
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient syntheses of macrocycles ranging from 22–28 atoms through spontaneous dimerization to yield bis-hydrazones†
Vishal R. Sharma,
Arshad Mehmood,
Benjamin G. Janesko and
Eric E. Simanek*
and
Eric E. Simanek*
Department of Chemistry & Biochemistry, Texas Christian University, Fort Worth, TX 76129, USA. E-mail: e.simanek@tcu.edu
First published on 20th January 2020
Abstract
Acid treatment of a triazine displaying both a tethered acetal and BOC-protected hydrazine group leads to spontaneous condensation to yield macrocyclic dimers in excellent yields and purity. The bis-triazinyl hydrazones that form are characterized by 1H-NMR, 13C-NMR, 1H-COSY spectroscopy, X-ray diffraction, and mass spectrometry. By varying the length of the tether—the condensation product of an amino acid and amino acetal—rings comprising 22–28 atoms can be accessed. Glycine and β-alanine were used for the amino acid. The amino acetal comprised 2, 3 or 4 carbon atoms in the backbone. High-performance liquid chromatography (HPLC) was employed to assess purity as well as to fingerprint the six homodimeric products. By combining the protected monomers and subjecting them to acid, mixtures of homodimers and heterodimers are obtained. When all six protected monomers are combined, at least 14 of the 21 theoretical dimeric products are observed by HPLC. Single crystal X-ray diffraction and solution NMR studies reveal the diversity of shapes available to these molecules.
Introduction
While macrocyclic drugs have been clinically useful for decades, a resurgence of interest in this class is fuelled by the belief that these larger molecules can offer therapeutic interventions through mechanisms that are inaccessible to small molecules.1 That is, macrocycles offer an opportunity to interfere with protein–protein interactions that are characterized by extended binding sites,2 while still benefiting from favourable administration routes and pharmacokinetics associated with small molecules.3 However, macrocycles are under-represented in both screening libraries and clinical trials primarily due to challenges of synthesis. Accordingly, the introduction of synthetic methods that can be generally applied to the creation of macrocycles is of great interest.4 The use of hydrazones and dynamic covalent chemistry offers a promising route to these molecules.5 To this end, hydrazones have been explored to make macrocycles of varying complexity,6 and elaborate cages as well.7Recently, we showed that macrocycles containing 24 atoms could be obtained through the spontaneous dimerization of protected monomers upon treatment with acid.8 Each monomer presents a BOC-protected hydrazine, a morpholine group, and a tethered acetal. By varying the choice of amino acid—using either glycine or β-alanine—as well as the length of the amino acetal, the tether length is readily manipulated to generate rings of varying sizes. Here, we further support the generality of this methodology by preparing macrocycles varying from 22 to 28 atoms (Scheme 1).
