
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSoybean straw biomass-derived Fe–N co-doped porous carbon as an efficient electrocatalyst for oxygen reduction in both alkaline and acidic media†
Yong Liu *a,
Miaojun Sua,
Dahuan Lia,
Shenshen Lia,
Xiying Lia,
Junwei Zhaoa and
Fujian Liu*b
*a,
Miaojun Sua,
Dahuan Lia,
Shenshen Lia,
Xiying Lia,
Junwei Zhaoa and
Fujian Liu*b
aHenan Key Laboratory of Polyoxometalate Chemistry, College of Chemistry and Chemical Engineering, Henan University, Kaifeng, 475004, P. R. China. E-mail: liuyong79@126.com
bNational Engineering Research Center of Chemical Fertilizer Catalyst (NERC-CFC), School of Chemical Engineering, Fuzhou University, Fuzhou, 350002, P. R. China. E-mail: fjliu@fzu.edu.cn
First published on 13th February 2020
Abstract
The development of highly efficient oxygen reduction reaction (ORR) catalysts is of great significance for the large-scale commercialization of fuel cells. In this work, honeycomb-like Fe–N co-doped porous carbon materials (Fe–N–PC) were prepared through a facile one-step pyrolysis strategy using soybean straw biomass as the precursor. The obtained Fe–N–PC catalyst exhibits excellent ORR performance with an onset potential of 0.989 V and a half-wave potential of 0.854 V in alkaline conditions, which positively shift only by 5 mV and 27 mV, respectively than those of the commercial Pt/C catalyst. Furthermore, the onset potential and the half-wave potential of the Fe–N–PC catalysts are up to 0.886 V and 0.754 V, respectively, under acidic conditions, which are superior to those of many other Fe, N-doped electrocatalysts. The ORR process can be regarded as a four-electron transfer process based on RRDE measurements. Moreover, the Fe–N–PC catalyst also shows greater stability and satisfactory methanol tolerance than the Pt/C catalyst. The superior electrocatalytic performance of Fe–N–PC may be attributed to the abundant nanoporous structure, large BET surface area, and Fe–N co-doping, which provide abundant and highly efficient active sites.
Introduction
The problems of environmental pollution and depletion of natural resources caused by the excessive use of fossil fuels are extremely detrimental to the sustainable development of our planet. Many researchers are eager to find new clean and pollution-free energy sources as substitutes for traditional fossil energy. Metal–air batteries and fuel cells have attracted the attention of researchers due to their high-energy and zero-pollutant emissions.1 However, the slow reaction kinetics and the diversity of the reaction pathways of the cathodic oxygen reduction reaction (ORR) severely limit their energy efficiency.2 At present, Pt and Pt-based catalysts are considered to be the best for ORR and have received increasing attention. However, the noble metal Pt has some disadvantages, such as high price and low natural reserves, which have limited its usage in the large-scale development of metal–air batteries and fuel cells.3 Therefore, it is very significant to find a high-performance low-cost non-precious metal ORR catalyst for promoting the development of new energy sources.4Carbon-based materials are considered as the most promising alternatives to Pt-based catalysts due to some of their advantages, including low cost, high stability and flexibility. Among them, nitrogen-doped carbon materials are often used as ORR catalysts because nitrogen doping may change the electron density of the carbon surface and enhance the electronegativity, which results in electron vacancies and thus produces more active sites.5,6 Nevertheless, many nitrogen-doped carbon materials are less than satisfactory compared with Pt/C in terms of current density and onset potential.7 On the other hand, it has been found that the combination of transition metals (especially Fe) and nitrogen-doped carbon results in excellent ORR performance. Li et al.8 synthesized a nitrogen-doped vesicle-like porous carbon material with dual iron-based catalytic sites; it exhibited outstanding ORR performance, which was even superior to that of the commercial Pt/C catalyst. Huang et al.9 used a porous carbon-supported melamine-formaldehyde resin as the precursor to prepare a Fe–N–C catalyst via the facile high-temperature pyrolysis method, which was used as ORR catalysts in both acid and alkaline media. We also reported that honeycomb Fe–N co-doped porous carbon could be successfully synthesized via one-step pyrolysis using iron-containing ionic liquids as precursors and showed superior ORR catalytic activity to that of the commercial Pt/C catalyst in an alkaline medium.10
Biomass is an abundant natural source of carbon and nitrogen. Recently, many research groups have used biomass, such as coffee waste,11 shrimp skin,12 leather,13 aquatic plants,14 as precursors to synthesize ORR catalysts. Among these sources, soybean straw is a perfect biomass precursor to synthesize nitrogen-doped carbon due to the presence of abundant plant proteins (10–12%).15 Northeast China is famous for soybean cultivation and produces abundant soybean straw, which is not properly utilized every year. Soybean straw has the advantages of abundant source, low cost and large output. Therefore, soybean straw can be used as a precursor to synthesize nitrogen-doped carbon materials. The catalyst cost will be greatly reduced, and the application value of waste soybean straw can be greatly improved. Lu et al.16 prepared a nitrogen and cobalt dual-doped porous electrocatalyst (CoNASS) by using soybean straw biomass as the precursor and used as an ORR catalyst in an alkaline medium.
Here, we present a simple method for the synthesis of a honeycomb-like Fe–N co-doped porous carbon material (Fe–N–PC) derived from the available and recyclable plant biomass, namely soybean straw. Fe–N–PC was applied as the electrocatalyst for ORR in both alkaline and acidic media, in which it exhibited excellent ORR catalytic activity and better stability and tolerance to methanol poisoning effects than the commercial Pt/C catalyst.
