
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Panax notoginseng saponins regulate VEGF to suppress esophageal squamous cell carcinoma progression via DVL3-mediated Wnt/β-catenin signaling†
Xiaoqi Chen‡
a,
Zhuan Lv‡b,
Chuanlei Zhanga,
Xinting Wanga,
Yunxia Zhaoc,
Xiao Wang *a and
Yuling Zheng*d
*a and
Yuling Zheng*d
aDepartment of Gastroenterology, The First Affiliated Hospital of Henan University of CM, No. 19 Renmin Road, Jinshui District, Zhengzhou, Henan, China. E-mail: yishilaiu7@126.com; Tel: +86-0371-66231632
bMedical Administration, The First Affiliated Hospital of Henan University of CM, China
cHenan University of Chinese Medicine, China
dGuoyitang, The First Affiliated Hospital of Henan University of CM, No. 19 Renmin Road, Jinshui District, Zhengzhou, Henan, China. E-mail: luji2835114@126.com; Tel: +86-0371-66231632
First published on 17th January 2020
Abstract
Panax notoginseng saponins (PNS) have recently attracted increasing attention for their anti-tumor activities. The aim of this study was to explore the functional role and underlying mechanisms of PNS on the progression of esophageal squamous cell carcinoma (ESCC). The mRNA levels of vascular endothelial growth factor (VEGF), β-catenin and dishevelled-3 (DVL3) were assessed using qRT-PCR. Western blot was performed to detect the expression levels of VEGF, β-catenin and DVL3. Cell viability and proliferation abilities were determined using MTT assay. Transwell assays were used to evaluate cell migration and invasion capacities. Our data revealed that PNS hampered the viability of ESCC cells. VEGF silencing weakened proliferation, migration and invasion in ESCC cells. Mechanistically, PNS time-dependently reduced VEGF expression and PNS hampered ESCC cell proliferation, migration and invasion through VEGF. Moreover, β-catenin and DVL3 were upregulated in ESCC tissues and cells and positively correlated with VEGF level in ESCC tissues. VEGF regulated DVL3 via the Wnt/β-catenin signaling pathway in ESCC cells. Furthermore, PNS repressed DVL3 expression through VEGF in ESCC cells. Our study suggested that PNS suppressed ESCC progression at least partly through repressing VEGF via the DVL3-mediated Wnt/β-catenin signaling pathway, indicating that PNS might be promising anti-tumor agents for ESCC treatment.
Introduction
Esophageal cancer (EC) is the seventh prevalent malignancy and sixth in cancer-related death, with an estimated 572![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new EC cases and 509000 deaths worldwide in 2018.1 Esophageal squamous cell carcinoma (ESCC) usually accounts for about 90% of all EC cases each year.2 Despite advances in diagnostic and therapeutic methods, the prognosis of ESCC patients remains very poor, with a 5 year survival rate of less than 30%.3,4 Therefore, it is imperative to identify new effective anti-cancer drugs for improving ESCC treatment.
000 new EC cases and 509000 deaths worldwide in 2018.1 Esophageal squamous cell carcinoma (ESCC) usually accounts for about 90% of all EC cases each year.2 Despite advances in diagnostic and therapeutic methods, the prognosis of ESCC patients remains very poor, with a 5 year survival rate of less than 30%.3,4 Therefore, it is imperative to identify new effective anti-cancer drugs for improving ESCC treatment.
Vascular endothelial growth factor (VEGF), one of the most prominent regulators of angiogenesis, often is upregulated in human cancers and functions as an oncogene involved in the progression of human cancers.5 Inhibition of VEGF has recently been postulated as a promising anti-angiogenic strategy during cancer therapy.6 Emerging evidence has demonstrated that VEGF is tightly relevant to tumor cell proliferation, invasion, migration and apoptosis in multiple human cancers, including osteosarcoma and hepatocellular carcinoma.7,8 Moreover, a recent document manifested that VEGF depletion induced by microRNA-(miR)-377 hindered tumor angiogenesis in ESCC, and thus weakened ESCC progression, highlighting its role as a potential molecular target for ESCC treatment.9
Panax notoginseng saponins (PNS) are the key active components isolated from the root of Panax notoginseng (Burk.) F. H. Chen (Araliaceae), which is well known as a Chinese medicinal herb for a long history.10 The pharmacological effects of PNS are thought to attribute to their anti-inflammatory, anti-diabetic, anti-oxidant, hepatoprotective, hypolipidemic, haemostatic, renoprotective and anti-cancer activities.11 Previous researches had reported that PNS suppressed tumor progression in breast cancer and lung cancer via regulating the expression of several cancer-related genes, illuminating their possibility as anti-cancer agents.12,13 The aim of this research was to explore the functions and underlying molecular mechanisms of PNS on ESCC progression.
Wnt/β-catenin signaling, a pivotal signaling pathway in development processes and homeostasis, has been widely accepted to participate in various types of human cancers, including ESCC.14,15 In this pathway, dishevelled proteins (DVL, including DVL1, DVL2 and DVL3) are key components to regulate its activation by repressing the downstream destruction complex to stabilize β-catenin.16,17 Increasing evidence has showed that DVL3-mediated Wnt/β-catenin signaling affects tumor growth and metastasis in several cancers, such as non-small cell lung cancer and cervical cancer.18,19 Herein, we aimed to explore whether DVL3-mediated Wnt/β-catenin signaling was involved in the regulation of PNS on ESCC progression.
In the present study, our data indicated that PNS or VEGF silencing weakened ESCC cell proliferation, migration and invasion. This striking resemblance prompted us to investigate VEGF as a potential molecular mediator of PNS in ESCC progression. Furthermore, we further explored whether DVL3-mediated Wnt/β-catenin signaling was involved in the regulatory mechanism of PNS on ESCC progression.
