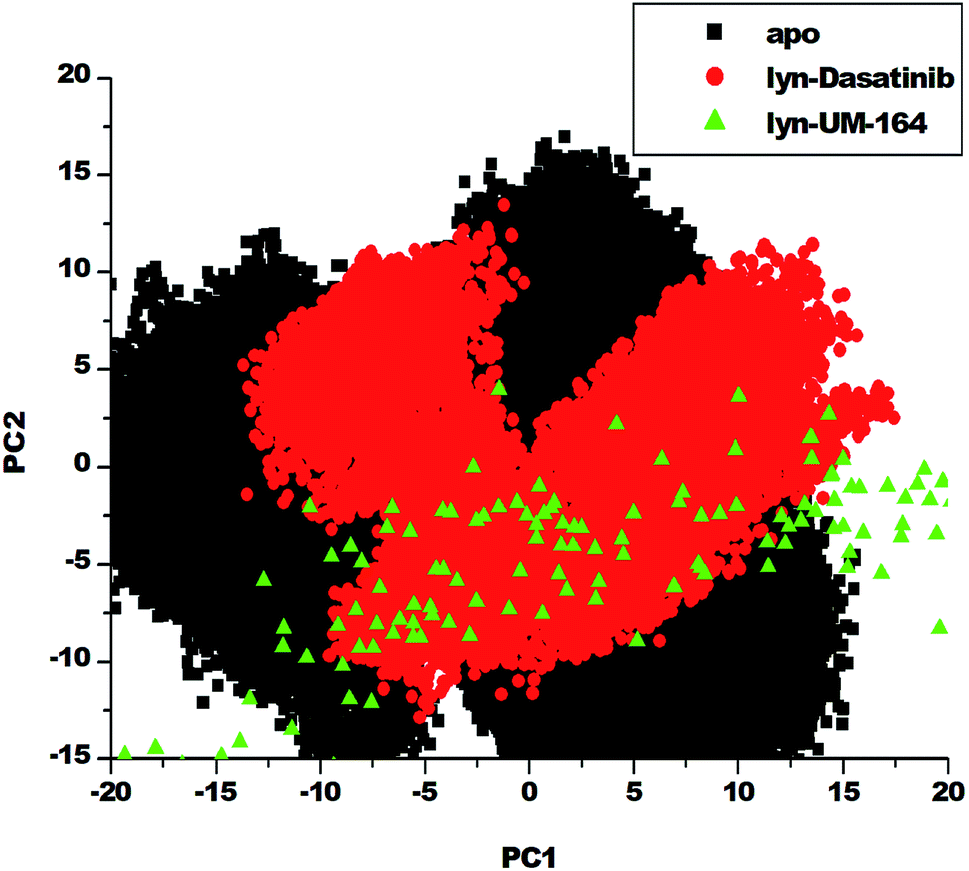 | ||
| Fig. 8 PCA projection of the motion of Cα atoms constructed by plotting the first two principal components (PC1 and PC2) in the conformational space with apo (black), lyn-Dasatinib (red) and lyn-UM-164 (green) colours, respectively. | ||
The eigenvalues can be used to provide adequate information on the percentage variability or the total mean square displacement of the atom's positional fluctuation captured in each dimension.30 This method of PCA was used to examine major conformational changes in apo, lyn-Dasatinib and lyn-UM-164 systems during a 300 ns simulations. For proper assessment of motions of systems with distinct molecular behaviour, the systems were projected along the direction of the first two principal components (PC1 vs. PC2) or eigenvectors. Fig. 8 depicts a scattered plot for apo, lyn-Dasatinib and lyn-UM-164 complexes. Notable from this plot is the significant difference in the three systems as evident from the features of the structures plotted along the direction of the two principal components.
There is a marked separation of motion from the three systems. However, a more correlated motion was observed in lyn-UM-164 along with two principal components PC1 and PC2 compared to lyn-Dasatinib and apo systems, which present relatively less correlated motions. The apo system appears to be more flexible than lyn-Dasatinib and lyn-UM-164 systems, suggesting that the binding of UM-164 to the protein (lyn) induces a conformational dynamics mirrored in PCs as a wave of motions. In contrast, the binding of Dasatinib to lyn protein impact a less dynamic effect on the protein compared to lyn-UM-164. A decrease in fluctuation observed in the lyn-UM-164 result from the impact of UM-164 binding on the lyn protein. This analysis agreed with experimental work of Williams et al. (2008) which observed that the binding of three distinct structural inhibitors of lyn including Dasatinib, caused only minor changes in overall structure from unliganded state.27
3.6. Dynamics cross-correlation matrices (DCCM) analysis
To assess conformational changes of lyn upon ligand binding, DCCM analysis was conducted on the position of Cα throughout the simulations to examine the dynamics and presence of correlated motions as seen in Fig. 9. Highly positive correlated motions of specific residues are represented as yellow-red (colour) regions, whereas anti-correlated motions (highly negative) of specific residues are represented as blue-black (colour) regions.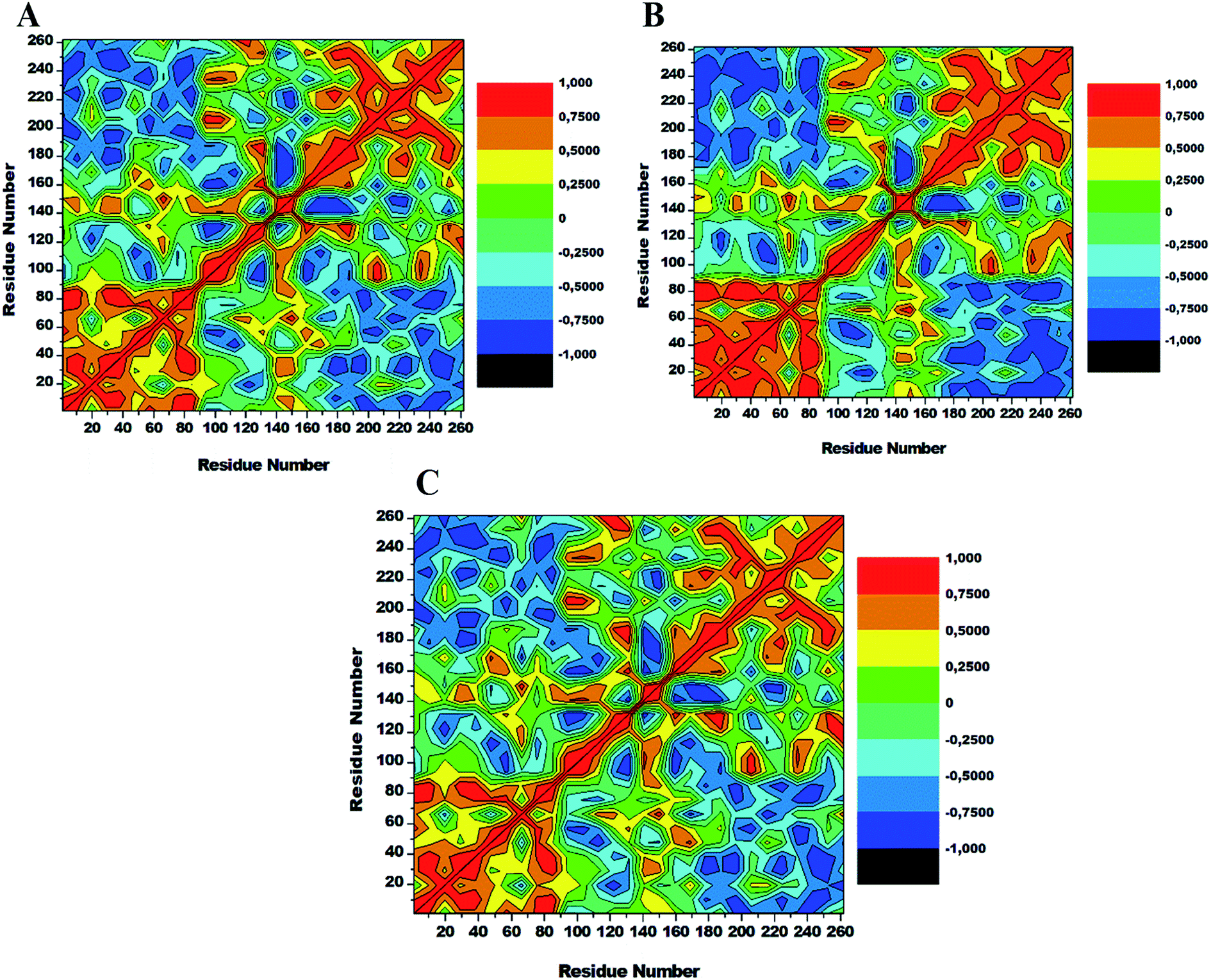 | ||
| Fig. 9 Cross-correlation matrices of the fluctuations of Cα atoms in apo (A), lyn-Dasatinib (B) and (C) lyn-UM-164. | ||