Experimental methods
Materials
Soybean straw was sourced from farmland in Qi County, Henan province, P. R. China. Melamine, ferric nitrate, and magnesia were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, P. R. China). All experimental reagents were of analytical grade and used without further purification.Preparation of catalysts
The honeycomb Fe–N doped porous carbon was synthesized by one-step pyrolysis using soybean straw, melamine, magnesium oxide and ferric nitrate. The synthesis process is shown in Scheme 1. 1.0 g soybean straw was placed in the vibration mill together with 1.0 g melamine, 2.0 g magnesium oxide, and 0.5 g ferric nitrate. After even grinding, the mixture was carbonized at a high temperature of 800 °C for 2 h at the heating rate of 3 °C min−1 under an N2 atmosphere in a tube furnace. The obtained black solid was treated with 1 M HCl in order to remove the magnesium oxide template and free iron/iron complex. Then, the samples were washed with distilled water until the solution pH reached 7 and dried at 60 °C in the oven. Thus, the porous Fe–N doped carbon (Fe–N–PC) was obtained. In order to investigate the effect of nitrogen and iron on the catalytic performance, other samples for comparison, including porous carbon (PC) using soybean straw as the precursor, N-doped carbon (N–PC) using soybean straw and melamine as precursors, and Fe-doped carbon (Fe–PC) using soybean straw and Fe(NO3)3 as precursors, were synthesized under the same conditions.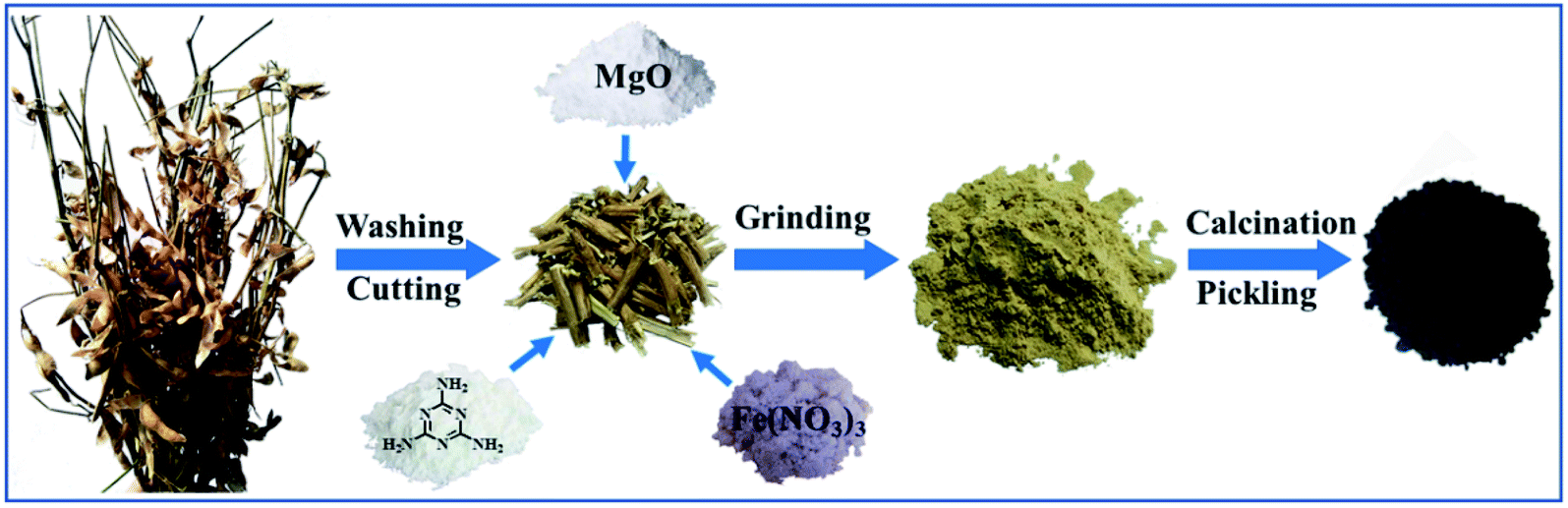 | ||
| Scheme 1 Schematic illustration of the fabrication procedure of Fe–N co-doped porous carbon. | ||
Characterization
The surface morphology of the materials was observed at 10 kV using a scanning electron microscope (SEM, JSM-7610F). Transmission electron microscopy (TEM, FEI Tecnai G2 F20 S-TWIN) was used to observe the internal structure of the materials. The analysis of the crystal structures of the carbon materials was carried out using an X-ray powder diffractometer (XRD, Bruker D8 Advance) from Bruker, Germany. N2 sorption-desorption was conducted on a Quadrasorb SI (Quantachrome, Boynton Beach, FL, USA) equipped with an automated surface area analyzer that employed the Brunauer–Emmett–Teller (BET) method. An X-ray photoelectron spectrometer (XPS, Escalab 250Xi) from American Thermo Scientific was used to analyze the elemental content and valence structure on the surface of the material. The metal content of the material was tested using an inductively-coupled plasma mass spectrometer (ICP, Optima 2100DV) from PerkinElmer, USA.Electrochemical measurements
The electrochemical performance of the catalyst was tested on a CHI760D electrochemical workstation (CH Instruments, China) using a standard three-electrode system. A saturated calomel electrode (SCE) was used as the reference electrode, a platinum plate as the counter electrode, and a modified rotating disk electrode (RDE) and a rotating ring disk electrode (RRDE) were the working electrodes. Cyclic voltammetry (CV), linear-sweep voltammetry (LSV) and chronoamperometry (CA) were performed on a rotating ring disk electrode instrument (RRDE-3A, ALS). The catalyst loading was 0.4 mg cm−2. All the resulting potentials were converted to those of a reversible hydrogen electrode (RHE) by the following formula:| E(RHE) = E(SCE) + 0.0591 × pH + 0.241 | (1) |
The RDE measurements were carried out at various rotating rates (400–2500 rpm). The electron transfer number (n) was determined by the Koutecky–Levich equation:17
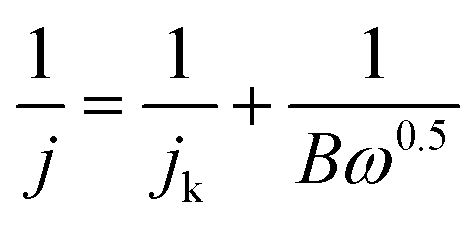 | (2) |
| B = 0.2nF(DO2)2/3v−1/6CO2 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 C mol−1), DO2 is the diffusion coefficient of O2 in 0.1 M KOH, υ is the kinetic viscosity (0.01 cm2 s−1), and CO2 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3).