Materials and methods
Cell culture, treatment and transfection
Human ESCC cell lines (KYSE450 and Eca109) were purchased from Shanghai aolushengwu Biotechnology Co., Ltd. (Shanghai, China) and human normal esophageal epithelium Het-1A cells were obtained from ATCC (Manassas, VA, USA). All cells were maintained under DMEM medium (Hyclone, Logan, UT, USA) plus 10% FBS (Biochrom AG, Berlin, Germany) in an incubator at 37 °C with 5% CO2 and saturated humidity.PNS were purchased from Kunming Pharmaceutical Co. (95% purity, Yunnan, China) were dissolved in 0.1% DMSO (we confirmed that 0.1% DMSO did not influence cell viability) following the instructions of manufacturers. To detect the impact of PNS on cell viability, cells were exposed to various concentrations (0, 0.05, 0.1 and 0.2 mg ml−1) of PNS for 48 h. In other experiments, cells were treated with 0.2 mg ml−1 of PNS for the indicated time. Additionally, cells were treated with 10 μM of IWR-1-endo (an inhibitor of Wnt signaling, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 24 h to retard the activation of Wnt/β-catenin signaling pathway, and vehicle control was used as negative control.
pcDNA3.1/CT-GFP-based VEGF overexpression plasmid (pcDNA-VEGF), siRNA against VEGF (si-VEGF) and corresponding negative control (pcDNA-con or si-con) were obtained from Ribobio (Guangzhou, China) and transfected into ESCC cells using Lipofectamine 3000 transfection reagent (Life Technologies, Bleiswijk, the Netherlands) in accordance with manufacturer's protocols.
Evaluating transfection efficiency using flow cytometry
KYSE450 and Eca109 cells were transfected with pcDNA-VEGF or pcDNA-con for 48 h. Then, the green fluorescence in the cells was detected by flow cytometry using the Accuri C6 flow cytometer (BD Biosciences, Heidelberg, Germany). The percentage of cells expressing green fluorescence was calculated as the transfection efficiency.Clinical tissues and ethical statement
ESCC tissues and corresponding noncancerous esophageal tissues were obtained from 50 ESCC patients who were diagnosed based on histopathological evaluation and underwent surgery at The First Affiliated Hospital of Henan University of CM from November 2015 to September 2017. No conventional chemoradiotherapy was implemented in these patients before surgical resection. All tissue specimens were stored at −80 °C until RNA extraction. Our research was approved by the committees of ethical review of research involving human subjects at The First Affiliated Hospital of Henan University of CM, and informed consent was signed by each participator.Quantitative real-time PCR
Total RNA from tissues and cells was isolated using MagMAX™-96 Total RNA Isolation Kit (Thermo Fisher Scientific, Darmstadt, Germany) following manufacturer's protocols. Total RNA (2 μg) was reverse-transcribed into cDNA using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real-time PCR (qRT-PCR) analysis was carried out on MyIQTM2 RT PCR Detection System (Bio-Rad, Marnes La Coquette, France) with the brilliant SYBR Green PCR Kit (Stratagene, Heidelberg, Germany) referring to manufacturer's guidance. Relative mRNA levels of VEGF, β-catenin and DVL3 were quantified using the 2−ΔΔCt method against GAPDH for normalization.Western blot
Cell lysates were prepared using RIPA lysis buffer (Beyotime Biotechnology, Beijing, China) with protease inhibitor cocktail (1×, Roche Diagnostics GmbH, Indianapolis, IN, USA). Total protein (60 μg) was fractionated by electrophoresis on a 10% SDS polyacrylamide gel, and then transferred to a PVDF membrane (Hybond P, GE Healthcare, Chalfont St Giles, UK). The membranes were probed with the following antibodies: anti-β-catenin (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-VEGF (1:500; Santa Cruz Biotechnology), anti-DVL3 (1:1000; Cell Signaling Technology) and anti-β-actin (1:1000; Cell Signaling Technology), and then were incubated with horseradish peroxidase (HRP)-coupled IgG antibody (1:3000; Santa Cruz Biotechnology) as secondary antibody. Protein bands were determined using the Enhanced Chemiluminescence Detection System (ECL plus, Amersham International PLC, Buckinghamshire, UK) and analyzed with BioMax Light Film (Carestream Health Inc., Tokyo, Japan) following manufacturer's guidance.Cell viability and proliferation assays