The three systems assessed in this study exhibited the overall correlated motions of residue relative to anti-correlated motions. Analysis of DCCM shows that the binding of UM-164 to lyn protein confers structural dynamics on the protein leading to conformational changes as reflected by the changes in the correlated motions. In the apo conformation (Fig. 9A) Phe50 correlates with leu60, similarly, Ile80 correlates with Ser89, while Glu140 slightly correlates with Leu 151. Anti-correlated motions of residue in apo occur between residues 170 to 260 compare to lyn-Dasatinib (Fig. 9B), and lyn-UM-164 systems with variant correlated and anti-correlated motions.
The lyn-UM-164 exhibit two notable correlated regions (Fig. 9C), this includes residues 1–90 the strongly correlated region, whereas residues 140–160 slightly correlated region. These regions take a host of most hydrophobic active site residues and it represents the most dynamic regions in the receptor. In a related development, the lyn-Dasatinib system relative to lyn-UM-164 system exhibits a decrease in correlation between residues 1–90 with a more pronounced anti-correlation motion between residues 170–261 (Fig. 9B). This phenomenal suggest that the binding of Dasatinib to lyn protein do not have much significant effect on the conformational state of the protein. Therefore, the potency of Dasatinib as lyn inhibitor may be a life dream compared to UM-164 that have demonstrated the characteristics of a highly potent agent on lyn protein.
3.7. Hydrogen bond formation between amino acid residues and the ligand
Hydrogen bond (H-bond) plays a central role in many chemical activities and are very important in biological systems, particularly in the maintenance of protein structural integrity.59 H-bond promotes protein–ligand interaction, catalysis and protein–ligand binding.59 Therefore, the formation of hydrogen bonds between amino acid residues is critical to the determination of conformational stability of amino acids. In line with this, we investigate the formation of hydrogen bonds during the simulation and represent in Fig. 10.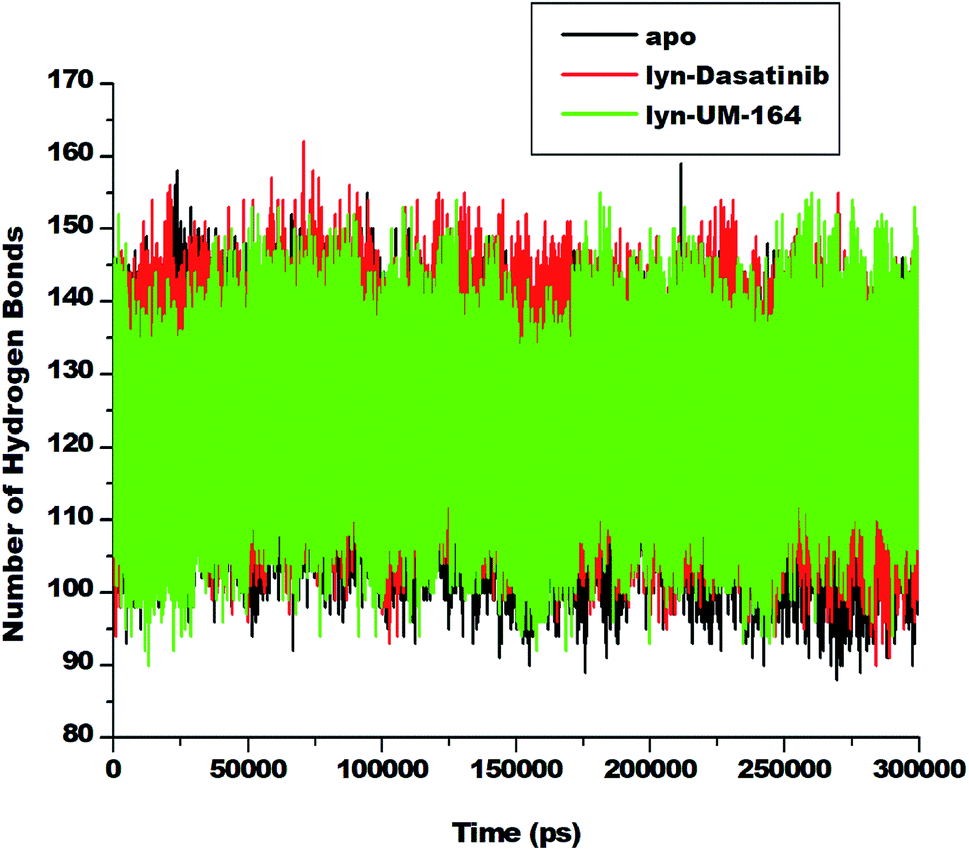 | ||
| Fig. 10 Number of hydrogen bond formation during simulation overtime between apo, lyn-Dasatinib and lyn-UM-164 systems. | ||
The apo system exhibits a lower average H-bond (120.39) formation during simulation compared to lyn-Dasatinib and lyn-UM-164 systems with average H-bond of 122.31 and 123.73, respectively. The relative decrease in H-bond formation observed in the lyn-Dasatinib system could be as a result of loop flexibility that ultimately affects the strength of drug binding. The decrease in H-bond formation leads to structural destabilization and conformational dynamics which consequently affects drug binding.59
To further assess the relative stability of lyn-UM-164, lyn-Dasatinib and apo, we monitored the H-bond distance and occupancy of the three respective systems throughout the simulation time and the results are presented in Table 1.