500 C mol−1), DO2 is the diffusion coefficient of O2 in 0.1 M KOH, υ is the kinetic viscosity (0.01 cm2 s−1), and CO2 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3).
In this work, the RRDE measurements were carried out in a 0.1 M KOH and 0.1 M HCIO4 solution under saturated oxygen conditions with a constant potential of 0.5 V (vs. SCE) on the ring electrode. The H2O2 yield and the number of electrons transferred during the ORR reaction were calculated using the following equations:18,19
 | (4) |
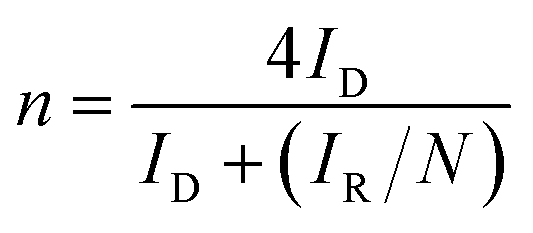 | (5) |
Results and discussion
Structure characterization
The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of Fe–N–PC are shown in Fig. 1a and b. Moreover, Fig. S1† shows the morphology of soybean straw mixed with MgO before and after calcination. The morphology of PC, N–PC and Fe–PC are shown in Fig. S2.† As seen in the SEM images, all samples were oriented to form a honeycomb-like porous structure using MgO as the template. From Fig. 1b and c, we found that some carbon-coated black particles, which may be iron/iron compounds, were present in the Fe–N–PC catalyst. | ||
| Fig. 1 (a) SEM image, (b) TEM image, (c–e) HRTEM images, (f) XRD pattern of Fe–N–PC, and (g) Raman spectra of the catalysts. | ||
The HRTEM images of Fe–N–PC are shown in Fig. 1c–e, in which the crystalline lattice can be observed. The lattice distance of black particles was calculated to be 0.21 nm and 0.16 nm, respectively, corresponding to the (111) crystal planes of the Fe3C phase (JCPDS 01-089-2005) and the (−222) crystal planes of the Fe3O4 phase (JCPDS 00-028-0491). The results are consistent with the XRD analysis in Fig. 1f, where 44.7° corresponds to Fe3C and 59.6° corresponds to Fe3O4. Notably, the diffraction peak appearing at 44.7° in Fig. 1f may be attributed to the binding of iron and nitrogen. It is speculated that the presence of iron–nitrogen bonds in the material might provide active sites for ORR.8 In addition, we could see that both Fe3C and Fe3O4 particles were encapsulated by the carbon layer, which might reduce the corrosion of the active reactive site on the catalyst and have a great effect on improving the electrochemical stability of the material.20 The XRD patterns of the other samples are shown in Fig. S3.† As revealed in Fig. S3,† the broad peaks at about 26.2° and 44.3° were attributed to the (002) and (101) planes of graphitic carbon (JCPDS 01-075-1621), respectively. The difference in the Fe–PC pattern (Fig. S3c†) was the peaks for iron oxides at 35.6° (JCPDS 01-033-0664) and Fe3C at 48.4° (JCPDS 01-035-0772). As shown in Fig. 1f, after the addition of a suitable nitrogen source to Fe–N–PC, some of the iron combined with nitrogen to form the iron nitride compound phase.
Raman spectroscopy was used to determine the degree of graphitization of the various catalysts prepared under different conditions. The Raman spectra are shown in Fig. 1g. All samples had two significant characteristic peaks: the D peak at about 1320 cm−1 corresponding to amorphous carbon and the G peak at about 1590 cm−1 corresponding to graphitized carbon. The ratio of the intensity of the D peak to the intensity of the G peak is an important index for evaluating the degree of graphitization of the material. It could be seen that with the introduction of iron and nitrogen, the value of ID/IG gradually increased, indicating that the doping of iron and nitrogen caused the formation of more defects.