Cell viability and proliferation abilities were determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. For the detection of cell viability, cells were seeded in 96-well plates and treated with various concentrations (0, 0.05, 0.1 and 0.2 mg ml−1) of PNS for 48 h. For the measurement of cell proliferation, MTT assay was performed at various time points (0, 24, 48 and 72 h) after transfection or/and PNS exposure. At the indicated time points, 20 μl of MTT solution (5 mg ml−1, Sigma-Aldrich, Madrid, Spain) was added to each well and the plate was incubated at 37 °C for 3 h. Afterward, the medium was discarded and 150 μl of DMSO was added into each well to dissolve formed formazan crystal. Lastly, the absorbance at an optical density of 490 nm was measured by a Varioskan Flash 3001 microplate reader (Thermo Fisher Scientific). The percentage of cell viability was calculated based on the negative control.Transwell assay for cell migration and invasion
Cell migration and invasion capacities were detected by transwell assay using a 24-Transwell plate (8 μm pore size; Corning Inc., Corning, Toledo, NY, USA) referring to the protocols of manufacturers. In migration assays, cells (5.0 × 104) in medium containing 1% FBS were seeded into the upper compartment of the chamber with non-precoated membrane. In invasion assays, 5.0 × 104 cells were loaded into the top compartment of the chamber with Matrigel-precoated membrane (Corning). In both assays, the medium containing 15% FBS was added into the lower as chemoattractant. 24 h later, the penetrated cells through the pores of membranes were fixed with 4% paraformaldehyde and stained with 0.2% crystal violet (Sigma-Aldrich), followed by the count under a light microscope (Olympus, Tokyo, Japan) in 10 random fields.Statistical analysis
All statistical analyses were carried out with SPSS 18.0 software (SPSS GmbH, Munchen, Germany) using a Student's t-test, Mann–Whitney U test or one-way ANOVA. The correlations between VEGF level and β-catenin expression or DVL3 expression in ESCC tissues were analyzed using Spearman rank test. These experimental data were presented as mean ± SD from three or more independent experiments. P values <0.05 were considered as significant for all tests.Results
PNS hampered the viability of ESCC cells
To determine the effect of PNS on ESCC cell viability, KYSE450 and Eca109 cells were treated with various concentrations (0, 0.05, 0.1 and 0.2 mg ml−1) of PNS for 48 h. As shown by MTT assay, little change was observed in the viability of Het-1A cells after PNS treatment (Fig. 1A). However, the viabilities of KYSE450 and Eca109 cells were significantly hindered by PNS in a dose-dependent manner (Fig. 1B and C). Remarkably, 0.2 mg ml−1 of PNS resulted in a more distinct repression in the viability of both KYSE450 and Eca109 cells (Fig. 1B and C). Hence, 0.2 mg ml−1 of PNS was used for further experiments. These data together indicated that PNC repressed the viability of ESCC cells.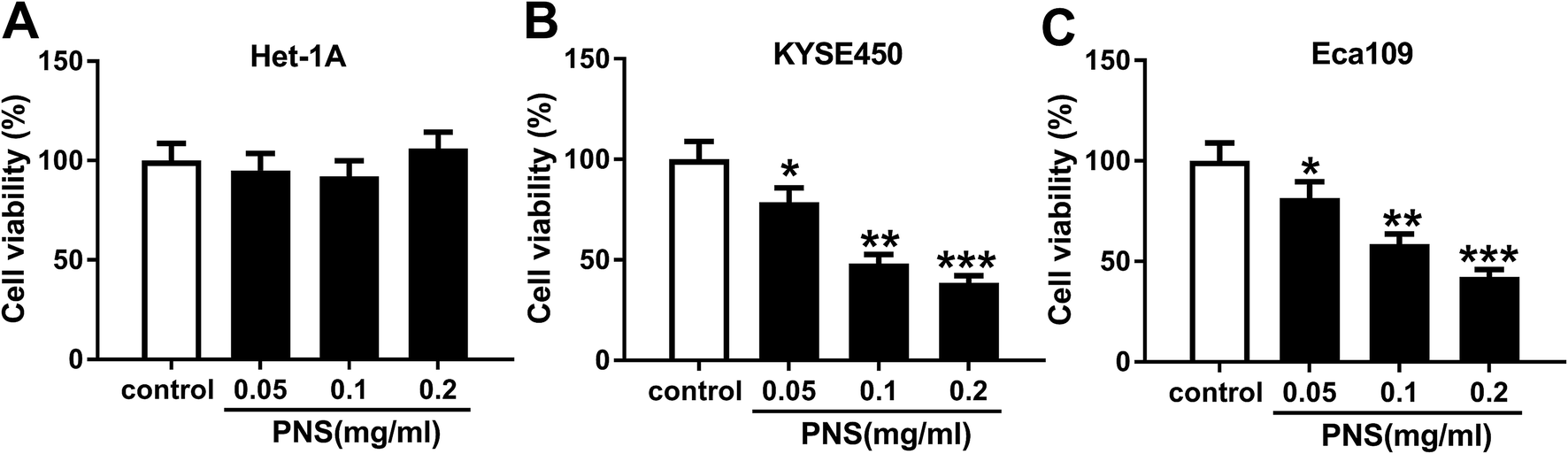 | ||
| Fig. 1 The effect of PNS on ESCC cell viability. Het-1A (A), KYSE450 (B) and Eca109 (C) cells were treated with various concentrations (0, 0.05, 0.1 and 0.2 mg ml−1) of PNS for 48 h, and then cell viability was assessed by MTT assay. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