| H-bond acceptor | H-bond donor | Frame number | Occupancy (%) | Average distance (Å) | Average angle (°) |
|---|---|---|---|---|---|
| lyn-Dasatinib | |||||
| LIG_262@O2 | ASP_148@H | 94357 |
2.2% | 2.81 | 153 |
| LIG_262@O1 | PHE_21@H | 29260 |
7.1% | 2.82 | 156 |
| LIG_262@N5 | ARG_132@HH22 | 12040 |
2.9% | 2.91 | 152 |
| LIG_262@N6 | ARG132@HH22 | 9837 | 2.3% | 2.92 | 148 |
| ILE_146@O | VAL_66@H | 236457 |
57% | 2.88 | 162 |
| ALA_36@O | THR_82@HG1 | 27618 |
6.7% | 2.80 | 156 |
| LYS_154@O | LEU_137@H | 341732 |
82% | 2.85 | 162 |
| MET_85@O | GLY_88@H | 76244 |
18% | 2.90 | 154 |
| LEU_90@O | PHE_93@H | 905 | 2.2% | 2.93 | 141 |
| ASP_92@O | SER_96@HG | 364 | 0.9% | 2.78 | 158 |
| TYR_120@H | LYS_24@H | 47 | 0.1% | 2.88 | 149 |
|
|||||
| lyn-UM-164 | |||||
| LIG_262@O1 | PHE_21@H | 29260 |
7.2% | 2.88 | 156 |
| LIG_262@N5 | ARG_132@HH22 | 12040 |
2.9% | 2.91 | 154 |
| LIG_262@N6 | ARG_132@HH22 | 9837 | 2.3% | 2.92 | 148 |
| LIG_262@O2 | GLY_150@H | 7136 | 1.7% | 2.90 | 142 |
| LIG_262@F1 | SER_96@HG | 252 | 0.6% | 2.84 | 152 |
| LIG_262@F2 | SER_96@HG | 261 | 0.6% | 2.85 | 152 |
| LIG_262@F3 | SER_96@HG | 249 | 0.6% | 2.86 | 151 |
| ILE_146@O | VAL_66@H | 236457 |
57% | 2.88 | 166 |
| ALA_36@O | THR_82@HG1 | 27618 |
6.8% | 2.80 | 156 |
| LYS_154@O | LEU_137@H | 341732 |
82% | 2.82 | 162 |
| MET_85@O | GLY_88@H | 76244 |
18% | 2.90 | 154 |
| TYR_120@O | LYS_24@H | 216500 |
52% | 2.87 | 155 |
In the lyn-UM-164 system, Lys154 exhibits high H-bond occupancy of 88% with an average H-bond distance of 2.82 Å. Similarly, lyn-Dasatinib exhibits the same H-bond occupancy but with slightly different average H-bond distance of 2.85 Å. This small difference in H-bond distance may be responsible for high bond strength in lyn-UM-164 complex compared to lyn-Dasatinib. Within the same systems, a maximum H-bond occupancy of 52% was observed between O atom of Tyr120 and H atom of Lys24 of lyn-UM-164, yet lyn-Dasatinib display a variable H-bond occupancy 0.1% and H-bond distance of 2.88 Å between the atoms of the same residues.
The high value of H-bond occupancy with relative low H-bond distance exhibited by Tyr120 in lyn-UM-164 signifies the importance Tyr120 to the conformational state of the complex. The most obvious difference observed between the two systems with respect to their H-bond angle, H-bond distance and H-bond occupancy is the relationship between fluoride atoms (F1, F2 and F3) of UM-164 and H atom of Ser96 which is completely absent in lyn-Dasatinib. This perhaps may be responsible for the magnificent behaviour of the UM-164 on lyn protein. Generally, the lyn-UM-164 system exhibits the highest H-bond occupancy compared lyn-Dasatinib system.
3.8. Binding free energy and energy decomposition analysis
The Molecular Mechanics/Generalized-Born Surface Area (MM/GBSA) method48 is the most acceptable technique used to estimate the binding free energy of small ligands to biological macromolecules.48 This method was used to estimate the total binding free energy of lyn-Dasatinib and lyn-UM-164 systems as presented in Table 2. Energy decomposition analysis shows that the lyn-UM-164 system has higher estimated binding free energy (−54.867 ± 5.961 kcal mol−1) compared to the lyn-Dasatinib system with an estimated binding free energy of −49.692 ± 3.885 kcal mol−1. The difference in the binding free energy between the lyn-Dasatinib and lyn-UM-164 is −5.175 ± 2.076 kcal mol−1, which is quite significant.| ΔGbind (kcal mol−1) | ΔEelec (kcal mol−1) | ΔEvdW (kcal mol−1) | ΔGgas (kcal mol−1) | ΔGsol (kcal mol−1) |
|---|---|---|---|---|
| a ΔEele = electrostatic energy; ΔEvdW = van der Waals energy; ΔGbind = calculated total binding free energy; ΔGsol = solvation free energy. | ||||
| lyn-Dasatinib | ||||
| −49.692 ± 3.885 | −36.605 ± 10.822 | −52.031 ± 4.074 | −88.636 ± 11.294 | 39.006 ± 9.403 |
|
||||
| lyn-UM-164 | ||||