The elemental composition of these catalysts was further studied by XPS. The surface survey XPS spectra of these catalysts are shown in Fig. 2a, and elemental analysis results are shown in Table 1. It could be seen that iron and nitrogen were successfully doped into the catalyst.21 Fig. 2b shows the N 1s spectra of Fe–N–PC, and it could be divided into four peaks at 398.41 eV, 399.6 eV, 400.75 eV and 402.7 eV, which correspond to pyridinic N, pyrrolic N, graphitic N and oxidized nitrogen, respectively.22 Notably, the peak at the binding energy of 398.4 eV might also present Fe–N binding due to the small difference between the binding energies of Fe–N and pyridinic N.23,24 It is generally believed that in addition to nitrogen oxides, pyridinic nitrogen, pyrrolic nitrogen and graphitic nitrogen are active sites for ORR and play an important role in increasing the ORR activity.25,26 Fig. 2c shows the high-resolution Fe 2p XPS spectrum of Fe–N–PC, which could be separated into four peaks at about 707.64, 710.8, 720.9 and 723.2 eV, corresponding to Fe2+ (2p3/2), Fe3+ (2p3/2), Fe2+ (2p1/2) and Fe3+ (2p1/2), respectively. In addition, the peaks at about 715.7 and 713.5 eV were attributable to the satellites.27 The figure also suggested the existence of Fe3O4 in the catalyst, which is consistent with the XRD analysis results.15 The peak at 710.8 eV indicated the probable formation of a Fe–N bond due to the combination of iron and nitrogen.28 The XPS spectra of other materials are shown in Fig. S4.† After the doping of iron, Fe–N bonds were formed in the catalyst, producing more active sites, which is extremely important for improving the ORR activity.29,30
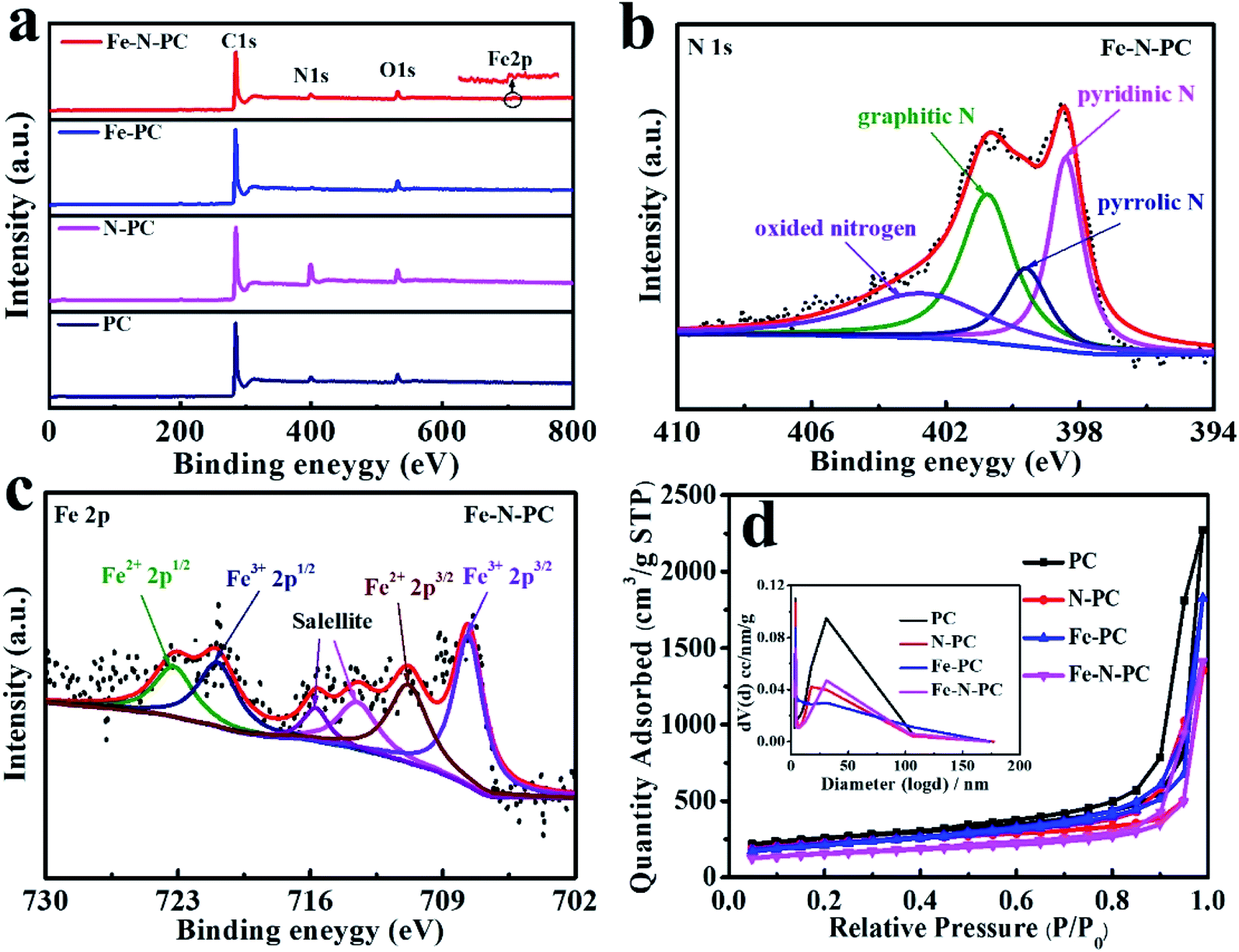 | ||
| Fig. 2 (a) XPS spectra of the series of catalysts; (b) N 1s spectra of Fe–N–PC; (c) Fe 2p spectra of Fe–N–PC; (d) nitrogen adsorption–desorption isotherms of the series of soybean straw catalysts, and the insets are the corresponding pore-size distributions of the catalysts. | ||
| Samples | C/wt% | O/wt% | N/wt% | Fe/wt% | BETc (m2 g−1) | Pore sizec (nm) | Pore volumec (cm2 g−1) |
|---|---|---|---|---|---|---|---|
| a Estimated from XPS results.b Calculated from ICP results.c Calculated from N2 adsorption–desorption isotherms. | |||||||
| PC | 87.54a | 6.48a | 5.98a | — | 846.2 | 3.8 | 3.5 |
| N–PC | 73.58a | 7.85a | 18.57a | — | 738.1 | 3.8 | 2.1 |
| Fe–PC | 90.09a | 6.4a | 2.15a | 1.36a/0.45b | 718.2 | 3.8 | 2.8 |
| Fe–N–PC | 86.04a | 5.93a | 6.24a | 1.79a/2.97b | 520.9 | 3.8 | 2.2 |
The N2 adsorption/desorption isotherms of the different catalysts are shown in Fig. 2d, which reflect the internal structural properties of the porous materials. The isotherm shape contained an obvious H3 hysteresis loop, indicating that these catalysts had mesoporous structures.31 The BET surface areas of different catalysts are shown in Table 1. It could also be seen from the pore size distribution in Fig. 2d (inset) that there were two peaks at about 3.8 nm and 30 nm, which could be attributed to the escape of unstable components during the calcination of the precursor and the removal of the MgO template after calcination, respectively. The large specific surface areas can expose more active sites and the porous structure can facilitate electron transport during the ORR process and enhance the reaction rate.32