VEGF was upregulated in ESCC tissues and cells
For a preliminary investigation for VEGF in EC progression, we determined the expression level of VEGF mRNA in ESCC tissues and adjacent normal tissues. As demonstrated by qRT-PCR, VEGF mRNA expression was significantly upregulated in ESCC tissues compared with normal controls (Fig. 2A). Moreover, in contrast to Het-1A cells, the mRNA levels of VEGF were prominently elevated in both KYSE450 and Eca109 cells (Fig. 2B). All these data suggested a potential involvement of VEGF in ESCC progression. | ||
| Fig. 2 Upregulation of VEGF in ESCC tissues and cells. The expression of VEGF mRNA was assessed by qRT-PCR in 50 pairs of ESCC tissues and adjacent noncancerous tissues (A), KYSE450, Eca109 and Het-1A cells (B). ***P < 0.001. | ||
VEGF silencing weakened proliferation, migration and invasion in ESCC cells
To explore the functional role of VEGF in ESCC progression, we performed “phenocopy” silencing by siRNA against VEGF (si-VEGF). Transient transfection of si-VEGF, but not a scrambled control sequence, significantly reduced the expression of VEGF in both KYSE450 and Eca109 cells (Fig. 3A). MTT assays revealed that VEGF knockdown triggered a conspicuous inhibition of cell proliferation in the two ESCC cells (Fig. 3B). Notably, transwell experiments showed that in comparison to negative control, VEGF depletion resulted in the significant repression of cell migration and invasion (Fig. 3C and D). These data together established a notion that VEGF silencing hindered the proliferation, migration and invasion of ESCC cells.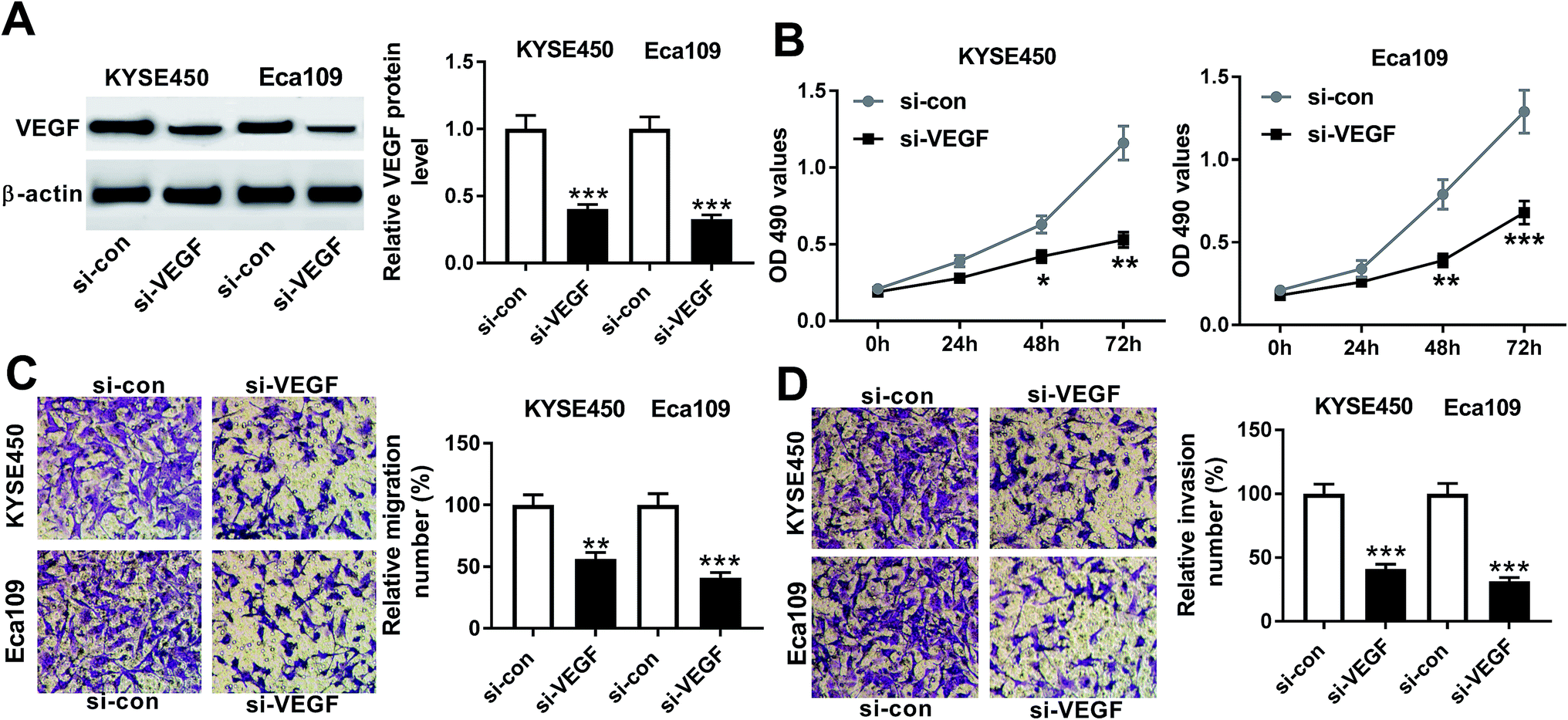 | ||
| Fig. 3 The effect of VEGF silencing on ESCC cell proliferation, migration and invasion. KYSE450 and Eca109 cells were transfected with si-VEGF or si-con. (A) 48 h after transfection, VEGF expression was detected by western blot. (B) After transfection for 0, 24, 48 and 72 h, cell proliferation was assessed by MTT assays. (C and D) After 24 h transfection, cell migration and invasion abilities were determined by transwell assays. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
PNS hampered ESCC cell proliferation, migration and invasion through VEGF
To further understand the effects of PNS and VEGF in ESCC progression, we next evaluated the impact of PNS on VEGF expression in KYSE450 and Eca109 cells. The results of qRT-PCR and western blot assays revealed that VEGF expression in the two cells was significantly elevated at both mRNA and protein levels by pcDNA-VEGF transfection compared with a corresponding negative control (Fig. 4A and B). Additionally, flow cytometry analysis showed high transfection efficiencies of pcDNA-VEGF in the two ESCC cells (ESI Fig. 1). In contrast to the control group, PNS treatment resulted in decreased VEGF expression in a time-dependent manner (Fig. 4C and D). Subsequently, the two ESCC cells were transfected with pcDNA-VEGF prior to PNS exposure. As demonstrated by western blot, introduction of pcDNA-VEGF, but not a negative plasmid, prominently abated PNS-mediated decreased VEGF expression in the two cells (Fig. 4E and F). MTT and transwell assays revealed that PNS treatment significantly suppressed the proliferation, migration and invasion in both KYSE450 and Eca109 cells (Fig. 4G–L). However, these effects were dramatically reversed by the transfection of pcDNA-VEGF (Fig. 4E–L). Together, these results implied that PNS suppressed the proliferation, migration and invasion in ESCC cells via downregulating VEGF.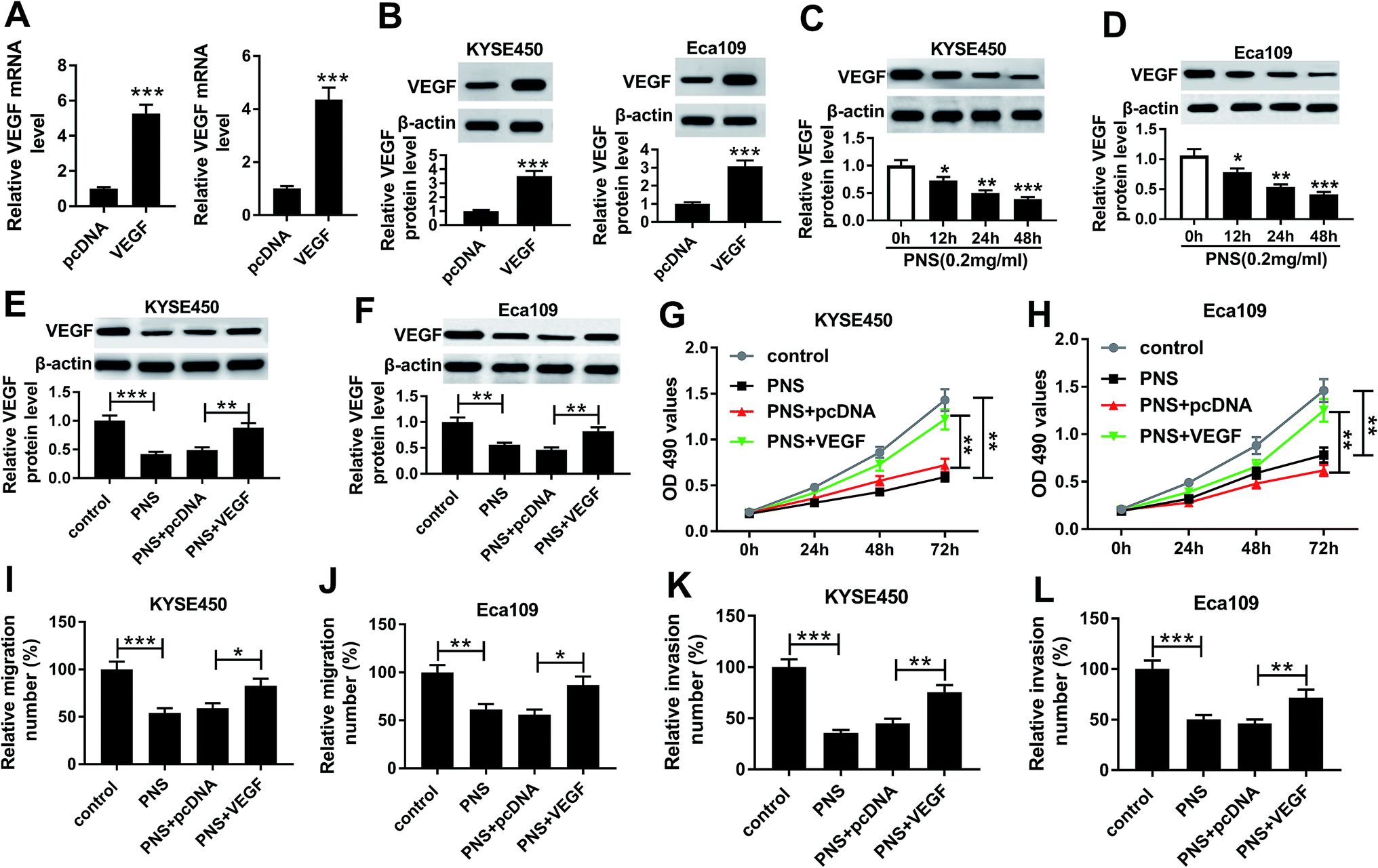 | ||