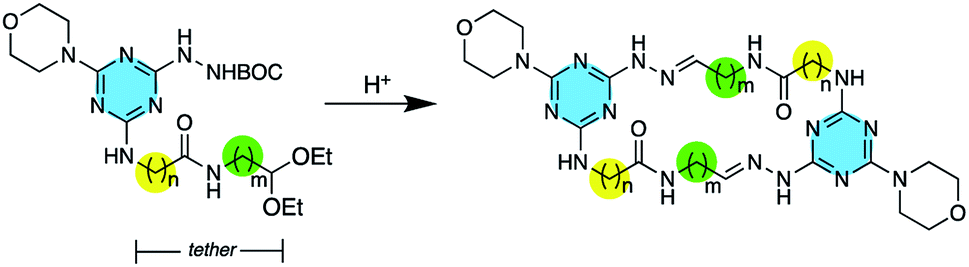 | ||
| Scheme 1 Upon treatment with acid, spontaneous dimerization yields macrocycles wherein n = 1–2 and m = 1–3. | ||
Result and discussion
For clarity of discussion, macrocycles are named for the monomers from which they are derived. That is, macrocycle 1·1 derives from dimerization of the protected monomer 1. Macrocycles are ordered based on size with 1·1 being the smallest and 6·6 being the largest. The monomers 1–6 are easily accessible through the stepwise substitution of triazine trichloride (Scheme 2). In the example shown, the synthesis of 1, the triazine was reacted first with the amino acid ester, followed by BOC-hydrazine and finally morpholine yield 7. The first two reactions commence at 0 °C and proceed with warming to room temperature. The last substitution occurs quickly at reflux. Saponification of 7 yields 8 that is further elaborated to 1 using an EDC-mediated coupling reaction with HOBT as an additive. These intermediates are characterized by 1H and 13C-NMR spectroscopy and mass spectrometry.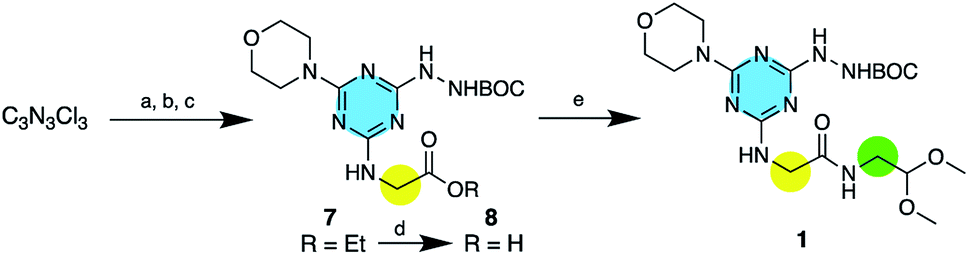 | ||
| Scheme 2 The synthesis of 1. (a) GlyOEt-HCl, DIPEA, THF, 0 °C-RT, 12 h. (b) BOCNHNH2, 0 °C-RT, 12 h. (c) Morpholine, reflux, 20 min. (d) MeOH/5% NaOH, 60 °C, 2 h. (e) HOBT, EDC-HCl, THF, 0 °C-RT, 12 h. | ||
Macrocycles are obtained by dissolving a monomer in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of trifluoroacetic acid and dichloromethane. While not extensively explored here, dimerization appears to be independent of concentration. That is, high dilution conditions are not required to prevent oligomerization. To isolate the macrocycle, the reaction solvent is allowed to evaporate. The resulting yellow solid is placed on filter paper and washed with dichloromethane and a small amount of methanol to remove the yellow color.9 Yields range from 83–100%.
:1 solution of trifluoroacetic acid and dichloromethane. While not extensively explored here, dimerization appears to be independent of concentration. That is, high dilution conditions are not required to prevent oligomerization. To isolate the macrocycle, the reaction solvent is allowed to evaporate. The resulting yellow solid is placed on filter paper and washed with dichloromethane and a small amount of methanol to remove the yellow color.9 Yields range from 83–100%.
Evidence for macrocyclization derives from a variety of sources. The 1H and 13C-NMR spectra are particularly noteworthy. Some of the chemical shift data appears in Table 1. Of primary import, signals corresponding to the imine are diagnostic in both the 1H and 13C spectra. The C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N appears at ∼7.5 ppm in 1H NMR spectra. The 13
N appears at ∼7.5 ppm in 1H NMR spectra. The 13![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HN appears at ∼150 ppm in 13C NMR spectra. Two additional models, 9 and 10, were also examined (Chart 1).