Electrochemical properties of catalysts
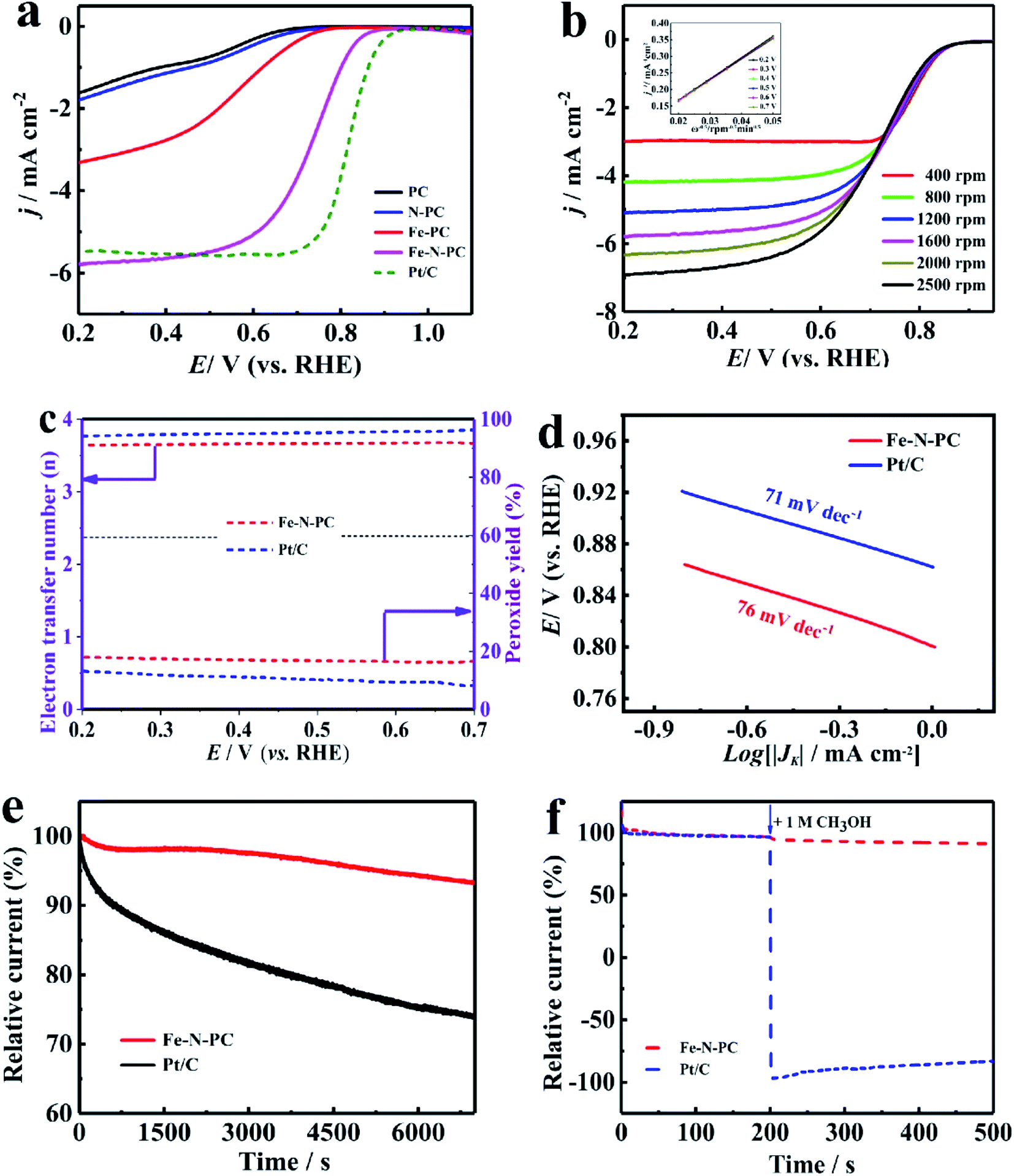
The CV tests were performed in an O2-saturated 0.1 M KOH aqueous solution at a scan rate of 50 mV s−1. As shown in Fig. S5a,† there was an obvious redox peak, which indicated that these catalysts had good ORR catalytic activity. The LSV curves of the various catalysts are shown in Fig. 3a, and their performance parameters are listed in Table S1.† It could be observed that the onset potential (Eonset) and the half-wave potential (E1/2) of the Fe–N–PC catalyst reached about 0.989 and 0.854 V, respectively, which showed higher catalytic activity compared with the other catalysts. Compared with the commercial Pt/C catalyst, the onset potential and half-wave potential of Fe–N–PC were positively shifted only by about 5 mV and 27 mV, respectively. PC had the lowest onset potential (0.882 V) and half-wave potential (0.755 V). The performance of the catalyst further increased with the introduction of nitrogen and iron, whereas the addition of one element alone did not significantly improve the ORR performance of the catalyst. In comparison with the other catalysts, the Fe–N–PC catalyst exhibits more excellent ORR activity, which is attributed that the combination of iron, nitrogen and carbon can form more active sites. Moreover, the presence of the honeycomb multi-stage pore structure also exposed abundant active sites and enhanced ORR performance.33 | ||
| Fig. 3 (a) LSV curves of the series of soybean straw catalysts at a rotation speed of 1600 rpm in a O2-saturated 0.1 M KOH solution; (b) LSV curves of Fe–N–PC at different rotation speeds in the O2-saturated 0.1 M KOH solution; (c) calculated H2O2 yields and electron transfer numbers from the ORRs of Fe–N–PC and Pt/C; (d) Tafel diagrams of Fe–N–PC and Pt/C in O2-saturated 0.1 M KOH; (e) chronoamperometric measurements of Fe–N–PC and Pt/C in the O2-saturated 0.1 M KOH solution; (f) methanol tolerance measurements of Fe–N–PC and Pt/C in the O2-saturated 0.1 M KOH solution. | ||
Fig. 3b shows the LSV curves of Fe–N–PC at different rotating speeds. It could be seen that the limit-current density increased with the increase in rotation rate. The increase in dissolved oxygen at high rotation rates promoted the contact between the composite and oxygen, leading to the enhancement of current density. The corresponding K–L curves of the Fe–N–PC catalyst showed an excellent linear relationship at each potential, which represented first-order reaction kinetics.34 The calculated average electron transfer number of Fe–N–PC, according to the K–L equation, was 3.97 at potentials ranging from 0.30–0.70 V (Fig. 3b, inset), which is close to that of the four-electron transfer process involved in ORR. In order to further reveal the ORR kinetics, the RRDE measurements were recorded, and the results are shown in Fig. 3c. It could be seen that a small amount of peroxide was generated during the reaction, and the electron transfer number was close to that of Pt/C, which suggested as a four-electron transfer process. Furthermore, the mechanistic and kinetic properties of the catalysts toward ORR were estimated from diffusion-corrected Tafel plots. As shown in Fig. 3d, the Tafel slope of Fe–N–PC was calculated to be 69 mV dec−1, which is close to that of Pt/C (67 mV dec−1).