| Fig. 4 VEGF was involved in the regulation of PNS on ESCC cell proliferation, migration and invasion. (A and B) VEGF expression was detected by qRT-PCR and western blot in KYSE450 and Eca109 cells transfected with pcDNA-VEGF or pcDNA-con. (C and D) KYSE450 and Eca109 cells were treated with 0.2 mg ml−1 of PNS for various time points (0, 12, 24 and 48 h), and then VEGF level was detected by western blot. KYSE450 and Eca109 cells were transfected with pcDNA-VEGF or pcDNA-con for 24 h before exposing to 0.2 mg ml−1 of PNS, followed by the detection of VEGF level by western blot after PNS treatment for 24 h (E and F), cell proliferation by MTT assays at 0, 24, 48 and 72 h after PNS exposure (G and H), cell migration (I and J) and invasion (K and L) by transwell assays after 24 h PNS exposure. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
β-Catenin and DVL3 were upregulated in ESCC tissues and cells and positively correlated with VEGF level in ESCC tissues
Wnt/β-catenin pathway is an essential signaling pathway that modulates cell proliferation, differentiation, migration and invasion, and its constitutive activation is being found to associate with the progression of many human cancers, including ESCC.20,21 Herein, we determined the expression levels of β-catenin and DVL3 in ESCC tissues and cells. As expected, the mRNA levels of β-catenin and DVL3 were manifestly increased in ESCC tissues and cells compared with their counterparts (Fig. 5A–D). More interestingly, β-catenin and DVL3 mRNA levels were positively correlated with VEGF expression, respectively, in ESCC tissues (Fig. 5E and F). All these results pointed that upregulated β-catenin and DVL3 were positively correlated with VEGF level in ESCC tissues. | ||
| Fig. 5 Upregulation of β-catenin and DVL3 were positively correlated with VEGF level in ESCC tissues. qRT-PCR for β-catenin (A and B) and DVL3 (C and D) mRNA levels in 50 pairs of ESCC tissues and adjacent noncancerous tissues, and KYSE450, Eca109 and Het-1A cells. The correlations between VEGF level and β-catenin expression (E) or DVL3 expression (F) in ESCC tissues using Spearman rank test. ***P < 0.001. | ||
VEGF regulated DVL3 via Wnt/β-catenin signaling pathway in ESCC cells
Our above data revealed that DVL3 and β-catenin were positively correlated with VEGF expression in ESCC tissues. We next investigated whether VEGF modulated their expressions in KYSE450 and Eca109 cells. In contrast to their counterparts, VEGF level was significantly elevated by transfection of pcDNA-VEGF in KYSE450 cells, while it was observably reduced after VEGF silencing in Eca109 cells (Fig. 6A and B). Notably, upregulated VEGF resulted in increased DVL3 and β-catenin levels, while VEGF silencing exerted opposite effects (Fig. 6A and B), eliciting a positive modulation of VEGF on the expression of DVL3 and β-catenin in ESCC cells. | ||
| Fig. 6 VEGF modulated DVL3 via Wnt/β-catenin signaling pathway in ESCC cells. The expression levels of VEGF, β-catenin and DVL3 in KYSE450 cells transfected with pcDNA-con or pcDNA-VEGF (A), and Eca109 cells transfected with si-con or si-VEGF (B) by western blot. (C) KYSE450 cells were transfected with pcDNA-con or pcDNA-VEGF for 24 h before treatment with 10 μM of IWR-1-endo or vehicle control (con) for 24 h, followed by the measurement of VEGF, β-catenin and DVL3 levels. NS: no significant. **P < 0.01 or ***P < 0.001. | ||
In order to further explore how VEGF regulated DVL3 in ESCC cells, we used IWR-1-endo, an inhibitor of Wnt/β-catenin response. Intriguingly, in comparison to control group, the increased effect of VEGF overexpression on DVL3 and β-catenin expression was remarkably abated by IWR-1-endo in KYSE450 cells (Fig. 6C). Moreover, VEGF expression did not been affected by IWR-1-endo, supporting VEGF as the upstream molecular of DVL3 in KYSE450 cells (Fig. 6C). Taken together, these data suggested that VEGF modulated DVL3 expression via Wnt/β-catenin signaling pathway in ESCC cells.