HN appears at ∼150 ppm in 13C NMR spectra. Two additional models, 9 and 10, were also examined (Chart 1).
| # | 1·1 | 2·2 | 3·3 | 4·4 | 5·5 | 6·6 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|
| a The observation of I does not preclude the existence of IV.b The observation of II does not preclude the existence of III. | ||||||||
| n | 1 | 1 | 2 | 1 | 2 | 2 | 2 | 1 |
| m | 1 | 2 | 1 | 3 | 2 | 3 | 2 | 3 |
| Size | 22 | 24 | 24 | 26 | 26 | 28 | NA | 26 |
| T1 | 161.7 | 161.3 | 161.3 | 161.6 | 161.7 | 161.7 | 161.6 | 161.0 |
| T2 | 155.0 | 154.6 | 155.3 | 154.5 | 154.7 | 154.7 | 154.7 | 154.7 |
| T3 | 154.8 | 154.0 | 154.3 | 154.1 | 154.6 | 154.3 | 154.3 | 154.2 |
| H N |
147.5 | 147.9 | 146.6 | 150.5 | 150.7 | 150.6 | 151.8 | 149.1 |
| CN |
7.63 | 7.51 | 7.55 | 7.62 | 7.57 | 7.60 | 7.55 | 7.60 |
| NN |
12.37 | 12.37 | 12.37 | 12.58 | 12.43 | 12.42 | 12.36 | NA |
| Ar–+ |
12.15 | 12.21 | 12.00 | 11.45 | 11.43 | 11.33 | 11.55 | 11.53 |
| Soln: | IV | Ia | IV | IIb | III | III | NA | Ia |
| X-ray: | I | I | I | I | NA | I | NA | NA |
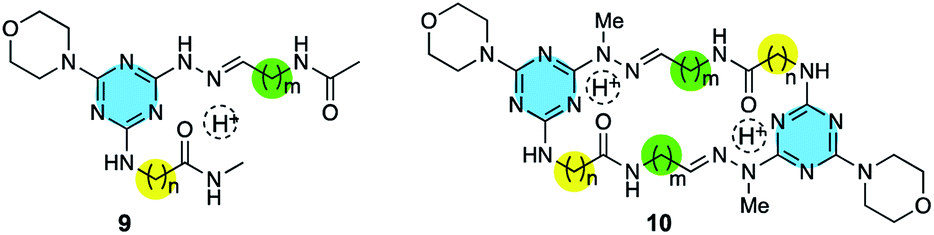 | ||
| Chart 1 Two additional models. | ||
The four potential rotational isomers available to a triazine are shown in Chart 2. Asymmetrically substituted triazines usually give 1H and 13C NMR spectra with signals that are numerous, broad and poorly resolved. Such is the case with the protected monomers. However, the spectra of the macrocycles—notably the 13C spectra—become sharp leading to one of two interpretations. The first is that rotation about the triazine–N bond is slow on the NMR timescale and a single conformational isomer is observed.10 Alternatively, rotation around the triazine–N bond is fast on the NMR timescale. A more thorough study of rotamer populations will be completed in due course.
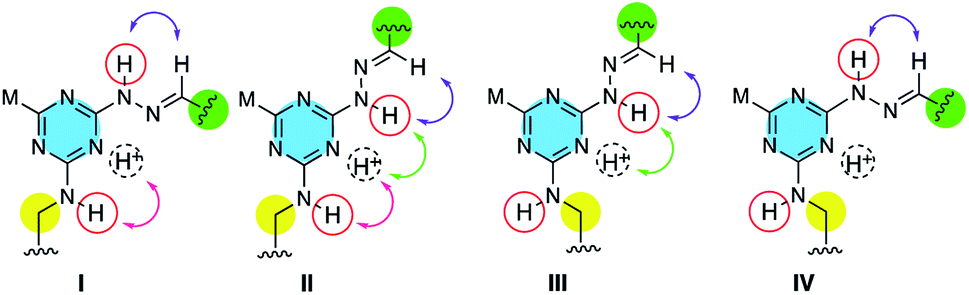 | ||