In addition, the catalytic stability and methanol tolerance were investigated by chronoamperometry measurements in an O2-saturated 0.1 M KOH solution. As shown in Fig. 3e, after 7000 s, the relative current of Pt/C decreased by 16.7%, while the relative current of Fe–N–PC decreased only by 8.8%. The durability of the catalyst was also characterized by cyclic CV tests from 0 to 1.2 V at a scan rate of 50 mV s−1 in O2-saturated 0.1 M KOH for 1000 cycles. It can be seen from Fig. S7a† that the E1/2 was negatively shifted by 13 mV for the Fe–N–PC catalyst after 10000 cycles in contrast to 46 mV for the Pt/C catalyst under the same conditions. The above results indicated that the Fe–N–PC catalyst had better stability than commercial Pt/C in ORR in the alkaline medium. This may be due to that the dissociation and aggregation of Pt nanoparticles during the ORR lead to low stability.34 Whereas, the formation of Fe–N–PC by covalent bond has good stability, which could avoid the dissolution of active sites. In addition, methanol tolerance is an important factor for ORR catalysts considered for fuel cells because methanol molecules easily cross through the membrane from the anode to the cathode, weakening the ORR activity of the cathode catalyst.35,36 The effect of methanol on the Fe–N–PC and Pt/C catalysts was examined by plotting their i–t curves in 0.1 M KOH without and with 1 M methanol. As shown in Fig. 3f, when methanol was added at 200 s, the i–t curve of Pt/C showed a sharp drop. However, the i–t curve of Fe–N–PC was substantially unchanged, indicating its excellent methanol tolerance.
Since Fe–N–PC has good ORR properties under alkaline conditions, we further studied its electrocatalytic properties in acidic electrolytes, which have great significance in the practical applications of ORR.37 The CV measurements showed that the redox peaks of all the catalysts were obvious in the O2 saturated 0.1 M HClO4 solution (Fig. S5b†), which indicated that they had significant ORR activities in the acidic medium. The LSV curves showed that Fe–N–PC exhibited good ORR performance with an E1/2 of 0.754 V, which was only 46 mV lower than that of the commercial Pt/C catalyst (0.80 V) (Fig. 4a). Moreover, it could be seen that the ORR performance of the Fe–N–PC catalyst was superior to those of many other Fe, N-doped electrocatalysts in the acidic medium (Table S2†). Fig. 4b shows the LSV curves of the Fe–N–PC catalyst at different rotating speeds. It could be seen that the limit-current density increased with the increase in rotation rate. The corresponding K–L curves are shown in Fig. 4b, and the calculated average electron transfer number of Fe–N–PC, according to the K–L equation, was 3.89 at potentials ranging from 0.20–0.70 V (Fig. 4b, inset), which is close to that of the four-electron transfer process involved in ORR. Moreover, RRDE measurements were carried out, and the results are shown in Fig. 4c. The electron transfer number was close to 3.65, which indicated a four-electron transfer process. As seen in Fig. 4d, the Tafel slope of Fe–N–PC was calculated to be 76 mV dec−1 in 0.1 M HClO4, which is slightly higher than that of Pt/C (71 mV dec−1), indicating their comparable catalytic performances in the acid medium.