| −54.867 ± 5.961 | −32.387 ± 11.763 | −71.319 ± 4.209 | −103.704 ± 12.191 | 48.837 ± 10.216 |
The force of interaction such as electrostatic (−32.387 ± 11.763 kcal mol−1) and van der Waals (−71.319 ± 4.209 kcal mol−1) greatly contributes to the total binding energy of UM-164 to lyn protein compared to lyn-Dasatinib. Hydrophobic packing contributes immensely to the binding free energy in lyn-UM-164 owing to a large number of aromatic and hydrophobic rings within the conformational space and also a set of hydrophobic residue around the binding pocket. However, a decrease in energy contribution from interacting forces particularly (ΔEvdW) in lyn-Dasatinib may result in a sharp decrease in potency of Dasatinib in lyn protein. This is probably because the binding of Dasatinib to lyn protein only cause minor changes to the conformal state of the protein.27
| Residues | ΔEvdW | ΔEele | ΔGpolar | ΔGnon-polar | ΔGbinding |
|---|---|---|---|---|---|
| a All energies are in kcal mol−1. ΔEele = electrostatic energy; ΔEvdW = van der Waals energy; ΔGpolar = polar solvation energy; ΔGnonpolar = nonpolar solvation energy; ΔGbinding = total binding free energy. | |||||
| lyn-Dasatinib | |||||
| Met85 | −0.281 ± 0.163 | −0.449 ± 0.515 | 0.590 ± 0.528 | −0.021 ± 0.020 | −0.161 ± 0.161 |
| Thr82 | −0.835 ± 0.563 | −2.902 ± 1.543 | 2.124 ± 0.640 | −0.126 ± 0.017 | −1.739 ± 1.116 |
| Phe84 | −0.189 ± 0.110 | 0.345 ± 0.193 | −0.302 ± 0.208 | −0.003 ± 0.006 | −0.150 ± 0.104 |
| Gly88 | −0.369 ± 0.120 | 0.662 ± 0.248 | −0.243 ± 0.248 | −0.087 ± 0.026 | −0.037 ± 0.201 |
| Glu98 | −0.002 ± 0.001 | −7.963 ± 0.520 | 7.965 ± 0.521 | 0.000 ± 0.000 | 0.001 ± 0.000 |
| Lys87 | −0.057 ± 0.013 | 9.822 ± 0.236 | −9.693 ± 0.221 | 0.000 ± 0.000 | 0.072 ± 0.022 |
| Tyr120 | −0.003 ± 0.001 | 0.125 ± 0.083 | −0.124 ± 0.082 | 0.000 ± 0.000 | −0.001 ± 0.003 |
| Phe93 | −0.018 ± 0.006 | 0.355 ± 0.039 | −0.350 ± 0.038 | 0.000 ± 0.000 | −0.013 ± 0.009 |
| Ser89 | −0.474 ± 0.218 | 0.964 ± 0.217 | −1.007 ± 0.205 | −0.071 ± 0.040 | −0.587 ± 0.224 |
| Asp92 | −0.168 ± 0.119 | −12.218 ± 1.14 | 12.765 ± 1.434 | −0.042 ± 0.038 | 0.338 ± 0.254 |
|
|||||
| lyn-UM-164 | |||||
| Met85 | −1.575 ± 0.305 | −2.662 ± 0.975 | 1.930 ± 0.589 | −0.090 ± 0.18 | −2.397 ± 0.823 |
| Thr82 | −0.878 ± 0.145 | −0.197 ± 0.153 | 0.495 ± 0.153 | −0.0678 ± 0.020 | −0.248 ± 0.139 |
| Gly88 | −1.541 ± 0.293 | −1.102 ± 0.606 | 1.035 ± 0.230 | −0.189 ± 0.029 | −1.796 ± 0.606 |
| Glu98 | −0.035 ± 0.025 | −5.254 ± 0.541 | 5.394 ± 0.560 | −0.003 ± 0.011 | 0.058 ± 0.069 |
| Lys87 | −1.631 ± 0.530 | 7.547 ± 0.653 | −6.685 ± 0.876 | −0.168 ± 0.069 | −0.936 ± 0.365 |
| Tyr120 | −0.002 ± 0.000 | −0.004 ± 0.047 | 0.010 ± 0.046 | 0.000 ± 0.000 | 0.004 ± 0.003 |
| Phe93 | −0.142 ± 0.082 | −0.146 ± 0.039 | −0.133 ± 0.037 | −0.016 ± 0.021 | −0.145 ± 0.102 |
| Ser89 | −0.891 ± 0.249 | −0.176 ± 0.408 | 0.294 ± 0.360 | −0.145 ± 0.032 | −0.918 ± 0.271 |
| Phe84 | −1.241 ± 0.351 | 0.140 ± 0.250 | 0.142 ± 0.271 | −0.104 ± 0.024 | −1.063 ± 0.336 |
| Asp92 | −0.206 ± 0.067 | −6.526 ± 0.491 | 7.015 ± 0.466 | −0.104 ± 0.024 | 0.255 ± 0.099 |
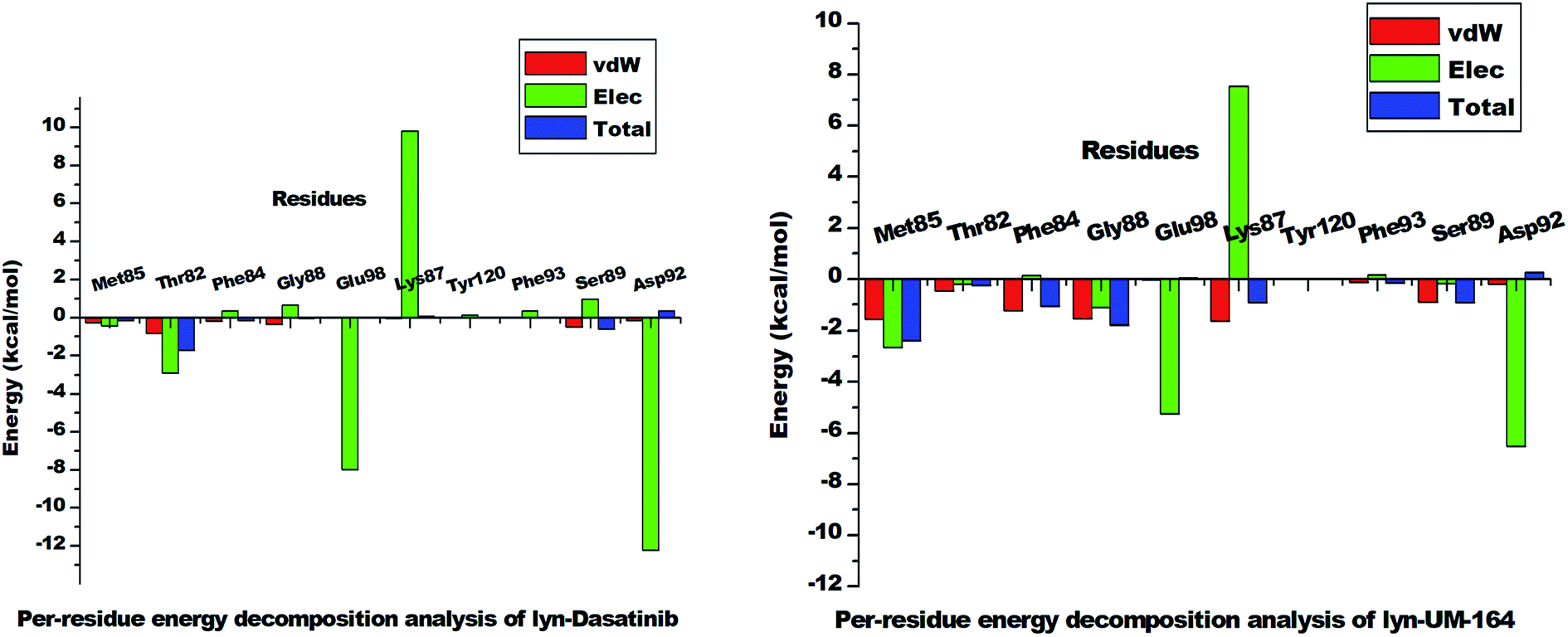 | ||
| Fig. 11 The per-residue energy decomposition analysis graph of lyn-Dasatinib and lyn-UM-164 systems. | ||
However, Met85 in the lyn-UM-164 system has the highest binding free energy contribution (−2.397 ± 0.823 kcal mol−1) relative to other active site residues in lyn-Dasatinib and lyn-UM-164 systems, respectively. Therefore, Met85 can be regarded as the stabilising factor in the binding of UM-164 to lyn protein. In a related development, there is a substantial reduction in per-residue energy contribution in the lyn-Dasatinib system except for Thr82 which has a higher total binding free energy of −1.739 ± 1.116 kcal mol−1 compared to a lower energy value (−0.248 ± 0.139 kcal mol−1) exhibited by Thr82 in the lyn-UM-164 system.