| Chart 2 Rotational isomers of the triazines. ‘M’ is morpholine. Arrows indicate a potential nOe. All macrocycles exist as trans-hydrazones (purple), but the rotamer can be assigned (Table 1) based on the presence and/or absence of the other nOes (green, red). | ||
In addition, the 1H NMR spectra show a pronounced pattern of hydrogen bonds. Instead of multiple, broad resonances between 5–7 ppm in DMSO-d6, the exchangeable hydrogen atoms show sharper signals that are farther downfield. Indeed, four different exchangeable protons are observed. To complement the three expected, a fourth corresponding to protonation of the triazine is observed in solution and the solid state. Symmetry in the solution state NMR suggests (and solid state structures corroborate) that protonation occurs on the nitrogen atom opposite the morpholine group indicated with a dashed ring and ‘M’, respectively, in Charts 1 and 2. The two signals between 13–11.5 ppm are attributed to the NN– and a protonated triazine which is consistent with what is observed in lattices of isocyanuric acid and melamine.11 The other two signals appear between 8–9 ppm, consistent with amide N shifts and data from 2D NMR.12
Ring size is reflected in the chemical shifts in the 1H and 13C NMR (Table 1). The HN shifts for smaller macrocycles (1·1, 2·2, and 3·3) ranges from 146–148 ppm. For larger macrocycles (4·4, 5·5, and 6·6), the range spans 150–152 ppm. The protonated triazine (Ar–+) for the smaller macrocycles appears between 12.0–12.2 ppm. For larger macrocycles, it appears between 11.3–11.5 ppm. A less pronounced clustering is observed for the NN– lines with smaller rings appearing at 12.37 ppm, and larger rings appearing between 12.42–12.58 ppm.
The most satisfying evidence for structure is derived from crystal structure data. In the five structures obtained, the macrocycle co-crystallizes with two molecules of trifluoroacetic acid. One nitrogen atom of the triazine ring—the one opposite the morpholine ring—is protonated. This proton engages in hydrogen bonding with either the trifluoroacetate counter ion (1·1, 2·2, and 3·3) or the carbonyl group of the juxtaposed monomer (4·4 and 6·6). Fig. 1 shows these structures as ball-and-stick and space filling models from the top and side. Rotamer I is observed throughout.