 | ||
| Fig. 4 (a) LSV curves of the series of soybean straw catalysts at a rotation speed of 1600 rpm in a O2-saturated 0.1 M HCIO4 solution; (b) LSV curves of Fe–N–PC at different rotation speeds in a O2-saturated 0.1 M HCIO4 solution; (c) calculated H2O2 yields and electron transfer numbers from the ORRs of Fe–N–PC and Pt/C; (d) Tafel diagrams of Fe–N–PC and Pt/C in O2-saturated 0.1 M HCIO4; (e) chronoamperometric measurements of Fe–N–PC and Pt/C in O2-saturated 0.1 M HCIO4; (f) methanol tolerance measurements of Fe–N–PC and Pt/C in O2-saturated 0.1 M HCIO4. | ||
Moreover, the catalytic stability and methanol tolerance were also studied by chronoamperometry measurements in an O2-saturated 0.1 M HCIO4 solution. As shown in Fig. 4e, after 7000 s, the relative current of Pt/C decreased by 30.22%, while the relative current of the Fe–N–PC catalyst decreased only by 6.85%. As shown in Fig. S7b,† the E1/2 was negatively shifted by 18 mV for the Fe–N–PC catalyst, and the E1/2 of the Pt/C catalyst was negatively shifted by 59 mV after 10000 cycles. These results indicated that Fe–N–PC had better stability than the commercial Pt/C catalyst for ORR in the acidic medium. The effect of methanol on the Fe–N–PC and Pt/C catalysts were examined by plotting their i–t curves in 0.1 M HCIO4 without and with 1 M methanol. As shown in Fig. 4f, when methanol was added at 200 s, the i–t curve of Pt/C showed a sharp drop. However, the i–t curve of Fe–N–PC was substantially unchanged, indicating that the Fe–N–PC catalyst retained very good methanol tolerance under acidic conditions as well.
Conclusion
In summary, a honeycomb-like Fe–N co-doped porous carbon material (Fe–N–PC) was synthesized by a one-step pyrolysis method using soy straw biomass as the precursor. The electrochemical measurement results showed that the Fe–N–PC catalyst exhibited excellent ORR catalytic performance in both alkaline and acidic media in comparison with other catalysts and the commercial Pt/C catalyst. Especially in the alkaline medium, the onset and half-wave potentials on Fe–N–PC were 0.989 V and 0.854 V, respectively, which showed a positive-shift by 5 mV and 27 mV than the commercial Pt/C catalyst, respectively. The onset potential and the half-wave potential were 0.886 V and 0.754 V, respectively, in the acidic medium. Furthermore, the ORR process on the Fe–N–PC catalyst could be considered as a four-electron transfer process based on the RRDE measurements in both alkaline and acidic solutions. In addition, Fe–N–PC showed better stability and methanol tolerance than the commercial Pt/C catalyst. The superior electrocatalytic performance of Fe–N–PC could be attributed to the formation of abundant active sites in the carbon material due to iron–nitrogen co-doping. The porous structure might also contribute to active site exposure, which accelerates the electrolyte and O2 transport rates. The results indicate that soybean straw biomass-derived Fe–N–PC is a promising and efficient ORR catalyst in both acid and alkaline media, and hence, is environmentally sustainable and important for the popularity of fuel cells.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21978052, 21573150, 21676072). The Natural Science Foundation of Henan Province (No. 182300410244), Foundation for University Youth Key Teachers from Henan Province (No. 2017GGJS018), the Program for Qishan Scholar of Fuzhou University (GXRC-18043), and the Program for Innovation Teams in Science and Technology in Universities of Henan Province (20IRTSTHN004).References
- J. Zou, B. Wang, B. Zhu, Y. Yang, W. Han and A. Dong, Nano Convergence, 2019, 6, 4 CrossRef PubMed.
- T. N. Tran, C. H. Shin, B. J. Lee, J. S. Samdani, J. D. Park, T. H. Kang and J. S. Yu, Catal. Sci. Technol., 2018, 8, 5368–5381 RSC.
- W. Wei, X. M. Shi, P. Gao, S. S. Wang, W. Hu, X. X. Zhao, Y. M. Ni, X. Y. Xu, Y. Q. Xu, W. S. Yan, H. X. Ji and M. H. Cao, Nano Energy, 2018, 52, 29–37 CrossRef CAS.
- X. R. Zhang, D. D. Lyu, Y. B. Mollamahale, F. Yu, M. Qing, S. B. Yin, X. Y. Zhang, Z. Q. Tian and P. K. Shen, Electrochim. Acta, 2018, 281, 502–509 CrossRef CAS.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- Q. Zhao, T. P. Fellinger, M. Antonietti and J. Y. Yuan, J. Mater. Chem. A, 2013, 1, 5113–5120 RSC.
- T. Zhou, Y. Zhou, R. Ma, Z. Zhou, G. Liu, Q. Liu, Y. Zhu and J. Wang, Nanoscale, 2016, 8, 18134–18142 RSC.
- Q. Li, G. Wan, H. Zhao, L. Pan, N. Wang, W. Zhao, X. Zhou, X. Cui and H. Chen, ChemSusChem, 2017, 10, 499–505 CrossRef CAS PubMed.