PNS repressed DVL3 expression through VEGF in ESCC cells
Given our data that PNS regulated ESCC cell progression by VEGF and VEGF modulated DVL3, we further observed whether PNS could regulate DVL3 in ESCC cells. As expected, PNS exposure led to the prominent suppression of VEGF, β-catenin and DVL3 levels in both KYSE450 and Eca109 cells (Fig. 7A and B). Moreover, this effect was markedly alleviated by VEGF overexpression in KYSE450 cells (Fig. 7A). More interestingly, simultaneous PNS treatment and VEGF silencing resulted in a more distinct inhibition of β-catenin and DVL3 expression in Eca109 cells (Fig. 7B). These data together strongly hinted that PNS repressed DVL3 expression through VEGF in ESCC cells (Fig. 8). | ||
| Fig. 7 DVL3 was regulated by PNS via VEGF in ESCC cells. (A) KYSE450 cells were transfected with or without pcDNA-con or pcDNA-VEGF and then exposed to 0.2 mg ml−1 of PNS for 24 h, followed by the determination of VEGF, β-catenin and DVL3 levels by western blot. (B) Eca109 cells were introduced with or without si-con or si-VEGF before exposure to 0.2 mg ml−1 of PNS for 24 h, and then VEGF, β-catenin and DVL3 levels were detected by western blot. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
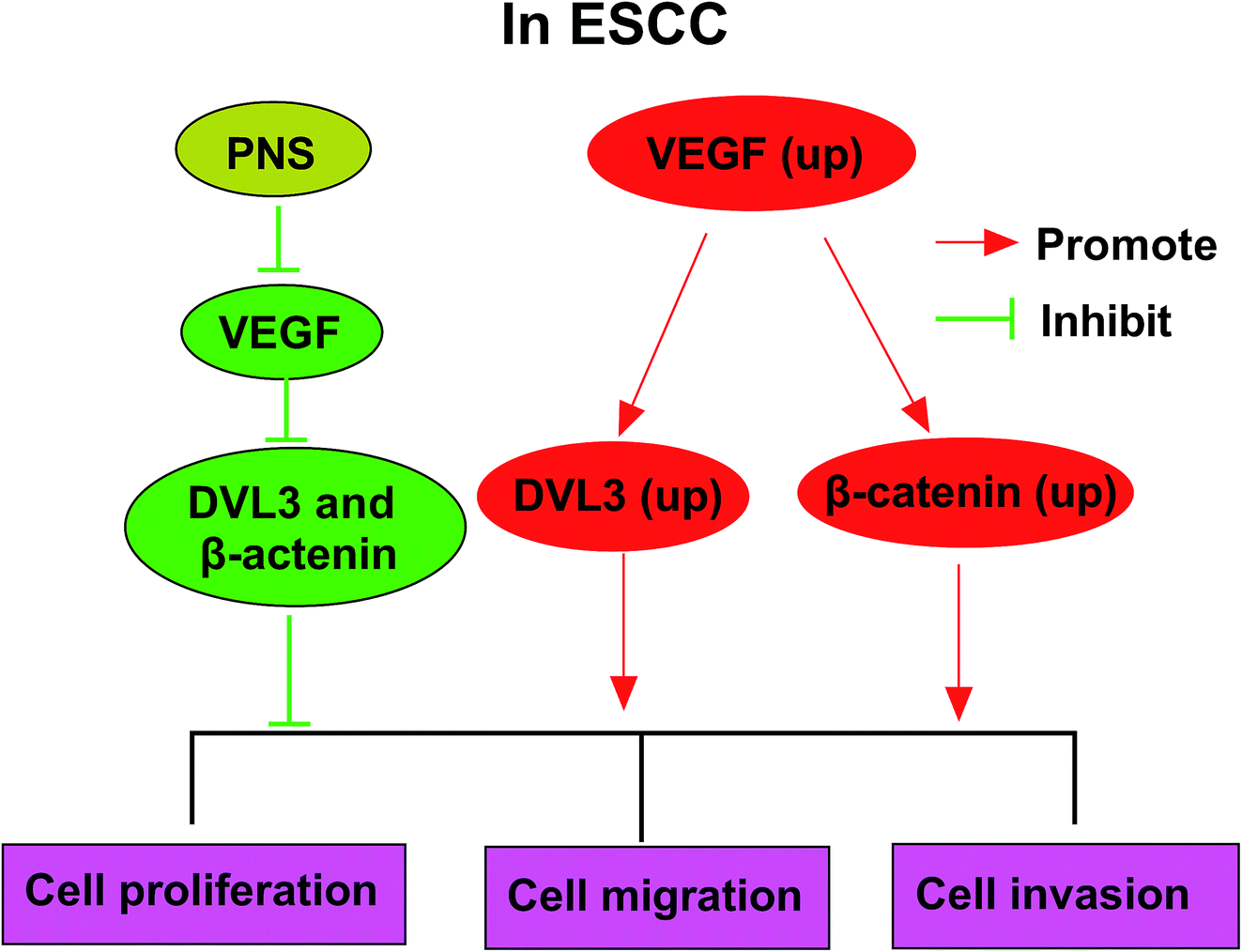 | ||
| Fig. 8 The mechanism schematic model by PNS/VEGF/DVL3/Wnt/β-catenin pathway in ESCC. (1) VEGF was upregulated in ESCC, and a high VEGF expression enhanced the expression levels of DVL3 and β-catenin. Then, cell proliferation, migration and invasion abilities were promoted. (2) PNS treatment reduced VEGF expression in ESCC cells. Afterward, down-regulated VEGF inhibited the expression of DVL3 and β-catenin in ESCC cells. Finally, cell proliferation, migration and invasion abilities were suppressed. | ||
Discussion
In recent years, PNS have attracted increasing attention for their anti-tumor activities. For example, Wang et al. demonstrated that PNS repressed migration and invasion of breast cancer cells partially through regulation of E-cadherin and vimentin.12 Yang et al. manifested that PNS weakened the tumor growth in lung cancer cells via regulating Met/miR-222 axis.13 Additionally, Wang et al. reported that PNS hampered cell proliferation and enhanced apoptosis, and promoted the chemosensitivity in colorectal cancer cells.22 Therefore, our study started from the hypothesis that PNS suppressed ESCC progression. To validate this, we firstly determined the impact of PNS on ESCC cell viability, and our data revealed that PNS weakened the viability of ESCC cells. Subsequently, we were first to demonstrate that PNS significantly hampered the proliferation, migration and invasion in ESCC cells. In a word, PNS performed anti-tumor effects in ESCC, highlighting their roles as potential therapeutic agents for improving ESCC treatment. However, the viability of normal esophageal cell line Het-1A was not been influenced by PNS, which possibly implied that ESCC cells could develop certain addiction to PNS.VEGF, a key mediator of angiogenesis, has been widely acknowledged to play an important role in the angiogenesis of many types of human cancers, such as ovarian cancer, colorectal cancer and lung cancer.23–25 VEGF was also manifested to implicate in the progression of several tumors, such as laryngeal cancer and osteosarcoma, through modulation of cancer cell proliferation, invasion and migration.7,26 Inhibitors of VEGF and VEGF receptor were illuminated to function as promising therapeutic options for renal cell carcinoma and glioblastoma.27,28 Previous researches described that high level of VEGF predicted poor prognosis in ESCC patients.29,30 Moreover, VEGF functioned as a predominant angiogenic regulator in ESCC.9,31 In this study, our data demonstrated a significant increase of VEGF level in ESCC tissues and cells, in accordance with earlier works.29,32 Furthermore, we substantiated that VEGF silencing hampered the proliferation, migration and invasion of ESCC cells.