Asp92 in lyn-Dasatinib system exhibit the highest electrostatic energy contribution −12.218 ± 1.14 kcal mol−1 compared to its contribution in lyn-UM-164, which is relatively lower (−6.526 ± 0.491 kcal mol−1). The van der Waals energy contribution of Asp92 in lyn-UM-164 is relatively higher −0.206 ± 0.067 kcal mol−1 compared to −0.168 ± 0.119 kcal mol−1 van der Waals exhibited by Asp92 in the lyn-Dasatinib system. This may be attributed to the orientation of UM-164 in the conformational space of the protein. The observed decrease in total binding free energy contribution in the lyn-Dasatinib system is a mirror image of the reduction in potency of Dasatinib as an inhibitor of lyn protein. Within the context of the total binding free energy contribution in the lyn-UM-164 system, UM-164 can be rated as a better potential inhibitor of lyn protein.
3.9. Ligand–residue interaction network profile
The interaction of active site residues with the ligand is an important element used in assessing the orientation of the ligand in the protein. Herein, interaction of active site residues of lyn protein with Dasatinib and UM-164 was examined to assess the level of ligand–residue interaction (Fig. 12). It was observed that orientation of UM-164 formed direct hydrogen bonds with Glu98, Met85, Phe21 and Gln20 in lyn protein. Similarly, the orientation of Dasatinib also formed direct hydrogen bonds with Met85, Thr82 and lys87. However, Dasatinib assumed three hydrogen bond formation relative to four hydrogen bonds formed by UM-164 and lyn residues. The loss of hydrogen bond in Gln20, Phe21 and Glu98 of lyn-Dasatinib directly affects the orientation of Dasatinib in the active site consequently affects the drug binding. Fig. 13 shows the conformational state of apo, lyn-Dasatinib and lyn-UM-164 superimposed on each other. The alignment shows the orientation of Dasatinib and UM-164 in lyn protein conformational space.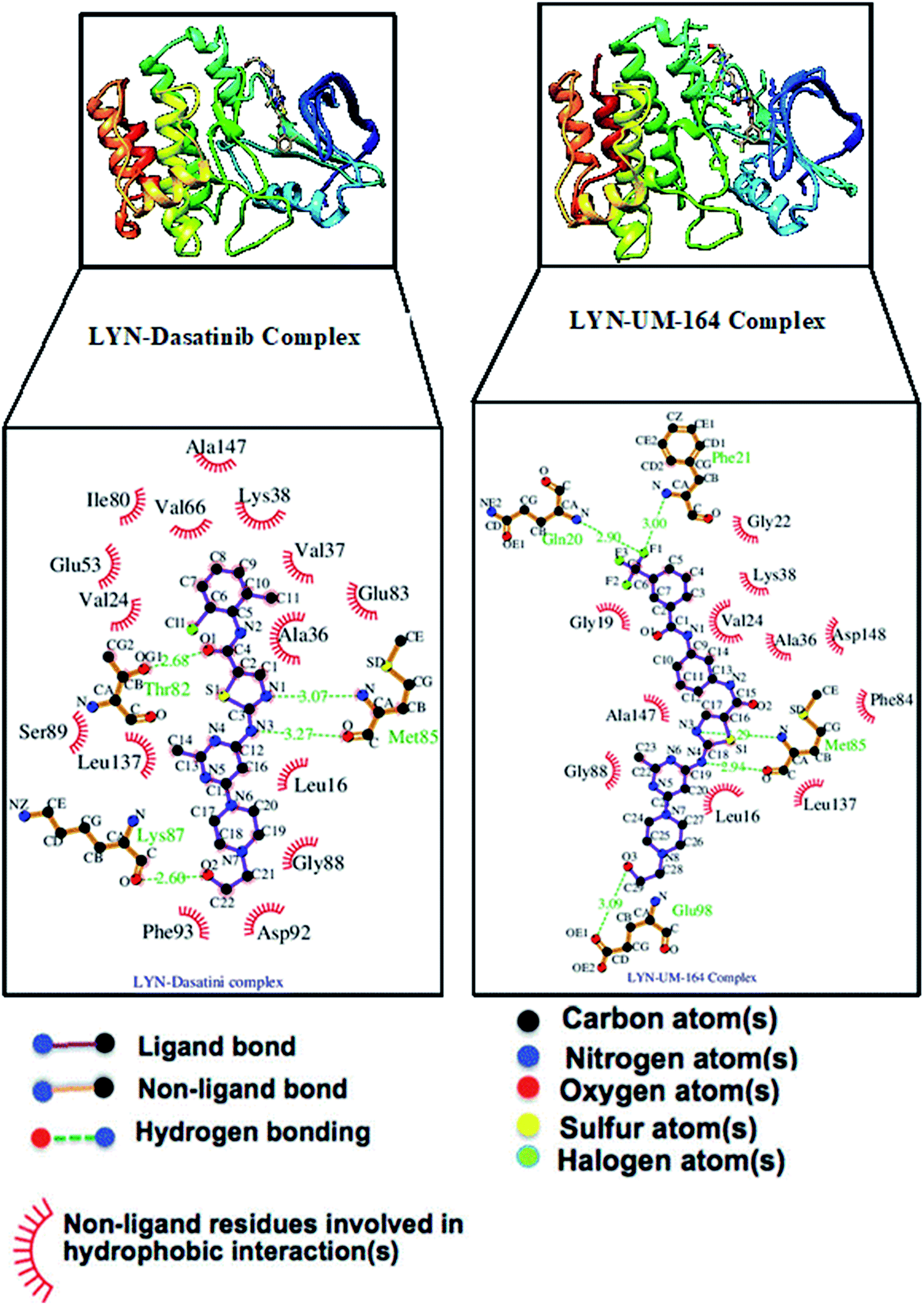 | ||
| Fig. 12 The interacting residues of lyn complexed with Dasatinib and UM-164. | ||
 | ||
| Fig. 13 3D structures of apo (light-brown), lyn-Dasatinib (light-blue) and lyn-UM-164 (light-pink) superimposed onto each other. | ||
The loss of α-amino acid (Phe21) in the lyn-Dasatinib system could sterically serve as an impediment in the binding of Dasatinib to lyn protein. This residue forms an essential hydrogen bond important for binding of UM-164 to the protein as depicted in Fig. 12.