- L. Huang, C. Y. Zhao, Y. F. Yao, Y. You, Z. W. Wang, C. P. Wu, Y. Sun, J. Tian, J. G. Liu and Z. G. Zou, Int. J. Hydrogen Energy, 2016, 41, 11090–11098 CrossRef CAS.
- Y. Liu, S. S. Li, X. Y. Li, L. Q. Mao and F. J. Liu, Ind. Eng. Chem. Res., 2018, 57, 15638–15646 CrossRef CAS.
- D. Y. Chung, Y. J. Son, J. M. Yoo, J. S. Kang, C. Y. Ahn, S. Park and Y. E. Sung, ACS Appl. Mater. Interfaces, 2017, 9, 41303–41313 CrossRef CAS PubMed.
- X. Zhang, R. R. Liu, Y. P. Zang, G. Q. Liu, S. W. Liu, G. Z. Wang, Y. X. Zhang, H. M. Zhang and H. J. Zhao, Inorg. Chem. Front., 2016, 3, 910–918 RSC.
- I. L. Alonso-Lemus, F. J. Rodriguez-Varela, M. Z. Figueroa-Torres, M. E. Sanchez-Castro, A. Hernandez-Ramírez, D. Lardizabal-Gutierrez and P. Quintana-Owen, Int. J. Hydrogen Energy, 2016, 41, 23409–23416 CrossRef CAS.
- H. Huang, X. j. Wei and S. Y. Gao, Electrochim. Acta, 2016, 220, 427–435 CrossRef CAS.
- Y. Y. Liu, J. M. Ruan, S. B. Sang, Z. C. Zhou and Q. M. Wu, Electrochim. Acta, 2016, 215, 388–397 CrossRef CAS.
- G. L. Lu, Z. Y. Li, W. X. Fan, M. Wang, S. C. Yang, J. Y. Li, Z. Y. Chang, H. Sun, S. Liang and Z. N. Liu, RSC Adv., 2019, 9, 4843–4848 RSC.
- G. A. Ferrero, A. B. Fuertes, M. Sevilla and M. M. Titirici, Carbon, 2016, 106, 179–187 CrossRef CAS.
- C. Han, X. J. Bo, J. Liu, M. Li, M. Zhou and L. P. Guo, J. Alloys Compd., 2017, 710, 57–65 CrossRef CAS.
- X. Chen, N. Wang, K. Shen, Y. Xie, Y. Tan and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 25976–25985 CrossRef CAS PubMed.
- H. Wang, W. Wang, M. Gui, M. Asif, Z. Wang, Y. Yu, J. Xiao and H. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 335–344 CrossRef CAS PubMed.
- Y. R. Li, C. Z. Guo, J. Q. Li, W. L. Liao, Z. B. Li, J. Zhang and C. G. Chen, Carbon, 2017, 119, 201–210 CrossRef CAS.
- J. C. Li, P. X. Hou, C. Shi, S. Y. Zhao, D. M. Tang, M. Cheng, C. Liu and H. M. Cheng, Carbon, 2016, 109, 632–639 CrossRef CAS.
- Z. Y. Wu, X. X. Xu, B. C. Hu, H. W. Liang, Y. Lin, L. F. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
- H. W. Liang, W. Wei, Z. S. Wu, X. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L. J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS PubMed.
- L. Lin, Q. Zhu and A. W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- B. L. Zhou, L. Z. Liu, P. W. Cai, G. Zeng, X. Q. Li, Z. H. Wen and L. Chen, J. Mater. Chem. A, 2017, 5, 22163–22169 RSC.
- Y. P. Zang, H. M. Zhang, X. Zhang, R. R. Liu, S. W. Liu, G. Z. Wang, Y. X. Zhang and H. J. Zhao, Nano Res., 2016, 9, 2123–2137 CrossRef CAS.
- Z. Li, G. Li, L. Jiang, J. Li, G. Sun, C. Xia and F. Li, Angew. Chem., Int. Ed., 2015, 54, 1494–1498 CrossRef CAS PubMed.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Am. Chem. Soc., 2012, 134, 19528–19531 CrossRef CAS PubMed.
- J. Xu, S. Lu, X. Chen, J. Wang, B. Zhang, X. Zhang, C. Xiao and S. Ding, Nanotechnology, 2017, 28, 485701 CrossRef PubMed.
- F. Tang, H. Lei, S. Wang, H. Wang and Z. Jin, Nanoscale, 2017, 9, 17364–17370 RSC.
- S. Lee, D. H. Kwak, S. B. Han, Y. W. Lee, J. Y. Lee, I. A. Choi, H. S. Park, J. Y. Park and K. W. Park, ACS Catal., 2016, 6, 5095–5102 CrossRef CAS.
- Z. W. Lu, B. C. Liu, W. L. Dai, L. Z. Ouyang and J. S. Ye, Appl. Surf. Sci., 2019, 463, 767–774 CrossRef CAS.
- H. Zhou, J. Zhang, I. S. Amiinu, C. Zhang, X. Liu, W. Tu, M. Pan and S. Mu, Phys. Chem. Chem. Phys., 2016, 18, 10392–10399 RSC.
- Z. W. Zhang, H. N. Li, J. Hu, B. Z. Liu, Q. R. Zhang, C. Fernandez and Q. M. Peng, J. Alloys Compd., 2017, 694, 419–428 CrossRef CAS.
- Q. X. Lai, L. R. Zheng, Y. Y. Liang, J. P. He, J. X. Zhao and J. H. Chen, ACS Catal., 2017, 7, 1655–1663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07539a |
| This journal is © The Royal Society of Chemistry 2020 |