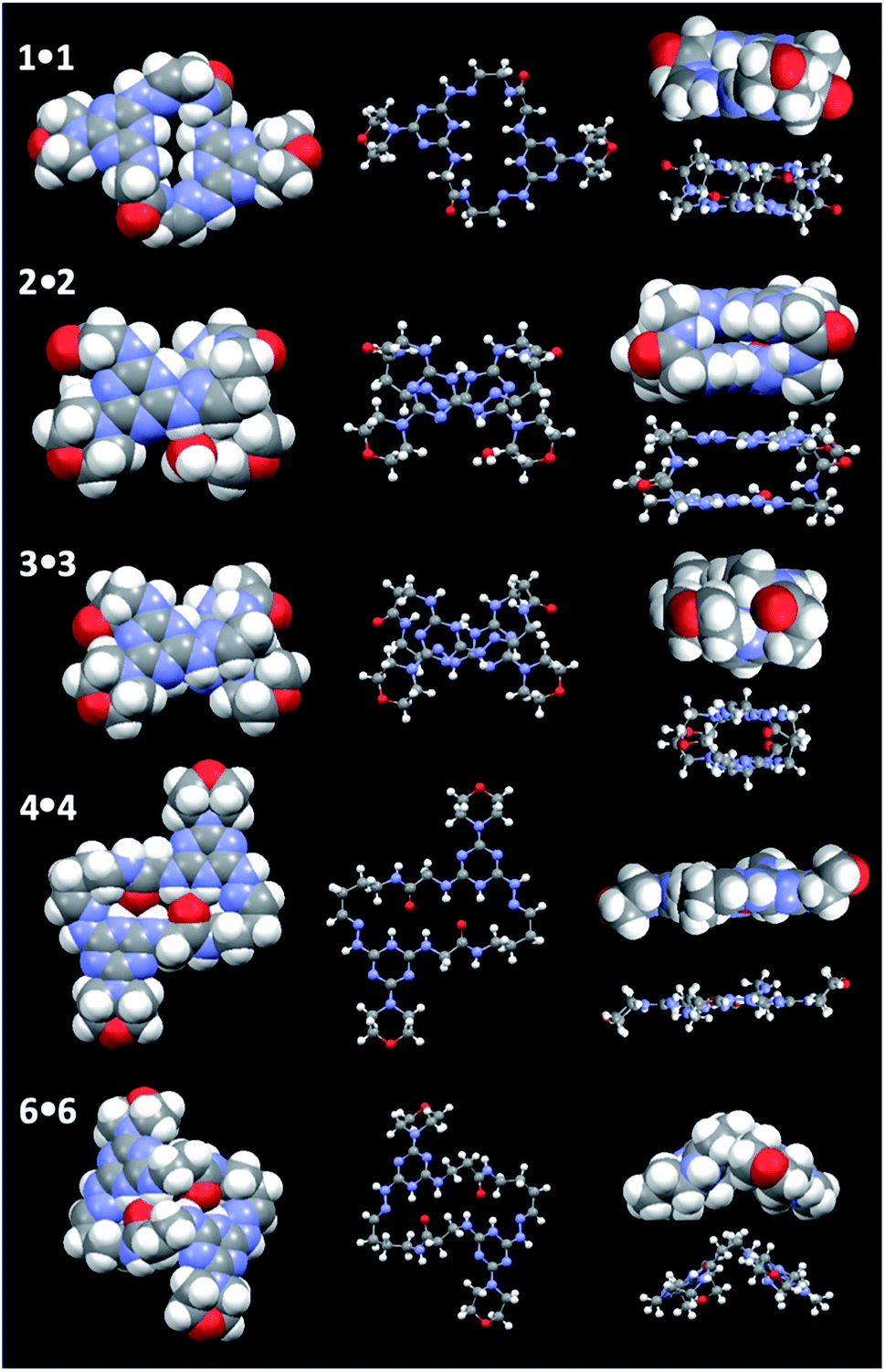 | ||
| Fig. 1 Crystal structures of 1·1, 2·2, 3·3, 4·4 and 6·6 from the top and edge. The protonated triazine is evident. | ||
This conservation of shape in the solid state led us to explore shape in solution as established by the presence or absence of nOes (Chart 2). In all cases, nOes were observed that are consistent with trans-hydrazones (purple arrow, Chart 2): cis-hydrazones were not observed. Trans-hydrazones were also observed in 9 and 10 (with the latter showing an nOe between the NC and 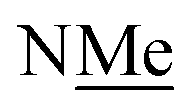 groups).
groups).
To identify which rotamer, I–IV, forms in solution 2D NOESY spectra were collected. An nOe between the protonated triazine Ar–H+ and –NN– (green arrow) was observed for larger macrocycles, 4·4, 5·5 and 6·6, but was absent in smaller ones, 1·1, 2·2 and 3·3. The analogous nOe (Ar–H+ and 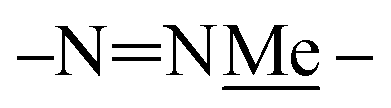 ) was also absent for 10. An nOe corresponding to the Ar–H+ and the –N– of the amino acid (red arrow) was observed in 2·2, 4·4, and 10 only. Table 1 shows the rotamer assignment.
) was also absent for 10. An nOe corresponding to the Ar–H+ and the –N– of the amino acid (red arrow) was observed in 2·2, 4·4, and 10 only. Table 1 shows the rotamer assignment.
These macrocycles are soluble in water and aqueous solutions of methanol. The HPLC traces obtained for the macrocycles reveal that elution times fall roughly into two categories (short and long) which loosely correlate with size, but more accurately with length of the acetal. That is, macrocycles 1·1, 2·2, 3·3, and 5·5 (m = 1 or 2) elute at ∼4 min, while macrocycles 4·4 or 6·6 (m = 3) elute at ∼14 min.