Fig. 14 shows Dasatinib and UM-164 oriented within the active site of the lyn protein. The orientation of UM-164 in lyn protein permits for hydrogen bond formation between N atoms of Gln20 and Phe21 of lyn protein and fluoride atom (F1) of UM-164. However, the characteristics orientation of Dasatinib in lyn protein impedes the formation of these hydrogen bonds, the loss of these bonds greatly contributes to the reduction in potency of Dasatinib relative to UM-164. In addition, the Arginine group (Arg132) of residue that forms a hydrogen bond with UM-164 is oriented at an angle of 148° (Table 1). Despite the existence of the interaction with same protein (lyn) the Arginine group of residue (Arg132) that forms a hydrogen bond with Dasatinib is oriented at an angle of 152° (Table 1). This remarkable differences between UM-164 and Dasatinib can result to steric effect that may affect drug binding. Most importantly, the addition of fluorinated benzenes ring in UM-164 created a niche for the molecule as a potent inhibitor of tyrosine kinase of all types.
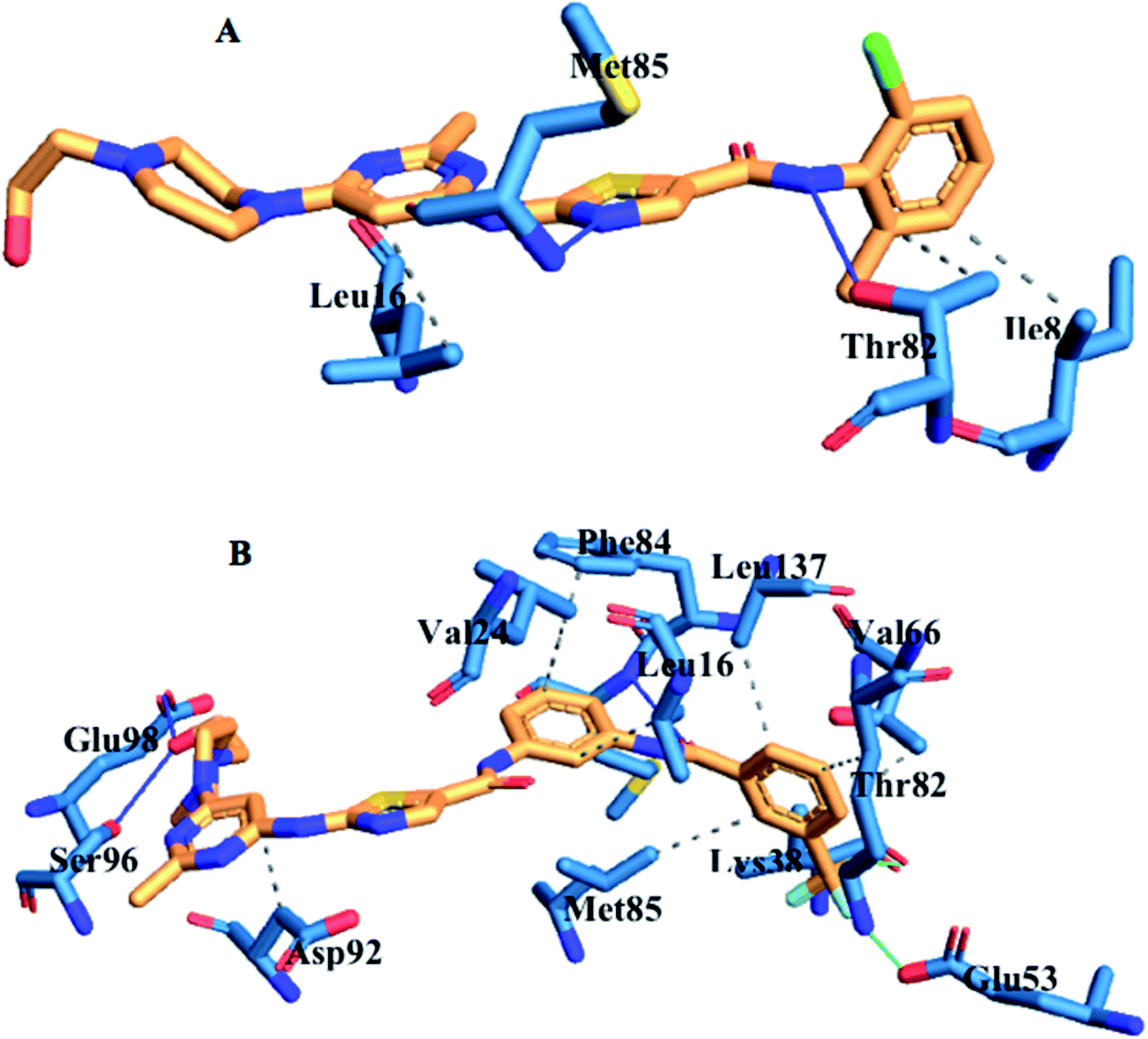 | ||
| Fig. 14 Ligand residue interaction network of [A] Dasatinib and [B] UM-164 complexed with lyn protein showing ligands orientation in the active site of the protein. | ||
4. Conclusion
Currently, molecular understanding of the impact of experimental drug UM-164 on lyn protein is lacking in literature. In this study, we adopted different computational approaches aimed at providing multidimensional insight into the effect of UM-164 binding on the conformational space of lyn protein and to establish the ligand binding landscape of UM-164 on lyn protein. Molecular docking and molecular dynamics simulations were performed. Post-MD analyses such as principal component analysis (PCA), dynamics cross-correlation matrices (DCCM) analysis, hydrogen bond occupancy, thermodynamics calculation and ligand–residue interaction guide us to the findings that provide adequate information on the impact of UM-164 binding with lyn protein.Outcome from the docking analysis revealed that UM-164 exhibited a higher binding affinity of −9.9 kcal mol−1 compared to Dasatinib with a binding affinity of −8.3 kcal mol−1. The MD simulations study enabled us to provide a better picture of the binding landscape of UM-164 to the lyn protein. Some of the crucial impacts of this binding leading to favourable inhibition of this protein include: decrease in the capacity of the loop to fluctuate; influence the ligand optimum orientation on the conformational space of lyn protein; increase the hydrogen bond formation in lyn-UM-164 complex.