Our above data suggested that PNS treatment or VEGF silencing retarded ESCC cell proliferation, migration and invasion. This striking resemblance prompted us to investigate VEGF as a potential molecular mediator of PNS treatment in ESCC progression. To confirm this, ESCC cells were treated with PNS for various time points, and our data revealed that PNS time-dependently reduced the expression of VEGF. After that, ESCC cells were synchronously subjected to PNS exposure and VEGF overexpression. Using qRT-PCR and western blot analyses, transient transfection of pcDNA-VEGF resulted in increased VEGF expression in the two ESCC cells, demonstrating the high transfection efficiency. We for the first time uncovered that VEGF mediated the suppressive effect of PNS on ESCC cell proliferation, migration and invasion. In short, PNS hampered the proliferation, migration and invasion in ESCC cells by VEGF. A previous report manifested that PNS played anti-arteriosclerotic activities through reducing VEGF expression in mice model of atherosclerosis.33 Previous studies had demonstrated that several other active components from Chinese herbs, such as curcumin and matrine, hindered cancer cell progression through inhibition of VEGF.34,35 Additionally, our data revealed that VEGF expression and cell migration and invasion abilities were prominently repressed, while cell proliferation did not been significantly changed after PNS treatment for 24 h. These findings might be attributed to the long lag phase of cell growth characteristic.
Aberrant activation of Wnt/β-catenin signaling pathway has been found in various types of human cancers. An increasing number of evidence has suggested that Wnt/β-catenin signaling may be activated by VEGF.36–38 Conversely, some other documents reported that Wnt/β-catenin pathway functioned as a crucial participator through regulating VEGF.39,40 These controversial conclusions may be attributed to different cell types or diversified regulatory mechanisms. Therefore, we continued our study from the assumption that Wnt/β-catenin signaling pathway was involved in the regulatory mechanism of PNS/VEGF on ESCC progression. Our data demonstrated that the levels of β-catenin and DVL3 were positively correlated with VEGF expression in ESCC tissues. More intriguingly, our results manifested that VEGF regulated DVL3 via Wnt/β-catenin signaling pathway in ESCC cells, suggesting that VEGF was an upstream molecular of Wnt/β-catenin signaling. DVL3, a crucial component for the activation of Wnt/β-catenin signaling, has been established to be involved in the progression of many human cancers, such as prostate cancer and breast cancer, via Wnt-DVL3-β-catenin signaling pathway.19,41 A recent document reported that DVL3 facilitated ESCC progression by elevating cancer cell proliferation and repressing cell apoptosis, providing possibility for DVL3 as diagnostic, therapeutic or prognostic biomarker for ESCC.42 In the present study, our data firstly substantiated that PNS suppressed DVL3 expression through VEGF in ESCC cells. These results indicated that DVL3-mediated Wnt/β-catenin signaling was involved in this regulatory mechanism of PNS on ESCC progression. The present study is limited to in vitro research, and more researches in vivo using xenograft model or ESCC mouse model will be implemented to validate this new mechanism in further work.
In conclusion, our study suggested that PNS hampered ESCC progression at least partly through repressing VEGF via DVL3-mediated Wnt/β-catenin signaling pathway. Our research provided a novel evidence for PNS as anti-tumor agents in ESCC.
Funding
(1) National Natural Science Foundation Youth Fund (No. 81804063).(2) Special Project of Traditional Chinese Medicine Research in Henan Province (No. 2018ZY2032).
(3) China Postdoctoral Science Foundation Grant (2019M662500).
Conflicts of interest
The authors declare that they have no financial conflicts of interest.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, CA, 2018, 68, 394–424 Search PubMed.
- C. C. Abnet, M. Arnold and W. Q. Wei, Gastroenterology, 2018, 154, 360–373 CrossRef PubMed.
- S. Jackie Oh, S. Han, W. Lee and A. C. Lockhart, Expert Opin. Invest. Drugs, 2016, 25, 667–677 CrossRef PubMed.
- K. D. Miller, R. L. Siegel, C. C. Lin, A. B. Mariotto, J. L. Kramer, J. H. Rowland, K. D. Stein, R. Alteri and A. Jemal, CA, 2016, 66, 271–289 Search PubMed.
- S. Karaman, V. M. Leppanen and K. Alitalo, Development, 2018, 145, dev151019 CrossRef PubMed.
- Y. Zhao and A. A. Adjei, Oncologist, 2015, 20, 660–673 CrossRef CAS PubMed.
- J. Liu, D. Kong, D. Sun and J. Li, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 2994–3003 CrossRef CAS PubMed.
- Q. Bi, S. Tang, L. Xia, R. Du, R. Fan, L. Gao, J. Jin, S. Liang, Z. Chen, G. Xu, Y. Nie, K. Wu, J. Liu, Y. Shi, J. Ding and D. Fan, PLoS One, 2012, 7, e40169 CrossRef CAS PubMed.
- B. Li, W. W. Xu, L. Han, K. T. Chan, S. W. Tsao, N. P. Y. Lee, S. Law, L. Y. Xu, E. M. Li, K. W. Chan, Y. R. Qin, X. Y. Guan, Q. Y. He and A. L. M. Cheung, Oncogene, 2017, 36, 3986–4000 CrossRef CAS PubMed.
- C. Xu, W. Wang, B. Wang, T. Zhang, X. Cui, Y. Pu and N. Li, J. Ethnopharmacol., 2019, 236, 443–465 CrossRef CAS PubMed.
- T. Ng, J. Pharm. Pharmacol., 2006, 58, 1007–1019 CrossRef CAS PubMed.
- P. Wang, J. Cui, X. Du, Q. Yang, C. Jia, M. Xiong, X. Yu, L. Li, W. Wang and Y. Chen, J. Ethnopharmacol., 2014, 154, 663–671 CrossRef CAS PubMed.
- Q. Yang, P. Wang, J. Cui, W. Wang, Y. Chen and T. Zhang, J. Ethnopharmacol., 2016, 193, 255–265 CrossRef CAS PubMed.
- R. Fodde and T. Brabletz, Curr. Opin. Cell Biol., 2007, 19, 150–158 CrossRef CAS PubMed.