In a series of final experiments, the opportunity to form heterodimers was explored using HPLC. That is, mixing 1 and 2 followed by deprotection gave a mixture of 1·1, 2·2 and 1·2, a ring comprising 23 atoms. Similarly, 3 and 5 gave 3·3, 5·5 and 3·5, a ring comprising 25 atoms. Subjecting a mixture of 1–6 yields a mixture of at least 14 of the expected 21 isomers as identified by LC-MS. Assignment and energetic analysis are the subjects of ongoing study.
Conclusions
The chemistry presented provides rapid access to dimeric products of varied ring sizes in high yields. Solution and solid state structural data suggests a range of conformations can be adopted. Mixtures of homodimers and heterodimers can be obtained readily by mixing the different protected monomers to yield mixtures containing even and odd-membered rings. A report on the use of solid-phase peptide synthesis of 26-atom triazine macrocycles offers proof-of-principle that these compounds, derived from dynamic covalent chemistry, might be reproduced as non-dynamic architectures.13Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Robert A. Welch Foundation (P-0008), Texas Christian University & the NIH (1R15 GM135900) for support. A. Yepremyan is thanked for preliminary synthetic efforts.Notes and references
- Macrocycles in Drug Discovery, ed. J. Levin, RSC Publishing, 2014, p. 525 Search PubMed.
- P. G. Dougherty, Z. Qian and D. Pei, Biochem. J., 2017, 474, 1109–1125 CrossRef CAS PubMed.
- (a) F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57(2), 278–295 CrossRef CAS PubMed; (b) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- (a) E. M. M. Abdelraheem, S. Shaabani and A. Domling, Synlett, 2018, 29, 1136–1151 CrossRef CAS PubMed; (b) S. Collins, S. Bartlett, F. Nie, H. F. Sore and D. R. Spring, Synthesis, 2016, 48, 1457–1473 CrossRef CAS; (c) V. Marti-Centelles, M. D. Pandey, I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef CAS PubMed; (d) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed; (e) X. Yu and D. Sun, Molecules, 2013, 18, 6230–6268 CrossRef CAS PubMed.
- (a) Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed; (b) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (c) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- (a) J. F. Reuther, J. L. Dees, I. V. Kolesnichenko, E. T. Hernandez, D. V. Ukraintsev, R. Guduru, M. Whiteley and E. V. Anslyn, Nat. Chem., 2018, 10, 45–50 CrossRef CAS PubMed; (b) Y. Zhang, X. Zheng, N. Cao, C. Yang and H. Li, Org. Lett., 2018, 20, 2356–2359 CrossRef CAS PubMed.
- M. Wierzbicki, A. A. Glowacka, M. P. Szymanski and A. Szumna, Chem. Commun., 2017, 53, 5200 RSC.
- A. Yepremyan, A. Mehmood, P. Asgari, B. Janesko and E. E. Simanek, ChemBioChem, 2019, 20, 241–246 CrossRef CAS PubMed.
- In more recent work, washing appears not to change NMR data and may be unnecessary for many applications..
- H. E. Birkett, R. K. Harris, P. Hodgkinson, K. Carr, M. H. Charlton, J. C. Cherryman, A. M. Chippendale and R. P. Glover, Magn. Reson. Chem., 2000, 38, 504–511 CrossRef CAS.
- E. E. Simanek, S. Qiao, I. S. Choi and G. M. Whitesides, J. Org. Chem., 1997, 62, 2619–2621 CrossRef CAS PubMed.
- D. S. Wishart, B. D. Sykes and F. M. Richards, J. Mol. Biol., 1991, 222, 311–333 CrossRef CAS PubMed.
- J. A. Zerkowski, L. M. Hensley and D. Abramowitz, Synlett, 2002, 4, 557–560 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, characterization data, crystallographic tables. CCDC 1957428–1957432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08056b |
| This journal is © The Royal Society of Chemistry 2020 |