In addition, an increase in drug binding energy (−5.175 ± 2.076 kcal mol−1) of UM-164 was noted in comparison to Dasatinib. It is quite fascinating to note that residue Met85 is an important stabilising factor in the binding of UM-164 to lyn protein. These findings can provide important insights that will serve as a baseline in the design of novel lyn inhibitors and stimulate further research into multidimensional approaches required to curb the influence of lyn protein in triple-negative breast cancer.
Conflicts of interest
The authors declare no financial and intellectual conflicts of interest.Acknowledgements
The authors acknowledge the Centre for High Performance Computing (CHPC, www.chpc.ac.za), Cape Town, South Africa, for computational resources and also appreciate Dr MM Lawal for her assistance with proof-reading.References
- G. N. Sharma, R. Dave, J. Sanadya, P. Sharma and K. K. Sharma, Various types and management of breast cancer: an overview, J. Adv. Pharm. Technol. Res., 2010, 1, 109–126 Search PubMed.
- F. Bray, J. Ferlay, and I. Soerjomataram, Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, 2018, pp. 394–424 Search PubMed.
- R. A. Gilani, S. Phadke, L. W. Bao, E. J. Lachacz, M. L. Dziubinski, K. R. Brandvold, M. E. Steffey, F. E. Kwarcinski, C. R. Graveel, M. Kidwell, S. D. Merajver and M. B. Soellner, Clin. Cancer Res., 2016, 22(20), 5087–5096 CrossRef CAS PubMed.
- V. G. Abramson, B. D. Lehmann, T. J. Ballinger and J. A. Pietenpol, Subtyping of triple-negative breast cancer: implications for therapy, Cancer, 2015, 121, 8–16 CrossRef PubMed.
- A.-A. Jitariu, A. M. Cîmpean, D. Ribatti and M. Raica, Triple negative breast cancer: the kiss of death, Oncotarget, 2017, 8, 46652–46662 CrossRef PubMed.
- Q. Jiao, A. Wu, G. Shao, H. Peng, M. Wang, S. Ji, P. Liu and J. Zhang, The latest progress in research on triple negative breast cancer (TNBC): risk factors, possible therapeutic targets and prognostic markers, J. Thorac. Dis., 2014, 6, 1329–1335 Search PubMed.
- U. Ndagi, N. N. Mhlongo and M. E. Soliman, The impact of Thr91 mutation on c-Src resistance to UM-164: molecular dynamics study revealed a new opportunity for drug design, Mol. BioSyst., 2017, 13, 1157–1171 RSC.
- B. P. Schneider, E. P. Winer, W. D. Foulkes, J. Garber, C. M. Perou, A. Richardson, G. W. Sledge and L. A. Carey, Triple-Negative Breast Cancer: Risk Factors to Potential Targets, Clin. Cancer Res., 2008, 14, 8010–8018 CrossRef CAS PubMed.
- E. Lee, R. McKean-Cowdin, H. Ma, D. V. Spicer, D. Van Den Berg, L. Bernstein and G. Ursin, Characteristics of Triple-Negative Breast Cancer in Patients With a BRCA1 Mutation: Results From a Population-Based Study of Young Women, J. Clin. Oncol., 2011, 29, 4373–4380 CrossRef PubMed.
- E. H. Lips, L. Mulder, A. Oonk, L. E. van der Kolk, F. B. L. Hogervorst, A. L. T. Imholz, J. Wesseling, S. Rodenhuis and P. M. Nederlof, Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers, Br. J. Cancer, 2013, 108, 2172–2177 CrossRef CAS PubMed.
- E. A. O'Reilly, L. Gubbins, S. Sharma, R. Tully, M. H. Z. Guang, K. Weiner-Gorzel, J. McCaffrey, M. Harrison, F. Furlong, M. Kell and A. McCann, The fate of chemoresistance in triple negative breast cancer (TNBC), BBA Clinical, 2015, 3, 257–275 CrossRef PubMed.
- J. L. Dillon, S. M. Mockus, G. Ananda, V. Spotlow, W. A. Wells, G. J. Tsongalis and J. D. Marotti, Somatic gene mutation analysis of triple negative breast cancers, Breast, 2016, 29, 202–207 CrossRef CAS PubMed.
- D. Meseure, S. Vacher, K. Drak Alsibai, M. Trassard, A. Nicolas, R. Leclere, F. Lerebours, J. M. Guinebretiere, E. Marangoni, R. Lidereau and I. Bieche, Biopathological Significance of TLR9 Expression in Cancer Cells and Tumor Microenvironment Across Invasive Breast Carcinomas Subtypes, Cancer Microenviron., 2016, 9, 107–118 CrossRef CAS PubMed.
- S. M. Davion, K. P. Siziopikou and M. E. Sullivan, Cytokeratin 7: a re-evaluation of the ‘tried and true’ in triple-negative breast cancers, Histopathology, 2012, 61, 660–666 CrossRef PubMed.
- L. Huo, Y. Gong, M. Guo, M. Z. Gilcrease, Y. Wu, H. Zhang, J. Zhang, E. Resetkova, K. K. Hunt and M. T. Deavers, GATA-binding protein 3 enhances the utility of gross cystic disease fluid protein-15 and mammaglobin A in triple-negative breast cancer by immunohistochemistry, Histopathology, 2015, 67, 245–254 CrossRef PubMed.
- P. Karihtala, S. Kauppila, Y. Soini and A. Jukkola-Vuorinen, Oxidative stress and counteracting mechanisms in hormone receptor positive, triple-negative and basal-like breast carcinomas, BMC Cancer, 2011, 11, 262 CrossRef PubMed.
- V. Ossovskaya, Y. Wang, A. Budoff, Q. Xu, A. Lituev, O. Potapova, G. Vansant, J. Monforte and N. Daraselia, Exploring molecular pathways of triple-negative breast cancer, Genes Cancer, 2011, 2, 870–879 CrossRef CAS PubMed.
- M. Bonzanini, L. Morelli, E. M. Bonandini, E. Leonardi, R. Pertile and P. Dalla Palma, Cytologic features of triple-negative breast carcinoma, Cancer Cytopathol., 2012, 120, 401–409 CrossRef PubMed.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS PubMed.
- A. Shindikar, A. Singh, M. Nobre and S. Kirolikar, Curcumin and Resveratrol as Promising Natural Remedies with Nanomedicine Approach for the Effective Treatment of Triple Negative Breast Cancer, J. Oncol., 2016, 2016, 1–13 CrossRef PubMed.
- G. Jerusalem, J. Collignon, H. Schroeder and L. Lousberg, Triple-negative breast cancer: treatment challenges and solutions, Breast Cancer: Targets Ther., 2016, 8, 93 Search PubMed.