- J. Li, J. Ying, Y. Fan, L. Wu, Y. Ying, A. T. Chan, G. Srivastava and Q. Tao, Cancer Biol. Ther., 2010, 10, 617–624 CrossRef CAS PubMed.
- S. Angers, C. J. Thorpe, T. L. Biechele, S. J. Goldenberg, N. Zheng, M. J. MacCoss and R. T. Moon, Nat. Cell Biol., 2006, 8, 348–357 CrossRef CAS PubMed.
- R. Habas and I. B. Dawid, J. Biol., 2005, 4, 2 CrossRef PubMed.
- Y. Zhao, Z. Q. Yang, Y. Wang, Y. Miao, Y. Liu, S. D. Dai, Y. Han and E. H. Wang, Mol. Carcinog., 2010, 49, 760–770 CrossRef CAS PubMed.
- H. T. Kwan, D. W. Chan, P. C. Cai, C. S. Mak, M. M. Yung, T. H. Leung, O. G. Wong, A. N. Cheung and H. Y. Ngan, PLoS One, 2013, 8, e53597 CrossRef CAS PubMed.
- K. B. Lee, S. Ye, M. H. Park, B. H. Park, J. S. Lee and S. M. Kim, Cancer Lett., 2014, 353, 124–132 CrossRef CAS PubMed.
- J. Lv, X. F. Cao, L. Ji, B. Zhu, D. D. Wang, L. Tao and S. Q. Li, Med. Oncol., 2012, 29, 151–160 CrossRef CAS PubMed.
- C. Z. Wang, J. T. Xie, B. Zhang, M. Ni, A. Fishbein, H. H. Aung, S. R. Mehendale, W. Du, T. C. He and C. S. Yuan, Internet J. Oncol., 2007, 31, 1149–1156 CAS.
- Y. Chen, L. Zhang, W. X. Liu and K. Wang, Cell. Mol. Biol. Lett., 2018, 23, 2 CrossRef PubMed.
- C. Ding, J. Luo, X. Fan, L. Li, S. Li, K. Wen, J. Feng and G. Wu, J. Exp. Clin. Cancer Res., 2017, 36, 56 CrossRef PubMed.
- H. Chen, Q. Cong, Z. Du, W. Liao, L. Zhang, Y. Yao and K. Ding, Cancer Lett., 2016, 382, 44–52 CrossRef CAS PubMed.
- T. Zhang, M. Liu, C. Wang, C. Lin, Y. Sun and D. Jin, Anticancer Res., 2011, 31, 3859–3863 CAS.
- R. Roskoski Jr, Pharmacol. Res., 2017, 120, 116–132 CrossRef PubMed.
- T. E. Peterson, N. D. Kirkpatrick, Y. Huang, C. T. Farrar, K. A. Marijt, J. Kloepper, M. Datta, Z. Amoozgar, G. Seano, K. Jung, W. S. Kamoun, T. Vardam, M. Snuderl, J. Goveia, S. Chatterjee, A. Batista, A. Muzikansky, C. C. Leow, L. Xu, T. T. Batchelor, D. G. Duda, D. Fukumura and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4470–4475 CrossRef CAS PubMed.
- J. Peng, N. Shao, H. Peng and L. Q. Chen, JBUON, 2013, 18, 398–406 Search PubMed.
- M. Chen, E. Cai, J. Huang, P. Yu and K. Li, Cancer Epidemiol., Biomarkers Prev., 2012, 21, 1126–1134 CrossRef CAS PubMed.
- W. XU, B. Li and A. Cheung, Gastrointest. Cancer: Targets Ther., 2015, 5, 79–88 Search PubMed.
- T. Mukherjee, A. Kumar, M. Mathur, T. K. Chattopadhyay and R. Ralhan, J. Cancer Res. Clin. Oncol., 2003, 129, 430–436 CrossRef CAS PubMed.
- Y. Qiao, P. J. Zhang, X. T. Lu, W. W. Sun, G. L. Liu, M. Ren, L. Yan and J. D. Zhang, Chin. J. Integr. Med., 2015, 21, 259–265 CrossRef CAS PubMed.
- S. S. Lin, K. C. Lai, S. C. Hsu, J. S. Yang, C. L. Kuo, J. P. Lin, Y. S. Ma, C. C. Wu and J. G. Chung, Cancer Lett., 2009, 285, 127–133 CrossRef CAS PubMed.
- P. Yu, Q. Liu, K. Liu, K. Yagasaki, E. Wu and G. Zhang, Cytotechnology, 2009, 59, 219–229 CrossRef CAS PubMed.
- L. Zhang, H. Wang, C. Li, Y. Zhao, L. Wu, X. Du and Z. Han, Cell. Physiol. Biochem., 2017, 44, 1251–1262 CrossRef CAS PubMed.
- C. Maes, S. Goossens, S. Bartunkova, B. Drogat, L. Coenegrachts, I. Stockmans, K. Moermans, O. Nyabi, K. Haigh and M. Naessens, EMBO J., 2010, 29, 424–441 CrossRef CAS PubMed.
- C. Wu, J. Chen, C. Chen, W. Wang, L. Wen, K. Gao, X. Chen, S. Xiong, H. Zhao and S. Li, Sci. Rep., 2015, 5, 16151 CrossRef CAS PubMed.
- Y. Wang, A. Sang, M. Zhu, G. Zhang, H. Guan, M. Ji and H. Chen, Mol. Vision, 2016, 22, 886–897 CAS.
- R. Giles, M. Lolkema, C. Snijckers, M. Belderbos, P. Van Der Groep, D. Mans, M. Van Beest, M. Van Noort, R. Goldschmeding and P. Van Diest, Oncogene, 2006, 25, 3065–3070 CrossRef CAS PubMed.
- V. C. Pai, C. C. Hsu, T. S. Chan, W. Y. Liao, C. P. Chuu, W. Y. Chen, C. R. Li, C. Y. Lin, S. P. Huang, L. T. Chen and K. K. Tsai, Oncogene, 2019, 38, 1340–1353 CrossRef CAS PubMed.
- X. Q. Chen, J. Jiang, X. T. Wang, C. L. Zhang, A. Y. Ji and X. J. Chen, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 7716–7725 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07830d |
| ‡ These authors contribute equally to this article. |
| This journal is © The Royal Society of Chemistry 2020 |