- M. Mirzania, Approach to the Triple Negative Breast Cancer in New Drugs Area, Int. J. Hematol. Oncol. Stem Cell Res., 2016, 10, 115–119 Search PubMed.
- U. Ndagi, N. N. Mhlongo and M. E. Soliman, Emergence of a Promising Lead Compound in the Treatment of Triple Negative Breast Cancer: An Insight into Conformational Features and Ligand Binding Landscape of c-Src Protein with UM-164, Appl. Biochem. Biotechnol., 2018, 185, 655–675 CrossRef CAS PubMed.
- M. Getlik, C. Grütter, J. R. Simard, S. Klüter, M. Rabiller, H. B. Rode, A. Robubi and D. Rauh, Hybrid Compound Design To Overcome the Gatekeeper T338M Mutation in cSrc#, J. Med. Chem., 2009, 52, 3915–3926 CrossRef CAS PubMed.
- G. Tornillo, C. Knowlson, H. Kendrick, J. Cooke, H. Mirza, I. Aurrekoetxea-Rodríguez, M. d. M. Vivanco, N. E. Buckley, A. Grigoriadis and M. J. Smalley, Dual Mechanisms of LYN Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells, Cell Rep., 2018, 25, 3674–3692.e10 CrossRef CAS PubMed.
- C. Liedtke, C. Mazouni, K. R. Hess, F. André, A. Tordai, J. A. Mejia, W. F. Symmans, A. M. Gonzalez-Angulo, B. Hennessy, M. Green, M. Cristofanilli, G. N. Hortobagyi and L. Pusztai, Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer, J. Clin. Oncol., 2008, 26, 1275–1281 CrossRef PubMed.
- N. K. Williams, I. S. Lucet, S. P. Klinken, E. Ingley and J. Rossjohn, Crystal structures of the Lyn protein tyrosine kinase domain in its Apo- and inhibitor-bound state, J. Biol. Chem., 2009, 284, 284–291 CrossRef CAS PubMed.
- R. A. Gilani, S. Phadke, L. W. Bao, E. J. Lachacz, M. L. Dziubinski, K. R. Brandvold, M. E. Steffey, F. E. Kwarcinski, C. R. Graveel, K. M. Kidwell, S. D. Merajver and M. B. Soellner, UM-164: A Potent c-Src/p38 Kinase Inhibitor with In Vivo Activity against Triple-Negative Breast Cancer, Clin. Cancer Res., 2016, 22, 5087–5096 CrossRef CAS PubMed.
- P. Boyle, Triple-negative breast cancer: epidemiological considerations and recommendations, Ann. Oncol., 2012, 23, vi7–vi12 CrossRef PubMed.
- C. C. David, and D. J. Jacobs, Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins, in Methods in molecular biology, ed. N. J. Clifton, 2014, pp. 193–226 Search PubMed.
- G. G. Maisuradze, A. Liwo and H. A. Scheraga, Principal Component Analysis for Protein Folding Dynamics, J. Mol. Biol., 2009, 385, 312–329 CrossRef CAS PubMed.
- W. Yu and A. D. MacKerell, Computer-Aided Drug Design Methods, Methods Mol. Biol., 2017, 1520, 85–106 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera? A visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455–461 CAS.
- R. R. Mittal, L. Harris, R. A. McKinnon and M. J. Sorich, Partial Charge Calculation Method Affects CoMFA QSAR Prediction Accuracy, J. Chem. Inf. Model., 2009, 49, 704–709 CrossRef CAS PubMed.
- H++ (Web-based computational prediction of protonation states and pK of ionizable groups in macromolecules), DOI:10.1021/jz501780a.
- Gaussian, 2009, available from http://gaussian.com/glossary/g09/.G09|Gaussian.com Search PubMed.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F.-Y. Dupradeau, R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments, Nucleic Acids Res., 2011, 39, W511–W517 CrossRef CAS PubMed.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- R. L. Davidchack, R. Handel and M. V. Tretyakov, Langevin thermostat for rigid body dynamics, J. Chem. Phys., 2009, 130, 234101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- P. Gonnet, P-SHAKE: A quadratically convergent SHAKE in, J. Comput. Phys., 2007, 220, 740–750 CrossRef.
- D. R. Roe and T. E. Cheatham, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- E. Seifert, OriginPro 9.1: Scientific Data Analysis and Graphing Software—Software Review, J. Chem. Inf. Model., 2014, 54, 1552 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- C. C. David, and D. J. Jacobs, Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins, 2014, pp. 193–226 Search PubMed.
- G. E. Arnold and R. L. Ornstein, Molecular dynamics study of time-correlated protein domain motions and molecular flexibility: cytochrome P450BM-3, Biophys. J., 1997, 73, 1147–1159 CrossRef CAS PubMed.
- M. Tavafoghi Jahromi and M. Cerruti, Amino Acid/Ion Aggregate Formation and Their Role in Hydroxyapatite Precipitation, Cryst. Growth Des., 2015, 15, 1096–1104 CrossRef CAS.
- N. Chaffey, Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. Molecular biology of the cell, 4th edn, Ann. Bot., 2003, 91, 401 CrossRef.
- R. H. Henchman, H.-L. Wang, S. M. Sine, P. Taylor and J. A. McCammon, Ligand-induced conformational change in the alpha7 nicotinic receptor ligand binding domain, Biophys. J., 2005, 88, 2564–2576 CrossRef CAS PubMed.
- E. Ahmad, G. Rabbani, N. Zaidi, M. A. Khan, A. Qadeer, M. Ishtikhar, S. Singh and R. H. Khan, Revisiting ligand-induced conformational changes in proteins: essence, advancements, implications and future challenges, J. Biomol. Struct. Dyn., 2013, 31, 630–648 CrossRef CAS PubMed.
- M. Tong and M. A. Seeliger, Targeting Conformational Plasticity of Protein Kinases, ACS Chem. Biol., 2015, 10, 39 CrossRef PubMed.
- H. Chen, Z. Huang, K. Dutta, S. Blais, T. A. Neubert, X. Li, D. Cowburn, N. J. Traaseth and M. Mohammadi, Cracking the Molecular Origin of Intrinsic Tyrosine Kinase Activity through Analysis of Pathogenic Gain-of-Function Mutations, Cell Rep., 2013, 4, 376–384 CrossRef CAS PubMed.
- H. Ur Rehman, N. Azam, J. Yao and A. Benso, A three-way approach for protein function classification, PLoS One, 2017, 12, e0171702 CrossRef PubMed.
- J. R. Brender and Y. Zhang, Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles, PLoS Comput. Biol., 2015, 11, e1004494 CrossRef PubMed.
- D. Chen, N. Oezguen, P. Urvil, C. Ferguson, S. M. Dann and T. C. Savidge, Regulation of protein-ligand binding affinity by hydrogen bond pairing, Sci. Adv., 2016, 2, e1501240 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |